

Abstract
Aberrant expression of the transcription factor double homeobox protein 4 (DUX4) can lead to a number of diseases including facio‐scapulo‐humeral muscular dystrophy (FSHD), acute lymphoblastic leukemia, and sarcomas. Inhibition of DUX4 may represent a therapeutic strategy for these diseases. By applying Systematic Evolution of Ligands by EXponential Enrichment (SELEX), we identified aptamers against DUX4 with specific secondary structural elements conveying high affinity to DUX4 as assessed by fluorescence resonance energy transfer and fluorescence polarization techniques. Sequences analysis of these aptamers revealed the presence of two consensus DUX4 motifs in a reverse complementary fashion forming hairpins interspersed with bulge loops at distinct positions that enlarged the binding surface with the DUX4 protein, as determined by crystal structure analysis. We demonstrate that insertion of specific structural elements into transcription factor binding oligonucleotides can enhance specificity and affinity.
Keywords: bulge loop, decoy, PAX7, PROP1, transcription factor
Abbreviations
- ßME
ß‐mercaptoethanol
- DNMT3b
DNA methyltransferase 3B
- DTT
dithiothreitol
- DUX4
double homeobox protein 4
- FSHD
facio‐scapulo‐humeral muscular dystrophy
- GST
glutathione S‐transferase
- HD
homeodomain
- His6
hexahistidine‐tag
- IPTG
isopropyl‐ß‐D‐thiogalactoside
- NGS
next generation sequencing
- PAX7
Paired box protein Pax7
- PEG
polyethylene glycol
- PROP1
Prophet of pit‐1
- ProT
propeller twist
- qPCR
quantitative PCR
- SB1
selection buffer
- SELEX
Systematic evolution of ligands by exponential enrichment
- SMCHD1
structural maintenance of chromosomes flexible hinge domain containing 1
- STAT3
Signal Transducers and Activators of Transcription
- TR‐FRET
time‐resolved fluorescence energy transfer
1. INTRODUCTION
Facio‐scapulo‐humeral muscular dystrophy (FSHD) is the third most common muscular dystrophy with an estimated prevalence of 12 in 100 000.1 The disease is characterized by a progressive weakness of selected muscle groups (facial muscles, periscapular muscles, abdominal wall muscles, and tibialis anterior).2 FSHD is inherited in an autosomal dominant manner.3 In the majority of cases, the disease is linked to contractions of the D4Z4 repeat array in the 4q35 subtelomeric region of chromosome 4. People with fewer than 11 of these repeats are at risk of developing FSHD1,3, 4, 5, 6 if, in addition, the D4Z4 contractions are located on a permissive chromosome containing a single nucleotide polymorphism creating a functional polyadenylation signal, leading to the expression of double homeobox protein 4 (DUX4),6, 7 which in turn is deleterious for skeletal muscle cells.4, 5, 6, 8, 9, 10, 11, 12, 13, 14 In rare cases, a digenic inheritance of a mutation in the structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) gene or in the DNA methyltransferase 3B (DNMT3b) gene and a permissive polyadenylation signal for DUX4 causes FSHD2, a genetic variant which is clinically indistinguishable from FSHD1.15, 16
Double homeobox protein 4 is a double homeodomain (HD) transcription factor present only in placental mammals.17 DUX4 contains two homeoboxes, located at the N‐terminus of the protein, which are responsible for binding to the DNA.18, 19, 20 The C‐terminal domain is responsible for target gene activation and contributes to the cellular toxicity of DUX4 by interacting with histone acetyl transferase p300 and transcriptional coactivator CBP.18, 21, 22, 23, 24, 25 DUX4 is benignly expressed in testis, thymus, and early germ line cells.9, 26, 27, 28 In early development, DUX4 acts as a pioneer transcription factor and activates many genes associated with the major wave of zygotic genome activation as well as activation of endogenous retroviral elements.27, 29, 30, 31
In addition to FSHD, DUX4 variants have been shown to be also pathogenic in other diseases. Translocations involving DUX4 causes subtypes of acute lymphoblastic leukemia,32, 33, 34, 35, 36 Ewing‐like sarcoma,37, 38, 39 and rhabdomyosarcoma.40 DUX4 might be involved in other cancer types as well.41
Currently there is no curative treatment available for FSHD.42 Therapeutic strategies under development include small molecular compounds,43, 44, 45 the use of RNAi gene therapy, antisense oligonucleotides, or CRISPR‐dCas9‐based methods.46, 47, 48 We aimed to directly target the DUX4 protein using nucleic acid aptamers.
Aptamers are short oligomers consisting of either amino acids, DNA, or RNA, which are designed to bind to a variety of different biomolecules or whole cells.49, 50, 51, 52 The utility of aptamers has previously been demonstrated in a number of applications such as in vivo biosensors, biomarker discovery, clinical drug discovery, and diagnostics (reviewed in 53, 54). The first FDA‐approved nucleotide aptamer based drug (Pegaptanib) was used for the treatment of neovascular age related macular degeneration.53, 56, 57 Meanwhile, several aptamer candidates have found their way into clinical trials. These include treatments of colorectal cancer, type 2 diabetes mellitus, and blood clotting diseases.53, 58, 59, 60 Aptamers are selected from a library of random oligonucleotide sequences in a process named Systematic Evolution of Ligands by EXponential Enrichment (SELEX).50
The SELEX‐originated aptamers against DUX4, which we identified in this work had pronounced conserved secondary structures. The importance of a sequence‐to‐structure synergy was shown by an optimized DNA aptamer variant which had a nanomolar affinity toward DUX4. We discuss how these structural elements, in particular, bulge loops could be applied to other transcription factor targeted oligonucleotides. Also the development of treatment strategies against FSHD and other DUX4‐mediated diseases might benefit from this study.61
2. MATERIALS AND METHODS
2.1. SELEX materials
A randomized library consisting of the sequence: 5′‐ATC CAG AGT GAC GCA (N45) TGG ACA CGG TGG CTT AGT‐3′, and corresponding primers containing the sequences: 5′‐biotin‐ACT AAG CCA CCG TGT CCA‐3′ and 5′‐ROX‐ATC CAG AGT GAC GCA GCA‐3′ as well as their equivalent untagged primers were purchased from Integrated DNA Technologies (IDT, Coralville, US‐IA).
For the negative selection and partitioning step, Ni‐NTA agarose beads were purchased from Qiagen (Hildesheim, Germany). The selection buffer (SB1) contained 50 mM of Tris‐HCl pH 7.5, 150 mM of NaCl, 5 mM of MgCl2, 0.1% of BSA, and 0.1% of Triton X‐100. Glutathione S‐Transferase (GST) was purchased from Sigma Aldrich (St. Louis, US‐MO) and dissolved in SB1. For the quantitative PCR (qPCR) and melting curve analysis, a 5x Hot Firepol Evagreen master mix plus (ROX) was used (Solis BioDyn, Tartu, Estonia). qPCR experiments were performed on a StepOnePlus qPCR thermocycler (Applied Biosystems, Foster City, US‐CA). For the strand separation step, Nanolink streptavidin magnetic beads were purchased from Solulink (San Diego, US‐CA). The beads were resuspended in strand separation buffer containing 50 mM of Tris‐HCl pH 7.5, 150 mM of NaCl, and 5 mM of MgCl2. Sanger sequencing and next generation sequencing (NGS) were performed by Microsynth (Balgach, Switzerland) on an Illumina HiSeq2000 sequencer. Fluorescence measurements were performed on a M1000 Infinite plate reader (Tecan, Männedorf, Switzerland). Fluorescein standard was purchased from Sigma Aldrich (St Louis, USA‐MO).
2.2. Recombinant full‐length DUX4 protein for SELEX
The coding sequence of DUX4 was amplified using human male genomic DNA (Promega, Madison, US‐WI) and the following primer: DUX4_for 5′‐GCT CGA ATT CAT GGC CCT CCC GAC ACC CTC‐3′ and DUX4_rev 5′‐ACC CCT CGA GCT AAA GCT CCT CCA GCA GAG CCC G‐3′. The PCR fragment was cloned into pGEX‐4T‐1 vector using the EcoRI and XhoI restriction sites. An Hexahistidine‐Tag (His6) was added by reamplification of the coding region using DUX4_for primer as described above and pGEX_DUX4_rev 5′‐ACC CCT CGA GCT AGT GGT GAT GGT GAT GAA GCT CCT CAA GCA GAG CCC G‐3′ primer. The PCR product was cloned into pGEX‐4T‐1 vector for the SELEX procedure. Using BL21‐RIPL cells, protein expression was induced with 0.1 mM of isopropyl‐ß‐D‐thiogalactoside (IPTG) at an OD600 = 0.8‐1, for 3‐4 hours at room temperature. Bacteria were pelleted at 12 000g for 10 minutes at 4°C. The supernatant was discarded and the pellet stored at −80°C. The pellet was thawed and lysed using lysis buffer (50 mM of Tris pH 8.0, 500 mM of NaCl, 1% of Triton X‐100, 1 mM of ß‐mercaptoethanol [ßME]) containing 1 mg/mL of lysozyme, 25 µg/mL of DNAse, Complete EDTA‐free protease inhibitor cocktail (Hoffmann‐La Roche, Basel, Switzerland), and 50 mM of imidazole. After lysis by sonication using six short ultrasound pulses (10 s at 30 W) the lysate was centrifuged at 4°C for 10 minutes at 12 000g and the supernatant, containing GST‐DUX4‐His6, was collected. GST‐DUX4‐His6 protein was bound to Ni‐NTA‐Agarose (Qiagen, Hildesheim, Germany) by incubation at 4°C for 1 hour. Beads were washed twice using lysis buffer containing 75 mM of imidazole and protein was eluted using lysis buffer containing 500 mM of imidazole. After binding of the eluate to glutathione sepharose 4B (GE Healthcare, #17‐0756‐01) for 1 hour at room temperature the beads were washed twice with wash buffer (50 mM of Tris‐HCl pH 8.0, 150 mM of NaCl, 1% of Triton X‐100, 1 mM of ßME) and eluted with wash buffer containing 40 mM of reduced glutathione. The eluate was dialyzed against wash buffer over night at 4°C. Protein concentration was determined with a NanoDrop spectrophotometer using a BCA assay (Sigma). The purity of GST‐DUX4‐His6 was evaluated by means of SDS‐PAGE. GST‐DUX4‐His6 aliquots were snap frozen in liquid nitrogen and stored at −80°C.
