

Abstract
Background
Myotonic dystrophy type 1 (DM1) is an inherited multi‐systemic disease involving the central nervous system (CNS) and is consequently characterized by a range of cognitive impairments. However, whether this cognitive profile progresses over time is still a matter of debate. The aim of this study was to longitudinally assess a DM1 sample, in order to compare, for the first time, this progression with that of a control group. Clinical and socio‐demographic predictive factors potentially implicated in this possible decline are analysed.
Method
Seventy‐five DM1 patients with childhood, juvenile, adult, and late‐onset, and 54 control participants were re‐assessed in an 11‐year follow‐up with a comprehensive neuropsychological battery. The analyses employed were mixed ANOVA for repeated measures to test intergroup comparisons over time and multiple linear regression for predictive variable analysis.
Results
Myotonic dystrophy type 1 patients significantly worsened in visuospatial/visuoconstructive abilities and visual memory compared with controls. Multiple linear regression revealed that progression of cognitive impairment measured by copy of the Rey–Osterrieth complex figure was predicted by muscular impairment, whilst on the block design test age predicted the change with a cut‐off at 31 years of age.
Discussion
A domain‐specific progressive cognitive decline was found in DM1, with visuospatial/visuoconstructive abilities showing the greatest vulnerability to the passage of time. In addition to important clinical implications, these results suggest the need for the scientific community to delve deeper into the potential mechanisms underlying early cognitive decline in this population.
Keywords: ageing, cognitive decline, longitudinal study, myotonic dystrophy type 1, visuoconstruction
Background
Myotonic dystrophy type 1 (DM1) is an autosomal dominant inherited disorder caused by an unstable CTG (cytosine–thymine–guanine) trinucleotide repeat expansion in the DM1 protein kinase (DMPK) gene on chromosome 19 (Brook et al., 1992). In the normal population, this CTG expansion does not exceed 50 repeats, whilst DM1 ranges from 50 to 2,500, and larger expansion sizes are associated with greater disease severity. This is the most common form of adult muscular dystrophy with a reported prevalence of 1/7,400 people worldwide (Harper, 2001). However, the prevalence is significantly higher in Gipuzkoa (North of Spain), reaching 300 cases per million inhabitants (López de Munain et al., 1993 and unpublished data).
Myotonic dystrophy type 1 is considered to be a progressive and chronic multisystem disorder causing muscular weakness (to both skeletal and smooth muscles) and impairment to organs such as the eyes, heart, endocrine system, and central nervous system (CNS) (Wenninger, Montagnese, & Schoser, 2018). CNS involvement has been studied through brain MRI studies, with several brain abnormalities reported in DM1 patients, such as increased prevalence of white matter hyperintensities, global and regional brain atrophy or alteration in the integrity of normal appearing white matter (Minnerop, Gliem, & Kornblum, 2018). Although the adult‐onset type is the most typical form of DM1, the disease can be classified into five typical phenotypes depending on age of onset: congenital, childhood, juvenile, adult, and late‐onset. Although each of them leads to distinct severity grades, disease course, and clinical features, including different forms of CNS involvement, the congenital form is considered to be a qualitatively distinct phenotype, and not merely a more severe form of the disease (Turner & Hilton‐Jones, 2010).
Whilst cognitive impairments in DM1 have been characterized in recent decades, studies have still yielded mixed results. A recent meta‐analysis on the cognitive profile of DM1 found larger effect sizes in global cognition, intelligence, visual memory, visuospatial and visuoconstructive abilities, psychomotor speed, and social cognition (Okkersen et al., 2017). The present study is preceded by a previous study (2005–2007) including what at that time was the largest sample under neuropsychological assessment. The results revealed a CTG correlating dysexecutive and visuoconstructive impairment, suggesting fronto‐parietal involvement (Sistiaga et al., 2010).
Beyond the recognized cognitive impairments in DM1, rather less is known about how the cognitive profile evolves over time in comparison with the progression of cognitive outcomes related to normal ageing. To the best of our knowledge, only one longitudinal study with congenital and childhood onset in a young population (up to 28 years old at follow‐up) (Lindeblad, Kroksmark, & Ekström, 2019) and five longitudinal studies in adult population have been carried out to date and the results have prompted differing conclusions. For instance, some studies have found no decline over time (Tuikka, Laaksonen, & Somer, 1993), whilst others have found a decrease in attention (Sansone et al., 2007), memory (Gallais, Gagnon, Mathieu, & Richer, 2017), executive functions (Modoni et al., 2008), language (Winblad, Samuelsson, Lindberg, & Meola, 2016), information processing speed (Gallais et al., 2017), and visuospatial abilities (Winblad et al., 2016). In spite of the fact that in general, the results suggest a decline over time, some of these studies have also found improvements, primarily in general cognitive measures (i.e., MMSE or IQ) or in certain cognitive domains, such as executive functions (Gallais et al., 2017), memory, and attention (Modoni et al., 2008). Moreover, the extent to which clinical severity markers (i.e., molecular defect, muscular impairment, age, and disease duration) relate to cognitive decline over time is unclear.
The variety of results yielded by previous studies could be due to their methodological differences and constraints, including small sample sizes, limited follow‐up duration, the exclusion of certain forms of disease (i.e., childhood or late‐onset), the absence of comparable control groups, and a lack of information regarding other potentially relevant clinical variables.
The extensive neuropsychological data collected more than 10 years ago on a large sample of DM1 and control participants allows us to analyse the evolution of the DM1 cognitive profile over a long period of time. The aim of the present study is to analyse the progression of cognitive outcomes in a DM1 sample in comparison with a control group, where an age‐related and domain‐dependent cognitive decline is expected in patients. Moreover, this study aims to determine the possible predictive factors of such a decline, where a main significant effect of age is expected, whilst other factors (socio‐demographic and clinical) will be considered in an exploratory manner.
