

Abstract
Roxadustat is an oral hypoxia‐inducible factor prolyl hydroxylase inhibitor developed to treat anemia in chronic kidney disease (CKD) patients. This Phase 3, randomized, open‐label, 24‐week study investigated the efficacy and safety of roxadustat in Japanese CKD patients with anemia on peritoneal dialysis (PD) who were previously treated or not treated with erythropoiesis stimulating agents (ESAs). Patients not previously receiving ESA (ESA‐Naïve group) were randomized to roxadustat at a starting dose of 50 or 70 mg three times weekly; patients previously receiving ESA (ESA‐Converted group) switched from ESA to roxadustat 70 or 100 mg three times weekly depending on the prior ESA dose. Outcomes included maintenance rate of average hemoglobin (Hb) level within 10–12 g/dL at weeks 18–24, cumulative response rate at end of treatment (Hb thresholds, 10.0 g/dL or 10.5 g/dL; Hb increase, ≥1.0 g/dL), and average Hb levels at weeks 18–24. Safety was assessed by occurrence of treatment‐emergent adverse events (TEAEs). Fifty‐six patients were enrolled (ESA‐Naïve, n = 13; ESA‐Converted, n = 43). Maintenance rates (weeks 18–24) were 92.3% (95% CI: 64.0–99.8; ESA‐Naïve) and 74.4% (95% CI: 58.8–86.5; ESA‐Converted). Cumulative response rate was 100.0% in the ESA‐Naïve group. Average Hb levels (weeks 18–24) were 11.05 g/dL (95% CI: 10.67–11.42; ESA‐Naïve) and 10.93 g/dL (95% CI: 10.73–11.13; ESA‐Converted). Common TEAEs included nasopharyngitis and back pain. Roxadustat was well tolerated and effective in maintaining target Hb levels in CKD patients on PD who were previously treated or not treated with ESA.
Keywords: Anemia, Chronic kidney disease, Clinical trial, Peritoneal dialysis, Roxadustat
Anemia is a common complication in patients with chronic kidney disease (CKD) and has been associated with increased risk of cardiovascular events, hospitalization, and mortality. 1 Anemia in CKD is associated with dysregulation of oxygen sensing by the malfunctioning kidneys, leading to inadequate synthesis of erythropoietin, functional iron deficiency, reduced erythrocyte survival, and inflammation. 2 The prevalence of anemia increases as CKD progresses and, in the United States, varies from 8.4% at stage 1 to 53.4% at stage 5 CKD. 3 Peritoneal dialysis (PD) is an effective modality for renal replacement therapy for patients with end‐stage renal disease (ESRD) that has the advantage of being able to be carried out at home, allowing patients to have greater control over the dialysis process, be more independent, and have a higher quality of life. 4, 5 It has also become evident that the clinical outcomes associated with PD are comparable, or in some cases, better than those associated with hemodialysis. 6 Current treatment to correct anemia in dialyzed patients includes recombinant human erythropoietin and its analogs, as well as iron supplementation. 7, 8 Although erythropoiesis stimulating agents (ESAs) can significantly improve hemoglobin (Hb) levels and quality of life for CKD patients, their use has been associated with increased safety risks, particularly in patients with cancer, diabetes, and cardiovascular disease. 9 Moreover, despite the advantage of PD as a home therapy, PD patients may still require dialysis center visits to receive ESA injections. These observations indicate that there is an unmet need for new therapeutic agents for the management of anemia in CKD, particularly in PD patients.
A new class of oral agents for the treatment of anemia in patients with CKD functions by inhibiting the activity of the hypoxia‐inducible factor prolyl hydroxylase enzymes. The transient inhibition of these enzymes mimics the body's natural response to hypoxia, activating erythropoiesis and altering iron transport, both of which increase Hb levels. 1 Roxadustat (ASP1517, FG‐4592, AZD9941) is an oral hypoxia‐inducible factor prolyl hydroxylase inhibitor that was approved in December 2018 in China for treatment of dialysis‐dependent CKD anemia and is currently in late‐stage clinical development in Japan, the United States, and Europe. Roxadustat has been shown to be well tolerated and effective in phase 2 studies of patients with dialysis‐dependent 10, 11, 12 and nondialysis‐dependent 11, 13, 14 CKD. Most recently, a 6‐month phase 3 study of roxadustat in China reported noninferiority of roxadustat to epoetin alfa in correcting and maintaining Hb levels in patients with CKD on hemodialysis. Furthermore, roxadustat significantly reduced hepcidin, a hormone that regulates iron homeostasis, and increased serum iron without the need for intravenous (IV) iron supplementation (manuscript submitted).
The objective of this phase 3 Japanese study was to investigate the efficacy and safety of roxadustat for the treatment of anemia exclusively in CKD patients on PD who had or had not previously received ESA treatment.
PATIENTS AND METHODS
Study design
This phase 3, multicenter, randomized, open‐label, noncomparative study (1517‐CL‐0302; http://ClinicalTrials.gov Identifier: NCT02780726) was conducted from 22 June 2016 to 2 August 2017 at 15 sites in Japan. Following eligibility assessments and screening, patients were enrolled into one of two cohorts based on previous treatment with ESA. Patients who were not previously receiving ESA (ESA‐Naïve group) were randomized (1:1) to receive oral roxadustat at a starting dose of 50 or 70 mg. Patients who were previously receiving ESA (ESA‐Converted group) were switched from ESA to oral roxadustat at a starting dose of 70 or 100 mg, depending upon the ESA dose prior to registration (Table S1, Supporting Information). Roxadustat was administered three times weekly for 24 weeks.
Study population
This study enrolled patients aged ≥20 years with ESRD who were receiving or scheduled to receive PD. Patients were not permitted to initiate hemodialysis during the study; however, daily PD solution glucose concentrations, total daily volume of PD, number and duration of exchanges, and type of PD fluid could be modified. Major inclusion criteria for patients in the ESA‐Naïve group were: (i) must not have received ESA treatment within 6 weeks before prescreening; (ii) Hb level < 10.5 g/dL (mean of two most recent Hb measurements taken at least a week apart before registration during the screening period with the absolute difference between the two measurements of ≤1.3 g/dL); and (iii) transferrin saturation (TSAT) ≥5% or serum ferritin ≥30 ng/mL during the screening period. Major inclusion criteria for patients in the ESA‐Converted group included: (i) must have received treatment with ESA at a dose approved in Japan for ≥8 weeks before prescreening and after the start of PD; (ii) Hb level ≥ 10 g/dL and ≤ 12 g/dL (mean of two most recent Hb measurements taken at least a week apart just before registration during the screening period); and (iii) TSAT ≥ 20% or serum ferritin ≥ 100 ng/mL during the screening period.
