

Summary
Genome‐scale reconstructions of metabolism are computational species‐specific knowledge bases able to compute systemic metabolic properties. We present a comprehensive and validated reconstruction of the biotechnologically relevant bacterium Pseudomonas putida KT2440 that greatly expands computable predictions of its metabolic states. The reconstruction represents a significant reactome expansion over available reconstructed bacterial metabolic networks. Specifically, iJN1462 (i) incorporates several hundred additional genes and associated reactions resulting in new predictive capabilities, including new nutrients supporting growth; (ii) was validated by in vivo growth screens that included previously untested carbon (48) and nitrogen (41) sources; (iii) yielded gene essentiality predictions showing large accuracy when compared with a knock‐out library and Bar‐seq data; and (iv) allowed mapping of its network to 82 P. putida sequenced strains revealing functional core that reflect the large metabolic versatility of this species, including aromatic compounds derived from lignin. Thus, this study provides a thoroughly updated metabolic reconstruction and new computable phenotypes for P. putida, which can be leveraged as a first step toward understanding the pan metabolic capabilities of Pseudomonas.
Introduction
The group Pseudomonas comprises a heterogeneous and large group (>100) of Gram‐negative, gamma‐proteobacterial species (Palleroni, 2010). They show a noteworthy metabolic versatility and adaptability, enabling colonization of diverse niches (Silby et al., 2011). Pseudomonas are of great interest because of their importance in human and plant diseases, e.g., P. aeruginosa (Gellatly and Hancock, 2013) and P. syringe (Morris et al., 2013), and due to their potential for promoting plant growth and biotechnological applications, e.g., P. fluorescens (Loper et al., 2012) and P. putida (Wu et al., 2011; Roca et al., 2013). Among this group, P. putida has been widely used as a model environmental bacterium free of undesirable biotechnological traits such as virulence factors (Udaondo et al., 2015). Pseudomonas putida strains can degrade a large array of chemicals, including xenobiotic compounds, while exhibiting a remarkable resistance to organic solvents and other environmental stresses, making P. putida strains highly valued biocatalysts (Nikel et al., 2014; Loeschcke and Thies, 2015; Franden et al., 2018; Kohlstedt et al., 2018). In addition, P. putida strains are amenable to genetic modification and are therefore seen by many as ideal workhorses for synthetic biology‐based cell factories (Nikel and de Lorenzo, 2018).
This high level of interest in P. putida has led to intense genome‐scale metabolic modelling efforts of strain KT2440; the best‐characterized strain and the first to be completely sequenced (Nelson et al., 2002). Four genome scale models (GEMs) for KT2440 have been previously published, formally known as iJN746 (Nogales et al., 2008), iJP850 (Puchalka et al., 2008), PpuMBEL1071 (Sohn et al., 2010) and iJP962 (Oberhardt et al., 2011). Recently, two new consensus models, formally iEB1050 (Belda et al., 2016) and PpuQY1140 (Yuan et al., 2017), have been published based on the genome reannotation of this strain and the integration of reactomes already present in previous P. putida GEMs respectively. Unfortunately, due to the nature of this approach, which only allows the inclusion of new metabolic capabilities based on computational evidence with scarce experimental validation, and/or from previous reconstructions, the available GEMs of P. putida still lack coverage of the known metabolism captured in decades of P. putida literature. Thus, these previous P. putida models fall into what we consider to be models of primary metabolism. Thus, as often occurs with current GEMs, their utility falls short of true and full genome‐scale studies (Monk et al., 2014).
We demonstrate here that all metabolic knowledge available for a single species, even a genus, can be manually collected and used for high‐quality metabolic modelling of a particular strain capable of addressing deep system‐wide inquiries. We deliver a complete and manually curated metabolic reconstruction of P. putida KT2440, named iJN1462. This detailed reconstruction not only largely captures the metabolic features of this strain, but it represents a computational scaffold for future semi‐automatic reconstruction of the Pseudomonas group. To demonstrate this potential, we built 82 strain‐specific model drafts of metabolism for P. putida using iJN1462 as a scaffold in order to analyse the shared metabolic capabilities of various P. putida strains. Overall, the strains all possess broad metabolic capabilities indicative of growth potential in a variety of environments and have several strain‐specific differences that could be areas of investigation to identify strains of interest for industrial applications.
Results
Reconstruction content and enhancements
iJN1462 represents a significant expansion over previous reconstructions of P. putida KT2440 and is comparable to other high‐quality E. coli models. (Table 1, SI1 Fig. S2). iJN1462 contains 1462 gene products (38% of the functionally annotated protein products in the KT2440 genome), 2929 reactions and 2155 non‐unique metabolites. The reconstruction includes 410 unique citations associated to reconstruction content and 2048 of the reactions have at least one citation supporting its inclusion (Table S1).
Table 1.
Comparison of iJN1462 with its antecessor iJN746 (Nogales et al., 2008), previous P. putida metabolic reconstructions iJP815 (Puchalka et al., 2008), PpuMBEL1071 (Sohn et al., 2010), iJP962 (Oberhardt et al., 2011), SEED (Henry et al., 2010) and the recently published P. putida consensus models iEB1050 (Belda et al., 2016) and PpuQY1140 (Yuan et al., 2017).
| iJN746 (2008) | iJP815 (2008) | PpuMBEL1071 (2010) | iJP962 (2011) | SEEDpput (2010) | iEB1050 (2016) | PpuQY1140 (2017) | iJN1462 (This study) | iML1515 (E. coli, 2017) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Metabolites | 911 | 888 | 1044 | 992 | 1227 | 1122 | 1104 | 2155 | 1877 | |
| Unique | 709 | 824 | 948 | 897 | 1106 | 1011 | 1009 | 1434 | 1169 | |
| Cytoplasmic | 697 | 824 | 946 | 897 | 1106 | 1008 | 1009 | 1341 | 1071 | |
| Periplasmic | 125 | – | – | – | – | – | – | 465 | 465 | |
| Extracellular | 90 | 67 | 106 | 95 | 121 | 114 | 95 | 349 | 341 | |
| Genes | 746 (14%) | 815 (15%) | 900 (16%) | 962 (17%) | 1081 (20%) | 1050 (19%) | 1140 (21%) | 1462 (27%) | 1516 (36%) | |
| Reactions | 950 | 877 | 1071 | 1070 | 1406 | 1256 | 1171 | 2929 | 2712 | |
| Metabolic | 667 (70%) | 799 (91%) | 958 (90%) | Nd | 1285 (91%) | 1004 (80%) | 958 (81%) | 1720 (59%) | 1542 (57%) | |
| Transport | 193 (20%) | 78 (9%) | 113 (10%) | Nd | Nd | 156 (12%) | 127 (11%) | 827 (28%) | 833 (31%) | |
| Exchange | 90 (9%) | 67 (7.5%) | Nd | 95 (9%) | 121 (9%) | 96 (8%) | 107 (9%) | 382 (13%) | 337 (12%) | |
| Orphan | 140 (17%) | 56 (6%) | 68 (6%) | 76 (6%) | 41 (3%) | 17a/70b (7%) | 80 (6%) | 92a/9b (3%) | 71a/21b (3%) | |
| Blocked | 108 (11%) | 289 (33%) | Nd | 436 (41%) | 777 (45%) | 457 (36%) | 389 (33%) | 247 (8%) | 260 (10%) | |
| Active | 842 (89%) | 588 (67%) | Nd | 634 (59%) | 629 (55%) | 799 (64%) | 782 (67%) | 2682 (92%) | 2452 (90%) | |
| Strain specific biomass | No | No | No | No | No | No | No | Yes | Yes | |
| Lipids | Complete | Lumped | Complete | Lumped | Lumped | Lumped | Lumped | Complete | Complete | |
| Peptidoglycan | Precursors | Precursors | Precursors | Precursors | Precursors | Complete | Precursors | Complete | Complete | |
| Lipopolysaccharide | Precursors | Precursors | Precursors | Precursors | Precursors | Precursors | Precursors | Complete | Complete | |
| Specific Cofactors/Vitamins | No | No | No | No | No | No | No | Yes | Yes | |
| PHA metabolism | Yes (7)c | No | Yes (7)c | No | No | No | No | Yes (24)c | NA | |
The last E. coli model iML1515 (Monk et al., 2017) was included as a reference for a high‐quality GEM.
Metabolic.
Transport.
Number of PHA monomers.
