

Abstract
Several bis(dimethylamino)‐substituted 1,4‐diaza‐2,3‐diborinines (DADBs) were synthesized with variable substituents at the backbone nitrogen atoms. By reaction with HCl or BX3 (X=Br, I), these species were successfully converted into their synthetically more useful halide congeners. The high versatility of the generated B−X bonds in further functionalization reactions at the boron centers was demonstrated by means of salt elimination (MeLi) and commutation (NMe2 DADBs) reactions, thus making the DADB system a general structural motif in diborane(4) chemistry. A total of 18 DADB derivatives were characterized in the solid state by X‐ray diffraction, revealing a strong dependence of the heterocyclic bonding parameters from the exocyclic substitution pattern at boron. According to our experiments towards the realization of a Dipp‐substituted, sterically encumbered DADB, the mechanism of DADB formation proceeds via a transient four‐membered azadiboretidine intermediate that subsequently undergoes ring expansion to afford the six‐membered DADB heterocycle.
Keywords: azadiboretidines; B,N-heterocycles; diazadiborinines; diboranes; ring expansion
Now established: Convenient access to 1,4‐diaza‐2,3‐diborinines (DADBs) with variable steric demand of the backbone has been developed. Conversion of the initial bis(dimethylamino)‐substituted DADBs into synthetically more useful halide derivatives offers almost indefinite possibilities for further functionalization of the boron centers aiming at the synthesis of cyclic bis(amino)diboranes(4) with specifically tuned properties.

Introduction
A recent review article by Marder and Westcott entitled diboranes(4) as synthetic workhorses.1 This is in fact true when considering the tremendous progress in the development of catalytic diboration and C‐borylation protocols including enantio‐selective variants.1, 2 Nowadays, diboranes(4) have become an indispensable tool in organic synthesis to prepare borylated substrates that serve as highly valuable building blocks for the construction of more complex molecules by subsequent Suzuki–Miyaura‐type cross‐coupling reactions.3 By far the most commonly applied diboron reagents are diboranes B2(OR)4 such as commercially available B2pin2 (pin=pinacolato) and B2cat2 (cat=catecholato), which provide the best compromise of availability, stability and reactivity.1, 2, 3 By contrast, the more reactive halide analogs B2X4 (X=Cl, Br, I) are difficult to handle and too labile for broad application,1, 4 whereas the more stable tetra(amino)diboranes B2(NR2)4, such as prototypical B2(NMe2)4, are often not sufficiently reactive to promote such processes. Actually, the effect of the amino groups on the reactivity of the B−B bond is dramatical, and only systems with a maximum of two amino groups were shown to participate in diboration/borylation reactions, that is, i) B2Cl2(NMe2)2,5 ii) strained [2]borametalloarenophanes,6 and (iii) unsymmetrical (OR)2B−B(NR2)2.7
Given the fundamental importance of this research area, however, it is rather surprising that the structural diversity of stable (amino)diboranes(4) is still rather low, and dominated by the B(NMe2) fragment.8 This shortcoming is primarily a consequence of the non‐trivial B−B bond formation process, which is conveniently accomplished on a large scale only for B2(NMe2)4, making it the reagent of choice in diborane(4) chemistry. Thus, the synthesis of B2(NMe2)4 by Brotherton in 19609 is generally recognized as the commencement of diborane(4) chemistry, and it has been established as a versatile and easy to handle reagent that is readily transformed into other diborane(4) species.4, 10 One strategy to significantly modify the structural appearance and properties of (amino)diboranes is the incorporation of the B‐N units into heterocyclic ring systems to generate for example 1,4‐diaza‐2,3‐diborinines (DADB, Figure 1).11 These cyclic (amino)diboranes(4) can be considered as B,N isosteres of benzene, in which two of the C=C bonds have been replaced by isoelectronic and isostructural B−N moieties. In sharp contrast to the well‐established chemistry of azaborinines,12 not much is known about the chemistry of DADBs so far, and only few examples have been realized. The first DADBs were isolated in 1963 as benzannulated derivatives by exchange amination of B2R2(NMe2)2 with aromatic amines (A, R=alkyl, Figure 1),13 while it was not until 1997 that a monocyclic oxygen‐bridged DADB dimer was reported as a byproduct (B, Figure 1).14 Very recently, a general and more selective route to monomeric DADBs (C, Figure 1) through salt elimination reactions of Li2[dab] salts (dab=1,4‐diazabutadiene) with dihalodiboranes(4) has been established independently by Sahin15 and by our group.16 So far, reactivity studies on type C DADBs are scarce and limited to the transformation of the NMe2‐substituted derivative 1 a to its dihydrido analog and subsequent ring contraction/expansion reactions.16, 17 In this contribution, we will demonstrate that this route is easily expanded to include other backbones, and more importantly that the NMe2 group is very well suited for functionalization reactions without affecting the B2N2C2 heterocyclic core structure. Thus, a wide range of DADBs becomes accessible with high potential for further applications.