Study drug administration
Roxadustat was administered as 20‐, 50‐, and 100‐mg tablets, and the starting dose and regimen were chosen based on findings from previous studies (data on file). Patients in the ESA‐Naïve group were randomized to oral roxadustat at a starting dose of 50 or 70 mg by a dynamic allocation method using a biased‐coin minimization approach with study site and body weight as assignment factors. Allocation of the investigational product and management of the patients were conducted by EPS Corporation (Tokyo, Japan). For patients in the ESA‐Converted group, a dose conversion between the average dose of ESA before enrollment and roxadustat was used for the starting dose (Table S1). Roxadustat dose adjustments were considered during even numbered weeks starting at week 4 and were made as needed according to the dose adjustment rule, which considered current Hb level and change in Hb level over the previous 4 weeks (Table S2), to maintain Hb levels within 10.0–12.0 g/dL. Any dose adjustment had to be maintained for ≥4 weeks before further adjustment and the maximum dose for any patient was to not exceed 3 mg/kg. Any concomitant phosphate binders were to be dosed at least 1 h before or after roxadustat. Administration of IV iron products was permitted at the discretion of the investigator only in cases of low TSAT (<5% for ESA‐Naïve; <20% for ESA‐Converted) or low serum ferritin (<30 ng/mL for ESA‐Naïve; <100 ng/mL for ESA‐Converted). There were no restrictions in the administration of concomitant oral iron products.
Study outcomes and assessments
Efficacy outcomes included the maintenance rate of a target Hb level (proportion of patients who achieved an average Hb level of 10.0–12.0 g/dL between weeks 18 and 24); cumulative response rates for the threshold Hb levels of 10.0 g/dL or 10.5 g/dL (calculated as the proportions of patients who achieved Hb ≥10.0 g/dL or Hb ≥10.5 g/dL and an increase in Hb from baseline to the end of treatment of ≥1.0 g/dL; ESA‐Naïve group only); average Hb levels between weeks 18 and 24; change in average Hb levels from baseline to weeks 18–24; rate of rise in Hb levels (g/dL/week) from week 0 to the earliest date of week 4, discontinuation, or dose adjustment; proportion of patients who achieved (and time to achievement) the lower limit of the target Hb level (i.e. 10.0 g/dL) (ESA‐Naïve group only); and serum iron, ferritin, transferrin, TSAT, and reticulocyte Hb content. An exploratory outcome was the change in mean serum hepcidin levels throughout the study. Safety was assessed by monitoring treatment‐emergent adverse events (TEAEs), vital signs (i.e. blood pressure and pulse rate), 12‐lead electrocardiogram (ECG), and laboratory tests.
Hemoglobin levels were measured every 2 weeks and the average Hb levels between weeks 18 and 24 were calculated for efficacy assessments. Serum iron, ferritin, transferrin, TSAT, and reticulocyte Hb content were assessed at prescreening, every 2 weeks from week 0 to week 4, and every 4 weeks from week 4 to week 24. Serum hepcidin was measured at weeks 0, 4, 12, and 24 using a liquid chromatography–tandem mass spectrometry method. A schedule of the assessments is shown in Table S3.
Statistical methods
A sample size of 50 patients was determined, considering the feasibility of the study, with the intention to yield sufficient exposure for regulatory purposes; no formal sample size or statistical power calculations were performed. The analysis of efficacy endpoints was performed on the full analysis set (FAS), which comprised patients who received at least one dose of roxadustat and for whom there were available data for at least one efficacy measurement after the start of the study treatment. The analysis of safety was conducted on the safety analysis set (SAF), which comprised patients who received at least one dose of roxadustat. Because the starting dose of roxadustat in the ESA‐Converted group was not randomized, but determined based on previous ESA dose, pooled data are reported for the ESA‐Converted group (all ESA‐Converted). Data are presented using summary statistics, and no inferential statistical analyses were conducted. No comparison between treatment groups was conducted.
RESULTS
Patient disposition
Of 69 PD patients screened, 13 discontinued due to screening failure (considered ineligible by the investigator [n = 5], did not meet Hb inclusion criteria [n = 4], others [n = 3], withdrawal by patient [n = 1]), 56 were randomized (ESA‐Naïve, n = 13) or registered (ESA‐Converted, n = 43), and 49 completed the study. All 56 patients were included in the SAF and FAS. In the ESA‐Naïve group, six patients were randomized to roxadustat 50 mg and seven were randomized to roxadustat 70 mg; all patients completed the study. In the ESA‐Converted group, 23 patients were registered to roxadustat 70 mg and 20 patients were registered to roxadustat 100 mg; seven patients discontinued the study (Fig. 1). Patients in the ESA‐Converted group (n = 43) were previously treated with and converted from recombinant human erythropoietin (n = 2; mean [SD] dose, 3500.00 [707.11] IU/week), darbepoetin alfa (n = 19; mean [SD] dose, 29.75 [24.10] μg/week), or epoetin beta pegol (n = 22; mean [SD] dose, 104.50 [61.27] μg/4 weeks).
Figure 1.
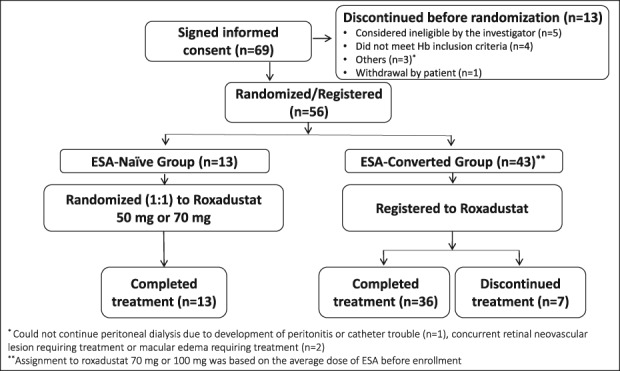
Disposition of patients. Abbreviations: ESA, erythropoiesis stimulating agent; Hb, hemoglobin.