2.3. Recombinant full‐length DUX4 protein for fluorescence assays (StrepII‐SUMO‐DUX4‐His6)
The coding sequence of DUX4‐His6 was subcloned into pTS2 vector (a gift from Yu‐Zhu Zhang,62 Addgene plasmid #87798) using EcoRI and XhoI restriction sites, adding a StrepII‐SUMO tag to the N‐terminus of DUX4‐His6. Protein expression was induced with 0.1 mM of IPTG in BL21‐RIPL cells at an OD600 = 0.6‐0.8 over night at room temperature. Bacteria were pelleted and lysed as described above using lysis buffer containing 10 µg/mL of avidin from egg white (Sigma Aldrich) without sonication. StrepII‐SUMO tagged DUX4‐His6 protein was bound to Strep‐Tactin sepharose beads (IBA Lifescience, Göttingen, Germany, #2‐1201) in columns for gravity flow purification. Beads were washed five times using buffer containing 100 mM of Tris‐HCl pH 7.5 and 150 mM of NaCl. Protein was eluted with washing buffer containing 5 mM of desthiobiotin (Sigma Aldrich). The eluate was purified with Ni‐NTA sepharose beads (Qiagen, Hilden, Germany) packed in columns for gravity flow purification. Beads were washed three times using buffer containing 25 mM of imidazole, 50 mM of Tris‐HCl pH 8, 500 mM of NaCl, 1% of Triton X‐100, and two times using buffer without Triton X‐100. StrepII‐SUMO‐DUX4‐His6 was eluted using buffer containing 500 mM of imidazole, 50 mM of Tris‐HCl pH 8, 500 mM of NaCl, 10 mM of EDTA. The eluate was dialyzed twice against 3 L buffer containing 50 mM of Tris‐HCl pH 8, 150 mM of NaCl at 4°C. The dialyzed sample was concentrated 10x using Slide‐A‐Lyzer Dialysis Cassettes 10 000 MWCO (Thermo Scientific, Waltham, US‐MA).
2.4. Selection of aptamers (SELEX)
For the selection of aptamers, a competitive binding approach was utilized to select aptamers against DUX4 and remove nonspecific GST binding aptamers. A 50 µL of DNA library (100 µM) was refolded by heating at 94°C for 10 minutes and cooling at a rate of 0.5°C. A negative selection was performed whereby 50 µL of NTA‐beads were equilibrated in SB1 for 1 hour. The refolded random DNA library was mixed with 24 µg of GST in excess to a final volume of 200 µL in SB1 and incubated with intermittent shaking at room temperature for 1 hour. For the partitioning step, the GST‐DUX4‐His6 target transcription factor (50‐250 nM) was added to the solution containing the refolded random DNA library, GST, and the mixture was left to reach equilibrium for 1 hour with intermittent shaking. The solution was incubated with 50 µL of equilibrated NTA beads to capture the GST‐DUX4‐His6‐aptamer complexes for 1 hour with shaking. The beads were then washed with 1 to 7 times using 1 mL of SB1. Bound DNA sequences were then eluted by adding 100 µL of 7 M urea to the beads. The eluted DNA sequences were purified using a chloroform‐phenol extraction followed by ethanol precipitation.
The recovered bound DNA library was reconstituted into 50 µL nuclease free water. A 20 µL PCR reactions were prepared containing 4 µL of 5x Hot Firepol Evagreen master mix plus (ROX), the forward and reverse primer (250 nM), 1‐5 µL of the recovered bound DNA library.
The PCR mixtures were amplified using a two‐step cycle of denaturing step at 94°C for 15 s and an annealing step at 68°C for 1 minute. A preparative PCR step to monitor the extent of enrichment was completed first using two samples and a negative control with a total of 35 cycles performed. The PCR reactions were then scaled up (16‐32 samples) and the same cycling conditions used with the exception that the method was stopped at the point when the fluorescence signal appears to decrease. All samples including the negative controls were analyzed using remelting point analysis and consisted of a denaturing step at 94°C for 1 minute and, an initial annealing step at 55°C with a fluorescence signal measurement at every +0.5°C increment to 95°C. A 16 PCR reactions were pooled together and the ssDNA library was regenerated by strand separation. A 100 µL of magnetic streptavidin beads were incubated with the biotin tagged dsDNA PCR product for 1 hour with intermittent shaking. The forward strand was then separated by heating to 87°C for 3 minutes with shaking. The supernatant was retained by magnetic separation.
A total of 10 rounds was performed until an enriched library was obtained as determined from qPCR analysis and remelting analysis. The negative selection step was also incorporated into round 5 as described previously. The pools from the random library, rounds 3, 6, and 7 were amplified using the unlabeled primers and Vent polymerase (New England Biolabs, Ipswich, US‐MA) followed by gel purification. NGS was performed by Microsynth on an Illumina HiSeq2000 instrument.
2.5. Analyzing aptamer sequences
The raw data from round 7 of SELEX gained from the NGS was processed according to the workflow depicted in Figure 1A. In short, the paired and demultiplexed sequences were uploaded into the aptamer analyzing software collection AptaSUITE V0.9.463 (running on Java v1.8.0_92). After removing primer sequences by the software, the aptamers were clustered by implementing AptaCluster using the following parameters: randomized region size = 45, locality sensitive hashing dimension = 33, locality sensitive hashing iterations = 5, K‐mer size = 3, edit distance = 5, K‐mer cutoff iterations = 10 000. The top 350 list of aptamer clusters was selected according to cluster size of at least 10 reads. The aptamer families were analyzed for common motifs using MEME V5.0.5 with parameters for motifs occurring at any number of repetitions within the sequences, a gap opening penalty of 1, a gap extension penalty of 5, and an E‐value limit of <0.05.
Figure 1.
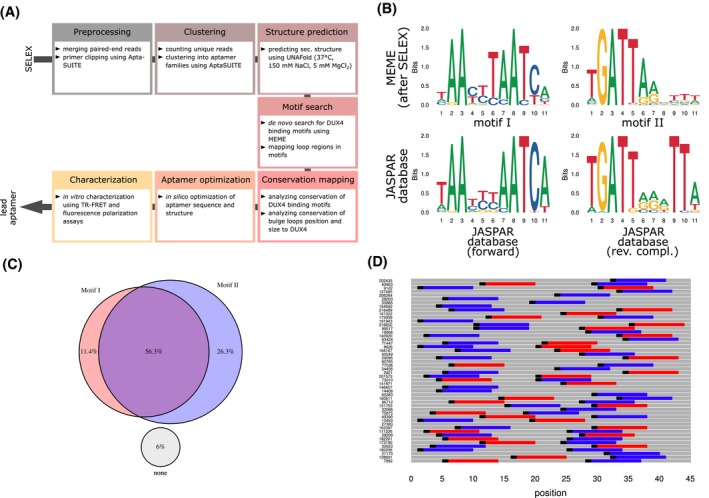
Characterizing results from the SELEX approach. A, Bioinformatical and experimental pipeline starting from SELEX NGS data to yield an optimized lead aptamer sequence. B, Sequence logos of the two motifs (top panel, motif I left and motif II right) found by MEME in SELEX NGS data. For comparison, the human DUX4 binding motif from JASPAR database (ID MA0468.1) was depicted (bottom panel, left) together with the reverse complementary logo (bottom panel, right). C, Venn diagram shows the mainly pairwise occurrence of the two SELEX motifs in percent across the total number of examined aptamer sequences. D, Visualization of the order (motif I in red, motif II in blue), orientation (motif beginning as black indicators), and distribution of motif I and II among the top 50 list of aptamers according to cluster family size
The sequences were subjected to DNA secondary structure prediction software UNAfold V3.8 adjusting to the following settings: 37°C, 150 mM of NaCl, and 5 mM of MgCl2.64 Bioinformatical analysis and visualizations were performed using R V3.5.2 together with several R packages. The reproducible R code is supplied as R Markdown document in the supplement material (Supplementary R codes).
2.6. Fluorescence polarization assays
Different concentrations of human recombinant StrepII‐SUMO‐DUX4‐His6 protein (as described above) was diluted in dialysis buffer and added to 15 nM of FAM labeled test aptamer in 20 µL total volume with reaction buffer containing 50 mM of Tris‐HCl pH 7.5, 150 mM of NaCl, 5 mM of MgCl2, 0.1% of Triton X‐100, 1 mM of dithiothreitol (DTT) in black 384 well microplates (Greiner Bio‐one, Kremsmünster, Austria). Fluorescence polarization was quantified on a TECAN Spark microplate reader (SparkControl software V2.3, Tecan Group Ltd., Männedorf, Switzerland) by optimizing the gain and Z position. G factor was calibrated using Fluorescein (Sigma, St. Louis, US‐MO). Measurements were performed in four independent technical replicates using different recombinant protein batches. To calculate the fraction of bound FAM‐labeled aptamer, a protocol described elsewhere was applied.65 The KD was determined from nonlinear four‐parameter regression analysis of the data using R package “drc” (V3.0‐1).66, 67
2.7. TR‐FRET competition assays
Aptamers were compared against 5′‐Atto647N labeled aptamers as probes with sequences described in in Supplemental Table T1. The labeled aptamers were incubated with 1 nM of either StrepII‐SUMO‐DUX4‐His6, 1 nM of PAX7‐His6 (Creative BioMart, Shirley US‐NA), or 15 nM of PROP1‐cMyc‐DDK(FLAG) (OriGene Technologies, Inc, Rockville, US‐MD) recombinant proteins and various concentrations of test aptamers (Microsynth, Balgach, Switzerland) in an assay buffer containing 50 mM of Tris‐HCl pH 7.5, 150 mM of NaCl, 5 mM of MgCl2, 0.1% of BSA, 0.1% of Triton X‐100, 1 mM of DTT, and 0.35 nM of MAb anti‐6His Terbium cryptate Gold antibody (cisbio, Codolet, France) in white 384 well microplates (Greiner Bio‐one, Kremsmünster, Austria) for 24 hours at 4°C. Donor fluorescence (340 nm/620 nm) and acceptor fluorescence (340 nm/665 nm) was quantified on a TECAN Spark microplate reader with a lag time of 100 µs and integration time of 300 µs at 37°C. Bioinformatical analysis was performed using the R package “drc”.66, 67
2.8. Crystal structure of DUX4 together with DNA aptamers
DUX4(15‐155) was purified as reported previously.20 Two types of DNA substrates were used; a hairpin DNA aptamer (5′‐GCT AAT CTA ATC AAC CGC AGG TTG ATT AGC CCA TTA GC‐3′) and a blunt‐ended counterpart (5′‐GCG TAA TCT AAT CAA CA‐3′ annealed to 5′‐TGT TGA TTA GCC CAT TAC GC‐3′). These oligonucleotides were obtained from IDT. Purified DUX4(15‐155) was mixed with ~1.5‐fold molar excess of each DNA substrate at an approximate protein concentration of 7 mg/mL and dialyzed overnight against 20 mM of Tris‐HCl pH 7.4, 0.1 M of NaCl, 1 mM tris(2‐carboxyethyl)phosphine at 4°C to assemble DUX4‐DNA complexes. Crystals of the DUX4‐DNA complexes were obtained by the sitting drop vapor diffusion method at 20°C, mixing 100 nL each of the sample and well solution to form the drops. The well solution consisted of 0.1 M imidazole‐MES buffer, pH 6.0, 12% of Ethylene glycol, 20% of Glycerol, and 10% of polyethylene glycol (PEG) 4000 for the complex with blunt‐ended DNA, and 0.2 M magnesium acetate, 20% PEG8000 and 0.1 M sodium cacodylate buffer pH 6.5 for the CCC‐bulge/hairpin‐loop aptamer complex. The crystals were cryoprotected by transferring to the cognate well solution supplemented by 25% of ethylene glycol and flash‐cooled in liquid nitrogen. X‐ray diffraction data were collected at the NE‐CAT beamline 24‐ID‐E of the Advanced Photon Source (Argonne National Laboratory, Lemont, IL) using the wavelength of 0.979 Å. Datasets were processed using XDS.68 Both crystal species suffered from severe anisotropy; the complex with the blunt‐ended DNA containing a CCC insertion diffracted to 3.1, 2.7, and 2.3 Å along the a*, b*, c* axis, respectively, and the CCC‐bulge/hairpin‐loop aptamer/DUX4 complex diffracted to 3.2, 4.0, and 3.6 Å. The datasets were subsequently subjected to an ellipsoidal truncation and anisotropic scaling.69 Molecular replacement calculation was done with PHASER70 using the crystal structure of DUX4(15‐155) bound to the canonical target DNA20 (PDB ID: 6E8C). Model building was performed using COOT71 and refinement using PHENIX.72 The data collection and model refinement statistics are summarized in Supplemental Table T2. Supplemental Figure S6 shows the two structures fitting with their corresponding 2mFo‐DFc map at 1σ contour level.