Method
Participants
For this follow‐up study, only participants who had attended neuropsychological assessment at baseline (DM1: N = 145, Controls: N = 76) and who still met the same inclusion criteria were eligible to take part. Exclusion criteria included congenital forms of the disease, a history of major psychiatric or somatic disorder (in accordance with DSM‐IV criteria), acquired brain damage, and alcohol or drug abuse. Inclusion criteria were being aged 16 years or above, molecular confirmation of DM1 diagnosis, and being able to complete a neuropsychological assessment. Figure 1 displays a flow chart representing participants who were lost at follow‐up and those who were re‐tested. The retention rate for DM1 patients was 51.72% of those assessed at baseline, reaching 73.53% once decease‐related drop out had been excluded. For controls, the retention rate was 71.05% of those assessed at baseline and 72.97% once deceased‐related drop out had been excluded.
Figure 1.
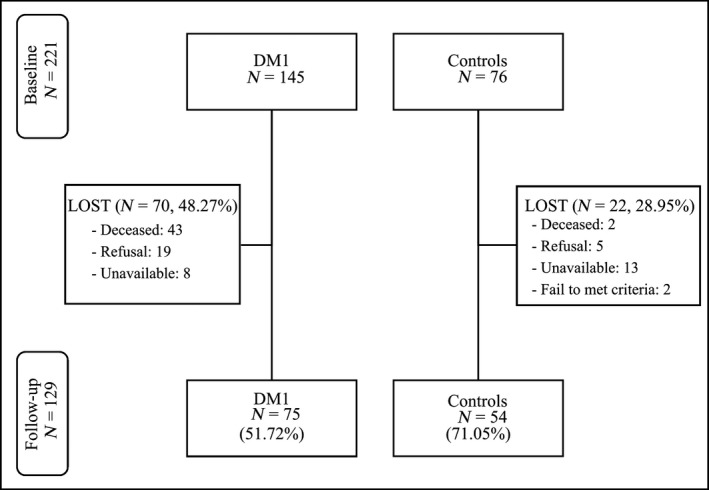
Flow chart showing the initial and follow‐up samples.
Considering the risk of bias due to selective attrition in cognitive longitudinal studies (Yao, Stawski, Hultsch, & Macdonald, 2016), the characteristics of patients and controls who failed to follow‐up and those who were re‐tested were statistically checked for equivalence, and only the same participants at baseline and follow‐up were used for all other analyses (i.e., complete case analysis). Hence, participants with both baseline and follow‐up assessment included in the statistical analysis comprised 75 DM1 patients (childhood [age of onset 1–10]: n = 7, 9.3%; juvenile [10–20]: n = 20, 26.7%; adult [20–40]: n = 35, 46.7%; late [>40 years]: n = 13, 17.3%) and 54 control participants (healthy relatives: n = 32, 59.3%; healthy non‐relatives accompanying patients: n = 10, 18.5%; limb‐girdle muscular dystrophy type 2: n = 12, 22.2%). The limb‐girdle muscular dystrophy type 2 group was selected as being a neuromuscular disease in which CNS involvement has been ruled out (Miladi, Bourguignon, & Hentati, 1999). All participants were recruited from the outpatient service of the Neurology Department and gave written informed consent. The study was approved by the Ethics Committee of the Hospital.
Neuropsychological assessment
All patients were examined by two experienced neuropsychologists who were blind to the patient's clinical condition (CTG expansion size, clinical form, and muscular impairment). Neuropsychological assessment included the following subtests from the Wechsler Adult Intelligence Scale III (WAIS III) (Wechsler, 1999): Block design, Digit span, and Vocabulary. An estimated IQ score was calculated from a two subtest short form (block design and vocabulary) with high reliability (r xx = .93) and validity (r = .87) based on Sattler and Ryan (Sattler & Ryan, 2001). Other cognitive tests used were as follows: Stroop test (Golden, 2001), California Computerized Assessment Package (CALCAP) (Miller, 1990), Rey Verbal Learning Test (RVLT) (Lezak, Howieson, & Loring, 2004), phonemic (P) and semantic (animals) verbal fluency test (Casals‐Coll et al., 2013; Peña‐Casanova et al., 2009), Rey–Osterrieth complex figure test (ROCF) (Rey, 2009), and Raven's progressive matrices (Raven, Court, & Raven, 2001).
Muscular impairment assessment
Muscular impairment was recorded by an experienced neurologist through the Muscular Impairment Rating Scale (MIRS) (Mathieu, Boivin, Meunier, Gaudreault, & Bégin, 2001) both at baseline and at follow‐up. This scale evaluates muscular impairment severity according to five grades: (1) no muscular impairment, (2) minimal signs, (3) distal weakness, (4) mild to moderate proximal weakness, and (5) severe proximal weakness.
Genetic assessment
Cytosine–thymine–guanine expansion size was obtained through genetic assessment of the DMPK gene isolated from circulating leucocyte DNA. PCR was used to measure repeat length in DMPK alleles up to approximately 100 CTG repeats and Southern blot analysis for larger expansions. At both baseline and follow‐up, genetic assessment was conducted only for the patients who had no recent data (>5 years) on CTG expansion size. Accordingly, up to 97.33% of the patients repeated the assessment at follow‐up.
