

Abstract
Iron acquisition mediated by siderophores, high‐affinity chelators for which bacteria have evolved specific synthesis and uptake mechanisms, plays a crucial role in microbiology and in host–pathogen interactions. In the ongoing fight against bacterial infections, this area has attracted biomedical interest. Beyond several approaches to interfere with siderophore‐mediated iron uptake from medicinal and immunochemistry, the development of high‐affinity protein scavengers that tightly complex the siderophores produced by pathogenic bacteria has appeared as a novel strategy. Such binding proteins have been engineered based on siderocalin—also known as lipocalin 2—an endogenous human scavenger of enterobactin and bacillibactin that controls the systemic spreading of commensal bacteria such as Escherichia coli. By using combinatorial protein design, siderocalin was reshaped to bind several siderophores from Pseudomonas aeruginosa and, in particular, petrobactin from Bacillus anthracis, none of which is recognized by the natural protein. Such engineered versions of siderocalin effectively suppress the growth of corresponding pathogenic bacteria by depriving them of their iron supply and offer the potential to complement antibiotic therapy in situations of acute or persistent infection.
Keywords: anticalin, iron chelator, lipocalin, protein engineering, siderophore
A reshaped version of the endogenous siderocalin (right) binds petrobactin from B. anthracis (left) instead of enterobactin. Thus, it can effectively suppress the growth of corresponding pathogenic bacteria by depriving them of their iron supply and has the potential to complement antibiotic therapy in situations of acute or persistent infection.
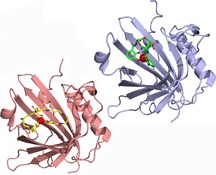
1. The Role of Siderophores in Pathogen–Host Interactions
Iron is an integral component of redox chains and many enzymes and, thus, is indispensable for metabolism, energy transfer and other processes.1 In fact, iron is an essential element not only for higher organisms but also for most microbes. However, the availability of free iron ions in a typical bacterial environment is rather low, simply due to the very limited solubility of FeIII of just 10−18 m in water at neutral pH.2 Bacterial pathogens that proliferate inside a host organism face the same problem; in fact, in vertebrates iron is specifically bound to transport proteins such as transferrin or lactoferrin in body fluids or to intracellular proteins such as hemoglobin and ferritin. On the one hand, this complexation circumvents the low solubility of free FeIII; on the other hand, it protects FeIII from reduction to FeII, which can induce the formation of toxic hydroxyl radicals in the Fenton reaction.
Apart from this, the tight regulation of iron availability can be perceived as an antimicrobial strategy as it restricts potential pathogens′ access to this essential element, a concept that was named “nutritional immunity”.3 In human plasma, for example, the concentration of free FeIII is only about 10−24 m, essentially due to the complexation by transferrin.4 However, to ensure an iron supply, microbes have evolved an elaborate strategy:5 They secrete so‐called siderophores, small organic molecules that chelate FeIII with extremely high affinity. For example, enterobactin (Ent) and bacillibactin (BB), which are produced by commensal bacteria including Escherichia coli, complex iron with extraordinary formation constants (K f) of 1049 and 1047 m −1, respectively.6
Both Ent and BB belong to the catecholate family of siderophores and contain a cyclic trilactone backbone with three 2,3‐dihydroxybenzoyl side chains that can tightly coordinate a central ferric ion (Figure 1).7 BB differs from Ent mainly by the additional glycyl spacer that links each catecholate moiety to the cyclic backbone (apart from extra methyl groups there). Other siderophore complexes have lower formation constants (e.g., aerobactin, K f≈1028 m −1),8 but this is still sufficient to acquire iron in human plasma considering that transferrin has a lower K f of 1022 m −1.4 Once formed, ferric siderophore complexes are imported via specific receptors into the bacterial cell, where iron is finally released by enzymatic reduction of FeIII to FeII or degradation of the siderophore.7
Figure 1.