3. RESULTS
3.1. SELEX approach with recombinant DUX4
In order to identify aptamers binding to DUX4 with high affinity, we conducted a SELEX experiment using recombinant DUX4. The SELEX procedure was performed using GST‐ and His6‐tagged DUX4 and a library of single stranded oligodeoxynucleotides with randomized sequences in the center, flanked by constant primer regions on both sides. For the selection of aptamers against DUX4, we chose NTA‐beads in the partitioning step. Negative selection steps in rounds 0 and 5, where the library was incubated with the beads only, was used to remove nonspecific binders. GST protein was added in excess to the binding buffer to prevent the selection of GST tag specific aptamers. The stringency of selection was increased from round 5 by decreasing the concentration of GST‐DUX4‐His6. In total, 10 rounds of selection were performed which was monitored by qPCR and remelting analysis.
The DNA pools from round 3, 5, 6, and 7 were sequenced using NGS. The aptamer sequences were fed into AptaSUITE software for preprocessing. As an increasing number of rounds of selection were performed, a decrease in the number of unique sequences were observed. This shows successful enrichment of high‐affinity aptamers. The clustering of round 7 resulted in 350 cluster families containing more than 10 reads, while the most abundant cluster contained >11 000 reads.
3.2. SELEX approach selected for aptamers with bulge loops
The aptamer cluster families were subjected to further bioinformatical analysis (Figure 1A). MEME, an ungapped de novo motif discovery software, was used for this purpose. In the motif search, the well‐known DUX4 binding motif (Figure 1B) was detected as one motif.73, 74 A second motif was discovered and identified as the reverse complementary counter motif.
We compared the two motifs from the SELEX approach with the published DUX4 logos from JASPAR database and its corresponding reverse complementary logo.75 The main differences were observed in the forward motif at the position T6 (5′‐TAAYYTAATCA‐3′), which is only slightly conserved in the JASPAR logo. The reverse SELEX motif (motif II) showed a weak conservation after position 7 (5′‐TGATTARVTTW‐3′) compared to the JASPAR logo.
The two motifs occurred pairwise in almost 60% of the cluster families (Figure 1C,D). The tandem‐wise occurrence and the reverse complementary character of the two motifs led us to conclude that hairpin‐like DNA structures might be formed. The sequences were analyzed in more detail concerning their secondary structure using UNAFold software. The predicted structures of the first nine aptamers with the highest cluster size are shown in Figure 2A. The structure elements included bulge loops, hairpin loops, unpaired ends, interior loops, and stem parts (Figure 2B,C). A pronounced accumulation of hairpin loops at the center flanked by bulge loops and interior loops was observed. Paired stem part are scattered homogeneously along the aptamers except for the areas close to the ends. Here, mainly unpaired ends were present.
Figure 2.
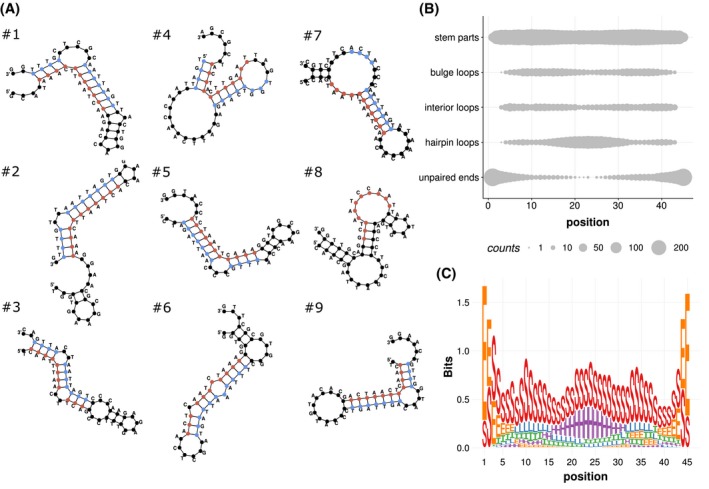
Predicting secondary structure of aptamers. A, Predicted secondary structures of the top nine list of aptamers (#1‐9) with ranking according to AptaSUITE cluster family size. Structures were predicted using UNAFold software. Manually mapped DUX4 motifs are highlighted in red (forward) and blue (reverse). B, The amount of structure elements bulge loops, hairpin loops, interior loops, stem parts, and unpaired ends were depicted by their occurrence on aptamer position 1 to 45 of the selected 350 aptamers. C, Visualization of the amount of structural elements as logo: E unpaired ends, H hairpin loops, I interior loops, L bulge loops, S stem parts
By manually mapping the DUX4 target motif onto the structures, we observed that the motifs occurred predominantly on base‐paired regions. We hypothesized that single stranded structures like bulge and internal loops within the motif may impede motif analysis by MEME software. To confirm this, the unpaired regions (bulge loops, interior loops, unpaired ends, and hairpin loops), according to UNAFold software results, were excluded from the aptamer sequences. Subsequently, a second MEME search was performed with a stricter search profile. Now, up to two motifs with a length of 11 bases were allowed, based on the first search result. From the multiple foldings per aptamer cluster, generated by UNAFold software, only the foldings with the lowest ΔG value were considered for analysis. With that prerequisite, motif and bulge loop conservation was summarized. The corresponding R script applied for analysis can be found in the supplemental material (Supplemental R codes).
The motif conservation changed after excluding unpaired regions from the motif search by MEME (Figure 3A). From the former 11 bases motif, only 10 bases remained conserved. With minor changes on the remaining motif, the conservation of the last two dT in 5′‐NTGATTARDTT‐3′ increased strikingly by the removal of unpaired regions. In addition, position 4 of the forward motif gained a higher conservation of pyrimidines (5′‐AAHYTAATCAN‐3′).
Figure 3.
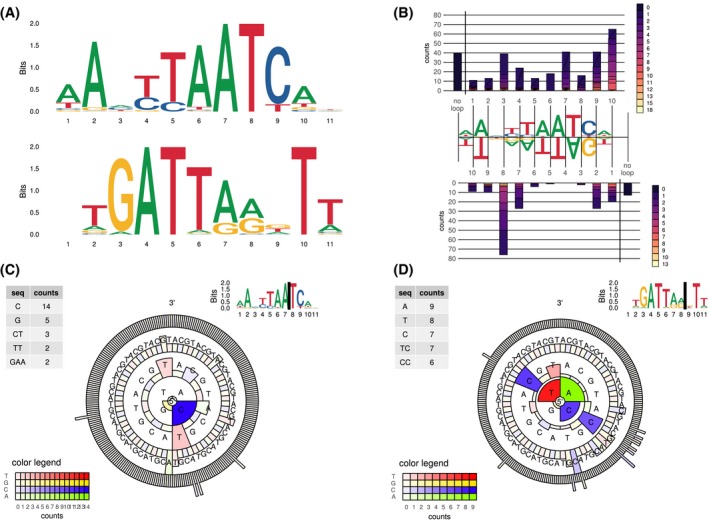
DUX4 preferentially binds to aptamers with bulge loops at certain positions. A, Sequence logos found by MEME after removing unpaired regions. MEME search parameters were adjusted to find two motifs of the size of 11 bases: forward DUX4 motif (top panel) and reverse DUX4 motif (bottom panel). B, Abundance and size of loop structures (bulge loops and interior loops) within the two motifs is depicted as counts. “No loop” bars represent aptamers in which the corresponding motif lacked unpaired structures. (C + D) Loop sequence abundance of only loops with up to four bases in size are displayed as circular plot. The sequences are read from the inside (5′‐end) to the outside (3′‐end). Bars indicate the last letter of the sequence. Examined loop position is located in the forward motif between base position 7 and 8 (C), and in the reverse motif between base 8 and 9 (D) (indicated as black bar in logos at top right corner). Counts are indicated as color hue according to the legend (bottom left corner). Circles indicate size category of the loop with lowest circle single base loops and outmost circle as four base loop. Bar size indicates relative abundance across the corresponding loop size category with highest bars represent most abundant sequence of that loop size. List of top five sequences (Seq) irrespective of size category found at the loop location is depicted as table at the right top corner
We asked whether secondary structures like bulge loops were enriched at certain positions of the DUX4 motif. The data showed that the bulge loop distribution within the forward and reverse motifs differed strikingly (Figure 3B). The forward motif allowed the occurrence of predominantly single nucleotide loops. Nevertheless, a large portion of aptamers did not contain loops in this motif. The reverse complementary motif had a distinct preference for loops between position 8 and 9 (5′‐NTGATTAR[loop]DTT‐3′). The size of the loops is mostly from one to four nucleotides. In contrast to the forward motif, only a minority of aptamers are lacking loops in the reverse motif, indicating the importance of the loops especially in the reverse DUX4 motif.