Statistical analysis
Data were analysed using the SPSS (IBM SPSS Statistics 24, IBM, Madrid, Spain) statistical package. Intergroup comparisons, using contingency analysis (chi‐square), parametric (t test), or non‐parametric (Mann–Whitney U test) statistical methods where appropriate, were conducted to compare those that failed to re‐test at follow‐up and those who were re‐tested. Analyses were carried out separately for controls and DM1 patients in order to rule out the possibility of there being a higher functioning sample in the re‐tested groups (selective attrition).
In order to compare intergroup socio‐demographic characteristics between controls and patients, we carried out contingency analysis (chi‐square) for categorical data and a parametric t test for interval data. To assess possible intra‐group differences between baseline and follow‐up in socio‐demographic (years of education) and clinical (CTG expansion size and MIRS) variables, the Wilcoxon signed‐rank test was carried out.
Intergroup non‐parametric comparisons (data not normally distributed) of neuropsychological outcomes at baseline were carried out in order to characterize the cognitive profile of the studied sample.
To compare the development of differences at follow‐up between controls and DM1 patients on neuropsychological measures, a mixed ANOVA for repeated measures was conducted. Variables for the main effects were labelled as (1) ‘group’ (with two levels: DM1 patients and controls); (2) ‘time’ (with two levels: baseline and follow‐up); and (3) time × group interaction. However, the main effect of time was not analysed here, since this was not considered to be informative for the purposes of this study. Pairwise comparisons with Bonferroni adjustment were used to interpret significant interactions.
In order to construct a regression model, only in DM1 patients were Pearson Correlation analyses were carried out between potential predictive factors at baseline and delta scores of the cognitive variables in which the time × group interaction had already been found. Sex, age, years of education, disease form, inheritance pattern (maternal or paternal), CTG expansion size, and MIRS score were selected as potential predictive factors. From the potentially predictive variables, only those with a significant correlation with the delta scores were finally included in the model, in order to determine their predictive capacity. Listwise deletion was used to deal with missing values.
Effect sizes were calculated and interpreted, r was calculated when non‐parametric tests were used, and were interpreted as small (.10), medium (.30), and large (.50); Cohen's d was used when t test was used and was interpreted as small (.20), medium (.50), and large (.80) (Cohen, 1988).
Results
There was no difference between those controls that failed to re‐test at follow‐up (excluded controls) and those who were re‐tested (included controls) in age, sex, and IQ. Statistically significant differences were found between the excluded and the included controls in years of education (U = 376; p = .01), with the excluded controls (mean rank = 28.59) scoring lower than the included controls (mean rank = 42.54), with a small effect size (r = .28). Similarly, there was no difference between the excluded and included DM1 patients in sex, years of education, inheritance pattern, IQ, and CTG expansion size. Statistically significant differences were found between the excluded and the included DM1 patients in age, excluded DM1 older: t(143) = 4.45; p = < .01; Cohen's d = .73, and MIRS score (excluded DM1 greater muscular impairment: U = 742.5; p = .00; r = .51).
In the final re‐tested sample (75 DM1 and 54 controls), there were no statistically significant intergroup differences in the follow‐up in terms of sex (DM1: 45.33% male, controls: 38.88% male; x 2 (1, N = 129) = 0.53; p = .46; V = .06), or age, t(127) = 1.41; p = .16; Cohen's d = .25, whilst there was a statistically significant difference—although with a small effect size—in years of education, t(127) = 2.12; p = .04; Cohen's d = .37. Descriptive data and intergroup and intra‐group follow‐up comparisons for the main socio‐demographic and clinical variables are displayed in Table 1. Only CTG expansion size showed a statistically significant difference in repeated measures. The mean duration between baseline and follow‐up was 11.62 years (SD = 0.81).
Table 1.
Baseline and follow‐up data on age and comparisons for years of education, CTG expansion size, and MIRS outcome per group
| Baseline | Follow‐up | Z | p | |
|---|---|---|---|---|
| Mean (SD) | Mean (SD) | |||
| Age | ||||
| Control | 42 (13.59) | 53.44 (13.51) | – | – |
| DM1 | 38.65 (11.105) | 50.40 (11.00) | – | – |
| Years of education | ||||
| Control | 16.28 (4.45) | 16.35 (4.99) | −0.34 | .73 |
| DM1 | 14.45 (3.95) | 14.55 (4.59) | −0.03 | .98 |
| CTG | ||||
| DM1 | 531.54 (437.26) | 651.12 (531.21) | −4.39*** | .00 |
| MIRS | ||||
| DM1 | 2.34 (0.883) | 2.51 (1.00) | −1.40 | .16 |
CTG = triplet expansion size; DM1 = myotonic dystrophy type 1; MIRS = Muscular Impairment Rating Scale; SD = standard deviation.
***p < .001.
The intergroup comparisons of cognitive outcome (baseline) of the re‐tested sample revealed statistically significant differences in block design (U = 1307.00; p = .00; r = .29), Raven total score (U = 1021.50; p = .00; r = .27), all measures of the Stroop test (word: U = 1184.00; p = .00; r = .27; colour: U = 1212.00; p = .00.; r = .25; word‐colour: U = 1039.00; p = .00; r = .34; interference: U = 1267.00; p = .01; r = .23), vocabulary (U = 1510.00; p = .02; r = .20), two measures of the CALCAP (Election RT: U = 904.50; p = .01; r = .23; Sequential 1 RT: U = 952.00; p = .04; r = .20) and IQ estimate (U = 1228.50; p = 00.; r = .32) (see Table S1 for full results).
Cognitive follow‐up in DM1 vs. controls
Table 2 shows that when comparing DM1 and control participants as groups (taking into account both baseline and follow‐up), the intergroup main effect reached significance for the block design test, vocabulary, estimated IQ, RAVEN's progressive matrices, copy of the ROCF, and all Stroop test variables.