Reasons for discontinuation in the ESA‐Converted group were TEAEs (n = 4), withdrawal of consent (n = 2), and need to start hemodialysis (n = 1). Mean (SD) baseline Hb levels were 9.35 g/dL (0.75) and 10.85 g/dL (0.54) in the ESA‐Naïve and ESA‐Converted groups, respectively. Patients' demographics and prior and concomitant iron use are reported in Tables 1 and 2, respectively. Of the 56 patients in the FAS, 36 (64.3%) were male and 20 (35.7%) were female. The mean (SD) body mass index was 23.10 (1.67) and 24.92 (4.09) kg/m2 in the ESA‐Naïve and ESA‐Converted groups, respectively. The mean (SD) levels of iron at baseline were similar in the ESA‐Naïve (102.8 [25.6] μg/dL) and ESA‐Converted (98.8 [44.3] μg/dL) groups. The proportion of patients who were fully iron replete (ferritin ≥100 ng/mL and TSAT ≥ 20%) was higher in the ESA‐Naïve (84.6%) than in the ESA‐Converted (44.2%) group. The mean (SD) duration of anemia associated with CKD, assessed by the investigator based on a patient report and/or a medical record, was 57.57 (35.86) months and 35.70 (31.03) months in the roxadustat 50 mg and roxadustat 70 mg ESA‐Naïve groups, respectively, and 48.90 (29.54) months in the ESA‐Converted group. Patients in the ESA‐Converted group had a higher rate of nephrosclerosis as the primary disease of CKD compared with those in the ESA‐Naïve group. The proportion of patients who had a ferritin level < 100 ng/mL was lower in the ESA‐Naïve group (TSAT < 20%, 0%; TSAT ≥ 20%, 15.4%) than in the ESA‐Converted group (TSAT < 20%, 2.3%; TSAT ≥ 20%, 44.2%).
Table 1.
Subject demographics (safety analysis set) and baseline efficacy variables (full analysis set)†
| ESA‐Naïve group | ESA‐Converted group | |||
|---|---|---|---|---|
| Parameter | Roxadustat 50 mg (n = 6) | Roxadustat 70 mg (n = 7) | All ESA‐Naïve (n = 13) | All ESA‐Converted (n = 43) |
| Subject demographics (safety analysis set) | ||||
| Sex, n (%) | ||||
| Male | 4 (66.7) | 6 (85.7) | 10 (76.9) | 26 (60.5) |
| Female | 2 (33.3) | 1 (14.3) | 3 (23.1) | 17 (39.5) |
| Age, years | ||||
| Mean (SD) | 64.5 (11.4) | 67.4 (11.2) | 66.1 (10.9) | 63.7 (10.1) |
| Median | 68.5 | 71.0 | 69.0 | 66.0 |
| Range | 43–73 | 53–83 | 43–83 | 39–87 |
| Age, n (%) | ||||
| <65 years | 2 (33.3) | 3 (42.9) | 5 (38.5) | 19 (44.2) |
| ≥65 years | 4 (66.7) | 4 (57.1) | 8 (61.5) | 24 (55.8) |
| Weight, kg | ||||
| Mean (SD) | 59.25 (8.68) | 63.76 (6.22) | 61.68 (7.50) | 64.52 (12.30) |
| Median | 56.05 | 61.20 | 61.10 | 62.70 |
| Range | 48.4–70.2 | 55.6–74.0 | 48.4–74.0 | 42.1–104.5 |
| Height, cm | ||||
| Mean (SD) | 163.30 (12.08) | 163.24 (5.47) | 163.27 (8.71) | 160.63 (7.68) |
| Median | 165.00 | 162.60 | 162.60 | 160.30 |
| Range | 148.2–180.3 | 157.6–174.0 | 148.2–180.3 | 145.6–175.2 |
| BMI, kg/m2 | ||||
| Mean (SD) | 22.18 (1.71) | 23.88 (1.24) | 23.10 (1.67) | 24.92 (4.09) |
| Median | 21.82 | 24.44 | 23.17 | 24.09 |
| Range | 19.6–24.2 | 22.4–25.5 | 19.6–25.5 | 19.9–42.9 |
| Duration of anemia in CKD‡, months | ||||
| n | 4 | 7 | 11 | 39 |
| Mean (SD) | 57.57 (35.86) | 35.70 (31.03) | 43.65 (32.94) | 48.90 (29.54) |
| Median | 64.76 | 26.15 | 32.30 | 45.01 |
| Range | 8.1–92.6 | 5.6–97.5 | 5.6–97.5 | 4.0–121.7 |
| Peritoneal dialysis vintage, months | ||||
| Mean (SD) | 62.01 (41.94) | 14.54 (13.67) | 36.45 (37.86) | 38.05 (38.14) |
| Median | 60.88 | 12.02 | 17.28 | 33.02 |
| Range | 9.1–131.7 | 1.5–42.5 | 1.5–131.7 | 3.0–189.8 |
| Primary disease of CKD, n (%) | ||||
| Chronic glomerular nephritis | 4 (66.7) | 4 (57.1) | 8 (61.5) | 11 (25.6) |
| Diabetic nephropathy | 2 (33.3) | 1 (14.3) | 3 (23.1) | 10 (23.3) |
| Chronic pyelonephritis | 0 | 0 | 0 | 0 |
| Polycystic kidney | 0 | 0 | 0 | 0 |
| Nephrosclerosis | 0 | 1 (14.3) | 1 (7.7) | 15 (34.9) |
| Other | 0 | 1 (14.3) | 1 (7.7) | 7 (16.3) |
| hsCRP, mg/L | ||||
| Mean (SD) | 1.8620 (2.7286) | 2.5354 (3.0493) | 2.2246 (2.8060) | 5.0287 (12.6600) |
| Median | 0.9860 | 1.4500 | 1.2900 | 0.8800 |
| Range | 0.134–7.310 | 0.180–9.060 | 0.134–9.060 | 0.050–63.100 |
| Baseline efficacy variables (full analysis set) | ||||
| Hemoglobin§, g/dL | ||||
| Mean (SD) | 9.57 (0.71) | 9.17 (0.78) | 9.35 (0.75) | 10.85 (0.54) |
| Median | 9.65 | 9.10 | 9.50 | 10.80 |
| Range | 8.3–10.5 | 7.7–10.0 | 7.7–10.5 | 9.9–11.9 |
| Iron, μg/dL | ||||
| Mean (SD) | 111.0 (30.8) | 95.9 (20.0) | 102.8 (25.6) | 98.8 (44.3) |