Concerning the sequence of the bulges, from the aptamers containing the reverse motif, the single base loops between position 8 and 9 lacked guanosines (Figure 3D). Larger loops consisted mainly of pyrimidines. In the less conserved loop in the forward motif between position 7 and 8 (5′‐AAHYTAA[loop]TCAN‐3′) also mainly pyrimidines were found (Figure 3C).
The top nine list of SELEX aptamers, according to the cluster size calculated by AptaSUITE, has been tested in time‐resolved fluorescence energy transfer (TR‐FRET) assays to confirm their ability to bind to DUX4. SELEX aptamers #1, #2, and #5 had the lowest IC50 in competition assays against a template aptamer as probe (Figure 4).
Figure 4.
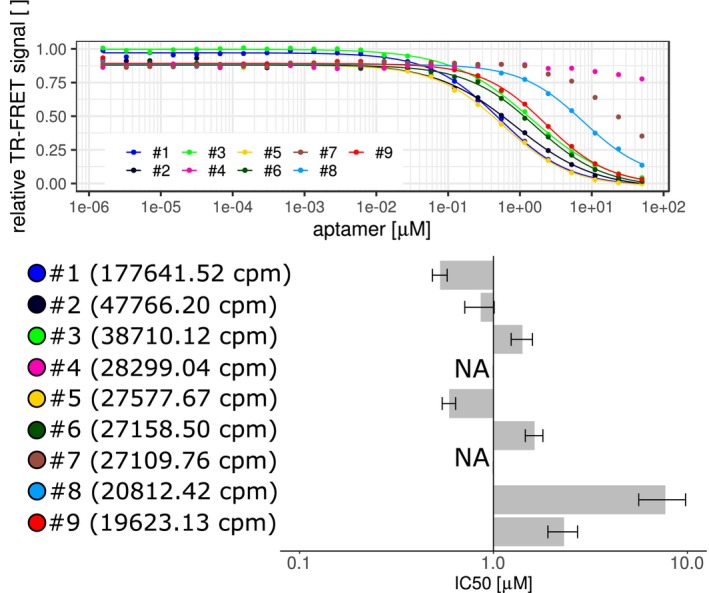
Testing the binding of SELEX‐originated aptamers. Comparing top nine list of SELEX generated aptamers in TR‐FRET assays. Aptamers were compared by competing against the 5′‐end Atto647N‐labeled template aptamer after incubation with StrepII‐SUMO‐DUX4‐His6. The fitted curves are displayed together with the IC50 values in micromolar from single experiments with ±95% confidence interval (bar plot). The SELEX aptamers were sorted according to their cluster size in counts‐per‐million (cpm) based on AptaSUITE software preprocessing steps (values in brackets). NA are not‐fittable data points indicating IC50 values higher than 10 µM
In summary, the SELEX procedure selected for aptamers that contained two motifs that potentially base‐pair each other in a hairpin structure. The double stranded motif confirmed the well documented DUX4 target motif. Furthermore, the aptamers included bulge loops at distinct positions. Loops between position 8 and 9 of the reverse complementary DUX4 motif were most conserved and consisted predominantly of pyrimidines.
3.3. Bulge loop conservation for the optimization of aptamer backbone
In order to confirm the findings from the SELEX approach and to obtain a putative high‐affinity aptamer sequence by applying the findings of the SELEX, a new set of DNA aptamers was tested (see Supplemental Table T1 for sequences). TR‐FRET competition assays were performed to compare the affinity of different aptamers. The labeled probe included the consensus DUX4 binding motif74 in a hairpin backbone.76
The optimal bulge loop position was examined. With a loop containing two unpaired deoxycytidines, every position in the 11 base pair long forward and reverse JASPAR DUX4 binding motifs was tested (Figure 5A,B). We observed that in the forward motif, positions 2‐6 were not allowing any bulges from the given size and sequence without substantial loss of affinity. Bulge loops were only tolerated at position 8. However, the introduction of bulge loops in that position did not further increase the affinity in comparison to the backbone aptamer without loops. In the reverse motif, only loops at position 7 were allowed and led to a pronounced decrease in the IC50 compared to the backbone aptamer without loops (Figure 5B).
Figure 5.
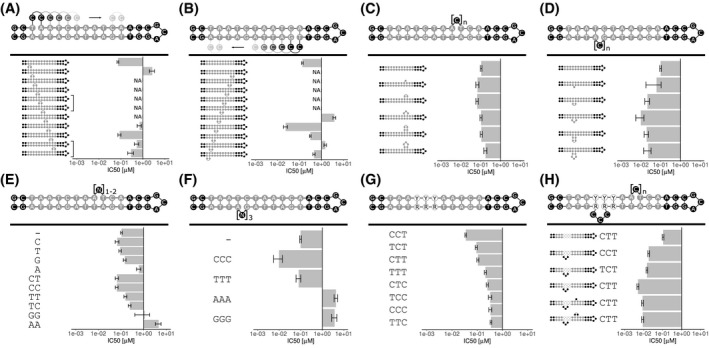
Comparing aptamer variants as part of an optimization process using TR‐FRET assays. Different rationally designed aptamers were compared against a labeled template aptamer. IC50 values ±95% confidence interval gained by four‐parameter logistic curve fitting of different data points are displayed (curves displayed in Supplemental Figures S1 and S2). NA: IC50 not measurable because data points indicated an IC50 > 10 µM. A, The position of a double‐dC loop was varied on the forward DUX4 motif. Brackets indicate identical aptamers. B, The position of a double‐dC loop was screened on the reverse DUX4 target motif. C, Loop size of a dC loop was varied from a single base to 5x dC on the forward motif. D, Loop size of a dC loop was varied from a single base to 5x dC on the reverse motif. E, Selected sequences of single base and double base loops were tested on the forward motif between A8 and T9. F, Base composition of a triple base loop between G7 and A8 of the reverse motif was examined. G, The triplet sequence after A3 of the forward motif was varied by permuting dC and dT. The reverse motif was altered correspondingly to retain base pairing. H, Bulge occurrence at two positions of the DUX4 motif in combination with three selected base triplets after A3 on the forward motif were tested
Regarding the optimal size of the bulge loops, position 8 in the forward or position 7 in the reverse motifs were tested with bulge loops from one to up to five unpaired deoxycytidines in size. In the forward motif, one to four bases large loops were tolerated (Figure 5C). In contrast to that, for the reverse motif, every loop size led to a lower IC50 value compared to the template aptamer without loops, with an optimal length of three deoxycytidines leading to a 10‐fold increase in affinity (Figure 5D).
Variations in the bulge loop sequence were analyzed. Deoxycytidines and deoxythymidines were superior to deoxyadenosines and deoxyguanosines in both motifs (Figure 5E,F). The forward motif benefited from both dC, dG, and dT loops of size of one nucleotide (Figure 5E). The dinucleotide loop dCdC as well as the mixed variant dCdT were tolerated. A sequence consisting of dTdC and other permutations decreased the affinity. In the reverse motif, a dT triplet was tolerated but triple‐dC were more advantageous concerning affinity (Figure 5F). Triple‐dG and triple‐dA increased the IC50 values.
The second triplet of the DUX4 motif (5′‐TAATTTAATCA‐3′) is not well conserved.26, 74 Because, we found a conservation of pyrimidines in the middle position of the triplet in the SELEX aptamers as well as a pronounced conservation of T in the last position (5′‐AAHYTAATCAN‐3′), we wanted to test if this triplet had an impact on the affinity. First, we found––by varying the sequence by permuting pyrimidines and the corresponding purines in the reverse motif––that there was a sequence preference toward CCT (Figure 5G). CTT and TCT were comparable in this regard and displayed the next best sequence variants with roughly 3‐fold difference to CCT in their IC50 values.
The synergy of bulges at position 8 in the forward, at position 7 in the reverse motif, and sequence variations in the second triplet of the motif were assessed (Figure 5H). Starting from the backbone aptamer, the introduction of an unpaired base triplet at position 7 of the reverse motif gave a substantial affinity boost of 20‐fold, but only in combination with a CTT mid‐section triplet. The same loop with a TCT or CCT triplet gave rise to aptamers with lower affinity. The addition of a second bulge of one or two deoxycytidines in the forward motif at position 8 decreased the affinity independently of the size of the loop slightly by almost 2‐fold.
In summary, the introduction of bulge loops exclusively in the reverse motif at a distinct position is advantageous and was in accordance to the findings of the SELEX result. Small loops in the forward DUX4 motif were tolerated but decreased the affinity benefit of loops in the reverse motif slightly. Also the non‐conserved mid‐section triplet had a strong impact on the affinity boost of the loops in the reverse motif. In total, the mid‐section triplet and bulge loop can influence each other mutually.
3.4. Sequence variants mediated changes in the affinity are proportional to DNA shaping parameter alterations
In order to explain the higher affinity of aptamers with certain mid‐section triplets, DNA shaping was examined. The average intra‐base pair (shear, stretch, stagger, buckle, propeller twist [ProT], opening), inter‐base pair parameters (shift, slide, rise, tilt, roll, helix twist), and minor groove width were predicted per nucleotide position of selected aptamers using the R package DNAshapeR.77 The shape values per position and aptamer were correlated against the IC50 values of the corresponding aptamers (Supplemental Figure S5). The aptamer selection was taken from the triplet permutation experiment in Figure 5F. The parameter ProT at aptamer position 9, as well as base tilting between positions 7 and 8 were proportional to the IC50 values of the corresponding aptamers (Figure 6A,B). The changes in the predicted shape parameters between the CCT‐ and the TTC‐triplet containing aptamers are 3.95° for inter‐base pair tilt between positions 7 and 8 and 0.66° for intra‐base pair ProT at position 9.
Figure 6.