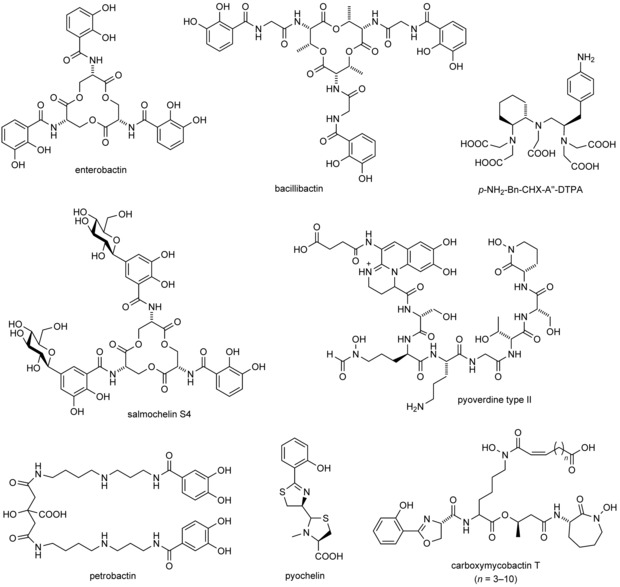
Chemical structures of selected siderophores: enterobactin (E. coli), bacillibactin (B. cereus), carboxymycobactin T (M. tuberculosis), salmochelin S4 (S. enterica, uropathogenic E. coli strains), pyoverdine type II (P. aeruginosa strain ATCC27853), pyochelin (Pseudomonas spp.), petrobactin (B. cereus, B. anthracis) and, for comparison, an artificial derivative of diethylenetriaminepentaacetic acid (DTPA) as a metal chelator with applications in nuclear medicine.
Thus, bacterial iron acquisition through siderophores can be very effective, and their secretion often significantly contributes to virulence.9 Conversely, due to their critical role for pathogens, siderophores serve as targets for the innate immune system to suppress bacterial growth.5 In this context, one of the most important proteins is siderocalin (Scn), also known as lipocalin 2 (Lcn2) or neutrophil gelatinase‐associated lipocalin (NGAL).10 Scn is an abundant human plasma protein that tightly binds several siderophores from the catecholate‐type family with sub‐nanomolar K D values, in particular Ent⋅FeIII and BB⋅FeIII. Furthermore, it recognizes some of the carboxymycobactins secreted by Mycobacterium tuberculosis, which complex iron in a different manner by using a phenyloxazoline and two hydroxamate groups (Figure 1).11
It is generally assumed that the acute‐phase protein Scn acts as an antimicrobial agent in human plasma and effectively prevents the systemic spreading of bacteria that depend on corresponding siderophores. Indeed, Lcn2‐deficient transgenic mice showed a strongly decreased resistance against a clinical strain of Ent‐dependent E. coli as compared to wild‐type mice, whereas application of recombinant Lcn2 to the acute‐phase serum of the knock‐out mice suppressed bacterial growth.12 Interestingly, iron depletion by Lcn2 could be bypassed by application of ferrichrome, a siderophore that is not produced by E. coli (or bound by Lcn2) but can be imported by this bacterium. The role of human Scn in the defense against bacterial infections was further corroborated in a recent study with HIV patients as well as healthy individuals infected with M. tuberculosis (whose virulence depends on iron acquisition through carboxymycobactin) by detecting strong up‐regulation of serum Scn in active tuberculosis for both groups.13 Apart from that, human tear lipocalin (Tlc), also known as lipocalin 1 (Lcn1), is another member of the lipocalin family with antimicrobial activity.14 Tlc is abundant in tear fluid as well as on surfaces of the respiratory tract and binds a broad spectrum of siderophores, including Ent, aerobactin, ferrioxamine B and several fungal siderophores, yet with lower affinity than Scn.15
2. Pathogenic Bacteria and Their Stealth Siderophores
From an evolutionary point of view, the competition for iron has had a strong impact on pathogen–host interactions and constantly forces adaptation by both sides.16 The appearance of an Scn‐based defense mechanism in higher organisms provided those pathogens that produce siderophores that are not recognized by Scn with an advantage. Such “stealth siderophores”11b exhibit structural features that preclude binding to the ligand pocket of this lipocalin. Salmochelins, for example, are derivatives of Ent that carry one or two C‐linked glucose substituents on the catechol groups (Figure 1)17 and cannot form a complex with Scn due to steric hindrance.18 Thus, corresponding strains of Salmonella spp., Klebsiella pneumoniae and E. coli evade the human innate immune system simply by making an ordinary siderophore more bulky. Interestingly, monoglucosylated (but not diglucosylated) salmochelin is bound by the extracellular fatty‐acid binding protein (Ex‐FABP), which is found in chicken and also binds Ent⋅FeIII with its enlarged pocket, providing sufficient space to accommodate the protruding glucose substituent.19 It has been hypothesized that this protein feature evolved from the specific interaction between chicken and their pathogens, in particular Salmonella enterica.20