NA, not applicable.
The major enhancements of iJN1462 over previous P. putida reconstructions are found in its strain‐specific metabolism (Fig. 1A). Some of these subsystems demonstrate well‐known metabolic features of P. putida, such as diverse growth sources for both carbon and nitrogen, or tolerance to heavy metals and some industrially relevant solvents. Additional subsystems, such as cell envelope biosynthesis or fatty acid metabolic pathways, were improved based on recent literature and experimental efforts (Table S1). Altogether, these expansions help better understand the capabilities of P. putida and define an accurate biomass reaction for in silico experiments.
Figure 1.
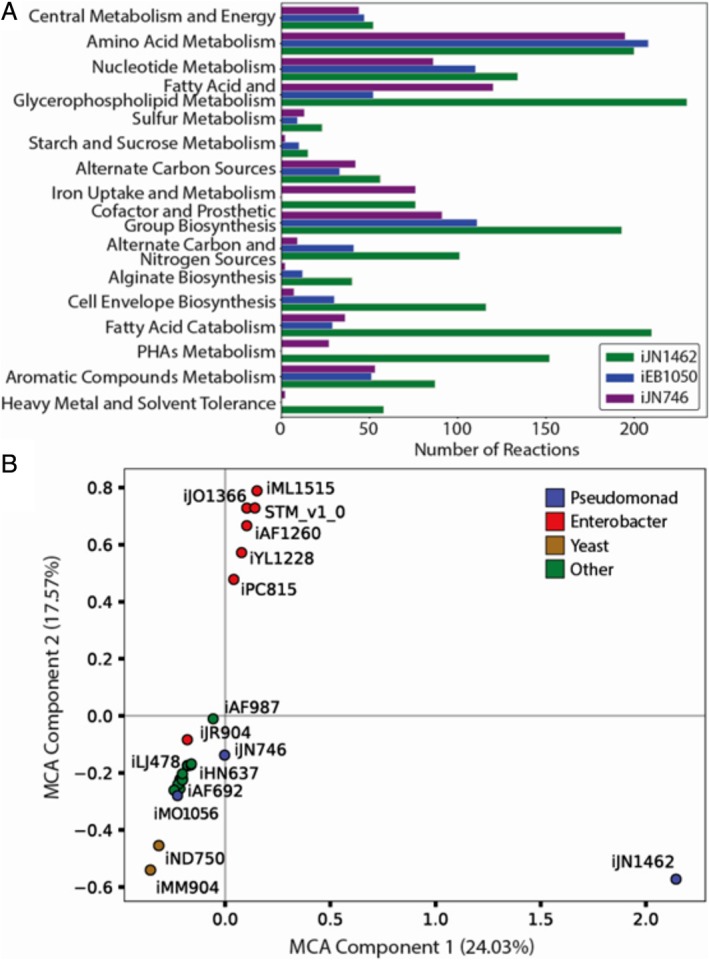
Comparison of iJN1462 to previous metabolic reconstructions. A. Comparison of the number of reactions in a subsystem for three P. putida metabolic reconstructions. Subsystems belonging to primary and strain‐specific metabolisms are shaded in green and orange respectively. B. A multi‐correspondence analysis (MCA) scatter plot showing the multiple correspondence analysis of the metabolic content, in terms of reactions and metabolites, of available metabolic reconstructions (see Monk et al., 2014). The amount of explained variance represented by each component is shown in parentheses. Reconstructions that are close to each other in the diagram are likely to have similar metabolic content. Most of the reconstructions analysed cluster around the origin. Reconstructions for Yeast (iND750 and iMM904) are significantly different, including specific metabolic content. Reconstructions of Enterobacterial strains form a clearly differentiated group. Finally, iJN1462 is located far away from the origin, showing very different reactome.
Pseudomonas putida is interesting due to its ability to grow in a variety of environments. New catabolic pathways were included in iJN1462, with several also being experimentally validated. These captured the metabolic versatility of P. putida (Jiménez et al., 2010) as new subsystems, such as aromatic compound metabolism, and several alternate carbon and nitrogen source subsystems were included. For instance, the complete modelling of the sarcosine, 2,5‐dioxopentanoate, polyamines and isovaleryl‐CoA metabolism has been included based on legacy data and completely validated by growth and gene knockout analysis (SI1, Fig. S3‐6). We also performed a detailed reconstruction of alginate biosynthesis, a Pseudomonas polysaccharide with high biotechnological and clinical interest. Furthermore, Pseudomonas has a robust iron uptake metabolism that plays a major role in niche colonization and pathogenesis (Cornelis, 2010; Wiens et al., 2014). Accordingly, the iron metabolism has been modelled, including the biosynthetic pathway for pyoverdine (a non‐ribosomal peptide acting as siderophore) of P. putida KT2440 based on structural studies (Matthijs et al., 2009).
Pseudomonas putida catabolizes a large variety of fatty acids (de Waard et al., 1993). Subsequently, the metabolism of fatty acids has been extensively expanded. In addition to saturated fatty acids, the catabolism of triacylglycerides, mono and poly‐unsaturated fatty acids, phenylacyl and thioacyl fatty acids has been reconstructed. The metabolism of unsaturated fatty acids, present in other bacterial models such as iML1515 (Monk et al., 2017), has also been revisited and extended by the inclusion of a NADPH‐dependent 2,4‐dienoyl‐CoA reductase, which is required for the β‐oxidation of polyunsaturated fatty acids and substrate‐specific cis‐3‐trans‐2‐enoyl‐CoA isomerase reactions (de Waard et al., 1993). As a direct consequence, the potential substrates for polyhydroxyalkanoate (PHA) synthesis via β‐oxidation have experienced a significant increase and 24 different PHA monomers can be synthetized by iJN1462 (Fig. S7). Even though the production of PHA is one of the most prominent biotechnological capabilities of P. putida, the PHA metabolism is absent in most of the previous GEMs except for iJN746 and PpuMBEL1071 (Table 1).
Other areas of significant expansion involved cell envelope biosynthesis and cofactor and prosthetic group biosynthesis (Fig. 1A). Within cell envelope biosynthesis, specific peptidoglycans from P. putida and the complete lipopolysaccharide biosynthesis pathway have been modelled in great detail based on available data (Quintela et al., 1995; Rodríguez‐Herva et al., 1999). Modelling of the cellulose, rhamnose and trehalose metabolism have also been included. Biosynthesis of most cofactors and prosthetic groups known to be present in Pseudomonas was revisited in iJN1462. Some of them, such as biosynthesis of pyrroloquinoline quinone (PQQ), are modelled here for the first time. These updates allowed for the assignment of the correct electron carrier to quinoproteins of Pseudomonas and a very accurate and strain‐specific biomass reaction.
A detailed P. putida‐specific biomass objective function (BOF) based on existing experimental data was constructed to enable in silico experiments. The BOF includes macromolecular composition (van Duuren et al., 2013), glycerophospholipid content (Rühl et al., 2012), murein composition (Quintela et al., 1995), lipopolysaccharide (King et al., 2009) and species‐specific soluble metabolites such as pyoverdine (Matthijs et al., 2009) and PQQ. A new value for non‐growth associated growth maintenance was also included based on recent findings (Ebert et al., 2011). This highly strain‐specific BOF contrasts with those present in previous reconstructions which lack P. putida's specific lipids, lipopolysaccharides, peptidoglycans and most cofactors and vitamins (Fig. S8). In addition, we formulated a core biomass reaction including metabolites that are completely essential for growth according to experimental reports (Feist et al., 2007). The core BOF indeed describes a viable cellular composition under rich nutritional conditions. In fact, many of the metabolites missing from the CORE biomass have been related to metabolic robustness but not essentiality in P. putida. Among others, specific lipids such as cardiolipin (involved in organic solvent resistant), pyoverdine (responsible for providing iron under limitation), PQQ (cofactor essential for the catabolism of certain alcohols), several vitamins and cofactors (involved in specific carbon source catabolisms), and so forth. Therefore, a strain having the cellular composition described by the core biomass, although extremely fragile from a metabolic robustness point of view, it should be likely a viable cell. Finally, the availability of both biomass functions in P. putida models represents an interesting update and the possibility to provide more accurate predictions when compared with models having only one. This is because while the core biomass function makes sense for analyses such as the assignment of gene essentiality under rich conditions or for strain designing endeavours, the full biomass catches better carbon flux distributions. Details of the new biomass reactions and their formulation are depicted in SI 1 and Table S3.