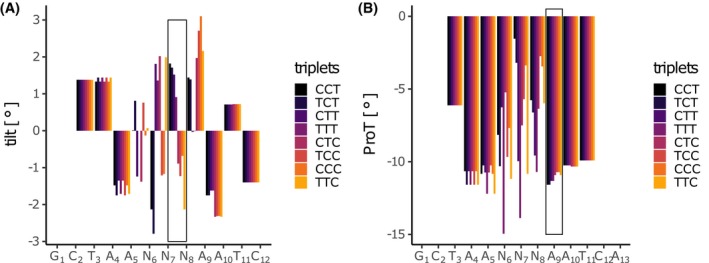
DNA shaping parameter tilt and propeller twist (ProT) are proportional to mid‐section triplet (5′‐TAANNNAATCA‐3′) alterations. The two DNA shaping parameter base tilt (A) and ProT (B) were displayed for the forward motif of the DUX4 aptamer while permuting the pyrimidines of the second base triplet of the motif (CCT, TCT, CTT, TTT, CTC, TCC, CCC, TTC). The sequences were sorted by increasing IC50 values (dark blue to yellow). The DNA shape values were predicted using the R package DNAshapeR
3.5. Simple optimization steps led to an aptamer with nanomolar affinity
The KD value of the aptamer with the lowest IC50 value of the tested aptamer set, 5′‐GCT AAC TTA ATC AAC CGC AGG TTG ATT AGC CCA TTA GC‐3′, was quantified by using fluorescence polarization assays. After correcting for nonspecific binding of the DUX4, a KD value of approximately 20 nM for this particular aptamer was calculated (Figure 7). The validity of the signal was further confirmed by experiments with a scrambled variant of the aptamer, reaction buffer containing 6 M of urea, or fluorescein without aptamer as negative control (Supplemental Figure S3A). The affinity was further affirmed by gel shift assays (Supplemental Figure S3B,C).
Figure 7.
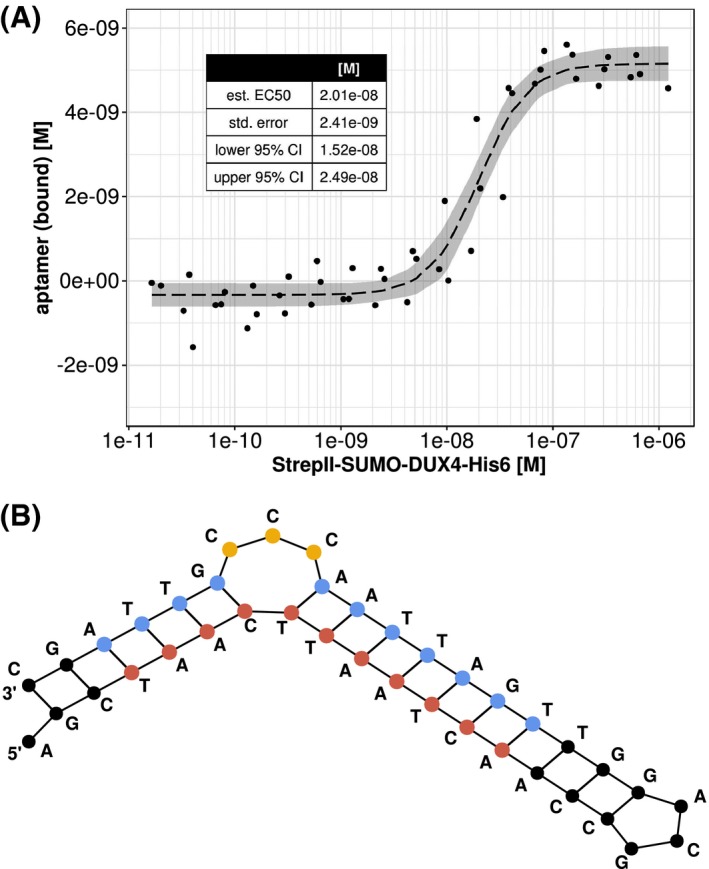
Calculating KD value of strongest aptamer from the optimization procedure. A, KD value of aptamer 5′‐GCTAA CTT AATCAACCGCAGGTTGATTAG CCC ATTAGC‐3′ was assessed by means of fluorescence polarization assay. A four‐parameter logistic was used to fit the calculated bound aptamer concentration (dashed line). Data are based on independent technical replicates using four different recombinant protein batches. Data are shown with 95% confidence band in gray. Fitting results are displayed in the table at the upper left corner. B, Corresponding predicted secondary structure of the aptamer is depicted. DUX4 binding motif is highlighted in red (forward motif) and blue (reverse motif). Affinity bulge loop is highlighted in yellow
3.6. DUX4 aptamers can also bind to PROP1 but spare PAX7
To validate the specificity of the DUX4 aptamer, different transcription factors were tested for their ability to bind to the aptamer. Prophet of pit‐1 (PROP1) and paired box protein Pax7 (PAX7) were chosen as representative transcription factors (Figure 8D). PROP1 has a comparable DNA binding motif as DUX4 with a base difference at position 10.78 PAX7 is known to act as a competing transcription factor in skeletal muscle. Overexpression of PAX7 can prevent DUX4 toxicity in C2C12 cells.18 In FSHD cells PAX7 target genes are repressed.79, 80 Replacing the HDs of DUX4 with the corresponding domains of PAX7 lead to a fully toxic DUX4 mutant.18 The binding motif of PAX7 is only 10 bases long and contains a major difference at position 9 in comparison to the analogous position in the DUX4 motif. Different aptamer variants (Figure 8E) were compared in TR‐FRET assays with the recombinant proteins PAX7, PROP1, and DUX4 together with labeled hairpin aptamers, containing the corresponding target motifs, as probes.
Figure 8.

Specificity of DUX4 aptamers. A, Different aptamers were tested against the consensus PROP1 binding sequence inserted into the aptamer backbone by means of a TR‐FRET competition assays. Single experiment data is shown as IC50 values of various aptamers ± standard error of the data fitting to a four‐parameter logistic using R package “drc” (curves are provided in Supplemental Figures S4A‐C). B, TR‐FRET competition assay was performed to compare the binding of different aptamers to PAX7. C, TR‐FRET competition assay was performed to compare the binding of different aptamers to DUX4. D, DNA target sequence logos of PAX7 (JASPAR ID: MA0680.1) and PROP1 (JASPAR ID: MA0715.1) extracted from JASPAR database are displayed. E, DNA sequences of all aptamers used for this experiment are listed. Target motifs are depicted in bold font. Special sequence alterations are given in red font and bulge loops in blue font. F, The affinity gain caused by the affinity bulge loops in the DUX4 aptamer were compared in the presence of different transcription factors. The IC50 ratios (ordinate) between aptamers DUX4_CTT and DUX4_CTT_loop from TR‐FRET competition assays were compared. Different concentrations of aptamers were incubated with the recombinant proteins DUX4, PAX7, and PROP1 (abscissa) and corresponding Atto647N‐labeled probes
PROP1 aptamer bound by PROP1 was replaced by the unlabeled PROP1 aptamer in the nanomolar range (Figure 8A). Poor curve fitting was obtained for the PAX7 aptamer due to its low affinity. DUX4 aptamers without loops had an IC50 in the micromolar range, independent of the two tested mid‐section triplet variants. DUX4 aptamer with CTT triplet and a loop between position 7 and 8 of the reverse motif increased the affinity to a lesser extent than in a corresponding aptamers with a TCT mid‐section triplet. An additional loop at position 8 of the forward motif did not alter the affinity compared to aptamers with loops only in the reverse motif.
The PAX7 aptamer as probe bound by PAX7 protein was displaced by the unlabeled PAX7 aptamer at nanomolar concentrations (Figure 8B). DUX4 and PROP1 aptamers had IC50 values in the micromolar range. The tested aptamers also included PAX7 aptamers with single base exchanges to bases matching the DUX4 target motif. These aptamer variants should show if the positions could cause higher specificity for DUX4 by decreasing the affinity for PAX7. It was tested whether T4 of the PAX7 motif (5′‐TAATCGAATTA‐3′) can be replaced by a dC, which is in accordance to the productive CTT DUX4 mid‐section triplet. The exchange of T9 of PAX7 motif (5′‐TAATCGAATTA‐3′) by dC would resemble the second homeobox binding site of DUX4 (5′‐TAACTTAATCA‐3′). Both base exchanges dramatically decreased the affinity to PAX7. Also the PROP1 aptamer had an IC50 in the micromolar range although, except for the length and the linker sequence, it had identical homeobox binding site sequences as the PAX7 aptamer.
In order to assess the specificity of DUX4 to other sequences, an aptamer with bulge loop at the beneficial position and the advantageous mid‐section triplet CTT were used as probe (Figure 8C). The tested Loop‐containing DUX4 aptamers had IC50 values in the low nanomolar range. PROP1 aptamers, any PAX7 aptamers, and DUX4 aptamers without bulge loops had IC50 values in the micromolar range.
Because bulge loops on DUX4 aptamers also increased the affinity for PROP1, we summarized the bulge loop effect for different TR‐FRET assays of this work (Figure 8F). The differences of the IC50 values between CTT‐aptamers with and without loops were compared in experimental TF‐FRET assays with different recombinant proteins. In summary, the affinity gain caused by the bulge loop was highest for DUX4 with about 21‐fold and only 5‐fold for PROP1.
In conclusion, bulge loops can influence the specificity of aptamers toward one transcription factor or the other. The affinity loop in the DUX4 aptamer can increase specificity in favor of DUX4 and disfavor of PAX7 and PROP1.
3.7. The loop sequence is not essential for the affinity effect
It was examined if the bulge loop of the DUX4 aptamers can be replaced by different types of spacers (Figure 9). TR‐FRET competition assays were used to compare aptamers containing the TCT triplet without loop, with triple‐dC loop, abasic loop, or aliphatic C3 spacers between the phosphates of the DNA backbone. Both abasic and C3 spacer loops had only marginally higher IC50 values compared to the triple‐dC loop containing aptamer.
Figure 9.
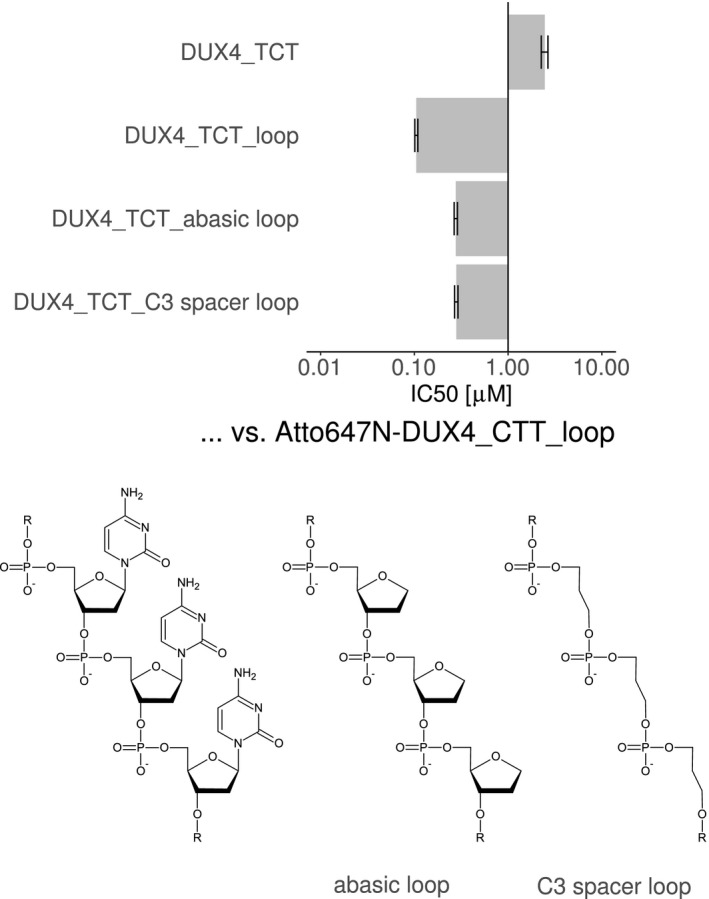
TR‐FRET competition assay was performed to compare the affinity of a template DUX4 aptamer with chemical loop modifications. An unmodified triple‐dC loop and an aptamer without loops were compared to aptamers with loop lacking the nucleobases (abasic) and aptamer with loops having aliphatic C3 spacer (no deoxyribose) between the phosphates. The chemical structures of the loops are depicted below. Single experiment data is shown as IC50 ± standard error of fitting to a four‐parameter logistic using R package “drc”. Curves are provided in Supplemental Figures S4D
The nucleobases of the loop sequence are not essential, and therefore, expendable for the affinity effect caused by this bulge loop.