Another stealth siderophore is petrobactin (PB), which was originally isolated from Marinobacter hydrocarbonoclasticus, an oil‐degrading microbe.21 More importantly, PB is also produced by pathogenic strains of Bacillus cereus and Bacillus anthracis.22 As infection with B. anthracis can be fatal, a better understanding of the PB‐based iron‐acquisition system is of great interest.23 PB contains two 3,4‐dihydroxy catecholates instead of the 2,3‐dihydroxy catecholates found in Ent and BB; together with the bridging citryl moiety and spermidine spacer groups these lead to a completely different geometry of the ferric complex (Figure 1).24 Thus, PB⋅FeIII is not bound by Scn and escapes the innate immune system in humans.11b Accordingly, the synthesis of PB is crucial for the virulence of these bacteria.25 The affinity of PB for iron is similar to that of BB (estimated K f=1043 vs. 1047 m −1),26 whereas it seems that PB is more efficient in abstracting iron from diferric transferrin.27
Strains of Pseudomonas also produce two types of siderophore that are not recognized by Scn.28 One of them is pyochelin (Figure 1), which contains a thiazoline and a phenolate moiety and belongs to the mixed‐type siderophores. Furthermore, these bacteria secrete pyoverdines, a class of highly diverse siderophores that comprise three parts: a conserved fluorescent dihydroquinoline moiety, an acyl side chain attached to its amino group, and a variable peptide chain with 6–12 amino acid residues. More than 50 pyoverdines have been identified from different strains of Pseudomonas.29 Pyoverdines secreted by Pseudomonas aeruginosa, an opportunistic human pathogen, can be grouped into three types each carrying a distinct peptide chain.29, 30 Furthermore, the siderophores from each type vary in their acyl chain, thus giving rise to a remarkable diversity in this iron‐uptake system. Notably, certain strains of P. aeruginosa produce one type of pyoverdine but can also import ferric complexes of siderophores secreted by other bacteria; this kind of “iron piracy” adds another component to the biological competition for iron.31
3. Iron‐Acquisition Systems as Targets for Antimicrobial Strategies
Bacterial pathogens constitute a serious threat to mankind, and this problem is aggravated by a growing resistance to available antibiotics.32 Thus, there is an increasing need for novel antimicrobial targets and therapies. In this context, iron‐acquisition systems have attracted considerable attention in recent years.33 In a so‐called “Trojan horse” strategy that makes use of siderophore–antibiotic conjugates, bacterial siderophore import systems have been employed to deliver toxic agents specifically to microbes.34 In principle, this strategy only targets bacteria that express the corresponding transporter, leaving unrelated bacteria as well as host cells unaffected. Other approaches aim to develop chemical inhibitors of siderophore biosynthesis.35 Baulamycins, for example, are a recently discovered class of antibiotics that were shown in vitro to inhibit a particular step in the biosynthesis of petrobactin, including the structurally related staphyloferrin.36 A subsequent study, however, came to the conclusion that the antibiotic effect of these amphiphilic compounds is mainly due to an induction of cell lysis.37
A different strategy involves siderophore‐based immunization. For example, Ent was conjugated to the cholera toxin subunit B as an immunogenic carrier protein and applied in a mouse model to elicit IgA antibodies both against the Ent hapten and the structurally related salmochelins.38 Indeed, such immunized mice contained fewer Salmonella in their intestines when infected with this bacterial pathogen, apparently due to the neutralization of the salmochelin stealth siderophore. Similarly, a vaccine based on yersiniabactin and aerobactin was shown to protect against a pathogenic strain of E. coli in a mouse model of urinary tract infection.39 Hence, immunization with siderophores (or their conjugates) might offer a promising way to protect hosts from infectious bacteria.