Figure 1.

DADBs known in the literature (R=alkyl, Ar=aryl).
Results and Discussion
To this end, we initially developed a more convenient protocol for the synthesis of mesityl‐substituted DADB 1 a (Ar=Mes=2,4,6‐Me3‐C6H2), which does not require the isolation and purification of Li2[Mesdab] (Scheme 1).16 Through this, DADB 1 a was readily obtained in moderate yields of 42 % even on a larger scale. To prove the generality of this approach, we also prepared related colorless DADBs 1 b–d with xylyl (1 b; 32 %; Ar=Xyl=2,6‐Me2‐C6H3), p‐tolyl (1 c; 54 %; Ar=Tol=4‐Me‐C6H4), and tert‐butyl (1 d; 26 %; Ar=tBu) groups attached to the backbone nitrogen atoms (Scheme 1).
Scheme 1.

Synthesis of DADBs 1 a–d, and conversion into halide derivatives 2 a–c, 3 a–c, and 4 a. Reaction conditions: (i) 4 equiv. HCl, Et2O, −78 °C; (ii) 2 equiv. BBr3, pentane, −78 °C; (iii) 1 equiv. BI3, pentane, −78 °C.
Compounds 1 b–d are readily identified by their 11B NMR chemical shifts of δ=33.5 (1 b: 11B{1H}; ω 1/2=1027 Hz), 34.1 (1 c: 11B{1H}; ω 1/2=941 Hz), and 36.7 ppm (1 d: ω 1/2=347 Hz) in [D6]benzene solutions, which strongly resemble those found for 1 a (δ=33.5 ppm)16 and the 2,4‐xylyl derivative of Sahin (δ=32 ppm).15 In the solid state however, these species feature marked differences with respect to the planarity of the B2N2C2 heterocyclic core, which are caused by the nature and the sterics of the groups attached to the backbone nitrogen atoms. Thus, the molecular structure of 1 b (Figure S62, Supporting Information) is virtually identical to that of 1 a,16 both showing almost planar B2N2C2 rings (N1‐C1‐C2‐N2: 1 a 2.7°; 1 b 2.2(3)°. N1‐B1‐B2‐N2: 1 a 6.2°; 1 b 5.0(3)°). By contrast, the B2N2C2 rings of 1 c (Figure 2) and 1 d (Figure S64) deviate significantly from planarity (N1‐C1‐C2‐N2: 1 c 6.7(2)°; 1 d 8.8(2)°. N1‐B1‐B2‐N2: 1 c 45.0(2)°; 1 b 52.0(1)°), similar to the data reported by Sahin (N1‐C10‐C9‐N2: −5.5°. N1‐B1‐B2‐N2: −41.7°).15 For 1 a and 1 b, steric repulsion between the NMe2 and Mes/Xyl units entails twisting of the aryl groups to adopt an orthogonal arrangement with respect to the heterocyclic core structure, which generates sufficient space for the NMe2 groups in a planar B2N2C2 environment. In 1 d, repulsive steric interactions between the NMe2 and the tBu groups within a planar geometry would be inevitable, thus the B2N2C2 ring displays a flattened chair conformation. For 1 c and Sahin's DADB, the planar and chair configurations seem to be close in energy, and the deviations from planarity observed in the solid state might already be caused by crystal packing effects favoring a coplanar arrangement of the aryl rings that necessarily provokes repulsion of the NMe2 units.
Figure 2.

Two alternative views of the molecular structure of 1 c in the solid state. The ellipsoids of the hydrogen and of some carbon atoms are omitted for clarity.