| Median | 104.0 | 92.0 | 97.0 | 92.0 |
| Range | 77–148 | 74–130 | 74–148 | 28–254 |
| Ferritin, ng/mL | ||||
| Mean (SD) | 215.40 (116.51) | 316.00 (131.13) | 269.57 (130.30) | 145.35 (111.20) |
| Median | 255.00 | 323.00 | 262.00 | 115.00 |
| Range | 51.4–340.0 | 122.0–501.0 | 51.4–501.0 | 22.1–500.0 |
| TSAT, % | ||||
| Mean (SD) | 49.45 (19.03) | 42.60 (9.55) | 45.76 (14.46) | 35.87 (14.17) |
| Median | 54.80 | 38.50 | 41.70 | 33.90 |
| Range | 25.9–69.0 | 34.1–56.3 | 25.9–69.0 | 17.0–93.7 |
| Iron repletion, n (%) | ||||
| Ferritin < 100 ng/mL and TSAT <20% | 0 | 0 | 0 | 1 (2.3) |
| Ferritin < 100 ng/mL and TSAT ≥20% | 2 (33.3) | 0 | 2 (15.4) | 19 (44.2) |
| Ferritin ≥ 100 ng/mL and TSAT <20% | 0 | 0 | 0 | 4 (9.3) |
| Ferritin ≥ 100 ng/mL and TSAT ≥20% | 4 (66.7) | 7 (100.0) | 11 (84.6) | 19 (44.2) |
| Reticulocyte Hb, pg | ||||
| Mean (SD) | 35.20 (1.37) | 36.61 (1.55) | 35.96 (1.59) | 34.02 (1.81) |
| Median | 35.50 | 35.60 | 35.60 | 34.10 |
| Range | 32.9–36.5 | 35.1–38.5 | 32.9–38.5 | 28.8–37.5 |
| Transferrin, g/L | ||||
| Mean (SD) | 1.828 (0.558) | 1.680 (0.240) | 1.748 (0.406) | 2.070 (0.420) |
| Median | 1.790 | 1.630 | 1.740 | 2.020 |
| Range | 0.91–2.43 | 1.41–1.99 | 0.91–2.43 | 1.16–3.27 |
Data are presented as n (%) unless otherwise noted.
Assessed by the investigator based on a patient report and/or a medical record.
Defined as the mean of three Hb values: two latest Hb values prior to registration and one Hb value at week 0 (when the Hb value at week 0 was the same value and on the same date as the latest Hb value prior to registration, baseline Hb was defined as the mean of the two latest Hb values prior to registration).
BMI, body mass index; CKD, chronic kidney disease; ESA, erythropoiesis stimulating agent; Hb, hemoglobin; hsCRP, high sensitivity C‐reactive protein; SD, standard deviation; TSAT, transferrin saturation.
Table 2.
Prior† and concomitant iron use (safety analysis set)
| ESA‐Naïve group | ESA‐Converted group | |||
|---|---|---|---|---|
| Parameter | Roxadustat 50 mg (n = 6) | Roxadustat 70 mg (n = 7) | All ESA‐Naïve (n = 13) | All ESA‐Converted (n = 43) |
| Prior oral iron use, n (%) | 1 (16.7) | 0 | 1 (7.7) | 8 (18.6) |
| Prior IV iron use, n (%) | 0 | 0 | 0 | 1 (2.3) |
| Concomitant oral iron use, n (%) | 2 (33.3) | 0 | 2 (15.4) | 11 (25.6) |
| Concomitant IV iron use, n (%) | 0 | 0 | 0 | 1 (2.3) |
Period between 42 days before prescreening to the end of screening.
ESA, erythropoiesis stimulating agent; IV, intravenous.
Roxadustat exposure and compliance
The overall roxadustat compliance was ≥95% in all treatment groups. In the ESA‐Naïve group, the mean (SD) dose of roxadustat at week 22 was 45.8 (22.7) mg (roxadustat 50 mg, 38.3 [14.7] mg; roxadustat 70 mg, 53.3 [28.0] mg). In the ESA‐Converted group, the mean (SD) dose of roxadustat at week 22 was 71.1 (36.0) mg. The mean number of times that the study drug dose was changed during the 24‐week treatment period ranged from 2.6 to 3.2 per patient across all study groups.
Efficacy outcomes
In the ESA‐Naïve group, mean Hb levels increased until week 8 and then remained stable within the target level through the end of the treatment; in the ESA‐Converted group, a slight increase was followed by stabilization within the target range (Fig. 2).
Figure 2.
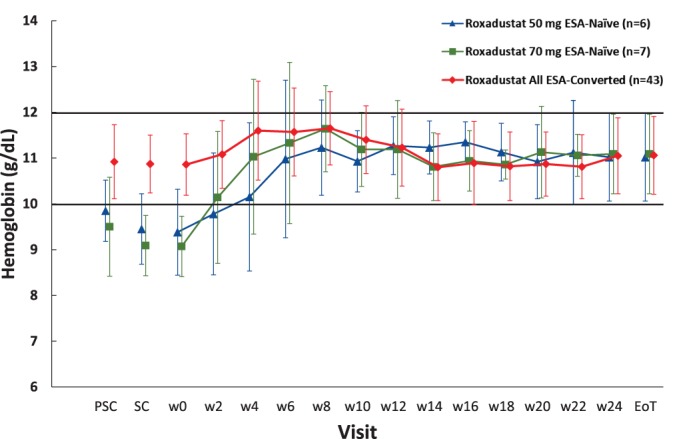
Mean (SD) hemoglobin concentrations (full analysis set). Abbreviations: EoT, end of treatment; ESA, erythropoiesis stimulating agent; PSC, prescreening; SC, screening; SD, standard deviation; w, week. [Color figure can beviewed at http://wileyonlinelibrary.com]
The maintenance rate of target Hb levels (proportion of patients who achieved an average Hb level of 10.0–12.0 g/dL between weeks 18 and 24) was 92.3% (95% CI: 64.0–99.8) in the ESA‐Naïve group and 74.4% (95% CI: 58.8─86.5) in the ESA‐Converted group. Of note, six patients in the ESA‐Converted group did not have a measurable Hb level during weeks 18–24, and therefore were not considered to have maintained the target Hb level. Among those patients who had at least one Hb measurement between weeks 18 and 24, the proportion who achieved an average Hb level within the target of 10.0–12.0 g/dL was 92.3% (95% CI: 64.0–99.8) in the ESA‐Naïve group and 86.5% (95% CI: 71.2–95.5) in the ESA‐Converted group (Table 3).