The metabolic expansion of the reactome represented by iJN1462 became evident when its content was compared with 22 pre‐existing GEMs by means of multiple correspondence analyses (Tenenhaus and Young, 1985). While the previous P. putida reconstruction iJN746 is located close to the centre of coordinates together with most of the current GEMs, iJN1462 is placed far away from the origin, thus suggesting its higher and more organism‐specific metabolic content (Fig. 1B). Highlights this fact, iJN1462 includes 1501 new reactions not found in any of the other models examined (Fig. 1B, Table S6). The largest portion of these reactions come from expanding P. putida's metabolic capabilities toward new substrates, with 255 new carbohydrate metabolism reactions and 367 new transport reactions. Lipid metabolism with 231 new reactions is the only other subsystem responsible for a large number of new reactions. Most other subsystems had between 25 and 100 new reactions unique to iJN1462. These new reactions demonstrate the uniqueness of iJN1462 and just how much previously non‐modelled information is contained.
Reconstruction validation
A large GEM with many genes and reactions does not always equate to a high‐quality GEM. In order to validate iJN1462 as a predictive model of P. putida, in silico test results were compared to several in vivo experiments. Comparisons included phenotypes on different nutrient sources, growth rates and measured carbon flux, as well as gene essentiality as determined by knockout strains and Bar‐Seq experiments. Additionally, new standards are being developed to help ensure newly developed GEMs are standardized and of high quality. The Memote tool was used to evaluate the model as compared to other models (Lieven et al., 2018).
To assess the ability of iJN1462 to predict phenotypes, we first evaluated all the potential carbon, nitrogen, sulphur, phosphorus and iron sources supporting in silico growth (Table S2). iJN1462 was able to use a significantly higher number of nutrients compared to previous reconstructions (Fig. 2). iJN1462 can grow on 142 and 71 new carbon and nitrogen sources respectively, many of which have never been experimentally reported as nutrients in P. putida (Table S2). We experimentally validated the accuracy of the growth predictions by performing growth screens (see Methods) with special emphasis on those nutrients that have not been tested thus far in P. putida (SI1 Table S2). The overall accuracy of growth predictions was high, predicting 79% and 84% (p‐values of Fisher's exact test were less than 10−12, and overall Matthews correlation coefficient of 0.608) of the growth phenotypes observed for carbon and nitrogen sources respectively (Fig. 2, Table S2). Therefore, iJN1462 largely captures the metabolic versatility of Pseudomonas.
Figure 2.
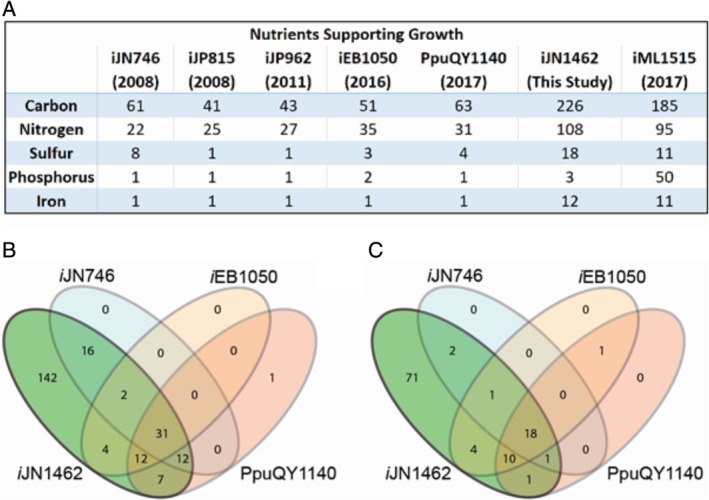
Identification and validation of nutrients supporting iJN1462 growth. A. The number of nutrients supporting growth in previous GEMs of P. putida, iJN1462, and the latest GEM of E. coli iML1515. B and C. A qualitative comparison of the growth‐supporting carbon and nitrogen sources respectively, as calculated using the iJN1462, iJN746, iEB1050 and PpuQY1140 reconstructions.
Comparisons of growth rate predictions and PHA production rates (Table 2) with experimental values provided further validation of the model. The prediction accuracy of iJN1462 significantly exceeds that of previous P. putida GEMs. However, iJN1462 grew faster than KT2440, suggesting an incomplete adaptation of KT2440 to these sugars as carbon sources and/or certain overflow of metabolism. When the observed secretion rates for gluconate and 2‐ketogluconate were included in the model as additional constraints, iJN1462 fits the experimental growth rate on glucose. iJN1462 also had a high level of accuracy concerning growth rate and production rate of PHA when grown on octanoate with limited nitrogen and oxygen (Table 2). These experiments demonstrate good capabilities to predict at least growth phenotypes and rates.
Table 2.
Comparison of growth performance of iJN1462 with previous GEMs of P. putida.
| Carbon source | (mmol gDW−1 h−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Uptake rate | Secretion rate (Gluconate) | Secretion rate (2‐Ketogluconate) | Growth rate/PHA production rate (PHAC6 + PHAC8) (h−1)/(mmol gDW−1 h−1) | References | ||||||
| iJN746 | iEB1050 | PpuQY1140 | iJN1462 | In vivo | ||||||
| Gluconate | 5.1 | NA | NA | 0.58/NA | 0.67/NA | 0.37/NA | 0.47/NA | 0.43/NA | del Castillo et al. (2007) | |
| Glucose | 6.3 | NA | NA | 0.76/NA | 0.91/NA | 0.50/NA | 0.61/NA | 0.56/NA | del Castillo et al. (2007) | |
| Glucose | 7.3 | NA | NA | 0.86/NA | 1.05/NA | 0.59/NA | 0.71/NA | 0.73/NA | Ebert et al. (2011) | |
| Glucose | 10.9 | 2.8 | 2.6 | 0.70/NA | 0.81/NA | 0.49/NA | 0.57/NA | 0.57/NA | Blank et al. (2008) | |
| Octanoate | 3.4 | NA | NA | 0.31/1.9 | NA/NA | NA/NA | 0.2912/1.11 | 0.29/1.5 | Escapa et al. (2012) | |
Constraints used are underlined. NA, not applicable. iEB1050 and PputQY1140 models lack of Octanoate and PHA metabolisms. For growth on octanoate as carbon source, nitrogen and oxygen uptake were constrained to 3.1 and 13.5 mmol gDW−1 h−1 respectively.
Since accurate predictions of growth rates alone cannot guarantee the quality of GEMs, we compared flux predictions on glucose to experimentally reported values (Blank et al., 2008). We found a good correlation between predicted and experimental values, with Kendall's τ = 0.80, significantly higher than for the iEB1050 and PpuQY1140 models, τ = 0.53 and τ = 0.68 respectively (Fig. 3, Fig. S12). A well‐known trait of Pseudomonas is the activation of the pyruvate shunt as a main source of oxaloacetate bypassing to the malate dehydrogenase (MDH) (del Castillo et al., 2007; Blank et al., 2008). Despite this alternative pathway being less efficient from an energetic point of view, this feature of Pseudomonas guarantees a high level of NADPH, which is critical in providing metabolic robustness, including tolerance to oxidative stress (Blank et al., 2008; Chavarría et al., 2012; Berger et al., 2014). iJN1462 fails to predict the activation of the pyruvate shunt as an alternative source of oxaloacetate since flux balance analysis excludes suboptimal flux distributions (Orth et al., 2010). We therefore performed a sensitivity analysis of flux predictions as a function of the flux through pyruvate carboxylase (PC). In good agreement with experimental results, increasing PC flux leads to a large flux decrease through MDH, a significant increase in the flux through Malic Enzyme (ME2), and a slight increase through the TCA cycle, pyruvate dehydrogenase and pyruvate kinase. When the experimental flux through PC was used as an additional constraint, the accuracy in the flux distribution prediction increased significantly (τ = 0.92) (Fig. 3). In summary, the flux predictions demonstrate the high accuracy of iJN1462, as well as the likely role of the mechanisms fuelling metabolic robustness such as the pyruvate shunt, as one of the main mechanisms disturbing the linearity of genotype–phenotype relationships. iJN1462 can thus predict growth capabilities, growth rates and flux distributions for KT2440 with high accuracy, at a comparable level to the well‐developed E. coli model.
Figure 3.