Table 2.
Baseline and follow‐up comparison between groups (controls and DM1 patients)
| N | Baseline | Follow‐up | Group | Time × group | |||
|---|---|---|---|---|---|---|---|
| Mean (SD) | Mean (SD) | F | p | F | p | ||
| WAIS III | |||||||
| Block design | |||||||
| Control | 54 | 38.78 (13.29) | 36.3 (13.68) | 17.35*** | .00 | 4.25* | .04 |
| DM1 | 73 | 31.1 (11.86) | 25.93 (11.93) | ||||
| Vocabulary | |||||||
| Control | 54 | 41.22 (11.48) | 42.06 (9.49) | 6.65* | .01 | 0.94 | .33 |
| DM1 | 73 | 36.77 (12.09) | 36.56 (11.04) | ||||
| Digit span | |||||||
| Forward | |||||||
| Control | 35 | 8.43 (1.75) | 7.91 (1.94) | 3.90 | .05 | 0.16 | .69 |
| DM1 | 52 | 7.62 (2.12) | 7.27 (1.85) | ||||
| Backward | |||||||
| Control | 35 | 5.97 (1.74) | 5.77 (2.00) | 1.20 | .28 | 0.07 | .79 |
| DM1 | 52 | 5.63 (1.97) | 5.33 (1.74) | ||||
| Total | |||||||
| Control | 35 | 14.4 (3.11) | 13.69 (3.38) | 2.78 | .10 | 0.01 | .92 |
| DM1 | 52 | 13.25 (3.70) | 12.6 (3.25) | ||||
| IQ estimate | |||||||
| Control | 54 | 102.85 (14.74) | 108.18 (13.06) | 21.99*** | .00 | 1.09 | .30 |
| DM1 | 73 | 91.55 (16.59) | 95.26 (14.84) | ||||
| RAVLT | |||||||
| Immediate | |||||||
| Control | 51 | 6.41 (1.96) | 6.12 (1.86) | 0.01 | .98 | 0.01 | .90 |
| DM1 | 75 | 6.44 (2.05) | 6.11 (2.10) | ||||
| Total (1–5) | |||||||
| Control | 51 | 51.33 (9.51) | 48.24 (9.67) | 0.14 | .71 | 0.66 | .42 |
| DM1 | 75 | 50.17 (10.15) | 48.09 (10.93) | ||||
| Delayed | |||||||
| Control | 51 | 10.55 (2.67) | 9.39 (3.03) | 0.01 | .98 | 0.10 | .75 |
| DM1 | 75 | 10.63 (3.01) | 9.33 (3.33) | ||||
| RAVEN | |||||||
| Control | 45 | 46.36 (8.49) | 43.38 (9.92) | 12.50*** | .00 | 0.21 | .64 |
| DM1 | 67 | 39.54 (12.00) | 36.04 (11.36) | ||||
| CALCAP | |||||||
| Simple RT | |||||||
| Control | 38 | 318.68 (54.65) | 393.82 (79.07) | 3.65 | .06 | 0.22 | .64 |
| DM1 | 65 | 340.25 (74.12) | 426.11 (118.22) | ||||
| Election RT | |||||||
| Control | 38 | 438 (92.96) | 486.13 (66.19) | 2.46 | .12 | 0.60 | .44 |
| DM1 | 65 | 467.98 (97.06) | 502.26 (73.61) | ||||
| Sequential 1 RT | |||||||
| Control | 38 | 557.61 (107.91) | 614.21 (100.70) | 3.76 | .05 | 1.27 | .26 |
| DM1 | 65 | 607.26 (116.42) | 643.26 (106.72) | ||||
| Sequential 2 RT | |||||||
| Control | 38 | 649.05 (104.56) | 662.21 (107.87) | 2.28 | .13 | 1.04 | .31 |
| DM1 | 65 | 667.74 (137.2) | 705.43 (104.52) | ||||
| ROCF | |||||||
| Copy | |||||||
| Control | 50 | 31.13 (4.4) | 32.79 (3.83) | 6.96** | .01 | 14.64*** | .00 |
| DM1 | 74 | 30.12 (5.31) | 28.96 (6.76) | ||||
| Delayed recall | |||||||
| Control | 49 | 17.15 (5.94) | 19.11 (5.03) | 2.01 | .16 | 9.74** | .00 |
| DM1 | 74 | 17.34 (6.62) | 15.99 (6.95) | ||||
| FLUENCY | |||||||
| Semantic | |||||||
| Control | 50 | 24.16 (5.90) | 23.12 (5.78) | 2.08 | .15 | 0.05 | .82 |
| DM1 | 74 | 22.85 (6.23) | 21.55 (6.69) | ||||
| Phonetic | |||||||
| Control | 50 | 16.06 (6.57) | 15.50 (4.82) | 0.38 | .54 | 1.15 | .29 |
| DM1 | 74 | 15.03 (5.50) | 15.38 (5.59) | ||||
| STROOP | |||||||
| Word | |||||||
| Control | 48 | 108.52 (17.40) | 109.92 (18.59) | 12.13*** | .00 | 3.63 | .06 |
| DM1 | 71 | 99.63 (16.91) | 96.94 (18.20) | ||||
| Colour | |||||||
| Control | 48 | 72.02 (12.70) | 72.50 (12.21) | 10.03** | .00 | 1.35 | .25 |
| DM1 | 70 | 65.76 (13.55) | 64.27 (13.20) | ||||
| Word‐Colour | |||||||
| Control | 48 | 45.40 (10.75) | 44.98 (10.22) | 16.31*** | .00 | 0.01 | .95 |
| DM1 | 70 | 37.86 (10.90) | 37.34 (11.53) | ||||
| Interference | |||||||
| Control | 48 | 2.27 (6.84) | 1.46 (6.47) | 6.83* | .01 | 0.74 | .39 |
| DM1 | 70 | −1.56 (8.64) | −1.06 (7.89) | ||||
CALCAP = California Computerized Assessment Package; DM1 = myotonic dystrophy type 1; IQ = Intelligence Quotient; RAVLT = Rey Auditory Verbal Learning Test; ROCF = Rey–Osterrieth complex figure; SD = standard deviation; WAIS III = Wechsler Adult Intelligence Scale III.