3.8. Preference of bulge loop position can be explained by the protein structure
In an attempt to explain why bulged DNA aptamers are superior to simple hairpin‐aptamers containing only the DUX4 target sequence, it was hypothesized that maybe the bulge loop causes a pre‐bending or kinking of the DNA at the site of the bulge loops as observed for 5x‐dA bulge loops.81, 82 Although the bending might be only subtle for small triple‐dC loops, as seen by molecular dynamics predictions with dT‐bulges, it has to be examined if the overall conformation of the DNA is changed during binding to DUX4.83
In order to further analyze why bulge insertions into the DUX4 binding motif enhance binding affinity, we performed structural analysis of the DUX4‐aptamer complex. A crystal structure of DUX4 double HDs in complex with a DNA aptamer, which contains the CCC insertion in the DUX4 target motif and a GCA hairpin loop connecting the complementary DNA strands, was determined at 3.2 Å resolution (Figure 10A). The overall structure of the DUX4‐aptamer complex is very similar to that of the DUX4‐canonical target DNA complex reported previously.20 However, as expected, the DNA strand at the site of CCC insertion deviates significantly from the canonical B‐form and forms a bulge that is extruded from the double helix. The CCC bulge projects out from the major groove that accommodates the recognition helix (α3) of the first homeodomain (HD1) (Figure 10B) and makes unique contacts with the C‐terminal end of the α3 helix; Arg79 and Arg80 interact with the extrahelical cytosine base at the first and third position of the 5′‐CCC‐3′ bulge, respectively. In addition, Arg76 is hydrogen‐bonded to a backbone phosphate group at the 3′ end of the bulge, which has the non‐bridging oxygen atoms facing inward of the double‐helix and toward α3 due to the backbone distortion. Furthermore, van der Waals contacts between deoxyribose moieties appear to stabilize the sharply bent, compact structure of the DNA bulge. These structural features may explain the enhanced affinity conferred by a position‐specific bulge insertion.
Figure 10.
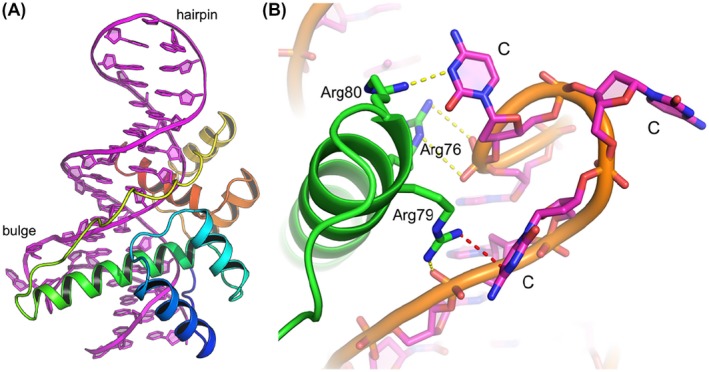
Crystal structure of DUX4 double homeodomain bound to the DNA aptamer. A, Crystal structure of DUX4 double homeodomain bound to the DNA aptamer containing a trinucleotide (‐CCC‐) bulge and a GCA hairpin loop. DNA is shown in magenta. DUX4 is colored in a gradient of blue to red from the N‐ to C‐terminus, respectively. B, A close‐up view showing the DUX4 interactions with the CCC bulge. The guanidinium group of Arg79 stacks on the first C base of the bulge (the van der Waals or cation‐π‐contact indicated by a red dotted line). Arg80 forms a salt bridge with the third C base and Arg76 is hydrogen‐bonded to a DNA backbone phosphate at the 3′‐end of the bulge (yellow dotted lines)
We also determined a crystal structure at 2.3 Å resolution of DUX4‐HDs bound to an aptamer variant, which contains the CCC insertion as above but lacking the GCA hairpin loop, assembled from two DNA strands. This structure again shows overall high similarity to the canonical DUX4 HDs‐DNA complex. However, the CCC insertion unexpectedly serves as a cross‐over linker and allowed the DNA strands to be swapped between two complexes, generating a Holliday junction DNA (Supplemental Figure S7). The result suggests flexibility of the CCC bulge motif and dynamic nature of the DUX4‐DNA interaction.
4. DISCUSSION
Here, we demonstrate that DUX4‐binding DNA aptamers generated by SELEX, using fully randomized single stranded oligonucleotides, form hairpin aptamers that present the consensus DUX4 binding motif and its reverse complementary sequence in the double stranded stem part. Motif analysis of this double stranded part revealed high similarity to the known consensus motif identified in several other studies.26, 27, 74 Interestingly, we identified an enrichment of bulge loops at distinct positions within this motif, which enhances the binding activity of the aptamers to DUX4. Binding experiments with rationally designed aptamers revealed the importance of certain sequence and structural elements on binding affinity. The integration of bulge loops at distinct positions within the binding motif can also enhance specificity as demonstrated by using transcription factors with related binding motifs.
4.1. Bulge loops enhance binding affinity by increasing interaction surface
The crystal structure revealed that the bulge loop interacts with DUX4 protein by enlarging the interaction surface leading to an enhanced affinity. The bulge loop is pointing outward of the DNA helix and is forming additional contact sites with the C‐terminal end of the α3 helix. The interaction involves base‐specific as well as DNA backbone contacts. The importance of base‐independent interaction was confirmed by replacing the triple‐dC loop with aliphatic spacers, which showed that the interaction of DUX4 with the bulge loops––in large part––is dependent on a sequence‐unrelated interaction with the phosphate backbone.
4.2. Binding motif sequence
At least one variant of the DUX4 consensus motif was found in 96% of the examined aptamer clusters. Apparently, the SELEX selected aptamers bind preferentially to the DNA binding site of DUX4 and not to other epitopes of the protein.Even though the provided library consisted of single stranded DNA oligomers without pre‐specified structure or sequence.
The identified sequence motif resembles the known consensus DUX4 binding motif26, 27, 74 with a minor exception, in that the base T6 (5′‐TAATTTAATCA‐3′) showed a high degree of conservation. We could show that this position had a pronounced effect on the affinity. Aptamers with certain triplets, like CCT, TCT, and CTT, had higher affinity than other permutations, as previously observed.74 In our study, the DUX4 motifs are presented in a hairpin structure in comparison to high‐throughput SELEX, where the motifs are present on a double stranded duplex structure. Slight differences in the helical structure of these two oligonucleotide variants may be the reason for the observed discrepancy in sequence conservation. In addition, the prediction of DNA shape parameters of different variants of this triplet, shows that some parameters of certain base positions are proportional to the affinity of the aptamers, potentially leading to a beneficial conformation of the DNA for binding to DUX4.
Surprisingly, bulge loops and the triplets acted synergistically on the affinity. The highest affinity gain of a loop insertion at position 7 of the reverse motif was in combination with a CTT triplet. The resulting aptamer had an affinity at the low nanomolar range as found by fluorescence polarization assays and confirmed by gel shift assays. This affinity is comparable to the EC50 of the FDA approved RNA‐aptamer Pegaptanib that is already in clinical use.56, 57
Concerning specificity of this triplet and the affinity loop containing aptamer, we tested two representative transcription factors, PROP1 and PAX7, with similar target motifs as DUX4. The PROP1 target motif contains a T instead of a C (in DUX4) at position 10.78 PROP1 is a homeobox transcription factor that is expressed early during development and exclusively in the pituitary gland.84 Mutations that affect the DNA binding of PROP1 are associated with pituitary hormone deficiencies.85, 86 This small difference in the target sequences between PROP1 and DUX4 caused a difference in the IC50 values of almost two orders of magnitude of the corresponding motif‐containing aptamers. Therefore, PROP1 has a low affinity for our DUX4 aptamer. Bulge loop insertions increased the affinity of aptamers for PROP1 especially pronounced in combination with a TCT triplet, but to a lesser extent than seen for DUX4. Therefore, the CTT triplet is beneficial in terms of affinity and specificity toward DUX4.
PAX7 is considered to be a counter‐player of DUX4 that is able to compete against the same target genes.10, 79, 80, 87 Furthermore, PAX7 target gene repression can be considered as a biomarker for FSHD and overexpression of PAX7 can rescue DUX4 cellular toxicity in DUX4 expressing cells.10, 79, 80, 87 Therefore, it was expected that PAX7 is able to bind DUX4 aptamers with comparable affinity keeping in mind that the PAX7 binding site is one base shorter than that of DUX4. However, in contrast to PROP1, we could show that PAX7 is binding only weakly to DUX4 aptamers.
It would be worthwhile to test whether the affinity gain toward DUX4 caused by loops at certain regions would sufficiently outcompete the affinity for other transcription factors as seen for PROP1 and PAX7, as representative transcription factors with highly similar binding sequences. PAX7 is not only one of the most important beneficial counter players of DUX4 concerning function and tissue expression, but is also essential for muscle regeneration, and therefore, potentially relevant to disease.18, 79, 88 In this regard, the aptamer developed in this work provided sufficient specificity and affinity to DUX4.