Short exocyclic B−N (1 a: 1.436(2), 1.433(2); 1 b: 1.449(3), 1.430(3); 1 c: 1.409(2); 1 d: 1.406(2), 1.412(2) Å) and rather long B−B bond lengths (1 a: 1.719(2); 1 b: 1.719(3); 1 c: 1.708(2); 1 d: 1.704(2) Å; calculated parent DADB B2N2C2H6 1.6514 Å)18 are reminiscent of strong conjugative effects arising from the pendant NMe2 groups, observations that have been interpreted earlier for 1 a in terms of aromaticity loss.16
We next wondered if the rather unreactive NMe2‐substituted DADBs can be converted into synthetically more useful species, and turned our attention to long‐established (amino)borane chemistry.4, 10 It is well documented that NMe2 groups attached to boron atoms are easily replaced by halides through reaction with HX or through commutation with BX3. Thus, we initially reacted 1 a with either ethereal HCl, or with BBr3 and BI3 in pentane solutions (Scheme 1). Monitoring the reaction mixtures by NMR spectroscopy indicated smooth conversion into the halide DADB derivatives 2 a, 3 a, and 4 a with concomitant formation of either [H2NMe2]Cl, or (amino)boranes (NMe2)nBX3−n. Compounds 2 a (Cl, 59 %), 3 a (Br, 81 %), and 4 a (I, 28 %) were isolated as colorless materials in moderate to good yields depending on the nature of the halide substituent. Similarly, 1 b and 1 c afforded the related chloro (2 b, 36 %; 2 c, 60 %) and bromo (3 b, 39; 3 c, 21 %) DADBs under the same reaction conditions (Scheme 1). In solution, 2–4 are characterized by broad 11B NMR resonances in a narrow range between δ=40.5 and δ=42.0 ppm (2 a/c, 3 a–c: 11B{1H}; ω 1/2=462–1061 Hz. 2 b, 4: ω 1/2=822, 1027 Hz). The observed shift of approximately 8 ppm to higher frequencies compared with the starting materials 1 a–c meets the expectations associated with the replacement of the electron‐rich NMe2 groups by halide atoms. X‐ray diffraction studies served to validate the structural composition of 2–4 as suggested by NMR spectroscopy (Figure 3 and Figures S65–S71, Supporting Information). Accordingly, removal of the strongly electron‐donating NMe2 groups of 1 a‐c entails marked structural changes of the B2N2C2 heterocyclic core structures. First of all, the smaller size of the halide substituents dramatically reduces the steric pressure within DADBs 2–4, thus all species feature quasi planar B2N2C2 rings with torsion angles N1‐C1‐C2‐N2 between 0.3(3)° and 2.1(3)°, and torsion angles N1‐B1‐B2‐N2 between −1.8(8)° and 6.0(9)°. More importantly, the missing conjugation of the exocyclic NMe2 groups causes adjustment of the central B−B (1.653(3)–1.667(5) Å), B−N (1.375(6)–1.411(4) Å), and C−C (1.337(2)–1.352(6) Å) bond lengths of 2–4 to those calculated for parent B2N2C2H6 (B−B 1.6514 Å; B−N 1.4034 Å; C−C 1.3677 Å),18 indicating an enhancement in aromaticity of the heterocycles going from 1 to 2–4. Furthermore, the aryl units are arranged orthogonal to the B2N2C2 rings in all halide‐substituted DADBs 2–4 prepared in this study, which is supportive for our argumentation that the coplanar arrangement found for 1 c is more likely due to crystal packing than due to sterics.
Figure 3.
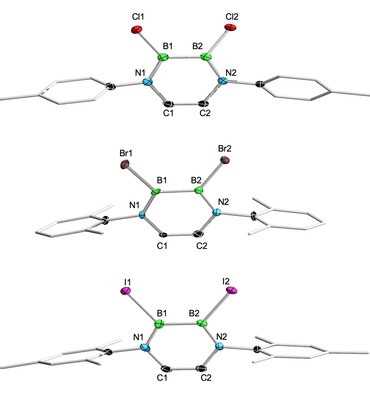
Molecular structures of 2 c (top), 3 b (middle), and 4 a (bottom) in the solid state as representative examples for halide DADBs 2–4. The ellipsoids of the hydrogen and of some carbon atoms are omitted for clarity.