4. Neutralization of Siderophores by Using Engineered Binding Proteins Based on Scn
Our laboratory has pioneered the engineering of novel binding proteins based on the lipocalin scaffold, including Scn/Lcn2, yielding so‐called Anticalin® molecules (registered trademark of Pieris Pharmaceuticals GmbH).40 Lipocalins are small proteins with a β‐barrel fold made of eight antiparallel strands that are arranged in a circular manner. These β‐strands are connected pairwise by four loops, thus creating a ligand pocket at the open end. The shape of this binding site, which specifically accommodates Ent and BB in the case of Scn, as explained above, varies considerably among the many different members of the lipocalin family.14, 41 In fact, this structurally variable region resembles the hypervariable loop region of antibodies.42 By employing targeted random mutagenesis together with combinatorial selection techniques (phage display, colony screening, high‐throughput ELISA, and the like), Anticalins with novel specificities and high affinities can be generated against a series of molecular targets, not only haptens but also peptides and proteins.40a, 43 For example, in the context of metal chelators, we have previously developed variants of Scn with picomolar affinity for lanthanide metal complexes of CHX‐A′′‐DTPA, a synthetic chelator that is of interest for positron emission tomography (PET) imaging and radioimmunotherapy (Figure 1).44
Based on these encouraging results, we recently set out to reshape Scn in order to achieve binding activity for FeIII complexes of stealth siderophores that play a role in infectious diseases. As a model target we chose PB⋅FeIII, which is of particular biomedical relevance.45 This siderophore is secreted by B. anthracis,46 as explained above, a highly infectious microbe that also represents a biothreat.47 Of note, B. anthracis produces BB as a second siderophore beside PB; however, BB is sequestered by natural Scn. Consequently, additional neutralization of PB by a cognate engineered lipocalin should fully block iron acquisition by this bacterial pathogen, thus inhibiting growth (Figure 2).
Figure 2.
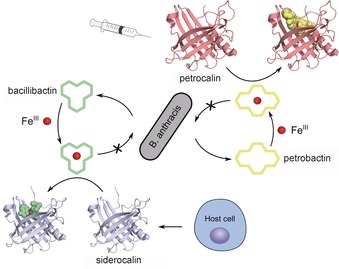
Sequestration of siderophores as an antimicrobial strategy. B. anthracis secretes two siderophores, BB and PB. Scn, which is produced by neutrophils and other human cell types, binds BB and its ferric complex, thus depleting it as a microbial source of iron. Additional administration of Pcn, an engineered variant of Scn that binds ferric PB with high affinity, blocks the pathogen's second route to iron and results in effective growth inhibition.
To select a binding protein specific for PB⋅FeIII, we used a previously described gene library of Scn (Lcn2) with 20 randomized amino acid positions that surround the ligand pocket at the open end of the β‐barrel.48 The chemical synthesis of PB, and also of a biotinylated derivative that was required for the selection process, was carried out according to published procedures.49 Selection from the naïve randomized lipocalin library was performed by using phagemid display as well as a filter sandwich colony screening assay.43b The Lcn2 variant with the best PB⋅FeIII‐binding activity resulting from this procedure was subjected to two rounds of affinity maturation by partially randomizing the major coding region followed by selection via phagemid display and bacterial surface display.45 Finally, one mutant was identified that not only exhibited a remarkable affinity towards PB⋅FeIII (K D=21±2 pm) but also had lost the original binding activity of Scn towards Ent⋅FeIII. Therefore, this engineered lipocalin was dubbed petrocalin (Pcn). Pcn has distinct ligand‐binding characteristics from Scn: it does not recognize Ent⋅FeIII, smaller catecholate⋅FeIII complexes, or metal‐free PB, thus exhibiting an entirely novel substrate specificity.