4.3. DUX4 aptamers and their potential for development of therapeutic strategies
To our knowledge, this work is the first to describe the impact of bulge loops within transcription factor binding sites contained in DNA aptamers. However, the insertion of bulge loops to increase the affinity of aptamers towards a target protein is not new. It has been shown, for instance, that the bulge loops in DNA aptamers against streptavidin are critical for the affinity.89 Bulge loops can also occur naturally in structural elements of RNA which are known to be recognized by RNA‐binding proteins.90 In genomic DNA, bulge loops are considered as erroneous structures and trigger repair mechanisms.91, 92 For instance the tumor suppressor protein p53, a hub and activator of stress response for many types of stress including DNA damage, is known to bind preferentially to triple‐dC bulged DNA structures.93, 94 Apart from that, bulge loops can also occur naturally at certain genomic repeat regions.95
One possible strategy to impede transcription factor functions is the use of double stranded oligodeoxynucleotide decoys. By mimicking a DNA binding site, these molecules selectively bind to their corresponding transcription factor and prevent it from binding genomic DNA and regulating its target genes.96 Clinical trials have been started initially to evaluate the therapeutic potential of several decoys; for example, NF‐κB decoys for the treatment of facial atopic dermatitis or Signal Transducers and Activators of Transcription (STAT3) decoys to treat head and neck squamous cell carcinoma.96, 97, 98 The spectrum of modification of these decoys was mainly limited to chemically modified nucleotides or circularization to form dumbbell shaped molecules in order to increase resistance against nuclease degradation.99, 100, 101 The insertion of structural elements like bulge loops into decoys may lead to superior affinity to compete with natural binding sites within the genome.
In summary, we identified hairpin aptamers with bulge loops at distinct positions within the DUX4 binding site. We successfully improved the affinity as well as specificity of these DNA aptamers by optimizing the bulge loop length and sequence as well as the sequence of the stem part. This approach can also be applied to oligonucleotide‐based strategies targeted against other transcription factors, especially if oligonucleotide decoys are developed for therapeutic applications.
5. CONCLUSION
The bioinformatic analysis of SELEX‐generated DNA aptamers against the transcription factor DUX4 revealed that new biochemical features can be modeled onto transcription factor binding sites. We have shown that a bulge loop at a distinct position within the binding motif strongly increases the affinity of the aptamer. Specificity can be customized by small changes of the motif at non‐conserved regions with mild consequences on the affinity toward DUX4. Such sequence and structural alterations may improve the impact of current therapeutic strategies like transcription factor oligodeoxynucleotide decoys. In addition, the bulge loops can potentially act as platforms for chemical and biological modifications that could be conducive not only to study protein‐DNA interaction, but also for therapeutic purposes.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTIONS
C. Klingler, J. Ashley , M. Sinnreich, and J. Kinter conceived and designed aptamer experiments. K. Shi and H. Aihara conceived and designed crystallization experiments. C. Klingler, J. Ashley , A. Stiefvater, and J. Kinter collected aptamer data. K. Shi collected crystallization data. C. Klingler, J. Shi, and K. Shi analyzed and interpreted aptamer data. C. Klingler contributed R code. K. Shi and H. Aihara analyzed and interpreted crystallization results. C. Klingler wrote the main manuscript text. J. Ashley , K. Shi, A. Stiefvater, M. Kyba, H. Aihara, M. Sinnreich, and J. Kinter revised and edited manuscript and approved final version. M. Sinnreich secured funding.
Supporting information
ACKNOWLEDGMENTS
We thank Tracy Dinh, Nicholas Moeller, and Kayo Orellana for help with protein purification and crystallization, and the staff of the NE‐CAT beamline for assistance with x‐ray data collection. This research used resources of the Advanced Photon Source (APS), a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science under Contract No. DE‐AC02‐06CH11357. The Northeastern Collaborative Access Team (NE‐CAT) beamlines are funded by the National Institutes of Health (NIH) (P30 GM124165). The Eiger 16M detector on 24‐ID‐E beam line is funded by a NIH grant (S10OD021527). This work was supported by NIH grant GM118047 (to H. A.). We thank CSL Behring AG for an unrestricted research grant.
Klingler C, Ashley J, Shi K, et al. DNA aptamers against the DUX4 protein reveal novel therapeutic implications for FSHD. The FASEB Journal. 2020;34:4573–4590. 10.1096/fj.201902696
DATA AVAILABILITY STATEMENT
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank (PDB) with accession codes 6U81 and 6U82.
REFERENCES
- 1. Deenen JC, Arnts H, van der Maarel SM, et al. Population‐based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056‐1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mul K, Lassche S, Voermans NC, Padberg GW, Horlings CG, van Engelen BG. What's in a name? The clinical features of facioscapulohumeral muscular dystrophy. Pract Neurol. 2016;16:201‐207. [DOI] [PubMed] [Google Scholar]
- 3. Himeda CL, Jones PL. The genetics and epigenetics of facioscapulohumeral muscular dystrophy. Annu Rev Genomics Hum Genet. 2019;20:265‐291. [DOI] [PubMed] [Google Scholar]
- 4. Jones TI, Chen JC, Rahimov F, et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet. 2012;21:4419‐4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gabriels J, Beckers MC, Ding H, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25‐32. [DOI] [PubMed] [Google Scholar]
- 6. Dixit M, Ansseau E, Tassin A, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci U S A. 2007;104:18157‐18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650‐1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kowaljow V, Marcowycz A, Ansseau E, et al. The DUX4 gene at the FSHD1A locus encodes a pro‐apoptotic protein. Neuromuscul Disord. 2007;17:611‐623. [DOI] [PubMed] [Google Scholar]
- 9. Snider L, Geng LN, Lemmers RJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bosnakovski D, Lamb S, Simsek T, et al. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp Neurol. 2008;214:87‐96. [DOI] [PubMed] [Google Scholar]
- 11. Mitsuhashi H, Mitsuhashi S, Lynn‐Jones T, Kawahara G, Kunkel LM. Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy. Hum Mol Genet. 2013;22:568‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vanderplanck C, Ansseau E, Charron S, et al. The FSHD atrophic myotube phenotype is caused by DUX4 expression. PLoS ONE. 2011;6:e26820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rickard AM, Petek LM, Miller DG. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum Mol Genet. 2015;24:5901‐5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Snider L, Asawachaicharn A, Tyler AE, et al. RNA transcripts, miRNA‐sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet. 2009;18:2414‐2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lemmers RJLF, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD‐permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370‐1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van den Boogaard ML, Lemmers RJ, Balog J, et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am J Hum Genet. 2016;98:1020‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leidenroth A, Hewitt JE. A family history of DUX4: phylogenetic analysis of DUXA, B, C and Duxbl reveals the ancestral DUX gene. BMC Evol Biol. 2010;10:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bosnakovski D, Toso EA, Hartweck LM, et al. The DUX4 homeodomains mediate inhibition of myogenesis and are functionally exchangeable with the Pax7 homeodomain. J Cell Sci. 2017;130:3685‐3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y, Wu B, Liu H, et al. Structural basis for multiple gene regulation by human DUX4. Biochem Biophys Res Commun. 2018;505:1161‐1167. [DOI] [PubMed] [Google Scholar]
- 20. Lee JK, Bosnakovski D, Toso EA, et al. Crystal structure of the double homeodomain of DUX4 in complex with DNA. Cell Rep. 2018;25:2955‐2962.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitsuhashi H, Ishimaru S, Homma S, et al. Functional domains of the FSHD‐associated DUX4 protein. Biol Open. 2018;7(4):bio033977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Corona ED, Jacquelin D, Gatica L, Rosa AL. Multiple protein domains contribute to nuclear import and cell toxicity of DUX4, a candidate pathogenic protein for facioscapulohumeral muscular dystrophy. PLoS ONE. 2013;8:e75614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choi SH, Gearhart MD, Cui Z, et al. DUX4 recruits p300/CBP through its C‐terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016;44:5161‐5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bosnakovski D, da Silva MT, Sunny ST, et al. A novel P300 inhibitor reverses DUX4‐mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci Adv. 2019;5:eaaw7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bosnakovski D, Xu Z, Gang EJ, et al. An isogenetic myoblast expression screen identifies DUX4‐mediated FSHD‐associated molecular pathologies. EMBO J. 2008;27:2766‐2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Young JM, Whiddon JL, Yao Z, et al. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet. 2013;9:e1003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geng LN, Yao Z, Snider L, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 2012;22:38‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Das S, Chadwick BP. Influence of repressive histone and DNA methylation upon D4Z4 transcription in non‐myogenic cells. PLoS ONE. 2016;11:e0160022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Iaco A, Planet E, Coluccio A, Verp S, Duc J, Trono D. DUX‐family transcription factors regulate zygotic genome activation in placental mammals. Nat Genet. 2017;49:941‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hendrickson PG, Dorais JA, Grow EJ, et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage‐stage genes and MERVL/HERVL retrotransposons. Nat Genet. 2017;49:925‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen Z, Zhang Y. Loss of DUX causes minor defects in zygotic genome activation and is compatible with mouse development. Nat Genet. 2019;51:947‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lilljebjorn H, Fioretos T. New oncogenic subtypes in pediatric B‐cell precursor acute lymphoblastic leukemia. Blood. 2017;130:1395‐1401. [DOI] [PubMed] [Google Scholar]
- 33. Liu YF, Wang BY, Zhang WN, et al. Genomic profiling of adult and pediatric B‐cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang J, McCastlain K, Yoshihara H, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48:1481‐1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lilljebjorn H, Henningsson R, Hyrenius‐Wittsten A, et al. Identification of ETV6‐RUNX1‐like and DUX4‐rearranged subtypes in paediatric B‐cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7:11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yasuda T, Tsuzuki S, Kawazu M, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48:569‐574. [DOI] [PubMed] [Google Scholar]