We subsequently tried to assess the synthetic utility of the B−X bonds of DADBs 2–4 by studying their reactivity in salt elimination and commutation reactions. When reacted with two equivalents of MeLi in diethyl ether at low temperatures, the bromo derivatives 3 a and 3 b are smoothly converted into their methylated analogs 5 a and 5 b, respectively (Scheme 2), which are isolated as colorless solids in moderate yields of 52 % (5 a) and 41 % (5 b) after standard workup. Both species are characterized by broad 11B NMR resonances in solution (5 a: δ=49.0 ppm; ω 1/2=1220 Hz. 5 b: δ=48.5 ppm; ω 1/2=1112 Hz) with chemical shifts at higher frequencies than their NMe2‐ (1 a,16 1 b: δ=33.5 ppm) and Br‐substituted (3 a: δ=42.0; 3 b: δ=41.4 ppm) counterparts. The solid‐state structures (X‐ray diffraction) of 5 a (Figure 4) and 5 b (Figure S73, Supporting Information) reveal similar metrical parameters and strongly resemble those of their precursor molecules, thus accounting for the similar steric demand of the bromine (3) and methyl (5) substituents. Hence, 5 a and 5 b exhibit conjugated (B−B: 5 a 1.688(5); 5 b: 1.684(3). B−N: 5 a 1.392(4), 1.426(5); 5 b: 1.400(2), 1.422(2). C−C: 5 a 1.335(5); 5 b: 1.331(3) Å) and planar (N1‐C1‐C2‐N2: 5 a −1.5(5)°; 5 b 0.7(3)°. N1‐B1‐B2‐N2: 5 a −4.7(4)°; 5 b −2.5(2)°) B2N2C2 heterocycles, also reminiscent of the parent diazadiborinine B2N2C2H6 (see above).18 The B−C bonds to the methyl groups are well within the expected range for such single bonds, and the average B−Me distance of DADBs 5 is measured to be 1.577 Å.19
Scheme 2.

Reactivity of bromo‐substituted DADBs 3. Left: Salt elimination reactions of 3 a and 3 b with MeLi to afford methylated 5 a and 5 b. Right: Commutation reactions with their chloro analogs to yield mixed species 6 a and 6 c as statistical equilibrium mixtures.
Figure 4.

Molecular structures of 5 a (top) and 7 c (bottom) in the solid state as representative examples for methylated (5) and mixed NMe2/X‐substituted (7/8) DADBs. The ellipsoids of the hydrogen and of some carbon atoms are omitted for clarity.
As outlined in Schemes 2 (right) and 3, the B−X bonds of DADBs 2 and 3 are also amenable to functionalization through classical commutation reactions. For instance, halide exchange is observed upon treatment of the bromo DADBs 3 a and 3 c with their chloro analogues 2 a and 2 c, respectively (Scheme 2, right). Here, 1H NMR spectroscopy clearly showed that statistical equilibrium mixtures are established containing the expected 1:1:2 ratio of chloro (2 a/c), bromo (3 a/c), and mixed chloro/bromo (6 a/c) DADBs. According to variable‐temperature (VT) NMR studies, these equilibria are temperature‐independent, and we were not able to separate 6 a and 6 c from the reaction mixtures. Similarly, equimolar reactions of halide derivatives 2 a–c and 3 a–c with their respective NMe2 counterparts 1 a–c selectively afforded the mixed NMe2/X DADBs 7 a–c (X=Cl) and 8 a–c (X=Br) through amide–halide exchange processes (Scheme 3). In all cases, the conversion proceeded quantitatively at room temperature, and colorless powders were isolated in yields up to 94 %. The solution NMR spectra of 7 a–c and 8 a–c are very similar, and all compounds feature two distinct 11B NMR resonances with chemical shifts characteristic for the B−NMe2 (δ=30.4–32.0 ppm; ω 1/2=821–1065 Hz) and B−X (δ=40.4–41.1 ppm; ω 1/2=873–1207 Hz) boron centers (see DADBs 1–4).
Scheme 3.
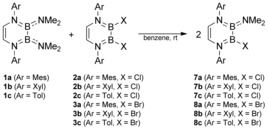
Commutation reactions between NMe2‐ (1) and halide‐substituted (2/3) DADBs to afford the corresponding mixed species 7 and 8.