The X‐ray crystal structure of Pcn in complex with PB⋅GaIII—a surrogate that is not light‐sensitive like the FeIII complex—yielded interesting insights into the binding mode (Figure 3);45 also, this was the first reported crystal structure of this siderophore. As anticipated from its chemical formula (cf. Figure 1), PB shows an extended, butterfly‐like conformation that allows octahedral coordination of GaIII (or FeIII) by the central carboxyl and hydroxy groups as well as the two 3,4‐dihydroxybenzoyl moieties that are attached via flexible spermidine linkers. This configuration leads to an overall larger complex than Ent⋅FeIII and, in agreement with this differing molecular geometry, Pcn has a wider ligand pocket than Scn. Nevertheless, there are also similarities in the mode of siderophore binding between Pcn and Scn (Figure 3). For example, the ligand pocket of Pcn has an overall positive charge, and two critical amino acid residues, Arg79 and Arg100, contact one of the catechol groups of PB⋅FeIII through cation–π interactions. This resembles the interaction between Ent⋅FeIII and Scn through the three basic side chains of Arg81, Lys125 and Lys134.11a
Figure 3.
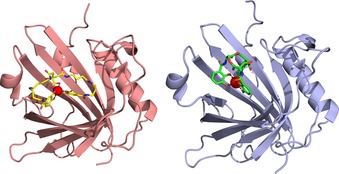
Comparison between the crystal structures of Pcn and Scn with bound siderophore ligands: (left) PB⋅GaIII (PDB ID: 6GR0) and (right) Ent⋅FeIII (PDB ID: 3CMP).
To investigate whether Pcn can suppress the growth of bacteria that depend on PB for iron acquisition, B. cereus F837/76 was chosen as a model strain. This less harmful strain is closely related to pathogenic B. anthracis 50 and, likewise, secretes both BB and PB.22 When B. cereus F837/76 was cultured in a low‐iron medium, growth was strongly inhibited when both the engineered Pcn and recombinant Scn were added to the culture. This effect was decreased by supplementation with PB, or PB⋅FeIII, in a concentration‐dependent manner. Notably, Pcn had no significant effect when applied to the bacterial culture without Scn, or when a related strain of B. cereus that only depends on BB was used. Taken together, these experiments provided convincing proof that Pcn exerts a bacterial growth‐inhibiting effect by neutralization of PB⋅FeIII.
5. The Potential of Engineered Lipocalins for Antibacterial Therapy
In conclusion, it has been demonstrated that human Scn, a natural protein from the innate immune system, can be reshaped by protein design to effectively sequester a siderophore that is critical for supplying iron to a bacterial pathogen. The growth‐limiting effect on relevant Bacillus strains observed in vitro suggests that it could be used to treat harmful bacterial infections, including anthrax, in a clinical setting. To this end, Pcn might be administered as a biopharmaceutical by infusion, exerting its antibacterial activity in synergy with the endogenous Scn.
This proof of concept opens wider applications of engineered lipocalins as siderophore scavengers to fight bacterial infections. In fact, siderophore‐specific Anticalin proteins have also been developed as potential therapeutic candidates to treat infections by P. aeruginosa.51 Four different Anticalins were selected with specificity for each of the three pyoverdine types (see above) as well as pyochelin and combined in a single fusion protein dubbed “Tetracalin”.52 In principle, this multispecific binding protein can neutralize all of the siderophores produced by clinically relevant strains of P. aeruginosa. The tetracalin was able to eradicate a chronic P. aeruginosa infection in a rat model. Pharmacokinetic studies showed good lung exposure of the intravenously administered substance and acceptable half‐life. These siderophore‐specific Anticalin proteins can possibly be used in the management of cystic fibrosis patients chronically infected by P. aeruginosa.53
Generally, reprogramming of lipocalins such as Scn offers an attractive strategy to scavenge stealth siderophores and, in this way, deprive bacteria of iron, a concept that might even be expanded to acquisition systems for other essential transition metals.54 Corresponding engineered lipocalins should be useful to treat bacterial infections as part of a therapeutic regimen, either alone, by complementing conventional antibiotics, or in combination with other antimicrobial compounds.33a
Conflict of interest
A.S. is founder and shareholder of Pieris Pharmaceuticals, Inc.
Biographical Information
Martin Dauner obtained a PhD in chemistry from the University of Konstanz, Germany, in 2012. From 2012 to 2017, he was a postdoctoral researcher in A.S.′s group at the Technical University of Munich, working on the development of proteins with novel binding activities. Since 2017, he has been a research associate in the group of Prof. Frank Bordusa at the Martin‐Luther‐Universität Halle–Wittenberg, Germany.

Biographical Information
Arne Skerra holds the Chair of Biological Chemistry at the Technical University of Munich. After chemical studies at the Technical University of Darmstadt, he received his PhD at Ludwig Maximilians University Munich, then worked as a post‐doctoral scientist at the MRC Laboratory of Molecular Biology in Cambridge (UK). He became group leader at the Max Planck Institute of Biophysics, Frankfurt am Main, and, from 1994 to 1998, was Professor of Protein Chemistry at the TU Darmstadt.

M. Dauner, A. Skerra, ChemBioChem 2020, 21, 601.
References
- 1. Wandersman C., Delepelaire P., Annu. Rev. Microbiol. 2004, 58, 611–647. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Andrews S. C., Robinson A. K., Rodríguez-Quiñones F., FEMS Microbiol. Rev. 2003, 27, 215–237; [DOI] [PubMed] [Google Scholar]
- 2b. Raymond K. N., Dertz E. A., Kim S. S., Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Skaar E. P., PLoS Pathog. 2010, 6, e1000949; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Skaar E. P., Raffatellu M., Metallomics 2015, 7, 926–928. [DOI] [PubMed] [Google Scholar]
- 4. Martin R. B., Savory J., Brown S., Bertholf R. L., Wills M. R., Clin. Chem. 1987, 33, 405–407. [PubMed] [Google Scholar]
- 5. Golonka R., Yeoh B. S., Vijay-Kumar M., J. Innate Immun. 2019, 11, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Loomis L. D., Raymond K. N., Inorg. Chem. 1991, 30, 906–911; [Google Scholar]
- 6b. Dertz E. A., Xu J., Stintzi A., Raymond K. N., J. Am. Chem. Soc. 2006, 128, 22–23. [DOI] [PubMed] [Google Scholar]
- 7. Miethke M., Marahiel M. A., Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harris W. R., Carrano C. J., Raymond K. N., J. Am. Chem. Soc. 1979, 101, 2722–2727. [Google Scholar]
- 9. Cassat J. E., Skaar E. P., Cell Host Microbe 2013, 13, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Breustedt D. A., Schönfeld D. L., Skerra A., Biochim. Biophys. Acta Proteins Proteomics 2006, 1764, 161–173; [DOI] [PubMed] [Google Scholar]
- 10b. Kjeldsen L., Cowland J. B., Borregaard N., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1482, 272–283. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Goetz D. H., Holmes M. A., Borregaard N., Bluhm M. E., Raymond K. N., Strong R. K., Mol. Cell 2002, 10, 1033–1043; [DOI] [PubMed] [Google Scholar]
- 11b. Abergel R. J., Wilson M. K., Arceneaux J. E. L., Hoette T. M., Strong R. K., Byers B. R., Raymond K. N., Proc. Natl. Acad. Sci. USA 2006, 103, 18499–18503; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Holmes M. A., Paulsene W., Jide X., Ratledge C., Strong R. K., Structure 2005, 13, 29–41; [DOI] [PubMed] [Google Scholar]
- 11d. Strong R. K. in Lipocalins (Eds.: B. Åkerström, N. Borregaard, D. R. Flower, J.-P. Salier), Landes Bioscience, Georgetown, 2006, pp. 83–98; [Google Scholar]
- 11e. Hoette T. M., Clifton M. C., Zawadzka A. M., Holmes M. A., Strong R. K., Raymond K. N., ACS Chem. Biol. 2011, 6, 1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flo T. H., Smith K. D., Sato S., Rodriguez D. J., Holmes M. A., Strong R. K., Akira S., Aderem A., Nature 2004, 432, 917–921. [DOI] [PubMed] [Google Scholar]
- 13. Varghese G. M., Turaka V. P., Janardhanan J., Yadav S., Lakshmi K. M., Vijayakumar T. S., Cherayil B., Int. J. Infect. Dis. 2019, 85, 132–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schiefner A., Skerra A., Acc. Chem. Res. 2015, 48, 976–985. [DOI] [PubMed] [Google Scholar]
- 15. Fluckinger M., Haas H., Merschak P., Glasgow B. J., Redl B., Antimicrob. Agents Chemother. 2004, 48, 3367–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barber M. F., Elde N. C., Trends Genet. 2015, 31, 627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Hantke K., Nicholson G., Rabsch W., Winkelmann G., Proc. Natl. Acad. Sci. USA 2003, 100, 3677–3682; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Bister B., Bischoff D., Nicholson G. J., Valdebenito M., Schneider K., Winkelmann G., Hantke K., Süssmuth R. D., BioMetals 2004, 17, 471–481. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Fischbach M. A., Lin H., Zhou L., Yu Y., Abergel R. J., Liu D. R., Raymond K. N., Wanner B. L., Strong R. K., Walsh C. T., Aderem A., Smith K. D., Proc. Natl. Acad. Sci. USA 2006, 103, 16502–16507; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Fischbach M. A., Lin H., Liu D. R., Walsh C. T., Nat. Chem. Biol. 2006, 2, 132–138. [DOI] [PubMed] [Google Scholar]
- 19. Correnti C., Clifton M. C., Abergel R. J., Allred B., Hoette T. M., Ruiz M., Cancedda R., Raymond K. N., Descalzi F., Strong R. K., Structure 2011, 19, 1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sia A. K., Allred B. E., Raymond K. N., Curr. Opin. Chem. Biol. 2013, 17, 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barbeau K., Zhang G., Live D. H., Butler A., J. Am. Chem. Soc. 2002, 124, 378–379. [DOI] [PubMed] [Google Scholar]
- 22. Koppisch A. T., Dhungana S., Hill K. K., Boukhalfa H., Heine H. S., Colip L. A., Romero R. B., Shou Y., Ticknor L. O., Marrone B. L., Hersman L. E., Iyer S., Ruggiero C. E., BioMetals 2008, 21, 581–589. [DOI] [PubMed] [Google Scholar]
- 23. Hagan A. K., Carlson P. E., Hanna P. C., Mol. Microbiol. 2016, 102, 196–206. [DOI] [PubMed] [Google Scholar]
- 24. Bergeron R. J., Huang G., Smith R. E., Bharti N., McManis J. S., Butler A., Tetrahedron 2003, 59, 2007–2014. [Google Scholar]
- 25. Cendrowski S., MacArthur W., Hanna P., Mol. Microbiol. 2004, 51, 407–417. [DOI] [PubMed] [Google Scholar]
- 26. Zhang G., Amin S. A., Küpper F. C., Holt P. D., Carrano C. J., Butler A., Inorg. Chem. 2009, 48, 11466–11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abergel R. J., Zawadzka A. M., Raymond K. N., J. Am. Chem. Soc. 2008, 130, 2124–2125. [DOI] [PubMed] [Google Scholar]
- 28. Cornelis P., Matthijs S., Environ. Microbiol. 2002, 4, 787–798. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Visca P., Imperi F., Lamont I. L., Trends Microbiol. 2007, 15, 22–30; [DOI] [PubMed] [Google Scholar]
- 29b. Cezard C., Farvacques N., Sonnet P., Curr. Med. Chem. 2015, 22, 165–186. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Meyer J.-M., Stintzi A., De Vos D., Cornelis P., Tappe R., Taraz K., Budzikiewicz H., Microbiology 1997, 143, 35–43; [DOI] [PubMed] [Google Scholar]
- 30b. Tappe R., Taraz K., Budzikiewicz H., Meyer J.-M., Lefèvre J. F., J. Prakt. Chem. 1993, 335, 83–87. [Google Scholar]
- 31. Cornelis P., Dingemans J., Front. Cell. Infect. Microbiol. 2013, 3, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fabbretti A., Gualerzi C. O., Brandi L., FEBS Lett. 2011, 585, 1673–1681. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Bilitewski U., Blodgett J. A. V., Duhme-Klair A.-K., Dallavalle S., Laschat S., Routledge A., Schobert R., Angew. Chem. Int. Ed. 2017, 56, 14360–14382; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14552–14575; [Google Scholar]
- 33b. Wilson B. R., Bogdan A. R., Miyazawa M., Hashimoto K., Tsuji Y., Trends Mol. Med. 2016, 22, 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mislin G. L. A., Schalk I. J., Metallomics 2014, 6, 408–420. [DOI] [PubMed] [Google Scholar]
- 35. Lamb A. L., Biochim. Biophys. Acta Proteins Proteomics 2015, 1854, 1054–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tripathi A., Schofield M. M., Chlipala G. E., Schultz P. J., Yim I., Newmister S. A., Nusca T. D., Scaglione J. B., Hanna P. C., Tamayo-Castillo G., Sherman D. H., J. Am. Chem. Soc. 2014, 136, 1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Steele A. D., Ernouf G., Lee Y. E., Wuest W. M., Org. Lett. 2018, 20, 1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sassone-Corsi M., Chairatana P., Zheng T., Perez-Lopez A., Edwards R. A., George M. D., Nolan E. M., Raffatellu M., Proc. Natl. Acad. Sci. USA 2016, 113, 13462–13467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mike L. A., Smith S. N., Sumner C. A., Eaton K. A., Mobley H. L. T., Proc. Natl. Acad. Sci. USA 2016, 113, 13468–13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.
- 40a. Richter A., Eggenstein E., Skerra A., FEBS Lett. 2014, 588, 213–218; [DOI] [PubMed] [Google Scholar]
- 40b. Rothe C., Skerra A., BioDrugs 2018, 32, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Skerra A., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1482, 337–350. [DOI] [PubMed] [Google Scholar]
- 42. Skerra A., Curr. Opin. Chem. Biol. 2003, 7, 683–693. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Skerra A., J. Biotechnol. 2001, 74, 257–275; [DOI] [PubMed] [Google Scholar]
- 43b. Gebauer M., Skerra A., Methods Enzymol. 2012, 503, 157–188. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Kim H. J., Eichinger A., Skerra A., J. Am. Chem. Soc. 2009, 131, 3565–3576; [DOI] [PubMed] [Google Scholar]
- 44b. Eggenstein E., Eichinger A., Kim H. J., Skerra A., J. Struct. Biol. 2014, 185, 203–214. [DOI] [PubMed] [Google Scholar]
- 45. Dauner M., Eichinger A., Lücking G., Scherer S., Skerra A., Angew. Chem. Int. Ed. 2018, 57, 14619–14623; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14829–14833. [Google Scholar]
- 46. Koppisch A. T., Browder C. C., Moe A. L., Shelley J. T., Kinkel B. A., Hersman L. E., Iyer S., Ruggiero C. E., BioMetals 2005, 18, 577–585. [DOI] [PubMed] [Google Scholar]
- 47. Friedlander A. M., Curr. Clin. Top. Infect. Dis. 2000, 20, 335–349. [PubMed] [Google Scholar]
- 48. Gebauer M., Schiefner A., Matschiner G., Skerra A., J. Mol. Biol. 2013, 425, 780–802. [DOI] [PubMed] [Google Scholar]
- 49.
- 49a. Bugdahn N., Peuckert F., Albrecht A. G., Miethke M., Marahiel M. A., Oberthur M., Angew. Chem. Int. Ed. 2010, 49, 10210–10213; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10408–10411; [Google Scholar]
- 49b. Bugdahn N., Oberthür M., Eur. J. Org. Chem. 2014, 426–435. [Google Scholar]
- 50. Böhm M.-E., Huptas C., Krey V. M., Scherer S., BMC Evol. Biol. 2015, 15, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Corvey C., Stump H., Kruip J., Calandra B., Rey A., Karst N., Mourez M., Fraisse L., Rothe C., Allersdorfer A., Wiedenmann A., Hinner M., Lunde B., Jensen K., Hülsmeyer M. (Sanofi), WO 2016/131804 A1.
- 52.“Pieris Pharmaceuticals′ collaborator presents promising preclinical data on tetraspecific infectious disease Anticalin program at ASM conference”, Press release, Pieris Pharmaceuticals Inc., Sept 15, 2015.
- 53.“Case study: Engineering of a tetraspecific protein biotherapeutics based on Anticalin scaffold to combat Pseudomonas aeruginosa infections by blocking iron uptake”, J. Beninga, The 2nd Biologics & Biosimilars Congress, 1–2 February 2016, Berlin, Germany.
- 54. Palmer L. D., Skaar E. P., Annu. Rev. Genet. 2016, 50, 67–91. [DOI] [PMC free article] [PubMed] [Google Scholar]