Table 3.
Maintenance rate of target Hb level† (full analysis set)
| ESA‐Naïve group | ESA‐Converted group | |||
|---|---|---|---|---|
| Parameter | Roxadustat 50 mg (n = 6) | Roxadustat 70 mg (n = 7) | All ESA‐Naïve (n = 13) | All ESA‐Converted (n = 43) |
| Maintenance rate for all patients in the FAS | ||||
| Maintenance rate, n (%) | 5/6 (83.3) | 7/7 (100.0) | 12/13 (92.3) | 32/43 (74.4) |
| 95% CI, % | 35.9–99.6 | 59.0–100.0 | 64.0–99.8 | 58.8–86.5 |
| Maintenance rate for those patients in the FAS who had at least one Hb measurement between weeks 18 and 24 | ||||
| Maintenance rate, n (%) | 5/6 (83.3) | 7/7 (100.0) | 12/13 (92.3) | 32/37 (86.5) |
| 95% CI, % | 35.9–99.6 | 59.0–100.0 | 64.0–99.8 | 71.2–95.5 |
Target Hb level, 10.0–12.0 g/dL.
CI, confidence interval; ESA, erythropoiesis stimulating agent; FAS, full analysis set; Hb, hemoglobin.
The cumulative response rate at both thresholds (i.e. Hb level ≥ 10.0 g/dL and ≥ 10.5 g/dL) was 100.0% in all patients in the ESA‐Naïve group. The average Hb levels between weeks 18 and 24 were 11.05 g/dL (95% CI: 10.67, 11.42) in the ESA‐Naïve group and 10.93 g/dL (95% CI: 10.73, 11.13) in the ESA‐Converted group. The mean change in average Hb levels from baseline to weeks 18–24 was 1.69 g/dL (95% CI: 1.06, 2.33) in the ESA‐Naïve group, and 0.14 g/dL (95% CI: −0.12, 0.39) in the ESA‐Converted group (Table 4).
Table 4.
Average Hb levels at weeks 18–24 and changes in Hb levels from baseline to weeks 18–24 (full analysis set)
| ESA‐Naïve group | ESA‐Converted group | |||
|---|---|---|---|---|
| Parameter | Roxadustat 50 mg (n = 6) | Roxadustat 70 mg (n = 7) | All ESA‐Naïve (n = 13) | All ESA‐Converted (n = 43) |
| n | 6 | 7 | 13 | 37 |
| Average Hb levels (g/dL) at weeks 18–24 | ||||
| Mean (SD) | 11.07 (0.81) | 11.03 (0.46) | 11.05 (0.62) | 10.93 (0.61) |
| Median | 11.30 | 11.10 | 11.20 | 10.90 |
| Range | 9.5–11.7 | 10.3–11.5 | 9.5–11.7 | 9.7–12.3 |
| Changes in average Hb levels (g/dL) from baseline to weeks 18–24 | ||||
| Mean (SD) | 1.50 (1.02) | 1.86 (1.13) | 1.69 (1.05) | 0.14 (0.76) |
| Median | 1.75 | 2.00 | 2.00 | 0 |
| Range | −0.2–2.7 | 0.3–3.5 | −0.2–3.5 | −1.1–1.9 |
ESA, erythropoiesis stimulating agent; Hb, hemoglobin; SD, standard deviation.
The mean (SD) rate of rise in Hb levels in the ESA‐Naïve group was 0.388 (0.368) g/dL/week (roxadustat 50 mg, 0.193 [0.203] g/dL/week; roxadustat 70 mg, 0.556 [0.408] g/dL/week). In the ESA‐Converted group, the rate of rise was 0.205 (0.268) g/dL/week (roxadustat 70 mg, 0.136 [0.217] g/dL/week; roxadustat 100 mg, 0.286 [0.302] g/dL/week). By the end of treatment, 92.3% of patients in the ESA‐Naïve group (roxadustat 50 mg, 83.3%; roxadustat 70 mg, 100.0%) achieved the lower limit of the target Hb level (10.0 g/dL); all patients achieved this target limit at least once by week 12.
Only one patient in the ESA‐Converted group received IV iron during the study. One (ESA‐Naïve) and six (ESA‐Converted) patients continued the prior use of oral iron during the study. The levels of iron, ferritin, TSAT, and reticulocyte Hb content remained clinically stable throughout the study. The mean transferrin levels increased through week 4 in all treatment groups and then remained stable until the end of treatment (Table 5 and Fig. S1). Mean serum hepcidin levels decreased through the end of treatment in all groups; the most pronounced decrease occurred at week 4 of treatment (Table 5 and Fig. S2).
Table 5.