We were also able to verify the unsymmetrical substitution pattern of the diborane moiety for all mixed NMe2/X DADBs in the solid state by X‐ray diffraction (Figures 4 and S74–S79, Supporting Information), and the molecular structure of 7 c is shown as representative example in Figure 4. Again, the key structural parameters of the quasi‐planar B2N2C2 heterocycles (N1‐C1‐C2‐N2: −0.4(6)–2.6(2)°; N1‐B1‐B2‐N2: 1.3(6)–6.6(3)°) are mainly determined by the electronics of the exocyclic substituents at boron. The presence of a strongly electron‐donating NMe2 group entails B−N conjugative interactions, which lower the aromaticity of the B2N2C2 ring compared to DADBs 3–5 and parent B2N2C2H6. Even though this effect is not as dramatic as for the bis(amino)‐substituted precursors 1 a–c, it is well measurable particularly for the B−B bond lengths (1.692(4)–1.699(2) Å), which show values in‐between those of the NMe2 (1 a–c: 1.708(2)–1.719(3) Å) and halide (2/3 a–c: 1.653(3)–1.667(5) Å) precursors (B2N2C2H6: 1.6514 Å).18 The unsymmetrical nature of 7 a–c and 8 a–c is also visible in the B1−N1 (1.396(7)–1.408(5) Å) and B2−N2 (1.453(6)–1.457(2) Å) bonds, which are significantly longer for B2 due to additional conjugation with the pendant NMe2 group. Similar findings have already been described for a mixed NMe2/H DADB (B−N: 1.398(6) and 1.449(6) Å).16
As part of our efforts to prepare the sterically demanding DADB 10 with bulky Dipp (Dipp=2,6‐iPr2‐C6H3) groups at the backbone nitrogen atoms, we caught unexpected insights into the mechanism of DADB formation (Figure 5). Accordingly, reactions of Li2[dab] with B2Cl2(NMe2)2 most likely do not directly afford the six‐membered B2N2C2 heterocyclic DADBs, but rather proceed via transient four‐membered azadiboretidine intermediates that subsequently undergo thermal ring‐expansion reactions to yield DADBs. Obviously, the immense steric demand of the Dipp group provides sufficient kinetic stabilization for azadiboretidine 9 to efficiently hamper DADB formation at room temperature, and to allow for the isolation of 9 as colorless crystals (26 %) (Figure 5, top). Compound 9 is not indefinitely stable at ambient conditions and slowly converts to DADB 10 both in solution and in the solid state. Nevertheless, NMR spectroscopic and X‐ray diffraction studies (Figure 5, bottom) clearly verified the classification of 9 as 1,2,3‐azadi‐boretidine, a very rare class of boron‐containing heterocycles. Although a small number of isomeric 1,2,4‐azadiboretidines are known in the literature,20 only one 1,2,3‐azadiboretidine has been mentioned so far, the identity of which, however, remains questionable.21 In solution, 9 exhibits two distinct 11B NMR signals at δ=33.1 (B1; ω 1/2=864 Hz) and δ=48.6 ppm (B2; ω 1/2=1049 Hz) with chemical shifts in the expected region for such substitution patterns.
Figure 5.

Experimental insights into the mechanism of DADB formation through salt elimination: Formation of azadiboretidine 9 and thermal ring expansion to DADB 10 (top). Molecular structure of 9 in the solid state (bottom). The ellipsoids of the hydrogen and of some carbon atoms are omitted for clarity.
In the solid state, the central four‐membered B2NC ring of 9 is almost perfectly planar with a torsion angle C1‐B2‐B1‐N1 of 1.8(1)°, which contrasts the butterfly type structures of 1,2,4‐azadiboretidines.20 The bonds B1−B2 (1.720(3) Å), B2−C1 (1.632(3) Å), and C1−N1 (1.494(2) Å) show values typically encountered for such single bonds. The B1−N1 bond length of 1.441(3) Å suggests some degree of conjugation, which effects a noticeable elongation of the exocyclic B1−N3 bond (1.401(3) Å) compared to the B2−N4 bond (1.370(3) Å). Unfortunately, bonding parameters of 9 cannot be discussed in relation to other 1,2,3‐azadiboretidines, for which structural information are absent. In line with azadiboretidine 9 being an intermediate on the way to DADB formation, 9 was converted quantitatively to DADB 10 by heating benzene solutions at 60 °C for two hours (Figure 5, top). The ring expansion reaction proceeded with high selectivity, thus analytically pure 10 was isolated by simple evaporation of the solvent. We have not been able to obtain single crystals of 10 for structural characterization in the solid state, however, NMR spectroscopy in solution (11B: δ=33.8 ppm; ω 1/2=1090 Hz. See 1 a‐d: δ=33.5–36.7 ppm) and elemental analysis convincingly substantiate its 1,4‐diaza‐2,3‐diborinine identity. According to quantum‐chemical DFT calculations, the conversion of 9 to 10 is highly exothermic (ΔG=31.0 kcal mol−1) and proceeds via concerted transition state TS, in which the B2−C1 and B2−N2 bonds are simultaneously broken and formed, respectively (Figure 6). With a calculated ΔG of 15.7 kcal mol−1, TS is thermally accessible, which is consistent with the observed lability of 9 even in the solid state.