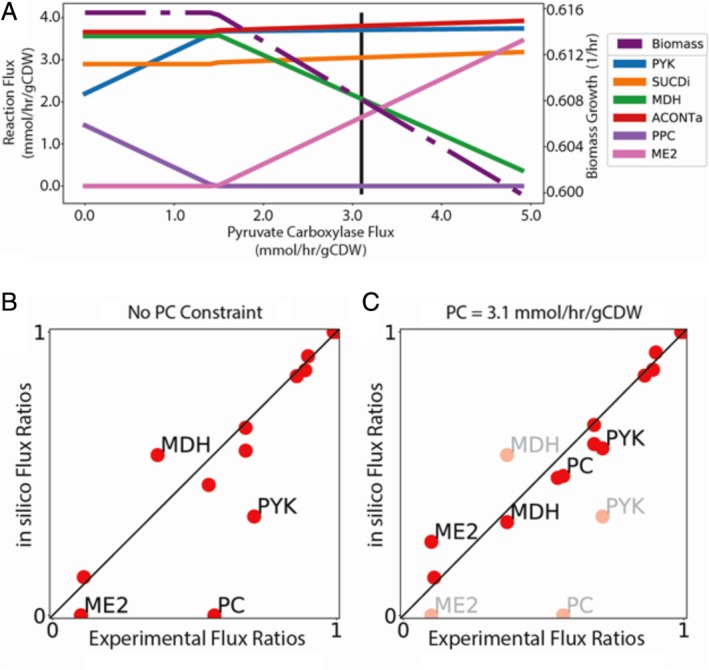
Validation of flux predictions and overall prediction accuracy. A. Robustness analysis of flux predictions in iJN1462 obtained by varying the flux through Pyruvate Carboxykinase (PC). The vertical line indicates reported flux for PC (3.1 mmol hr−1/gCDW). B and C. Comparisons between experimentally reported flux values (x‐axis) in the central metabolism of P. putida growing on glucose and predicted flux values obtained with iJN1462 (y‐axis). Reaction fluxes are normalized to the glucose uptake rate. Fluxes across the Pyruvate Carboxylase (PC), Malic enzyme (ME2), Pyruvate kinase (PYK) and Malate Dehydrogenase (MDH) are indicated. In (B) no constraints are placed on internal fluxes, while in C Pyruvate Carboxylase flux was constrained to be 3.1 mmol h−1/gCDW after having performed sensitivity analysis.
Gene essentiality data contextualization within iJN1462
The validation of GEMs through prediction of gene essentiality is a powerful way to assess and improve the accuracy of prediction while providing a suitable platform for the contextualization of knockout mutant studies at the genome‐scale (Förster et al., 2003; Covert et al., 2004; Oh et al., 2007). We performed a gene essentiality analysis in rich medium and then mapped the predicted essential genes with the knockouts available in the Pseudomonas Reference Culture Collection (PRCC) (del Castillo et al., 2007). This approach defined an accurate in silico LB (iLB) medium and a core BOF (See SI1, Table S3). A total of 114 essential genes were predicted under these conditions (Fig. 4, Table S4). Only eight gene knockouts (7%) predicted as essential were found to be not essential in PRCC, thus being false‐negative predictions. The accuracy of iJN1462 was further evaluated using glucose minimal media against an existing experimental data set (Molina‐Henares et al., 2010). Seventy‐eight conditionally essential genes in glucose minimal media were predicted after excluding those also essential in iLB. Forty‐seven of the 78 predicted glucose conditionally essential genes as well as seven predicted glucose conditionally non‐essential that were available from the PRCC were experimentally validated to assess the gene‐essentiality accuracy compared with previous models (Fig. 4B and C; Table S4). We found that iJN1462 was significantly more accurate than iJN746, iEB1050 and PpuQY1140 with 85% accuracy compared to 57%, 65% and 63% (two‐sided p‐values of Fisher's exact test was <10−3) respectively. The strain‐specific BOF of iJN1462 allowed the correct prediction of several genes involved in cofactor biosynthesis as essential in contrast to previous reconstructions. Interestingly, similar analysis performed with a GEM of P. aeruginosa matched in vivo essentiality for only 41% of in vivo essential genes while providing an overall accuracy of 83.9% (Oberhardt et al., 2011). Overall, the accuracy of iJN1462 is in the range of other high‐quality models such as E. coli, 93.4%, (Monk et al., 2017) and Bacillus subtilis, 93.4% (Tanaka et al., 2013).
Figure 4.
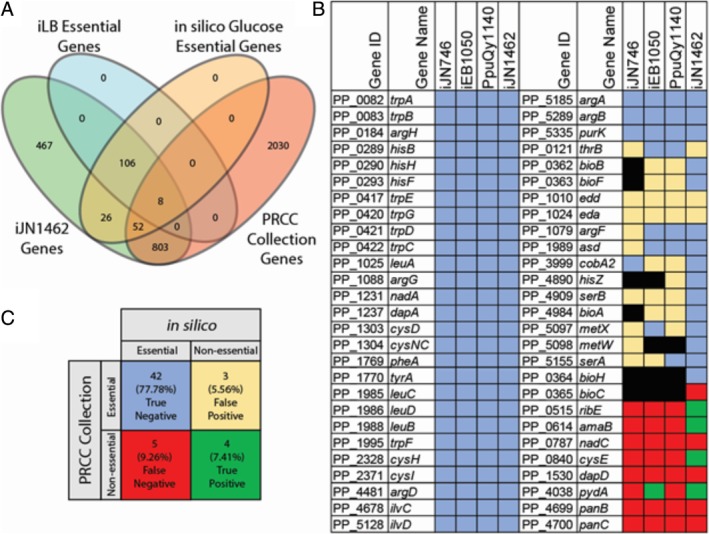
Gene essentiality analysis and validation. A. Genes predicted to be essential in the iLB medium (i.e., rich medium) were compared with the gene content of iJN1462 and single‐gene knockouts present in the PRCC screened in rich medium. Eight (7% of total predictions) false positives were predicted by iJN1462. B. The capabilities of iJN1462, iJN746, iEB1050 and PpuQY1140 for predicting essential genes in glucose minimal medium were compared to the experimental results from the PRCC Collection. C. Tabulated results for iJN1462 are given to demonstrate its accuracy. Gene essentiality prediction was correct for 85.2% of tested genes. Blue and green denote genes that were correctly predicted as essential and non‐essential respectively. Red and tan denote incorrectly predicted genes. Genes not included in a GENRE are shown in black.
Further model validation was performed using data from BarSeq experiments with P. putida using three different carbon sources: glucose, acetate and p‐coumaric acid in minimal media (Rand et al., 2017; Price et al., 2019; Thompson et al., 2019). Sensitivity analysis was performed to identify a proper cut‐off of gene fitness that corresponds with gene essentiality in the model (SI 1). The sensitivity analysis identified a range of possible cut‐offs that had similar Matthews correlation coefficients. Using cut‐offs ranging from −2.15 to −3.56 resulted in Matthews correlation coefficients that were at least 95% of the maximum value obtained using the selected cut‐off of −2.7. A cut‐off of −2.7 was also validated by comparing the conditionally essential genes determined by the BarSeq experiments and from the PRCC knockout collection. Thirty‐seven of the 44 possible genes were found to be conditionally essential in both data sets. Comparing computational results to the BarSeq data, the accuracy was 91% for all three carbon sources, with a Matthews correlation coefficient of 0.434, 0.438 and 0.409 for glucose, acetate and p‐coumaric acid respectively. The overall accuracy was 91% with a correlation coefficient of 0.426 (Table 3, Table S7). Similar comparisons using models of Rhodobacter spaeroides (Burger et al., 2017) and Synechococcus elongatus (Broddrick et al., 2016) only had 63% and 74% accuracy respectively. These experiments demonstrate that iJN1462 accurately predicted the genotype–phenotype relationships regarding gene essentiality in a variety of conditions.
Table 3.