Mean levels of iron, ferritin, transferrin, TSAT, reticulocyte hemoglobin, and hepcidin (full analysis set)†
| ESA‐Naïve group (n = 13) | ESA‐Converted group (n = 43) | |||||
|---|---|---|---|---|---|---|
| Parameter | Week 0 | EoT | Change from baseline to EoT | Week 0 | EoT | Change from baseline to EoT |
| Iron, μg/dL | 102.8 (25.6) | 85.6 (35.5) | −17.2 (35.7) | 98.8 (44.3) | 87.7 (26.5) | −11.1 (40.2) |
| Ferritin, ng/mL | 269.57 (130.30) | 159.12 (137.71) | −110.45 (80.94) | 145.35 (111.20) | 107.83 (82.76) | −37.52 (83.14) |
| Transferrin, g/L | 1.748 (0.406) | 2.204 (0.568) | 0.455 (0.330) | 2.070 (0.420) | 2.542 (0.548) | 0.472 (0.349) |
| TSAT, % | 45.76 (14.46) | 31.18 (13.29) | −14.58 (15.76) | 35.87 (14.17) | 27.78 (9.95) | −8.09 (14.10) |
| Reticulocyte Hb, pg | 35.96 (1.59) | 36.22 (2.00) | 0.25 (1.27) | 34.02 (1.81) | 35.30 (2.04) | 1.28 (1.81) |
| Hepcidin, ng/mL | 63.562 (31.253) | 31.780 (22.991) | −31.782 (39.416) | 45.864 (30.099) | 26.874 (20.345) | −18.990 (21.565) |
Data are presented as mean (SD).
EoT, end of treatment; Hb, hemoglobin; SD, standard deviation; TSAT, transferrin saturation.
Safety findings
Treatment‐emergent adverse events were reported in 10 (76.9%) patients in the ESA‐Naïve group and in 39 (90.7%) patients in the ESA‐Converted group (Table 6). Serious TEAEs were reported in three (23.1%) patients in the ESA‐Naïve group and five (11.6%) patients in the ESA‐Converted group. The most common serious TEAE was peritonitis, reported in two of 56 (3.6%) patients overall. Both cases of peritonitis were mild/moderate in severity and were resolved with antibiotic treatment. Both cases were considered by the investigator to be not related to roxadustat and neither resulted in discontinuation of roxadustat. Treatment‐emergent adverse events leading to withdrawal of treatment were reported in four (9.3%) patients in the ESA‐Converted group. One patient treated with a starting dose of 70 mg roxadustat was receiving concomitant pravastatin and experienced increased alanine aminotransferase, increased aspartate aminotransferase, and numbness in the forearms associated with increased lactate dehydrogenase and creatine kinase levels, which led to a diagnosis of rhabdomyolysis. The investigators believed that an interaction between pravastatin and roxadustat could have caused an increase in the blood level of pravastatin leading to rhabdomyolysis in this patient. However, since the patient continued to have high levels of creatine kinase and myoglobin after pravastatin was discontinued, treatment with roxadustat was discontinued even though a direct relationship between roxadustat and rhabdomyolysis was not established. This event was considered serious and the patient recovered from all events. The other TEAEs leading to withdrawal of treatment were palpitations and muscular weakness (n = 1), pruritus (n = 1), and dyspepsia (n = 1). The most commonly reported TEAEs were nasopharyngitis (25.0%), back pain (8.9%), catheter site infection (7.1%), diarrhea (7.1%), and vomiting (7.1%) (Table 6, Table S4). No clinically relevant changes were observed in clinical laboratory evaluations, hematology variables, 12‐lead ECG, or blood pressure, and no deaths occurred during the study. The mean (SD) total cholesterol in all patients (n = 56) was 4.774 (0.967) mmol/L at week 0, decreased to 3.783 (0.941) mmol/L at week 4, and then increased to 4.433 (1.091) mmol/L at week 12 and remained stable through the end of treatment.
Table 6.
Summary of treatment‐emergent adverse events (safety analysis set)†
| ESA‐Naïve group (n = 13) | ESA‐Converted group (n = 43) | Total (n = 56) | |
|---|---|---|---|
| Overall TEAEs | 10 (76.9) | 39 (90.7) | 49 (87.5) |
| Serious TEAEs | 3 (23.1) | 5 (11.6) | 8 (14.3) |
| Drug‐related serious TEAEs ‡ | 0 | 1 (2.3) | 1 (1.8) |
| TEAEs leading to withdrawal of treatment | 0 | 4 (9.3) | 4 (7.1) |
| TEAEs occurring in ≥5% of patients by MedDRA v19.0 system organ class and preferred term | |||
| Gastrointestinal disorders | 1 (7.7) | 18 (41.9) | 19 (33.9) |
| Diarrhea | 0 | 4 (9.3) | 4 (7.1) |
| Vomiting | 0 | 4 (9.3) | 4 (7.1) |
| Abdominal pain | 0 | 3 (7.0) | 3 (5.4) |
| Constipation | 0 | 3 (7.0) | 3 (5.4) |
| Nausea | 0 | 3 (7.0) | 3 (5.4) |
| Infections and infestations | 7 (53.8) | 21 (48.8) | 28 (50.0) |
| Nasopharyngitis | 0 | 14 (32.6) | 14 (25.0) |
| Catheter site infection | 0 | 4 (9.3) | 4 (7.1) |
| Conjunctivitis | 2 (15.4) | 1 (2.3) | 3 (5.4) |
| Musculoskeletal and connective tissue disorders | 3 (23.1) | 8 (18.6) | 11 (19.6) |
| Back pain | 3 (23.1) | 2 (4.7) | 5 (8.9) |
| Skin and subcutaneous tissue disorders | 3 (23.1) | 5 (11.6) | 8 (14.3) |
| Pruritus | 0 | 3 (7.0) | 3 (5.4) |
Data are presented as n (%).
Possible or probable as assessed by the investigator or records where relationship was missing.
ESA, erythropoiesis stimulating agent; MedDRA, Medical Dictionary for Regulatory Activities; TEAEs, treatment‐emergent adverse events.
DISCUSSION
Overall, the results of this study of 56 Japanese patients with PD‐dependent CKD are the first to indicate that self‐administration of oral roxadustat three times a week in ESRD patients who are on PD is feasible and associated with an acceptable safety profile. Target Hb levels (10.0–12.0 g/dL) were maintained in patients who were either not previously treated with ESA or who switched from ESA to roxadustat. The maintenance rate of a target Hb level (proportion of patients who achieved an average Hb level of 10.0─12.0 g/dL between weeks 18 and 24) among patients who had at least one Hb measurement between weeks 18 and 24 was 92.3% (12/13) and 86.5% (32/37) in the ESA‐Naïve and ESA‐Converted groups, respectively, and all patients in the ESA‐Naïve group achieved Hb ≥ 10.5 g/dL and an increase in Hb of ≥1.0 g/dL from baseline to the end of treatment. In the ESA‐Naïve group, the mean Hb levels increased during the first 8 weeks and then reached a plateau until the end of treatment, whereas, in the ESA‐Converted group, a slight increase was followed by stabilization within the target range. The mean average Hb level of weeks 18–24 and the mean change of the average Hb levels of weeks 18–24 from baseline were 11.05 g/dL (95% CI: 10.67, 11.42) and 1.69 g/dL (95% CI: 1.06, 2.33), respectively, in the ESA‐Naïve group; and 10.93 (95% CI: 10.73, 11.13) and 0.14 g/dL (95% CI: −0.12, 0.39), respectively, in the ESA‐Converted group, which shows that roxadustat increased the Hb levels to the target range in patients not treated with ESA, and maintained Hb in the target levels in patients who had been previously treated with or not treated with ESA.