Figure 6.

Calculated mechanism for the conversion of 9 to 10 via transition state TS. Energies are given as ΔG in kcal mol−1 at the ωB97xd/6‐31G* level of theory.
Conclusions
With our present study, we have significantly expanded the scope of known 1,4‐diaza‐2,3‐diborinines (DADBs), a class of cyclic (amino)diborane(4) molecules neglected for a long time. We have shown that the synthetic approach to enter DADB chemistry, the synthesis of doubly NMe2‐substituted DADBs 1 via one‐pot salt metathesis reactions of Li2[dab] with B2Cl2(NMe2)2, also provides the possibility to simple vary the steric demand of the DADB backbone. Most importantly, we succeeded in converting these rather unreactive molecules into their synthetically more useful dihalide derivatives 2–4 by convenient methods long‐established in diborane(4) chemistry. Subsequent salt elimination reactions with MeLi (5) and commutation reactions with NMe2 DADBs 1 (7/8) served to demonstrate the high versatility of the B−X bonds for further functionalization at the boron centers. Thus, the DADB system becomes highly relevant as a general structural motif in diborane(4) chemistry allowing for the targeted synthesis of (amino)diboranes(4) with highly specific properties for applications in borylation/diboration processes. Inspection of the solid‐state structures of a total of 18 DADBs established a strong dependence of the heterocyclic B2N2C2 bonding parameters and thus its aromaticity from the electronics of the boron‐bound substituents. With the isolation of the first structurally authenticated 1,2,3‐azadiboretidine 9, we were also able to shed some light onto the mechanism of DADB formation. Currently, we are forcefully exploring the scope of substituents that can be attached to the DADB boron centers via halide ADBs 2–4 aiming at creation of a large DADB database. Concomitantly, we are testing the new DADBs with respect to their suitability in catalytic processes involving diboranes(4).
Experimental Section
Synthesis and characterization of new compounds, NMR spectra, crystallographic details, and supplementary structures can be found in the Supporting Information. CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/chem.201905356 contain the supplementary crystallographic data for this paper. These data are provided free of charge by http://www.ccdc.cam.ac.uk/.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The Julius‐Maximilians‐Universität Würzburg and the Deutsche Forschungsgemeinschaft (DFG) are gratefully acknowledged for funding.
T. Thiess, M. Ernst, T. Kupfer, H. Braunschweig, Chem. Eur. J. 2020, 26, 2967.
References
- 1. Neeve E. C., Geier S. J., Mkhakid I. A. I., Westcott S. A., Marder T. B., Chem. Rev. 2016, 116, 9091–9161. [DOI] [PubMed] [Google Scholar]
- 2.Selected reviews:
- 2a. Irvine G. J., Lesley M. J. G., Marder T. B., Norman N. C., Rice C. R., Robins E. G., Roper W. R., Whittell G. R., Wright L. J., Chem. Rev. 1998, 98, 2685–2722; [DOI] [PubMed] [Google Scholar]
- 2b. Marder T. B., Norman N. C., Top. Catal. 1998, 5, 63–73; [Google Scholar]
- 2c. Han L.-B., Tanaka M., Chem. Commun. 1999, 395–402; [Google Scholar]
- 2d. Ishiyama T., Miyaura N., J. Organomet. Chem. 2000, 611, 392–402; [Google Scholar]
- 2e. Dembitsky V. M., Ali H. A., Srebnik M., Appl. Organomet. Chem. 2003, 17, 327–345; [Google Scholar]
- 2f. Ishiyama T., Miyaura N., Chem. Rec. 2004, 3, 271–280; [DOI] [PubMed] [Google Scholar]
- 2g. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931; [DOI] [PubMed] [Google Scholar]
- 2h. Hartwig J. F., Chem. Soc. Rev. 2011, 40, 1992–2002; [DOI] [PubMed] [Google Scholar]
- 2i. Hartwig J. F., Acc. Chem. Res. 2012, 45, 864–873; [DOI] [PubMed] [Google Scholar]