- 37. Kawamura‐Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up‐regulates PEA3 family genes in Ewing‐like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15:2125‐2137. [DOI] [PubMed] [Google Scholar]
- 38. Yoshimoto T, Tanaka M, Homme M, et al. CIC‐DUX4 induces small round cell sarcomas distinct from Ewing sarcoma. Can Res. 2017;77:2927‐2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Specht K, Sung YS, Zhang L, Richter GH, Fletcher CD, Antonescu CR. Distinct transcriptional signature and immunoprofile of CIC‐DUX4 fusion‐positive round cell tumors compared to EWSR1‐rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosom Cancer. 2014;53:622‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sirvent N, Trassard M, Ebran N, Attias R, Pedeutour F. Fusion of EWSR1 with the DUX4 facioscapulohumeral muscular dystrophy region resulting from t(4;22)(q35;q12) in a case of embryonal rhabdomyosarcoma. Cancer Genet Cytogenet. 2009;195:12‐18. [DOI] [PubMed] [Google Scholar]
- 41. Chew GL, Campbell AE, De Neef E, et al. DUX4 suppresses MHC class I to promote cancer immune evasion and resistance to checkpoint blockade. Dev Cell. 2019;50:658‐671.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu J, Yao Z, Yang Y, Zhang C, Zhang J, Zhang Y. Management strategies in facioscapulohumeral muscular dystrophy. Intractable Rare Dis Res. 2019;8:9‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cruz JM Jr, Hupper N, Wilson LS, et al. Protein kinase A activation inhibits DUX4 gene expression in myotubes from patients with facioscapulohumeral muscular dystrophy. J Biol Chem. 2018;293:11837‐11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi SH, Bosnakovski D, Strasser JM, Toso EA, Walters MA, Kyba M. Transcriptional inhibitors identified in a 160,000‐compound small‐molecule DUX4 viability screen. J Biomol Screen. 2016;21:680‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bosnakovski D, Choi SH, Strasser JM, Toso EA, Walters MA, Kyba M. High‐throughput screening identifies inhibitors of DUX4‐induced myoblast toxicity. Skelet Muscle. 2014;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Himeda CL, Jones TI, Jones PL. CRISPR/dCas9‐mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4‐fl in FSH muscular dystrophy. Mol Ther. 2016;24:527‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wallace LM, Liu J, Domire JS, et al. RNA interference inhibits DUX4‐induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol Ther. 2012;20:1417‐1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen JC, King OD, Zhang Y, et al. Morpholino‐mediated knockdown of DUX4 toward facioscapulohumeral muscular dystrophy therapeutics. Mol Ther. 2016;24:1405‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818‐822. [DOI] [PubMed] [Google Scholar]
- 50. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505‐510. [DOI] [PubMed] [Google Scholar]
- 51. Yoon S, Huang K‐W, Reebye V, et al. Targeted delivery of C/EBPalpha ‐saRNA by pancreatic ductal adenocarcinoma‐specific RNA aptamers inhibits tumor growth in vivo. Mol Ther. 2016;24:1106‐1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poolsup S, Kim CY. Therapeutic applications of synthetic nucleic acid aptamers. Curr Opin Biotechnol. 2017;48:180‐186. [DOI] [PubMed] [Google Scholar]
- 53. Kaur H, Bruno JG, Kumar A, Sharma TK. Aptamers in the therapeutics and diagnostics pipelines. Theranostics. 2018;8:4016‐4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pfeiffer F, Mayer G. Selection and biosensor application of aptamers for small molecules. Front Chem. 2016;4:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ku T‐H, Zhang T, Luo H, et al. Nucleic acid aptamers: an emerging tool for biotechnology and biomedical sensing. Sensors. 2015;15:16281‐16313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ng EW, Shima DT, Calias P, Cunningham ET Jr, Guyer DR, Adamis AP. Pegaptanib, a targeted anti‐VEGF aptamer for ocular vascular disease. Nat Rev Drug Discovery. 2006;5:123‐132. [DOI] [PubMed] [Google Scholar]
- 57. Siddiqui MA, Keating GM. Pegaptanib: in exudative age‐related macular degeneration. Drugs. 2005;65:1571‐1577; discussion 1578‐1579. [DOI] [PubMed] [Google Scholar]
- 58. Zboralski D, Hoehlig K, Eulberg D, Fromming A, Vater A. Increasing tumor‐infiltrating T cells through inhibition of CXCL12 with NOX‐A12 synergizes with PD‐1 blockade. Cancer Immunol Res. 2017;5:950‐956. [DOI] [PubMed] [Google Scholar]
- 59. Bae O‐N. Targeting von Willebrand factor as a novel anti‐platelet therapy; application of ARC1779, an anti‐vWF aptamer, against thrombotic risk. Arch Pharm Res. 2012;35:1693‐1699. [DOI] [PubMed] [Google Scholar]
- 60. Menne J, Eulberg D, Beyer D, et al. C‐C motif‐ligand 2 inhibition with emapticap pegol (NOX‐E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant. 2017;32:307‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dib C, Zakharova V, Popova E, et al. DUX4 pathological expression: causes and consequences in cancer. Trends in cancer. 2019;5:268‐271. [DOI] [PubMed] [Google Scholar]
- 62. Zhang Y, Fan Y. A mutant sumo facilitates quick plasmid construction for expressing proteins with native N‐termini after tag removal. Mol Biotechnol. 2017;59:159‐167. [DOI] [PubMed] [Google Scholar]
- 63. Hoinka J, Backofen R, Przytycka TM. AptaSUITE: a full‐featured bioinformatics framework for the comprehensive analysis of aptamers from HT‐SELEX experiments. Mol Ther Nucleic Acids. 2018;11:515‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3‐31. [DOI] [PubMed] [Google Scholar]
- 65. Rossi AM, Taylor CW. Analysis of protein‐ligand interactions by fluorescence polarization. Nat Protoc. 2011;6:365‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ritz C, Baty F, Streibig JC, Gerhard D. Dose‐response analysis using R. PLoS ONE. 2016;10:e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ritz C, Streibig JC. Bioassay analysis using R. J Stat Softw. 2005;2005(12):22. [Google Scholar]
- 68. Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:8060‐8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mathelier A, Zhao X, Zhang AW, et al. JASPAR 2014: an extensively expanded and updated open‐access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42:D142‐D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang Y, Lee JK, Toso EA, et al. DNA‐binding sequence specificity of DUX4. Skelet Muscle. 2016;6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open‐access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Varani G. Exceptionally stable nucleic acid hairpins. Annu Rev Biophys Biomol Struct. 1995;24:379‐404. [DOI] [PubMed] [Google Scholar]
- 77. Chiu TP, Comoglio F, Zhou T, Yang L, Paro R, Rohs R. DNAshapeR: an R/Bioconductor package for DNA shape prediction and feature encoding. Bioinformatics. 2016;32:1211‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nakayama M, Kato T, Susa T, Sano A, Kitahara K, Kato Y. Dimeric PROP1 binding to diverse palindromic TAAT sequences promotes its transcriptional activity. Mol Cell Endocrinol. 2009;307:36‐42. [DOI] [PubMed] [Google Scholar]
- 79. Banerji CRS, Panamarova M, Hebaishi H, et al. PAX7 target genes are globally repressed in facioscapulohumeral muscular dystrophy skeletal muscle. Nat Commun. 2017;8:2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Banerji CRS, Zammit PS. PAX7 target gene repression is a superior FSHD biomarker than DUX4 target gene activation, associating with pathological severity and identifying FSHD at the single‐cell level. Hum Mol Genet. 2019;28:2224‐2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dornberger U, Hillisch A, Gollmick FA, Fritzsche H, Diekmann S. Solution structure of a five‐adenine bulge loop within a DNA duplex. Biochemistry. 1999;38:12860‐12868. [DOI] [PubMed] [Google Scholar]
- 82. Gollmick FA, Lorenz M, Dornberger U, von Langen J, Diekmann S, Fritzsche H. Solution structure of dAATAA and dAAUAA DNA bulges. Nucleic Acids Res. 2002;30:2669‐2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schreck JS, Ouldridge TE, Romano F, Louis AA, Doye JP. Characterizing the bending and flexibility induced by bulges in DNA duplexes. J Chem Phys. 2015;142:165101. [DOI] [PubMed] [Google Scholar]
- 84. Sornson MW, Wu W, Dasen JS, et al. Pituitary lineage determination by the Prophet of Pit‐1 homeodomain factor defective in Ames dwarfism. Nature. 1996;384:327‐333. [DOI] [PubMed] [Google Scholar]
- 85. Davis SW, Ellsworth BS, Peréz Millan MI, et al. Chapter one—Pituitary gland development and disease: from stem cell to hormone production. In: Thomas P, ed. Current Topics in Developmental Biology. Vol. 106 San Diego, CA: Academic Press; 2013:1‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Correa FA, Nakaguma M, Madeira JLO, et al. Combined pituitary hormone deficiency caused by PROP1 mutations: update 20 years post‐discovery. Arch Endocrinol Metab. 2019;63:167‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Haynes P, Kernan K, Zhou S‐L, Miller DG. Expression patterns of FSHD‐causing DUX4 and myogenic transcription factors PAX3 and PAX7 are spatially distinct in differentiating human stem cell cultures. Skelet Muscle. 2017;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci U S A. 2013;110:16474‐16479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bing T, Yang X, Mei H, Cao Z, Shangguan D. Conservative secondary structure motif of streptavidin‐binding aptamers generated by different laboratories. Bioorg Med Chem. 2010;18:1798‐1805. [DOI] [PubMed] [Google Scholar]
- 90. Bevilacqua PC, Ritchey LE, Su Z, Assmann SM. Genome‐wide analysis of RNA secondary structure. Annu Rev Genet. 2016;50:235‐266. [DOI] [PubMed] [Google Scholar]
- 91. Lyons DM, O'Brien PJ. Efficient recognition of an unpaired lesion by a DNA repair glycosylase. J Am Chem Soc. 2009;131:17742‐17743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jiricny J. The multifaceted mismatch‐repair system. Nat Rev Mol Cell Biol. 2006;7:335‐346. [DOI] [PubMed] [Google Scholar]
- 93. Degtyareva N, Subramanian D, Griffith JD. Analysis of the binding of p53 to DNAs containing mismatched and bulged bases. J Biol Chem. 2001;276:8778‐8784. [DOI] [PubMed] [Google Scholar]
- 94. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53‐mediated tumour suppression. Nat Rev Cancer. 2014;14:359‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Volker J, Plum GE, Gindikin V, Klump HH, Breslauer KJ. Impact of bulge loop size on DNA triplet repeat domains: implications for DNA repair and expansion. Biopolymers. 2014;101:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hecker M, Wagner AH. Transcription factor decoy technology: a therapeutic update. Biochem Pharmacol. 2017;144:29‐34. [DOI] [PubMed] [Google Scholar]
- 97. Sen M, Thomas SM, Kim S, et al. First‐in‐human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2:694‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discovery. 2013;12:611‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Osako MK, Tomita N, Nakagami H, et al. Increase in nuclease resistance and incorporation of NF‐kappaB decoy oligodeoxynucleotides by modification of the 3′‐terminus. J Gene Med. 2007;9:812‐819. [DOI] [PubMed] [Google Scholar]
- 100. Hosoya T, Takeuchi H, Kanesaka Y, et al. Sequence‐specific inhibition of a transcription factor by circular dumbbell DNA oligonucleotides. FEBS Lett. 1999;461:136‐140. [DOI] [PubMed] [Google Scholar]
- 101. Lee SW, Sullenger BA. Isolation of a nuclease‐resistant decoy RNA that can protect human acetylcholine receptors from myasthenic antibodies. Nat Biotechnol. 1997;15:41‐45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank (PDB) with accession codes 6U81 and 6U82.