In the ESA‐Converted group, the levels of iron, ferritin, TSAT, and reticulocyte Hb content remained clinically stable throughout the study, and only one patient (2.3%) was treated with IV iron despite robust erythropoiesis. This stability in iron stores in the absence of IV iron supplementation suggests an increase in iron bioavailability via enhanced intestinal iron absorption mediated by the observed reduction in hepcidin levels. Although generally stable, the mean levels of iron, TSAT, and ferritin decreased slightly between baseline and EoT in the ESA‐Naïve group. This may reflect the higher amount of iron required to support the high degree of erythropoiesis induced by roxadustat in the ESA‐Naïve compared with the ESA‐Converted group because of the lower baseline Hb levels in patients who were ESA‐Naïve. Hepcidin is a major negative regulator of intestinal iron absorption, 15 thus contributing to functional iron deficiency. A decrease in hepcidin level, which occurred in both groups and was most pronounced in ESA‐Naïve patients, may reflect the increased erythropoiesis and improved iron mobilization. Although this study was not designed to compare the doses of roxadustat required to maintain Hb levels within the target range between patients in the ESA‐Naïve and ESA‐Converted groups, the mean (SD) roxadustat dose at week 22 was slightly higher in the ESA‐Converted (71.1 [36.0] mg) than in the ESA‐Naïve (45.8 [22.7] mg) group. This difference may be due, in part, to the higher proportion of iron replete patients observed in the ESA‐Naïve (84.6%) than in the ESA‐Converted (44.2) group. In accordance with the available safety data on roxadustat in CKD patients, roxadustat was generally safe and well tolerated in CKD patients on PD in this study, and the most commonly reported TEAEs were nasopharyngitis (25.0%) and back pain (8.9%). Serious TEAEs were reported in three patients in the ESA‐Naïve group and five patients in the ESA‐Converted group. No severe TEAEs, defined as those events that prevent patients from performing daily activities, or deaths occurred during the study.
This is the first study to describe the course of ESRD during treatment with roxadustat exclusively in patients on PD. Due to the small sample size, and concordant with this study design, no formal inferential analysis was conducted to determine statistical significance, which limits the interpretation of the results. Other study limitations include the open‐label study design and the absence of an untreated control or a parallel ESA‐Converted arm. The efficacy results of this study are consistent with previous findings that showed that roxadustat is effective in improving anemia in both non‐dialysis‐dependent 11, 13, 14 CKD and hemodialysis‐dependent 10, 11, 12 ESRD patients, including a recent phase 3 study in Chinese patients with CKD who were on hemodialysis or PD (manuscript submitted). This Chinese study reported that roxadustat increased Hb levels in patients on hemodialysis or PD by 0.75 g/dL over 23–27 weeks of treatment; this increase was noninferior to the increase of 0.46 g/dL observed in patients treated with epoetin alfa. Furthermore, in line with previous studies of roxadustat, 10, 11, 12, 13 a reduction in the levels of serum hepcidin from baseline to the end of treatment was observed in all treatment groups of the current study, suggesting an improvement in iron mobilization and absorption. This is also supported by the increase in transferrin observed in all treatment groups, which may indicate an increased capacity to deliver iron to the developing red blood cells.
Patients on PD are underrepresented in clinical trials of treatment for anemia in CKD, 16 which yields limited information regarding anemia management in this population. This is one of the few clinical studies of the treatment of anemia in CKD conducted exclusively in patients on PD, and is the first report of the use of roxadustat exclusively in this population.
CONCLUSION
Overall, this study shows that treatment with oral roxadustat three times a week increased and maintained Hb levels within the target range of 10.0–12.0 g/dL at all tested doses in Japanese peritoneal dialysis patients who had not received previous treatment with ESA. Similarly, roxadustat maintained the Hb levels within the target range in chronic kidney disease patients with anemia and on PD who were switched from erythropoiesis stimulating agent treatment to roxadustat. Since this study provides valuable information regarding roxadustat treatment in patients with PD, it warrants further investigation in larger populations of patients to investigate whether treatment with roxadustat in PD patients is an effective strategy for increasing Hb levels with an acceptable safety profile.
Conflict of Interest
Tadao Akizawa reports personal fees from Astellas, Bayer Yakuhin Ltd., GlaxoSmithKline, JT Pharmaceuticals, Kissei Pharmaceutical, Kyowa Hakko Kirin, and Chugai Pharmaceutical during the conduct of the study and reports personal fees from Ono Pharmaceutical, Fuso Pharmaceutical Industries, Nipro Corporation, and Torii Pharmaceutical outside of the submitted work. Tetsuro Otsuka and Mai Ueno are employees of Astellas Pharma and both own Astellas stock. Michael Reusch is an employee of Astellas Pharma Europe B.V.
Data Sharing Statement
Access to anonymized individual participant level data collected during the study, in addition to supporting clinical documentation, is planned for studies conducted with approved product indications and formulations, as well as compounds terminated during development. Studies conducted with product indications or formulations that remain active in development are assessed after study completion to determine if individual participant data can be shared. Conditions and exceptions are described under the sponsor‐specific details for Astellas on http://www.clinicalstudydatarequest.com. Study‐related supporting documentation is redacted and provided if available, such as the protocol and amendments, statistical analysis plan, and clinical study report. Access to participant level data is offered to researchers after publication of the primary manuscript (if applicable) and is available as long as Astellas has legal authority to provide the data. Researchers must submit a proposal to conduct a scientifically relevant analysis of the study data. The research proposal is reviewed by an Independent Research Panel. If the proposal is approved, access to the study data is provided in a secure data sharing environment after receipt of a signed Data Sharing Agreement.
Supporting information
FIG. S1. Mean (SD) concentrations of (a) iron, (b) ferritin, (c) TSAT, (d) reticulocyte Hb content, and (e) transferrin (full analysis set).
FIG. S2. Mean (SD) hepcidin concentrations (full analysis set).
TABLE S1. Dose conversion between average doses of ESA before study registration and roxadustat
TABLE S2. Dose‐adjusting criteria
TABLE S3. Schedule of assessments during the treatment period
TABLE S4. Summary of treatment‐emergent adverse events (safety analysis set)
Acknowledgments
Medical writing and editorial support were provided by OPEN Health Medical Communications and was funded by Astellas Pharma, Inc. Roxadustat is being developed by FibroGen, AstraZeneca, and Astellas. The authors would like to thank all principal investigators and staff from each participating clinical site: Hidetoshi Kanai (Kokura Memorial Hospital), Hirotake Kasuga (Kaikoukai Healthcare Corporation Nagoya Kyoritsu Hospital), Katsuhiko Tamura (Shinonoi General Hospital), Tomohiko Naruse (Kasugai Municipal Hospital), Dai Matsuo (Munakata Medical Association Hospital), Masahiro Takeda (Komatsu Municipal Hospital), Jun Minakuchi (Kawashima Hospital), Hidekazu Moriya (Shonan Kamakura General Hospital), Yuko Fujiwara (Inoue Hospital), Satoshi Suzuki (Kainan Hospital), Satoshi Ota (Toyama City Hospital), Hironori Kobayashi (Japanese Red Cross Asahikawa Hospital), Masayoshi Yamaha (Daiyukai Health System Daiyukai Dai‐ichi Hospital), Fumio Obara (Hakodate Goryoukaku Hospital), Makoto Hiramatsu (Okayama Saiseikai General Hospital Outpatient Center, present affiliation: Okayama Saiseikai Outpatient Center Hospital). This study was funded by Astellas Pharma, Inc. The funder of the study had a critical role in the study design, and was responsible for the collection, analysis and interpretation of data, writing of the report, and the decision to submit the paper for publication.
REFERENCES
- 1. Locatelli F, Fishbane S, Block GA, Macdougall IC. Targeting hypoxia‐inducible factors for the treatment of anemia in chronic kidney disease patients. Am J Nephrol 2017;45:187–99. [DOI] [PubMed] [Google Scholar]
- 2. Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol 2012;23:1631–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stauffer ME, Fan T. Prevalence of anemia in chronic kidney disease in the United States. PLoS One 2014;9:e84943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krediet RT, Abrahams AC, de Fijter CWH et al. The truth on current peritoneal dialysis: state of the art. Neth J Med 2017;75:179–89. [PubMed] [Google Scholar]
- 5. Li PK, Chow KM, Van de Luijtgaarden MW et al. Changes in the worldwide epidemiology of peritoneal dialysis. Nat Rev Nephrol 2017;13:90–103. [DOI] [PubMed] [Google Scholar]
- 6. Li PK, Chow KM. Peritoneal dialysis‐first policy made successful: perspectives and actions. Am J Kidney Dis 2013;62:993–1005. [DOI] [PubMed] [Google Scholar]
- 7. Tsubakihara Y, Nishi S, Akiba T et al. 2008 Japanese Society for Dialysis Therapy: guidelines for renal anemia in chronic kidney disease. Ther Apher Dial 2010;14:240–75. [DOI] [PubMed] [Google Scholar]
- 8. Akizawa T, Okumura H, Alexandre AF, Fukushima A, Kiyabu G, Dorey J. Burden of anemia in chronic kidney disease patients in Japan: a literature review. Ther Apher Dial 2018;22:444–56. [DOI] [PubMed] [Google Scholar]
- 9. Del Vecchio L, Locatelli F. An overview on safety issues related to erythropoiesis‐stimulating agents for the treatment of anaemia in patients with chronic kidney disease. Expert Opin Drug Saf 2016;15:1021–30. [DOI] [PubMed] [Google Scholar]
- 10. Besarab A, Chernyavskaya E, Motylev I et al. Roxadustat (FG‐4592): correction of anemia in incident dialysis patients. J Am Soc Nephrol 2016;27:1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen N, Qian J, Chen J et al. Phase 2 studies of oral hypoxia‐inducible factor prolyl hydroxylase inhibitor FG‐4592 for treatment of anemia in China. Nephrol Dial Transplant 2017;32:1373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Provenzano R, Besarab A, Wright S et al. Roxadustat (FG‐4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: a phase 2, randomized, 6‐ to 19‐week, open‐label, active‐comparator, dose‐ranging, safety and exploratory efficacy study. Am J Kidney Dis 2016;67:912–24. [DOI] [PubMed] [Google Scholar]
- 13. Besarab A, Provenzano R, Hertel J et al. Randomized placebo‐controlled dose‐ranging and pharmacodynamics study of roxadustat (FG‐4592) to treat anemia in nondialysis‐dependent chronic kidney disease (NDD‐CKD) patients. Nephrol Dial Transplant 2015;30:1665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Provenzano R, Besarab A, Sun CH et al. Oral hypoxia‐inducible factor prolyl hydroxylase inhibitor roxadustat (FG‐4592) for the treatment of anemia in patients with CKD. Clin J Am Soc Nephrol 2016;11:982–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003;102:783–8. [DOI] [PubMed] [Google Scholar]
- 16. Del Vecchio L, Cavalli A, Locatelli F. Anemia management in patients on peritoneal dialysis. Contrib Nephrol 2012;178:89–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIG. S1. Mean (SD) concentrations of (a) iron, (b) ferritin, (c) TSAT, (d) reticulocyte Hb content, and (e) transferrin (full analysis set).
FIG. S2. Mean (SD) hepcidin concentrations (full analysis set).
TABLE S1. Dose conversion between average doses of ESA before study registration and roxadustat
TABLE S2. Dose‐adjusting criteria
TABLE S3. Schedule of assessments during the treatment period
TABLE S4. Summary of treatment‐emergent adverse events (safety analysis set)