- 2j. Mlynarski N., Schuster C. H., Morken J. P., Nature 2014, 505, 386–390; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2k. Ros A., Fernández R., Lassaletta J. M., Chem. Soc. Rev. 2014, 43, 3229–3243; [DOI] [PubMed] [Google Scholar]
- 2l. Chow W. K., Yuen O. Y., Choy P. Y., So C. M., Lau C. P., Wong W. T., Kwong F. Y., RSC Adv. 2013, 3, 12518–12539; [Google Scholar]
- 2m. Fang L., Yan L., Haeffner F., Morken J. P., J. Am. Chem. Soc. 2016, 138, 2508–2511; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2n. Zhao F., Jia X., Li P., Zhao J., Zhao Y., Wang J., Liu H., Org. Chem. Front. 2017, 4, 2235–2255. [Google Scholar]
- 3.Selected books and reviews:
- 3a. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457–2483; [Google Scholar]
- 3b. Suzuki A., Brown H. C., Organic Syntheses via Boranes, Suzuki Coupling, Vol. 3, Aldrich Chemical Company, Milwaukee, 2003; [Google Scholar]
- 3c.A. Suzuki, Nobel Lecture, 2010;
- 3d. Han F.-S., Chem. Soc. Rev. 2013, 42, 5270–5298; [DOI] [PubMed] [Google Scholar]
- 3e. Applied Cross-Coupling Reactions (Ed.: Y. Nishihara), Springer, Heidelberg, 2013; [Google Scholar]; Lennox A. J. J., Lloyd-Jones G. C., Chem. Soc. Rev. 2014, 43, 412–443. [DOI] [PubMed] [Google Scholar]
- 4. Coyle T. D., Ritter J. J., Adv. Organomet. Chem. 1972, 10, 237–272. [Google Scholar]
- 5. Curtis D., Lesley M. J. G., Norman N. C., Orpen A. G., Starbuck J., J. Chem. Soc. Dalton Trans. 1999, 1687–1694. [Google Scholar]
- 6.
- 6a. Braunschweig H., Lutz M., Radacki K., Angew. Chem. Int. Ed. 2005, 44, 5647–5651; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5792–5796; [Google Scholar]
- 6b. Braunschweig H., Kupfer T., Lutz M., Radacki K., Seeler F., Sigritz R., Angew. Chem. Int. Ed. 2006, 45, 8048–8051; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 8217–8220; [Google Scholar]
- 6c. Braunschweig H., Kupfer T., J. Am. Chem. Soc. 2008, 130, 4242–4243; [DOI] [PubMed] [Google Scholar]
- 6d. Braunschweig H., Kaupp M., Adams C. J., Kupfer T., Radacki K., Schinzel S., J. Am. Chem. Soc. 2008, 130, 11376–11393; [DOI] [PubMed] [Google Scholar]
- 6e. Braunschweig H., Kupfer T., Acc. Chem. Res. 2010, 43, 455–465. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Borner C., Kleeberg C., Eur. J. Inorg. Chem. 2014, 2486–2489; [Google Scholar]
- 7b. Drescher W., Borner C., Tindall D. J., Kleeberg C., RSC Adv. 2019, 9, 3900–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Notable exceptions include the pyrrolidino group NC4H8 group, which has been studied occasionally. See for example:
- 8a. Nöth H., Wagner M., Chem. Ber. 1991, 124, 1963–1972; [Google Scholar]
- 8b. Loderer D., Nöth H., Pommerening H., Rattay W., Schick H., Chem. Ber. 1994, 127, 1605–1611; [Google Scholar]
- 8c. Braunschweig H., Koster M., J. Organomet. Chem. 1999, 588, 231–234; [Google Scholar]
- 8d. Braunschweig H., Koster M., Z. Naturforsch. B 2002, 57, 483–487. [Google Scholar]
- 9. Brotherton R. J., McCloskey A. L., J. Am. Chem. Soc. 1960, 82, 6242–6245. [Google Scholar]
- 10.See for example:
- 10a. Nöth H., Meister W., Z. Naturforsch. B 1962, 17, 714–718; [Google Scholar]
- 10b. Malhotra S. C., Inorg. Chem. 1964, 3, 862–864; [Google Scholar]
- 10c. Nöth H., Schick H., Meister W., J. Organomet. Chem. 1964, 1, 401–410; [Google Scholar]
- 10d.H. Schick, Dissertation, Ludwig-Maximilians Universität München 1966;
- 10e. Welch C. N., Shore S. G., Inorg. Chem. 1968, 7, 225–230; [Google Scholar]
- 10f. Moezzi A., Olmstead M. M., Power P. P., J. Chem. Soc. Dalton Trans. 1992, 2429–2434; [Google Scholar]
- 10g. Hommer H., Nöth H., Knizek J., Ponikwar W., Schwenk-Kircher H., Eur. J. Inorg. Chem. 1998, 1519–1527; [Google Scholar]
- 10h. Ishiyama T., Murata M., Ahiko T. A., Miyaura N., Org. Synth. 2000, 77, 176. [Google Scholar]
- 11.It should be noted that few isomeric 2,3-diaza-1,4-diborinines are also known.