*p < .05; **p < .01; ***p < .001.
In terms of cognitive change over time, the comparison of control and DM1 patients in terms of raw score progression in the repeated measures analysis revealed a statistically significant difference (time × group interaction) on the block design test, copy of the ROCF, and delayed recall of the ROCF. There were no statistically significant interactions between any other measures.
Explanatory factors of cognitive decline
Correlation analysis (Table 3) revealed no correlation between any of the delta criterion variables and sex and disease form. The statistically significant variables in the correlation analysis were introduced in the model for multiple linear regression analyses (Table 4), revealing a statistically significant effect of age and MIRS score in predicting the progressive decline in DM1 patients when measured by the block design test and copy of the ROCF, respectively. An increase in age predicted higher delta scores (greater decline) in block design, and increase in MIRS score predicted higher delta scores on ROCF copy.
Table 3.
Pearson correlation analysis between significant decline delta scores and potentially predictive variables in DM1 patients
| Delta scores | Sex | Years of education | Age | CTG | MIRS | Inheritance | Disease form |
|---|---|---|---|---|---|---|---|
| Block design | .05 | .00 | −.27* | .05 | −.14 | −.28* | −.09 |
| ROCF copy | .02 | .24* | −.11 | −.29* | −.50** | .02 | .15 |
| ROCF delayed recall | .15 | .23* | −.05 | −.11 | −.13 | −.11 | .11 |
CTG = triplet expansion size; DM1 = myotonic dystrophy type 1; MIRS = Muscular Impairment Rating Scale; ROCF = Rey–Osterrieth complex figure.
** p < .01; * p < .05.
Table 4.
Multiple linear regression analyses for delta scores in DM1 patients
| β | t | p | Adjusted R² | F | p | |
|---|---|---|---|---|---|---|
| Block design | ||||||
| Years of education | −.15 | −0.99 | .33 | .12 | 2.55* | .04 |
| Age | −.33 | −2.19 | .03 | |||
| CTG | −.13 | −0.75 | .45 | |||
| MIRS | −.13 | −0.88 | .38 | |||
| Inheritance | −.27 | −1.93 | .06 | |||
| ROCF copy | ||||||
| Years of education | .22 | 1.67 | .10 | .30 | 5.68*** | .00 |
| Age | −.23 | −1.67 | .10 | |||
| CTG | .02 | 0.12 | .91 | |||
| MIRS | −.45 | −3.37 | .00 | |||
| Inheritance | .01 | 0.07 | .94 | |||
| ROCF delayed recall | ||||||
| Years of education | .33 | 2.08 | .04 | .04 | 1.51 | .20 |
| Age | −.02 | −0.14 | .88 | |||
| CTG | −.00 | −0.01 | .99 | |||
| MIRS | −.10 | −0.63 | .53 | |||
| Inheritance | −.14 | −0.99 | .33 | |||
CTG = triplet expansion size; DM1 = myotonic dystrophy type 1; MIRS = Muscular Impairment Rating Scale; ROCF = Rey–Osterrieth complex figure.
*p < .05; ***p < .001.
Additionally, the effect of age on block design decline was further analysed using locally weighted scatterplot smoothing (LOWESS) (Figure 2). The graph indicates a cut‐off point at 31 years of age for greater decline.
Figure 2.
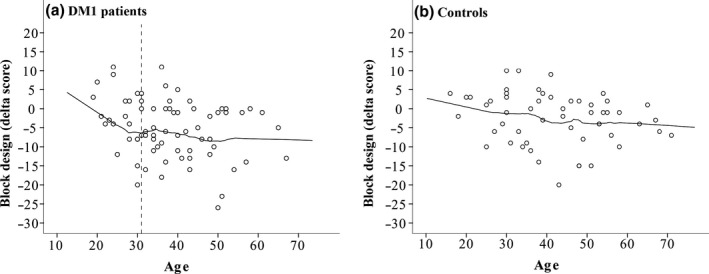
Locally weighted scatterplot smoothing for age on block design test delta scores in myotonic dystrophy type 1 patients (a) and controls (b).
Discussion
To the best of our knowledge, this is the first longitudinal study that compares DM1 and control subjects in neuropsychological terms. Although it was not the main aim of the present study to analyse the cognitive profile of DM1, a transversal comparison of both groups confirmed that the cognitive profile of our sample is in line with the pattern of dysexecutive and visuoconstructive deficits described previously in DM1, suggesting the involvement of fronto‐parietal areas (Peric et al., 2014).
From a longitudinal point of view, only some of the difficulties described in the cognitive profile of DM1 showed a decline over time greater than that expected for normal ageing. We failed to find a greater decline in patients compared with controls in attention, processing speed, verbal memory, language, executive functions, or IQ. These are the functions for which other studies have found statistically significant differences at follow‐up, although both decline and improvements have been reported, depending on the study. Whilst some authors found improvements on naming and fluency tests (Gallais et al., 2017), others have found a decline in language (Modoni et al., 2008). Similarly, some authors have found a decrease in verbal memory (Gallais et al., 2017) whilst others have found the opposite pattern of results (Modoni et al., 2008).
However, our results appear to suggest a domain‐specific pattern of cognitive decline in DM1. In particular, visuoconstructive functioning emerged as the only cognitive domain to be vulnerable to the passage of time. In our sample, there was a significant decline in block design and in the copy and delayed recall of the ROCF. Among the most recent four longitudinal studies, two of these employed the block design test and both found a decline over time (Gallais et al., 2017, non‐significant; Winblad et al., 2016), which is in line with the results found in this study. Although all the longitudinal studies mentioned used the ROCF, consensus regarding the outcomes of this test is rather less clear. Whilst Gallais et al. (2017) and Sansone et al. (2007) found a non‐significant decline in the copy subtest, Modoni et al. (2008) and Winblad et al. (2016) found no changes.
Several reasons could account for the discrepancies found between our results and those of others. Apart from the variations regarding sample sizes or the elapsed time between baseline and follow‐up, the fact that disease duration and mean age at baseline and at follow‐up are different across studies is of special interest. Thus, each study covers a different point in the life span, which could be directly related to the worsening (or absence of it) found. Moreover, DM1 is a neurologic condition that may affect the course of development of cognitive function and decline. Indeed, as already reported in DM1 patients (Gallais et al., 2017), it could be hypothesized that the decline in various cognitive functions occurs at different points during the life span.
Regarding the domain‐specific cognitive decline found in the present study, predictive factors were analysed in order to search for possible markers of this early deterioration in DM1 patients. The predictive model employed in this study was able to explain up to 12.4% of the variance in change in the performance on block design between the first and second neuropsychological test, with age emerging as the only statistically significant predictor. Further analysis of this effect revealed a cut‐off point at 31 years of age, suggesting that the decline in this cognitive ability in DM1 starts early in life and continues into the thirties. From this age onwards, this decline remains stable with the higher delta scores, which is in line with the hypothesis of an accelerated ageing process that has already been suggested in DM1 (Mateos‐Aierdi et al., 2015). Functional sequential studies with functional RM could help to further deepen knowledge regarding the selective vulnerability of certain neuronal circuits. Further, muscular impairment was the only significant predictor for the decline in ROCF copy, reaching 29.9% of the variance explained by the model. Conversely, the model was not able to significantly explain the decline in visual memory.
In line with the involvement of visuospatial/visuoconstructive impairment in DM1, a recent meta‐analysis on cognitive impairments in DM1 (Okkersen et al., 2017) found the largest effect sizes on visuospatial perception tasks and one of the largest effects on visuoconstructive tasks. This suggests that visuospatial/visuoconstructive deficits could be among the most sensitive outcomes in the DM1 cognitive profile. Taken together, from a translational point of view these results provide clinicians and researchers with relevant information for selecting neuropsychological assessment tools for this population. Thus, the inclusion of tests such as block design and ROCF could be suggested as a first‐line option, not only for describing the cognitive profile of this population, but also as possible markers of cognitive decline.
Moreover, although the use of time‐dependent and graphomotor tasks can be questioned in the assessment of neuromuscular disorders with distal muscular involvement, the results of a recent study support the use of block design in DM1, since speed difficulties derived from peripheral muscle weakness have been ruled out (Hamilton et al., 2018). Whilst ROCF has been extensively used in DM1, it is rather more difficult to rule out the possibility that distal muscle impairment could affect the copy, particularly when taking into account the fact that the MIRS score significantly predicted the change in performance between baseline and follow‐up in copy of the ROCF. However, beyond the visuoconstructive load in ROCF, there is a considerable organizational component that could be compromised in this population. Indeed, in other neurological conditions with motor affectation such as traumatic brain injury, executive functions have been found to account for a large proportion of the performance on both copy and memory of ROCF, whilst fine motor ability failed to correlate with these measures (Schwarz, Penna, & Novack, 2009). Using an alternative scoring approach such as the Boston Qualitative Scoring System for the ROCF (Stern et al., 1999) or the Developmental Scoring System for the ROCF (Bernstein & Waber, 1996) could help to better understand the underlying cognitive mechanisms involved.
Some limitations of the present study should be taken into account. Firstly—and inherent in all longitudinal studies—selective attrition must be considered. We believe we addressed this issue by employing all possible statistic control methods to minimize the effect. In any case, the intergroup differences found between those who failed to follow‐up and those who were re‐tested (the former were older and with greater muscular impairment) could be masking an even greater difference in decline between DM1 and controls. In fact, age and MIRS score were negatively correlated with target delta scores. Moreover, given that clinical heterogeneity is a hallmark of DM1, a larger sample size would allow for comparing patterns of decline between groups according to different variables such as inheritance pattern or disease form. Finally, IQ had to be estimated from a reduced set of subtests instead of administering the whole WAIS III scale due to time constraints and in order to avoid the well‐documented fatigue in patients (Kalkman et al., 2005).
Nonetheless, this study was based on a large and carefully selected control group with both assessments at baseline and follow‐up, which constitutes a notable strength of this work, since there are no previous DM1 longitudinal studies that include a control group. Further, the comprehensive and extensive neuropsychological battery employed complies with most of the latest recommended guidelines for cognitive assessment in DM1 (Gagnon et al., 2013). Finally, whilst a high mortality rate was observed among our DM1 sample (29.65% of patients at baseline died), a re‐assessment rate of almost 60% of the total sample is noteworthy, particularly for a follow‐up of such long duration.
Taken together, the results of the present study show evidence for visuoconstructive decline as a reliable cognitive marker of ageing in DM1. However, the possible explanatory factors for cognitive decline remain an issue that requires further analysis. Topography related to the suggested cognitive dysfunction could be taken to indicate the existence of circuits and neurons that are particularly vulnerable to ageing processes. To this end, the inclusion of neuropathological/neuroimaging data in longitudinal studies is strongly recommended in order to clarify the accelerated ageing process in DM1 and its possible cerebral biomarkers.
Supporting information
Table S1. Inter‐group comparison on neuropsychological outcomes at baseline.
Acknowledgements
The present study has been supported by funding from CIBERNED (Ref: 609), from the Institute of Health Carlos III co‐founded by Fondo Europeo de Desarrollo Regional‐FEDER (Ref: PI17/01231 and PI17/01841), and from the Basque Government (S‐PE13UN030). G. Labayru was supported by a predoctoral grant from the Basque Government (PRE_2016_1_0187).
References
- Bernstein, J. H. , & Waber, D. P. (1996). Developmental scoring system for the Rey‐Osterrieth Complex Figure: Professional manual. Lutz, FL: Psychological Assessment Resources Inc. [Google Scholar]
- Brook, J. D. , McCurrach, M. E. , Harley, H. G. , Buckler, A. J. , Church, D. , Aburatani, H. , … Housman, D. E. (1992). Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell, 68, 799–808. 10.1016/0092-8674(92)90154-5 [DOI] [PubMed] [Google Scholar]
- Casals‐Coll, M. , Sánchez‐Benavides, G. , Quintana, M. , Manero, R. M. , Rognoni, T. , Calvo, L. , … Peña‐Casanova, J. (2013). Spanish normative studies in young adults (NEURONORMA young adults project): norms for verbal fluency tests. Neurology, 28(1), 33–40. 10.1016/j.nrl.2012.02.010 [DOI] [PubMed] [Google Scholar]
- Cohen, J. (1988). Statistical power analysis for the behavioral sciences (2nd ed.). Hillsdate, NJ: LEA. [Google Scholar]
- Gagnon, C. , Meola, G. , Hébert, L. J. , Puymirat, J. , Laberge, L. , & Leone, M. (2013). Report of the first Outcome Measures in Myotonic Dystrophy type 1 (OMMYD‐1) international workshop. Clearwater, Florida, November 30, 2011. Neuromuscular Disorders, 23, 1056–1068. 10.1016/j.nmd.2013.07.004 [DOI] [PubMed] [Google Scholar]
- Gallais, B. , Gagnon, C. , Mathieu, J. , & Richer, L. (2017). Cognitive decline over time in adults with myotonic dystrophy type 1: A 9‐year longitudinal study. Neuromuscular Disorders, 27(1), 61–72. 10.1016/j.nmd.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Golden, C. J. (2001). STROOP. Color and word test (3rd ed.). Madrid, Spain: TEA Ediciones. [Google Scholar]
- Hamilton, M. J. , Mclean, J. , Cumming, S. , Ballantyne, B. , Mcghie, J. , Jampana, R. , … Farrugia, M. E. (2018). Outcome measures for central nervous system evaluation in myotonic dystrophy type 1 may be confounded by deficits in motor function or insight. Frontiers in Neurology, 9, 1–15. 10.3389/fneur.2018.00780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, P. S. (2001). Myotonic dystrophy (3rd ed.). London, UK: Saunders. [Google Scholar]
- Kalkman, J. S. , Schillings, M. L. , van der Werf, S. P. , Padberg, G. W. , Zwarts, M. J. , van Engelen, B. G. , & Bleijenberg, G. (2005). Experienced fatigue in facioscapulohumeral dystrophy, myotonic dystrophy, and HMSN‐I. Journal of Neurology, Neurosurgery and Psychiatry, 76, 1406–1409. 10.1136/jnnp.2004.050005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezak, M. , Howieson, D. B. , & Loring, D. (2004). Neuropsychological assessment (4th ed.). New York, NY: Oxford University Press. [Google Scholar]
- Lindeblad, G. , Kroksmark, A.‐K. , & Ekström, A. B. (2019). Cognitive and adaptive functioning in congenital and childhood forms of myotonic dystrophy type 1: A longitudinal study. Developmental Medicine and Child Neurology, 1–7. 10.1111/dmcn.14161 [DOI] [PubMed] [Google Scholar]
- López de Munain, A. , Blanco, A. , Emparanza, J. I. , Poza, J. J. , Martí Massó, J. F. , Cobo, A. , … Martínez Lage, J. M. (1993). Prevalence of myotonic dystrophy in Guipúzcoa (Basque Country, Spain). Neurology, 43, 1573–1576. [DOI] [PubMed] [Google Scholar]
- Mateos‐Aierdi, A. J. , Goicoechea, M. , Aiastui, A. , Torrón, R. F. , Garcia‐Puga, M. , Matheu, A. , & López de Munain, A. (2015). Muscle wasting in myotonic dystrophies: A model of premature aging. Frontiers in Aging Neuroscience, 7, 1–16. 10.3389/fnagi.2015.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu, J. , Boivin, H. , Meunier, D. , Gaudreault, M. , & Bégin, P. (2001). Assessment of a disease‐specific muscular impairment rating scale in myotonic dystrophy. Neurology, 56, 336–340. 10.1212/WNL.56.3.336 [DOI] [PubMed] [Google Scholar]
- Miladi, N. , Bourguignon, J. , & Hentati, F. (1999). Cognitive and psychological profile of a Tunisian population of limb girdle muscular dystrophy. Neuromuscular Disorders, 9, 352–354. [DOI] [PubMed] [Google Scholar]
- Miller, E. N. (1990). CalCAP: California Computerized Assessment Package. Los Angeles, CA: Norland Software. [Google Scholar]
- Minnerop, M. , Gliem, C. , & Kornblum, C. (2018). Current progress in CNS imaging of myotonic dystrophy. Frontiers in Neurology, 9, 1–21. 10.3389/fneur.2018.00646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modoni, A. , Silvestri, G. , Vita, M. G. , Quaranta, D. , Tonali, P. A. , & Marra, C. (2008). Cognitive impairment in myotonic dystrophy type 1 (DM1): A longitudinal follow‐up study. Journal of Neurology, 255, 1737–1742. 10.1007/s00415-008-0017-5 [DOI] [PubMed] [Google Scholar]
- Okkersen, K. , Buskes, M. , Groenewoud, J. , Kessels, R. P. C. , Knoop, H. , van Engelen, B. , & Raaphorst, J. (2017). The cognitive profile of myotonic dystrophy type 1: A systematic review and meta‐analysis. Cortex, 95, 143–155. 10.1016/j.cortex.2017.08.008 [DOI] [PubMed] [Google Scholar]
- Peña‐Casanova, J. , Quiñones‐Úbeda, S. , Gramunt‐Fombuena, N. , Quintana‐Aparicio, M. , Aguilar, M. , Badenes, D. , … Blesa, R. (2009). Spanish Multicenter Normative Studies (NEURONORMA Project): Norms for verbal fluency tests. Archives of Clinical Neuropsychology, 24, 395–411. 10.1093/arclin/acp042 [DOI] [PubMed] [Google Scholar]
- Peric, S. , Mandic‐Stojmenovic, G. , Stefanova, E. , Savic‐Paviceciv, D. , Pesovic, J. , Ilic, V. , … Rakocevic‐Stojanovic, V. (2014). Frontostriatal dysexecutive syndrome: A core cognitive feature of myotonic dystrophy type 2. Journal of Neurology, 262(1), 142–148. 10.1007/s00415-014-7545-y [DOI] [PubMed] [Google Scholar]
- Raven, J. C. , Court, J. H. , & Raven, J. (2001). Raven: Standard progressive matrices. Madrid, Spain: TEA Ediciones. [Google Scholar]
- Rey, A. (2009). Copy and memory reproduction test of complex geometric figures. Madrid, Spain: TEA Ediciones. [Google Scholar]
- Sansone, V. , Gandossini, S. , Cotelli, M. , Calabria, M. , Zanetti, O. , & Meola, G. (2007). Cognitive impairment in adult myotonic dystrophies: A longitudinal study. Neurological Sciences, 28(1), 9–15. 10.1007/s10072-007-0742-z [DOI] [PubMed] [Google Scholar]
- Sattler, J. M. , & Ryan, J. J. (2001). Wechsler Adult Intelligence Scale‐III (WAIS‐III): Description In Sattler J. M. (Ed.), Assessment of children. Cognitive applications (4th ed.). San Diego, CA: Jerome M Sattler Publisher, Inc. [Google Scholar]
- Schwarz, L. , Penna, S. , & Novack, T. (2009). Factors contributing to performance on the Rey Complex Figure Test in individuals with traumatic brain injury. The Clinical Neuropsychologist, 23(2), 255–267. 10.1080/13854040802220034 [DOI] [PubMed] [Google Scholar]
- Sistiaga, A. , Urreta, I. , Jodar, M. , Cobo, A. M. , Emparanza, J. , Otaegui, D. , … López de Munain, A. (2010). Cognitive/personality pattern and triplet expansion size in adult myotonic dystrophy type 1 (DM1): CTG repeats, cognition and personality in DM1. Psychological Medicine, 40(3), 487–495. 10.1017/S0033291709990602 [DOI] [PubMed] [Google Scholar]
- Stern, R. A. , Javorsky, D. J. , Singer, E. A. , Harris, N. G. S. , Somerville, J. A. , Duke, L. M. , … Kaplan, E. (1999). BQSS: The Boston Qualitative Scoring System for the Rey‐Osterrieth Complex Figure. Odessa, FL: Psychological Assessment Resources Inc. [Google Scholar]
- Tuikka, R. A. , Laaksonen, R. K. , & Somer, V. K. (1993). Cognitive function in myotonic dystrophy: A follow‐up study. European Neurology, 33, 436–441. [DOI] [PubMed] [Google Scholar]
- Turner, C. , & Hilton‐Jones, D. (2010). The myotonic dystrophies: Diagnosis and management. Journal of Neurology, Neurosurgery and Psychiatry, 81, 358–367. 10.1136/jnnp.2008.158261 [DOI] [PubMed] [Google Scholar]
- Wechsler, D. (1999). WAIS‐III: Wechsler Adult Intelligence Scale. Madrid, Spain: TEA Ediciones. [Google Scholar]
- Wenninger, S. , Montagnese, F. , & Schoser, B. (2018). Core clinical phenotypes in Myotonic Dystrophies. Frontiers in Neurology, 9, 1–9. 10.3389/fneur.2018.00303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad, S. , Samuelsson, L. , Lindberg, C. , & Meola, G. (2016). Cognition in myotonic dystrophy type 1: A 5‐year follow‐up study. European Journal of Neurology, 23, 1471–1476. 10.1111/ene.13062 [DOI] [PubMed] [Google Scholar]
- Yao, C. , Stawski, R. S. , Hultsch, D. F. , & Macdonald, S. W. S. (2016). Selective attrition and intraindividual variability in response time moderate cognitive change. Journal of Clinical and Experimental Neuropsychology, 38, 227–237. 10.1080/13803395.2015.1102869 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Inter‐group comparison on neuropsychological outcomes at baseline.