Results from comparing BarSeq in vivo fitness data with in silico simulated growth data utilizing different carbon substrates for growth.
| Carbon source | True positive | True negative | False positive | False negative | Accuracy | Matthews correlation coefficient |
|---|---|---|---|---|---|---|
| Glucose | 1011 | 37 | 13 | 87 | 91.3 | 0.434 |
| Acetate | 1011 | 37 | 12 | 88 | 91.3 | 0.438 |
| Coumarate | 1000 | 40 | 22 | 86 | 90.6 | 0.409 |
| Overall | 3022 | 114 | 47 | 261 | 91.1 | 0.426 |
Model evaluation using Memote
The completed iJN1462 model was also evaluated using the Memote tool (https://memote.io/) (Lieven et al., 2018) in order to define its completeness as a model and analyse potential flaws or shortcomings. The model's overall score was 91%, which suggests a very good model completeness (SI2). The main reason for the model not scoring higher was a lack of annotation to outside references for all genes, metabolites and reactions in the GEM. This indicates that the model might be somewhat difficult to use with certain automated tools or scripts, but its accuracy or usability should not be affected. The model scored 97% for the important category of consistency, which represents accuracy in stoichiometry, mass balances, charge balances, connectivity of metabolites and reaction cycles. The Memote analysis demonstrated that iJN1462 is a highly complete and detailed model and can be used as a reference for other GEM constructions.
Functional assignment of metabolic capabilities of P. putida based on multi‐strain modelling
Strain‐specific GEMs have been produced that take advantage of a highly curated reference strain to identify unique metabolic capabilities that can be used to study evolutionary histories and nutrient niches (Orth et al., 2011; Monk et al., 2013; Bosi et al., 2016; Seif et al., 2018). To show the potential of iJN1462 as a template for modelling the Pseudomonas group, we performed a reconstruction of 82 P. putida strains with publicly available, high depth genomes (See Methods, Table S5). This approach, although exclude the inclusion of the strain‐specific metabolic content, resulted in highly complete metabolic models. Analysis of these models paved the way for research into the metabolic abilities and diversity of different strains of P. putida. Furthermore, by keeping only those genes present in all P. putida strains, a core‐genome metabolic model of P. putida (PP_CORE) was obtained. PP_CORE possesses only the common metabolic capabilities of all the sequenced strains of this species and allows for comparison of where genes, reactions and subsystems are conserved across strains.
Through the multi‐strain reconstruction, we demonstrated that P. putida strains have diverse metabolic capabilities, yet they also maintain broad growth potential across the whole species. We evaluated the metabolic capabilities of each model by analysing the array of carbon sources supporting growth. We found that the strain‐specific models shared the high metabolic versatility of iJN1462. Eight percent of the carbon sources available in the models had identical growth phenotypes, with 165 carbon sources able to support growth for all strains. Similar multi‐strain reconstructions of 47 E. coli strains and 64 S. aureus strains had only 61% and 24% agreement in growth phenotypes across different carbon sources in aerobic conditions. Meanwhile, 61 of the 226 carbon sources in P. putida featured variation between models as shown in Fig. 5.
Figure 5.
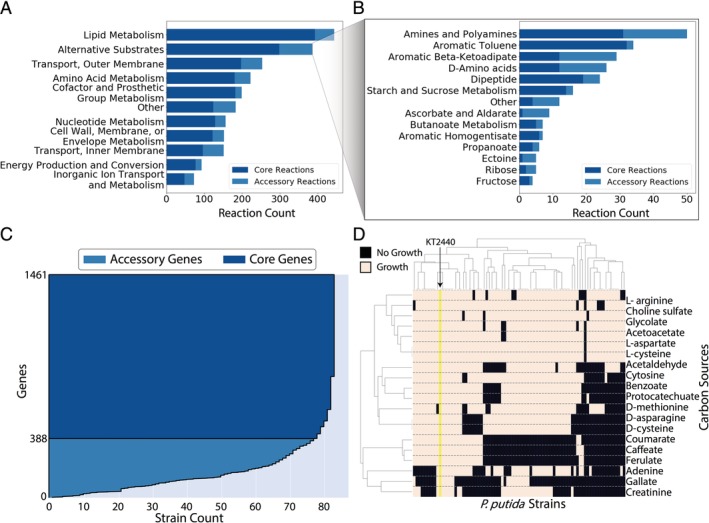
Multi‐strain comparison of P. putida species. A and B. The number of reactions predicted to be in the core‐genome vs. the pan‐genome of P. putida by grouped subsystems. Panel B provides a breakdown of subsystems involved in growth on alternative substrates. The polyhydroxyalkanoate metabolism subsystem is excluded from B and had 152 of 152 reactions in the core model. C. Dark blue shows genes in at least 95% of the strain models and considered to be part of the core genome. The lighter blue shows the accessory genes, which are responsible for the diversity between different strains. D. A clustered heatmap of growth capabilities of P. putida strains on select carbon sources. Clustered differences could be due to variances in environmental niches.
Analysis of the differences in metabolic capabilities between strains could help identify possible differences between the environmental niches that the individual strains fulfil. Figure 5C illustrates some of the differences in a subset of the carbon substrates on which P. putida can grow. Lignin‐derived monomers and other phenols show some of the largest diversities in catabolic capabilities between strains. Gallate is one of the least conserved substrates for growth (22% of strains with predicted growth), while ferulate (35%) and coumarate (35%) are equally not highly conserved. Gallate is normally derived from the syringyl component of lignin, while ferulate and coumarate are derived from the guaiacyl and p‐hydroxyphenol components. The makeup of lignin can vary significantly between plant species resulting in different abundances of these monomers when the lignin is broken down (Campbell and Sederoff, 1996). There are many strains capable of displaying growth using one group of compounds but not another, possibly indicating that they may have developed near varieties of plants that had different ratios of lignin monomers.
The PP_CORE model helped identify conserved subsystems that define the P. putida species. The core model identified 1073 genes found in at least 95% of the reconstructed strains, as seen in Fig. 5A. Most of the reactions catalysed by genes found in the core genome are involved in central cell growth and metabolism. The most conserved systems are Cofactor and Prosthetic Group Metabolism (88.9%), Lipid Metabolism (88.5%), Energy Production and Conversion (82.8%) and Nucleotide Metabolism (82.7%) as seen in Fig. 5B. In contrast, the Inner Membrane Transport, Outer Membrane Transport and Alternate Growth Substrate Systems were some of the least conserved systems at 63.6%, 77.9% and 74.8% respectively. This demonstrates that the species is likely to grow on a wide variety of substrates. Other strains of P. putida have demonstrated an ability to utilize substrates as diverse as 2,4,6‐trinitrotoluene and chlorinated aliphatic acids (Slater et al., 1979; Park et al., 2003). Overall, this analysis shows that metabolic versatility and broad growth capabilities are general features of the P. putida species, irrespective of the strain.
Discussion
A detailed metabolic model is a powerful tool for analysing the systems metabolic properties of its target organism (Nam et al., 2012; Nogales et al., 2012; Chang et al., 2013). The level of completeness and accuracy of iJN1462 makes it one of the largest and highest‐quality genome‐scale metabolic reconstructions built to date. The thorough reconstruction process allowed for detailed modelling of P. putida catabolism and anabolism beyond what was captured by previous models (Fig. 2). The high level of detail and accuracy in iJN1462 also enabled it to be used as a template for other model reconstructions of different P. putida strains, in order to explore the diversity of the species. This was a major contribution to the exploration of the broad metabolic capabilities and potential usefulness of P. putida strains. iJN1462 expands the metabolic reactome available for computation, including many of the unique metabolic pathways of Pseudomonas, a bacterial group with significant biotechnological and clinical interest (Silby et al., 2011; Nikel et al., 2014).
The accuracy of the iJN1462 model's predictions has been validated under experimental conditions and it features demonstrable improvements over previous models (Table 2). C13 flux analysis showed a very high correlation to in silico flux prediction. Adjustment of flux through the PC reaction resulted in markedly improved correlation (Fig. 3). This could be indicative of a metabolic cycle in P. putida that results in an improved ability to respond to changes in the environment. In laboratory and in silico steady‐state settings, the flux results in suboptimal growth, but in P. putida's native environments it could result in faster responses to environmental change.
Comparison with the PRCC and BarSeq knockout data also showed very high correlations for conditionally essential genes, although some areas for improvement were identified. The current level of accuracy suggests that the gene–protein relationships included are mostly accurate and well supported. BarSeq data could also be used for future improvements of the model by providing genome‐wide gene essentiality data for a wider variety of growth conditions. A single experiment can demonstrate the changes in fitness that every individual gene is responsible for in a given set of growth conditions. In the future, this could be used to identify missing or mischaracterized genes for this organism.
Aside from experimental validation, iJN1462 was also validated as a model using Memote. The Memote analysis demonstrated that it is a well‐characterized and well‐defined model. Although it did not receive a perfect score, it scored highly in the important category of stoichiometric consistency and was demonstrated to have very few flaws. The Memote analysis proved to be an effective way to quickly analyse the model and determine improvements that would make it more accurate and useful to other researchers. While there is still room for improvement of the P. putida model, particularly in linking model contents to external resources, we demonstrate here that iJN1462 serves as a highly accurate representation of our current understanding of P. putida KT2440 metabolism.
We demonstrated that iJN1462 is a useful tool for reconstructing other P. putida strains. Eighty‐two GEMs of P. putida strains were successfully created, using iJN1462 as a template. The reconstruction and analysis of the diversity of P. putida is comparable to what was previously done for the better characterized E. coli (Monk et al., 2013). The functional comparison between the P. putida strains highlighted that metabolic versatility and robustness are metabolic traits inherent to the whole P. putida species. Even as draft reconstructions, which still require careful manual curation and the addition of strain‐specific metabolic content, all the models demonstrated growth capabilities in a wide variety of conditions including lignin derivate metabolites. Despite lignin degradation not having been traditionally studied in microorganisms other than fungi, recent reports have highlighted the role of bacteria being able to break down lignin (Bugg et al., 2011; Huang et al., 2013). Interestingly, several P. putida strains, including KT2440, have been found displaying certain lignin degradation capabilities. Upon the increasing metabolic knowledge and synthetic biology tools available for this bacterial group (Franden et al., 2018; Kohlstedt et al., 2018; Nikel and de Lorenzo, 2018) these recent findings are driving important efforts toward the use of lignin and recalcitrant lignin‐derived metabolites as promising feedstock toward the sustainable production of important fine chemicals and industrial building blocks using P. putida (Linger et al., 2014; Johnson et al., 2017; Kohlstedt et al., 2018). This complex metabolism of aromatic compounds can now be optimized and redesigned with the aim of producing fine chemicals using the large computational arsenal provided by COBRA approaches within the context of well‐curated and strain‐specific P. putida GEMs. Thus, the collection of P. putida draft GEMs described here represents a first step toward the systematic analysis of the full space of biological revalorization of lignin and lignin‐derived monomers using P. putida strains.
Furthermore, the addition of known or theorized metabolic capabilities of different P. putida strains could greatly contribute to the pan‐genome of P. putida. It is likely that if complete genome reconstructions were to be carried out for all available strains of P. putida, its pan‐genome and metabolic capabilities would grow increasingly larger as the metabolic versatility of the P. putida species is revealed.
Having GEMs of different strains could aid in the identification of strains with potential for industrial applications. Even draft reconstructions can help identify which strains have pathways of interest and compare them with each other in order to identify highly efficient sets of enzymes. Identification of which GEMs have mechanisms for tolerance to solvents could also be used to identify strains that might serve as a good starting platform for the bio‐production of industrially relevant compounds. Altogether, the multi‐strain reconstruction offers an excellent starting point for identifying which strains might be of interest without having to perform wet‐lab experiments for every possible strain.
Material and methods
Metabolic reconstruction process of P. putida KT2440
The overall workflow for the reconstruction process is shown in SI1 (Fig. S1), and it is detailed in SI1. We followed a manual and iterative tri‐dimensional approach based on (i) genome annotation, (ii) biochemical legacy knowledge and (iii) phenotypic experimental validation. As a result, a more accurate assignment of function to 297 genes was achieved (Table S1).
Constraints‐based analysis
A detailed description of methods and constraints used for analysing the models can be found in SI1. iJN1462 was initially constructed on SimPheny and exported as an SBML file. Updates were made using Python and the Cobrapy package. COBRA Toolbox v2.0 (Schellenberger et al., 2011) within the MATLAB environment (The MathWorks Inc.) was used to analyse the models. Tomlab CPLEX and Gurobi were used for solving the linear programming problems.
Growth experiments on carbon and nitrogen sources
Procedures for growth experiments and knockouts analysis are found in SI1.
Gene essentiality predictions on iLB and glucose
The singleGeneDeletion function in the Cobra Toolbox (Schellenberger et al., 2011) with the minimization of metabolic adjustment algorithm (Segrè et al., 2002) were used to simulate knockouts. Additional constraints are available in SI1.
Pseudomonas putida multi‐strain genome‐scale modelling
The multi‐strain modelling was performed according to established procedures (Orth et al., 2011). We constructed a gene orthology matrix between KT2440 and the sequenced P. putida strains (Table S5). We then identified the genes present in iJN1462 for which no orthologous gene was found in each of the strains analysed and subsequently removed the corresponding GPR from iJN1462 to obtain the strain‐specific models. Gap filling was then performed to ensure growth capabilities on glucose. Additional details can be found in SI1.
Supporting information
Appendix S1. Supplementary Information (PDF)
Appendix S2. Memote Report (PDF)
Appendix S3. iJN1462 (.xml)
Table S1 Model and Manual Curation (.xlsx)
Table S2 Table S2_Nutrients and Carbon flux Validations (.xlsx)
Table S3 Biomass Formulation (.xlsx)
Table S4 Essentiality Analysis (.xlsx)
Table S5 Multi strains Modelling (.xlsx)
Table S6 MCA Models Matrix (.xlsx)
Table S7 BarSeq Analysis (.xlsx)
Acknowledgements
This work was supported by i) the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreements no 635536, 686585 and 814650, ii) the Spanish Ministry of Economy and Competitiveness through funding provided to projects RobDcode (BIO2014‐59528‐JIN) and MEPRIVA (RTI‐2018‐094370‐B‐I00), iii) the National Science Foundation Graduate Research Fellowship Program under Grant No. 1610400, iv) Novo Nordisk Fonden contract NNF10CC1016517. This work was part of the DOE Joint BioEnergyInstitute (http://www.jbei.org) supported by the U. S. Department of Energy, Office of Science, through contract DE‐AC02‐05CH11231 between Lawrence Berkeley National Laboratory and the U. S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a nonexclusive, paid‐up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of any of the agencies that funded their research. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. The authors thank Marc Abrams and Clive A. Dovefor critical reading of the manuscript and R. van Heck for providing the iEB1050 model.
References
- Belda, E. , van Heck, R.G.A. , José Lopez‐Sanchez, M. , Cruveiller, S. , Barbe, V. , Fraser, C. , et al (2016) The revisited genome of Pseudomonas putida KT2440 enlightens its value as a robust metabolic chassis. Environ Microbiol 18: 3403–3424. [DOI] [PubMed] [Google Scholar]
- Berger, A. , Dohnt, K. , Tielen, P. , Jahn, D. , Becker, J. , and Wittmann, C. (2014) Robustness and plasticity of metabolic pathway flux among uropathogenic isolates of Pseudomonas aeruginosa . PLoS One 9: e88368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank, L.M. , Ionidis, G. , Ebert, B.E. , Bühler, B. , and Schmid, A. (2008) Metabolic response of Pseudomonas putida during redox biocatalysis in the presence of a second octanol phase. FEBS J 275: 5173–5190. [DOI] [PubMed] [Google Scholar]
- Bosi, E. , Monk, J.M. , Aziz, R.K. , Fondi, M. , Nizet, V. , and Palsson, B.O. (2016) Comparative genome‐scale modelling of Staphylococcus aureus strains identifies strain‐specific metabolic capabilities linked to pathogenicity. Proc Natl Acad Sci U S A 113: E3801–E3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broddrick, J.T. , Rubin, B.E. , Welkie, D.G. , Du, N. , Mih, N. , Diamond, S. , et al (2016) Unique attributes of cyanobacterial metabolism revealed by improved genome‐scale metabolic modeling and essential gene analysis. Proc Natl Acad Sci U S A 113: E8344–E8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugg, T.D. , Ahmad, M. , Hardiman, E.M. , and Rahmanpour, R. (2011) Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep 28: 1883–1896. [DOI] [PubMed] [Google Scholar]
- Burger, B.T. , Imam, S. , Scarborough, M.J. , Noguera, D.R. , and Donohue, T.J. (2017) Combining genome‐scale experimental and computational methods to identify essential genes in Rhodobacter sphaeroides . mSystems 2: e00015‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, M.M. , and Sederoff, R.R. (1996) Variation in lignin content and composition (mechanisms of control and implications for the genetic improvement of plants). Plant Physiol 110: 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, R.L. , Andrews, K. , Kim, D. , Li, Z. , Godzik, A. , and Palsson, B.O. (2013) Structural systems biology evaluation of metabolic thermotolerance in Escherichia coli . Science 340: 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarría, M. , Kleijn, R.J. , Sauer, U. , Pflüger‐Grau, K. , and de Lorenzo, V. (2012) Regulatory tasks of the phosphoenolpyruvate‐phosphotransferase system of Pseudomonas putida in central carbon metabolism. MBio 3: e00028‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis, P. (2010) Iron uptake and metabolism in pseudomonads . Appl Microbiol Biotechnol 86: 1637–1645. [DOI] [PubMed] [Google Scholar]
- Covert, M.W. , Knight, E.M. , Reed, J.L. , Herrgard, M.J. , and Palsson, B.O. (2004) Integrating high‐throughput and computational data elucidates bacterial networks. Nature 429: 92–96. [DOI] [PubMed] [Google Scholar]
- de Waard, P. , van der Wal, H. , Huijberts, G.N. , and Eggink, G. (1993) Heteronuclear NMR analysis of unsaturated fatty acids in poly(3‐hydroxyalkanoates). Study of beta‐oxidation in Pseudomonas putida . J Biol Chem 268: 315–319. [PubMed] [Google Scholar]
- del Castillo, T. , Ramos, J.L. , Rodríguez‐Herva, J.J. , Fuhrer, T. , Sauer, U. , and Duque, E. (2007) Convergent peripheral pathways catalyze initial glucose catabolism in Pseudomonas putida: genomic and flux analysis. J Bacteriol 189: 5142–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert, B.E. , Kurth, F. , Grund, M. , Blank, L.M. , and Schmid, A. (2011) Response of Pseudomonas putida KT2440 to increased NADH and ATP demand. Appl Environ Microbiol 77: 6597–6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escapa, I.F. , García, J.L. , Bühler, B. , Blank, L.M. , and Prieto, M.A. (2012) The polyhydroxyalkanoate metabolism controls carbon and energy spillage in Pseudomonas putida . Environ Microbiol 14: 1049–1063. [DOI] [PubMed] [Google Scholar]
- Feist, A.M. , Henry, C.S. , Reed, J.L. , Krummenacker, M. , Joyce, A.R. , Karp, P.D. , et al (2007) A genome‐scale metabolic reconstruction for Escherichia coli K‐12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol 3: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster, J. , Famili, I. , Palsson, B.Ø. , and Nielsen, J. (2003) Large‐scale evaluation of in silico gene deletions in Saccharomyces cerevisiae . OMICS Int 7: 193–202. [DOI] [PubMed] [Google Scholar]
- Franden, M.A. , Jayakody, L.N. , Li, W.‐J. , Wagner, N.J. , Cleveland, N.S. , Michener, W.E. , et al (2018) Engineering Pseudomonas putida KT2440 for efficient ethylene glycol utilization. Metab Eng 48: 197–207. [DOI] [PubMed] [Google Scholar]
- Gellatly, S.L. , and Hancock, R.E.W. (2013) Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis 67: 159–173. [DOI] [PubMed] [Google Scholar]
- Henry, C.S. , DeJongh, M. , Best, A.A. , Frybarger, P.M. , Linsay, B. , and Stevens, R.L. (2010) High‐throughput generation, optimization and analysis of genome‐scale metabolic models. Nat Biotechnol 28: 977–982. [DOI] [PubMed] [Google Scholar]
- Huang, X.F. , Santhanam, N. , Badri, D.V. , Hunter, W.J. , Manter, D.K. , Decker, S.R. , et al (2013) Isolation and characterization of lignin‐degrading bacteria from rainforest soils. Biotechnol Bioeng 110: 1616–1626. [DOI] [PubMed] [Google Scholar]
- Jiménez, J.I. , Nogales, J. , García, J.L. , and Díaz, E. (2010) A genomic view of the catabolism of aromatic compounds in Pseudomonas In Handbook of Hydrocarbon and Lipid Microbiology, Timmis K. (ed). Springer: Berlin, Heidelberg, pp. 1297–1325. [Google Scholar]
- Johnson, C.W. , Abraham, P.E. , Linger, J.G. , Khanna, P. , Hettich, R.L. , and Beckham, G.T. (2017) Eliminating a global regulator of carbon catabolite repression enhances the conversion of aromatic lignin monomers to muconate in Pseudomonas putida KT2440. Metab Eng Commun 5: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, J.D. , Kocíncová, D. , Westman, E.L. , and Lam, J.S. (2009) Review: lipopolysaccharide biosynthesis in Pseudomonas aeruginosa . Innate Immun 15: 261–312. [DOI] [PubMed] [Google Scholar]
- Kohlstedt, M. , Starck, S. , Barton, N. , Stolzenberger, J. , Selzer, M. , Mehlmann, K. , et al (2018) From lignin to nylon: cascaded chemical and biochemical conversion using metabolically engineered Pseudomonas putida . Metab Eng 47: 279–293. [DOI] [PubMed] [Google Scholar]
- Lieven, C. , Beber, M.E. , Olivier, B.G. , Bergmann, F.T. , Babaei, P. , Bartell, J.A. et al. (2018) Memote: A community‐driven effort towards a standardized genome‐scale metabolic model test suite.
- Linger, J.G. , Vardon, D.R. , Guarnieri, M.T. , Karp, E.M. , Hunsinger, G.B. , Franden, M.A. , et al (2014) Lignin valorization through integrated biological funneling and chemical catalysis. Proc Natl Acad Sci U S A 111: 12013–12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeschcke, A. , and Thies, S. (2015) Pseudomonas putida—a versatile host for the production of natural products. Appl Microbiol Biotechnol 99: 6197–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loper, J.E. , Hassan, K.A. , Mavrodi, D.V. , Davis, E.W., II , Lim, C.K. , Shaffer, B.T. , et al (2012) Comparative genomics of plant‐associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet 8: e1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijs, S. , Laus, G. , Meyer, J.‐M. , Abbaspour‐Tehrani, K. , Schäfer, M. , Budzikiewicz, H. , and Cornelis, P. (2009) Siderophore‐mediated iron acquisition in the entomopathogenic bacterium Pseudomonas entomophila L48 and its close relative Pseudomonas putida KT2440. Biometals 22: 951–964. [DOI] [PubMed] [Google Scholar]
- Molina‐Henares, M.A. , De La Torre, J. , García‐Salamanca, A. , Molina‐Henares, A.J. , Herrera, M.C. , Ramos, J.L. , and Duque, E. (2010) Identification of conditionally essential genes for growth of Pseudomonas putida KT2440 on minimal medium through the screening of a genome‐wide mutant library. Environ Microbiol 12: 1468–1485. [DOI] [PubMed] [Google Scholar]
- Monk, J. , Nogales, J. , and Palsson, B.O. (2014) Optimizing genome‐scale network reconstructions. Nat Biotechnol 32: 447–452. [DOI] [PubMed] [Google Scholar]
- Monk, J.M. , Charusanti, P. , Aziz, R.K. , Lerman, J.A. , Premyodhin, N. , Orth, J.D. , et al (2013) Genome‐scale metabolic reconstructions of multiple Escherichia coli strains highlight strain‐specific adaptations to nutritional environments. Proc Natl Acad Sci U S A 110: 20338–20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk, J.M. , Lloyd, C.J. , Brunk, E. , Mih, N. , Sastry, A. , King, Z. , et al (2017) iML1515, a knowledgebase that computes Escherichia coli traits. Nat Biotechnol 35: 904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, C.E. , Monteil, C.L. , and Berge, O. (2013) The life history of Pseudomonas syringae: linking agriculture to earth system processes. Annu Rev Phytopathol 51: 85–104. [DOI] [PubMed] [Google Scholar]
- Nam, H. , Lewis, N.E. , Lerman, J.A. , Lee, D.‐H. , Chang, R.L. , Kim, D. , and Palsson, B.O. (2012) Network context and selection in the evolution to enzyme specificity. Science 337: 1101–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, K.E. , Weinel, C. , Paulsen, I.T. , Dodson, R.J. , Hilbert, H. , Martins dos Santos, V.A.P. , et al (2002) Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol 4: 799–808. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , and de Lorenzo, V. (2018) Pseudomonas putida as a functional chassis for industrial biocatalysis: from native biochemistry to trans‐metabolism. Metab Eng 50: 142–155. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , Martinez‐Garcia, E. , and de Lorenzo, V. (2014) Biotechnological domestication of pseudomonads using synthetic biology. Nat Rev Microbiol 12: 368–379. [DOI] [PubMed] [Google Scholar]
- Nogales, J. , Gudmundsson, S. , Knight, E.M. , Palsson, B.O. , and Thiele, I. (2012) Detailing the optimality of photosynthesis in cyanobacteria through systems biology analysis. Proc Natl Acad Sci U S A 109: 2678–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales, J. , Palsson, B. , and Thiele, I. (2008) A genome‐scale metabolic reconstruction of Pseudomonas putida KT2440: iJN746 as a cell factory. BMC Syst Biol 2: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhardt, M.A. , Puchałka, J. , Martins dos Santos, V.A.P. , and Papin, J.A. (2011) Reconciliation of genome‐scale metabolic reconstructions for comparative systems analysis. PLoS Comput Biol 7: e1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, Y.‐K. , Palsson, B.O. , Park, S.M. , Schilling, C.H. , and Mahadevan, R. (2007) Genome‐scale reconstruction of metabolic network in Bacillus subtilis based on high‐throughput phenotyping and gene essentiality data. J Biol Chem 282: 28791–28799. [DOI] [PubMed] [Google Scholar]
- Orth, J.D. , Conrad, T.M. , Na, J. , Lerman, J.A. , Nam, H. , Feist, A.M. , and Palsson, B.O. (2011) A comprehensive genome‐scale reconstruction of Escherichia coli metabolism–2011. Mol Syst Biol 7: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth, J.D. , Thiele, I. , and Palsson, B.O. (2010) What is flux balance analysis? Nat Biotechnol 28: 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palleroni, N.J. (2010) The Pseudomonas story. Environ Microbiol 12: 1377–1383. [DOI] [PubMed] [Google Scholar]
- Park, C. , Kim, T.H. , Kim, S. , Kim, S.W. , Lee, J. , and Kim, S.H. (2003) Optimization for biodegradation of 2,4,6‐trinitrotoluene (TNT) by Pseudomonas putida . J Biosci Bioeng 95: 567–571. [DOI] [PubMed] [Google Scholar]
- Price, M.N. , Ray, J. , Iavarone, A.T. , Carlson, H.K. , Ryan, E.M. , Malmstrom, R.R. , et al (2019) Oxidative pathways of deoxyribose and deoxyribonate catabolism. mSystems 4: e00297‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchalka, J. , Oberhardt, M. , Godinho, M. , Bielecka, A. , Regenhardt, D. , Timmis, K. , et al (2008) Genome‐scale reconstruction and analysis of the Pseudomonas putida KT2440 metabolic network facilitates applications in biotechnology. PLoS Comput Biol 4: e1000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintela, J. , Caparrós, M. , and de Pedro, M.A. (1995) Variability of peptidoglycan structural parameters in Gram‐negative bacteria. FEMS Microbiol Lett 125: 95–100. [DOI] [PubMed] [Google Scholar]
- Rand, J.M. , Pisithkul, T. , Clark, R.L. , Thiede, J.M. , Mehrer, C.R. , Agnew, D.E. , et al (2017) A metabolic pathway for catabolizing levulinic acid in bacteria. Nat Microbiol 2: 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca, A. , Pizarro‐Tobías, P. , Udaondo, Z. , Fernández, M. , Matilla, M.A. , Molina‐Henares, M.A. , et al (2013) Analysis of the plant growth‐promoting properties encoded by the genome of the rhizobacterium Pseudomonas putida BIRD‐1. Environ Microbiol 15: 780–794. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Herva, J.‐J. , Reniero, D. , Galli, E. , and Ramos, J.L. (1999) Cell envelope mutants of Pseudomonas putida: physiological characterization and analysis of their ability to survive in soil. Environ Microbiol 1: 479–488. [DOI] [PubMed] [Google Scholar]
- Rühl, J. , Hein, E.M. , Hayen, H. , Schmid, A. , and Blank, L.M. (2012) The glycerophospholipid inventory of Pseudomonas putida is conserved between strains and enables growth condition‐related alterations. J Microbial Biotechnol 5: 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger, J. , Que, R. , Fleming, R.M.T. , Thiele, I. , Orth, J.D. , Feist, A.M. , et al (2011) Quantitative prediction of cellular metabolism with constraint‐based models: the COBRA toolbox v2.0. Nat Protocols 6: 1290–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrè, D. , Vitkup, D. , and Church, G.M. (2002) Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci U S A 99: 15112–15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif, Y. , Kavvas, E. , Lachance, J.C. , Yurkovich, J.T. , Nuccio, S.P. , Fang, X. , et al (2018) Genome‐scale metabolic reconstructions of multiple Salmonella strains reveal serovar‐specific metabolic traits. Nat Commun 9: 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silby, M.W. , Winstanley, C. , Godfrey, S.A.C. , Levy, S.B. , and Jackson, R.W. (2011) Pseudomonas genomes: diverse and adaptable. FEMS Microbiol Rev 35: 652–680. [DOI] [PubMed] [Google Scholar]
- Slater, H.J.L. , Weightman, A.J. , Senior, E. , and Bull, A.T. (1979) The growth of Pseudomonas putida on chlorinated aliphatic acids and its dehalogenase activity. Microbiology 114: 125–136. [Google Scholar]
- Sohn, S.B. , Kim, T.Y. , Park, J.M. , and Lee, S.Y. (2010) In silico genome‐scale metabolic analysis of Pseudomonas putida KT2440 for polyhydroxyalkanoate synthesis, degradation of aromatics and anaerobic survival. Biotechnol J 5: 739–750. [DOI] [PubMed] [Google Scholar]
- Tanaka, K. , Henry, C.S. , Zinner, J.F. , Jolivet, E. , Cohoon, M.P. , Xia, F. , et al (2013) Building the repertoire of dispensable chromosome regions in Bacillus subtilis entails major refinement of cognate large‐scale metabolic model. Nucleic Acids Res 41: 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenhaus, M. , and Young, F.W. (1985) An analysis and synthesis of multiple correspondence analysis, optimal scaling, dual scaling, homogeneity analysis and other methods for quantifying categorical multivariate data. Psychometrika 50: 99–119. [Google Scholar]
- Thompson, M.G. , Blake‐Hedges, J.M. , Cruz‐Morales, P. , Barajas, J.F. , Curran, S.C. , Harris, N.C. , et al (2019) Massively parallel fitness profiling reveals multiple novel enzymes in Pseudomonas putida lysine metabolism. MBio 10: e02577‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udaondo, Z. , Molina, L. , Segura, A. , Duque, E. , and Ramos, J.L. (2015) Analysis of the core genome and pangenome of Pseudomonas putida . Environ Microbiol 18: 3268–3283. [DOI] [PubMed] [Google Scholar]
- van Duuren, J. , Puchalka, J. , Mars, A. , Bucker, R. , Eggink, G. , Wittmann, C. , and dos Santos, V.A. (2013) Reconciling in vivo and in silico key biological parameters of Pseudomonas putida KT2440 during growth on glucose under carbon‐limited condition. BMC Biotechnol 13: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens, J.R. , Vasil, A.I. , Schurr, M.J. , and Vasil, M.L. (2014) Iron‐regulated expression of alginate production, mucoid phenotype, and biofilm formation by Pseudomonas aeruginosa . MBio 5: e01010–e01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. , Monchy, S. , Taghavi, S. , Zhu, W. , Ramos, J. , and van der Lelie, D. (2011) Comparative genomics and functional analysis of niche‐specific adaptation in Pseudomonas putida. FEMS Microbiol Rev 35: 299–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, Q. , Huang, T. , Li, P. , Hao, T. , Li, F. , Ma, H. , et al (2017) Pathway‐consensus approach to metabolic network reconstruction for Pseudomonas putida KT2440 by systematic comparison of published models. PLoS One 12: e0169437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary Information (PDF)
Appendix S2. Memote Report (PDF)
Appendix S3. iJN1462 (.xml)
Table S1 Model and Manual Curation (.xlsx)
Table S2 Table S2_Nutrients and Carbon flux Validations (.xlsx)
Table S3 Biomass Formulation (.xlsx)
Table S4 Essentiality Analysis (.xlsx)
Table S5 Multi strains Modelling (.xlsx)
Table S6 MCA Models Matrix (.xlsx)
Table S7 BarSeq Analysis (.xlsx)