- 11a. Asgarouladi B., Full R., Schaper K. J., Siebert W., Chem. Ber. 1974, 107, 34–47; [Google Scholar]
- 11b. Schmidt H., Siebert W., J. Organomet. Chem. 1978, 155, 157–163; [Google Scholar]
- 11c. Siebert W., Full R., Schmidt H., Von Seyerl J., Halstenberg M., Huttner G., J. Organomet. Chem. 1980, 191, 15–25; [Google Scholar]
- 11d. Emslie D. J. H., Piers W. E., Parvez M., Angew. Chem. Int. Ed. 2003, 42, 1252–1255; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 1290–1293; [Google Scholar]
- 11e. Jaska C. A., Emslie D. J. H., Bosdet M. J. D., Piers W. E., Sorensen T. S., Parvez M., J. Am. Chem. Soc. 2006, 128, 10885–10896; [DOI] [PubMed] [Google Scholar]
- 11f. Lu Z., Quanz H., Ruhl J., Albrecht G., Logemann W., Schlettwein D., Schreiner P. R., Wegner A., Angew. Chem. Int. Ed. 2019, 58, 4259–4263; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4303–4307. [Google Scholar]
- 12.Selected reviews:
- 12a. Bosdet M. J. D., Piers W. E., Can. J. Chem. 2009, 87, 8–29; [Google Scholar]
- 12b. Wang X. Y., Wang J.-Y., Pei J., Chem. Eur. J. 2015, 21, 3528–3539; [DOI] [PubMed] [Google Scholar]
- 12c. Morgan M. M., Piers W. E., Dalton Trans. 2016, 45, 5920–5924; [DOI] [PubMed] [Google Scholar]
- 12d. Bélanger-Chabot G., Braunschweig H., Roy D. K., Eur. J. Inorg. Chem. 2017, 4353–4368; [Google Scholar]
- 12e. Giustra Z. X., Liu S.-Y., J. Am. Chem. Soc. 2018, 140, 1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nöth H., Fritz P., Z. Anorg. Allg. Chem. 1963, 324, 129–145. [Google Scholar]
- 14. Carmalt C. J., Clegg W., Cowley A. H., Lawlor F. J., Marder T. B., Norman N. C., Rice C. R., Sandoval O. J., Scott A. J., Polyhedron 1997, 16, 2325–2328. [Google Scholar]
- 15. Söyleyici H. C., Uyanik S., Sevincek R., Firinci E., Bursali B., Burgaz O., Aygün M., Sahin Y., Inorg. Chem. Commun. 2015, 61, 214–216. [Google Scholar]
- 16. Arrowsmith M., Braunschweig H., Radacki K., Thiess T., Turkina A., Chem. Eur. J. 2017, 23, 2179–2183. [DOI] [PubMed] [Google Scholar]
- 17. Thiess T., Mellerup S. K., Braunschweig H., Chem. Eur. J. 2019, 25, 13572–13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Massey S. T., Zoellner R. W., Inorg. Chem. 1991, 30, 1063–1066. [Google Scholar]
- 19.See for example:
- 19a. Englert U., Finger R., Paetzold P., Redenz-Stormanns B., Pawelec Z., Wojnowski W., Organometallics 1995, 14, 1507–1509; [Google Scholar]
- 19b. Grigsby W. J., Power P., Chem. Eur. J. 1997, 3, 368–375. [Google Scholar]
- 20.
- 20a. Glaser B., Mayer E. P., Nöth H., Z. Naturforsch. B 1988, 43, 449–456; [Google Scholar]
- 20b. Sahin Y., Ziegler A., Happel T., Meyer H., Bayer M. J., Pritzkow H., Massa W., Hofmann M., von Ragué Schleyer P., Siebert W., Berndt A., J. Organomet. Chem. 2003, 680, 244–256. [Google Scholar]
- 21. Maringgele W., Heine A., Noltemeyer M., Meller A., J. Organomet. Chem. 1994, 468, 25–35. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary