

Abstract
In nature, dynamic processes are ubiquitous and often characterized by adaptive, transient behavior. Herein, we present the development of a transient bowl‐shaped nanoreactor system, or stomatocyte, the properties of which are mediated by molecular interactions. In a stepwise fashion, we couple motility to a dynamic process, which is maintained by transient events; namely, binding and unbinding of adenosine triphosphate (ATP). The surface of the nanosystem is decorated with polylysine (PLL), and regulation is achieved by addition of ATP. The dynamic interaction between PLL and ATP leads to an increase in the hydrophobicity of the PLL–ATP complex and subsequently to a collapse of the polymer; this causes a narrowing of the opening of the stomatocytes. The presence of the apyrase, which hydrolyzes ATP, leads to a decrease of the ATP concentration, decomplexation of PLL, and reopening of the stomatocyte. The competition between ATP input and consumption gives rise to a transient state that is controlled by the out‐of‐equilibrium process.
Keywords: ATP, nanomotors, nanoreactors, out-of-equilibrium systems, stomatocytes
Imitation of life: ATP was utilized to mediate the transient behavior of stomatocyte nanosystems, the “opening” of which was regulated by competition between ATP input and consumption. This work is a step toward the design of man‐made adaptive nanosystems to mimic and understand the dynamic behavior found in nature.
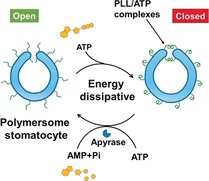
Introduction
One of the inherent and most interesting features of living systems is their adaptability to show complex behavior in response to a variety of signals. Among these behaviors, autonomous motion has been an important source of inspiration for scientists who, over the years, have created a variety of synthetic motor systems that imitate biological motility.1 For example, molecular motors,2 micro‐ and nanoscale sized Janus motors,3 self‐assembled polymeric motors,4 and movable tubules and rods5 have been developed. Regardless of the excellent performance of these motor systems, there is a fundamental difference in the way movement is regulated in synthetic and natural systems. Cellular autonomous motion (e.g., vesicular transport and motility),6 displays adaptive features as a result of competing transient activation and deactivation processes, which are governed by enzyme‐mediated energy input and consumption, and molecular interactions. Such dynamic processes are also referred to as out‐of‐equilibrium or dissipative; mimicking these behaviors in synthetic systems has recently drawn much attention from the scientific community.7 The introduction of transient behavior into synthetic molecular or nanoscaled systems has been demonstrated for active materials with unique properties such as dissipative fibres,8 transient peptide hydrogels, vesicles or microcapsules,9 temporally programmed “breathing” microgels and polymersomes,10 and non‐equilibrium molecular recognition and colloidal systems.11 In synthetic motor systems, switchable behavior has been introduced, for example by making nanomotors temperature responsive.3b Compared to the above‐mentioned transient systems, these nanomotors require a second stimulus to switch back to their initial states and thus do not operate in a transient fashion. Arguably, closest to transiently regulated motile systems are the DNA‐based molecular motors or walkers.12
Herein, we describe such transient behavior in a stomatocyte nanosystem that is mediated by the addition of the biomolecule adenosine‐5′‐triphosphate (ATP). Our design relies on our previously reported bowl‐shaped polymer vesicle, or stomatocyte,13 which is composed of poly(ethylene glycol)‐block‐polystyrene (PEG‐b‐PS) block copolymers and loaded with catalytically active species. In order to modulate the output of such stomatocytes with ATP, the stomatocyte surface was decorated with polylysine (PLL). These PLL moieties were introduced to participate in the dynamic non‐covalent complexation with ATP, to alter the size of the opening of the stomatocytes. As ATP contains a hydrophobic adenine group and four negative charges, we hypothesized that the formation of PLL/ATP complexes would introduce hydrophobic adenine groups along the PLL backbone. Thus, the opening of the stomatocyte would then be blocked to prevent access of substrates (for the nanoreactor) or fuel (for the nanomotor) in the stomatocyte, leading to a decreased activity or speed, respectively. To introduce transient behavior in the system, binding of ATP to polylysine should be counteracted. This can be achieved when potato apyrase is present in the system, which hydrolyzes ATP into AMP, resulting in disassembly and recovery of the original switched‐on output. Moreover, by refeeding ATP to the system, the transient behavior of the stomatocytes could be reinstalled, endowing it with non‐equilibrium behavior (Scheme 1).
Scheme 1.
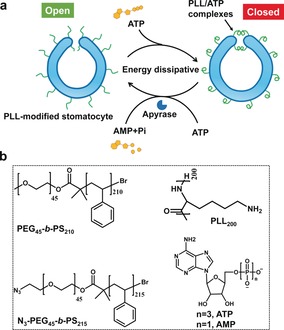
a) Schematic of the transient deactivation and activation of a stomatocyte nanosystem mediated by ATP. b) Chemical structure of mPEG45‐b‐PS210, N3‐mPEG45‐b‐PS215, PLL200, ATP, and AMP.
Results and Discussion
The morphology of the assemblies was characterized by transmission electron microscopy (TEM). Polymer stomatocytes with an almost closed opening (smaller than 5 nm) were observed (Figure 1 a). Most importantly, grafting the PLL brush did not affect the size and morphology of the stomatocytes, and the particles were stable for at least six months at room temperature (Figure S4, Supporting Information). These bowl‐shaped structures with small nanopores were further confirmed by scanning electron microscopy (SEM) (Figure 1 b) and cryo‐TEM (Figure S5, Supporting Information).
Figure 1.
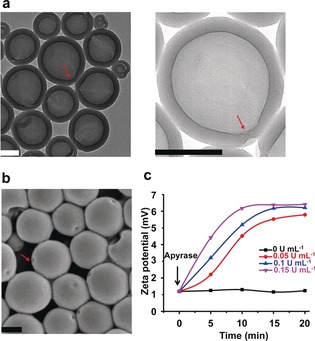
a) TEM and b) SEM images of PLL‐modified stomatocytes. The red arrows indicate the opening of the stomatocytes. Scale bars=200 nm. c) Zeta potential changes of the ATP‐complexed PLL‐stomatocytes as a function of time upon the addition of apyrase. The PLL‐stomatocytes were pretreated with ATP (62.5 μm, r=1). Experimental conditions: 0.5 mg mL−1 PLL‐stomatocytes, MES buffer (5 mm, pH 6.5).
Zeta potential (ζ) measurements were performed to indicate whether PLL was successfully coupled to the surface of the stomatocytes. After surface modification, the ζ value of the stomatocytes in a 2‐(N‐morpholino) ethanesulfonic acid (MES) buffer solution (5 mm, pH 6.5) increased from −2.2 mV to 6.1 mV due to the protonated primary amine groups at the stomatocyte surface (Figure S7, Supporting Information). To confirm that ATP was able to complex with PLL, the change of the stomatocyte surface charge was studied over a wide range of charge ratios (r), where r=(4ATP4−):(200PL200+). With an increase in r, the ζ value of the samples witnessed a continuous decrease from an original value of 6.1 mV (no ATP) to 1.2 mV (r=1). It is well known that the enzyme potato apyrase can effectively hydrolyze ATP into AMP and two phosphate (Pi) species.14 Owing to the decreased charge of AMP compared to ATP, this will lead to a much weaker interaction with PLL and recovery of the initial positive surface charge. As expected, the addition of apyrase resulted in a gradual increase in the zeta potential of the ATP complexed stomatocyte solution, which then remained constant over time (Figure 1 c). Interestingly, we found that the zeta potential recovery rates of the system are highly dependent on the concentration of apyrase. Control experiments demonstrated that the addition of AMP only led to a surface charge change of 0.4 mV, which means that the interaction between AMP and PLL is indeed very weak (Figure S7, Supporting Information).
To demonstrate that PLL–ATP complexation led to hydrophobic collapse and therefore to the closure of the narrow stomatocyte opening, we used this system to construct ATP‐mediated transient nanoreactors (Figure 2 a). For this purpose, horseradish peroxidase (HRP) as a model enzyme was encapsulated in the cavity of the stomatocytes to generate nanoreactors. The hybrid stomatocytes were purified by spin filtration, and size exclusion chromatography (SEC) data confirmed the complete removal of unencapsulated enzyme (Figure S8, Supporting Information). To determine enzyme loading efficiency, organic solvent was added after purification to the stomatocyte solution to reshape the stomatocytes back into spherical polymersomes, thereby releasing encapsulated enzymes (Figure S9, Supporting Information).15 The released enzymes were collected to analyze incorporation efficiency, which was calculated to be 9.5 % based on the bicinchoninic acid (BCA) assay.
Figure 2.
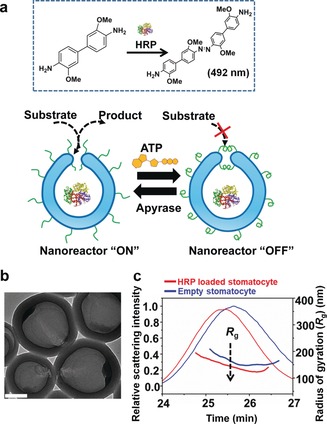
Adaptive stomatocyte nanoreactors. a) Scheme of the ATP‐mediated responsive nanoreactors. HRP was encapsulated inside the cavity of the stomatocyte, and DMB was utilized as a substrate to evaluate the enzymatic reaction. (b) TEM image of HRP loaded PLL‐stomatocytes. Scale bar=200 nm. c) Asymmetric flow field‐flow fractionation (AF4) fractograms and radius of gyration (R g) of empty PLL‐stomatocytes and HRP loaded PLL‐stomatocytes.
Enzyme incorporation was further indicated by the black aggregates at the bottom of the stomatocytes as observed by TEM and also by a combination of asymmetric flow field flow fractionation (AF4) and static light scattering (LS) analysis, which showed a significant decrease in the radius of gyration (R g) after enzyme encapsulation (Figure 2 b,c and Table S2, Supporting Information). HRP was used to catalyze the oxidation of 3,3′‐dimethoxybenzidine (DMB), and the product was monitored through the gradual increase in absorbance at 492 nm. Different ATP levels gave rise to different biocatalytic activity, and the more ATP was added, the lower the enzymatic reaction, as shown in Figure 3 a. In the absence of ATP, the nanoreactors showed the highest activity. This controllable catalytic behavior is attributed to the ATP‐induced collapse of the PLL chains on the surface of the stomatocytes, which hinders diffusion of the substrate into the cavity. The degree of collapse of the PLL chains is regulated by the concentration of ATP, thereby leading to modulated, and even switched‐off enzymatic activity at the highest ATP concentration. Next, we sought to re‐open the closed “mouth” of the stomatocytes by apyrase. As shown in Figure 3 b, in the absence of apyrase, the ATP‐gated stomatocyte nanoreactors demonstrated hardly any activity and were in the “OFF” state. However, in presence of apyrase, a noticeable increase in the enzymatic reaction was observed, which is caused by the reopening of the stomatocytes, allowing substrate diffusion into the cavity. Moreover, higher concentrations of apyrase led to faster catalysis, which means switching on the nanoreactors is dependent on the amount of apyrase. Importantly, temporal control of nanoreactors with out‐of‐equilibrium catalytic behavior was explored. When ATP was repeatedly applied to the system in the presence of apyrase, the HRP enzymatic reaction first stopped and subsequently recovered, which was caused by the transient complexation between PLL and ATP, highlighting the dissipative nature of our system (Figure S11, Supporting Information).
Figure 3.
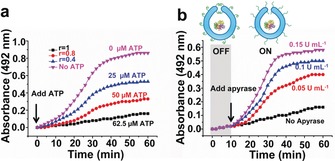
a) UV absorbance at 492 nm of the oxidation of DMB as a function of time upon the addition of different concentrations of ATP. b) Addition of apyrase switches nanoreactors back to the “ON” state. Experimental conditions: 0.5 mg mL−1 PLL‐stomatocytes, MES buffer (5 mm, pH 6.5), [HRP]=5 U mL−1 and [DMB]=400 μm.
After establishing the strong dependence of the activity of the nanoreactors on ATP and the responsiveness of the system toward apyrase, we adapted this responsive behavior to autonomous movement (Figure 4 a). As with many dynamic processes in nature, motility (at the nanoscale) is a critical mechanism for many biological processes. Despite the great efforts to incorporate life‐like behaviors into artificial structures, integration of motion with dynamic processes has so far remained elusive. Although previous work has shown that temperature can be used to tune the velocity of the stomatocyte nanomotors,16 the way motion was regulated is perpetual. Developing motile systems in which function is coupled to a dissipative process is an exciting way to create complex biomimetic systems that are inherently dynamic. In this study, PLL‐modified nanomotor systems were constructed by encapsulation of platinum nanoparticles (PtNPs) inside the stomatocyte; these particles are known to decompose hydrogen peroxide (H2O2) into oxygen, driving the nanomotor. TEM characterization confirmed the incorporation of PtNPs in the cavity of the stomatocytes (Figure 4 b and Figures S12 and S13, Supporting Information). Static light‐scattering analysis showed that after encapsulation of PtNPs, the R g value of the stomatocytes decreased from 151.5±9.8 nm to 136.8±13.4 nm, which is another sign that PtNPs were successfully loaded into the cavity of the stomatocytes (Figure 4 c).
Figure 4.
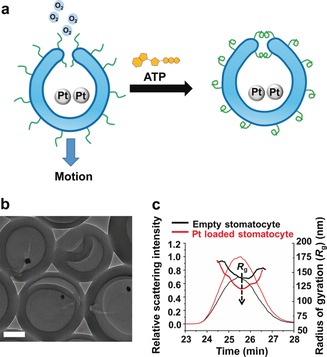
a) Schematic of ATP‐regulated PtNP‐loaded stomatocyte nanomotors. b) TEM image of PtNP‐loaded PLL‐stomatocytes. Scale bar=100 nm. c) Asymmetric flow field‐flow fractionation (AF4) fractograms and radius of gyration (R g) of empty PLL‐stomatocytes and PtNP‐loaded PLL‐stomatocytes.
Next, we performed measurements of motion with nanoparticle‐tracking analysis (NTA) in the presence of ATP (motion analysis is described in the Supporting Information). In the absence of H2O2, the motors demonstrated Brownian motion (Video S1, Supporting Information). As a control, upon applying H2O2, PLL‐modified motors in the absence of ATP demonstrated directional autonomous movement (Video S2, Supporting Information), in line with previous reports on the motion of stomatocyte nanomotors.4a, 15, 17 Indeed, increasing fuel concentration resulted in a transition of the motion mechanism from diffusive to ballistic, as evident by the acquired anomalous diffusion exponent α (alpha) values (Figure S17 and Table S4, Supporting Information). However, the PLL‐modified motors displayed slightly lower velocity compared to the unmodified ones, which implies that the attachment of PLL on the surface of the stomatocyte has only a limited effect on the movement of the motors (Figure 5 a). As shown in Figure 5 b, the mean squared displacement (MSD) of the stomatocyte nanomotors experienced a significant decrease upon the addition of ATP, and the more ATP was applied, the more the MSD decreased. Upon increasing ATP levels, the velocity of the motors decreased steadily from 35 μm s−1 (no ATP) to 7 μm s−1 (r=1), which agrees remarkably well with what we observed for the ATP‐modulated nanoreactors. Furthermore, PLL‐motors in absence of ATP showed consistently a five‐fold higher rate of motion than for the motors exposed to the highest ATP concentration. When 62.5 μm ATP (r=1) was added, the movement was reduced to levels close to Brownian motion (Video S3, Supporting Information), indicating a near complete closure of the opening. In addition, the measurement of the x and y trajectories of the particles at different ATP concentrations suggested that the motors could undergo a transition from directional movement to Brownian motion, as evidenced by the observed linearity of the MSD curves (Figure S19, Supporting Information). The formation of PLL/ATP complexes leads to collapsed hydrophobic domains around the surface of the stomatocytes, which thus hinders the penetration of fuel into the nanocavity, reducing the speed of the motors. Moreover, DLS results show that the increase in ATP concentration causes an increase in the apparent hydrodynamic size of the motors (Figure S16, Supporting Information), highlighting the effective binding of stomatocytes to ATP and thus, reducing the speed of these nanomotors. To demonstrate the importance of this hydrophobic domain formation, we employed a shorter polylysine (PLL100). When this polymer was used to decorate the surface of the stomatocytes, the velocity profiles did not noticeably change after the injection of ATP, even though the PLL chains were completely complexed with ATP (r=1), highlighting that the PLL length drastically affects the modulation (Figure S20, Supporting Information).
Figure 5.
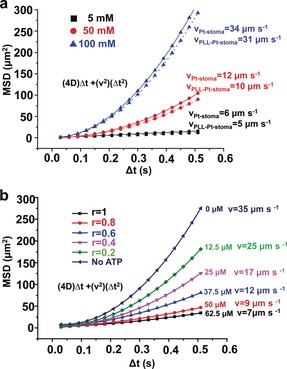
a) MSD and velocity of PtNP‐loaded stomatocytes (solid line) and PtNP‐loaded PLL‐stomatocytes (dotted line) in the presence of H2O2 at different concentrations. b) MSD and velocity of PtNP‐loaded PLL‐stomatocytes in the presence of ATP at different concentrations. The extracted MSDs were used as a qualitative indication of how active our motors are as a result of their transient binding or unbinding to ATP.
Finally, we investigated the ability of such nanomotors to transiently respond, with their output being regulated by an external dynamic process (Figure 6 a). To fulfill this goal, potato apyrase was introduced into the system to compete with PLL for the ATP that was added to the nanomotors, by decomposing ATP to AMP, the latter of which is unable to regulate the movement of the nanomotors (Figure S21, Supporting Information). To trigger the adaptive movement, 62.5 μm ATP (r=1) was added to the nanomotor system in the presence of apyrase, and the nanomotor movement was observed in real‐time. NTA showed that after the addition of ATP, two distinct regimes could be observed. In the first regime, the motion of the motors spontaneously decreased from the original 35 μm s−1 to a minimum value of 7 μm s−1 (Figure 6 b). The switched off state of the motors was, however, transient, and the motors slowly auto‐accelerated back to their initial state (second regime). The recovery process took much longer than the deceleration process, which was completed within 30 s. These results indicate that there is a reversible molecular complexing process inside this system. ATP induced the formation of ATP/PLL complexes to block the “mouth” of the stomatocytes, turning the motors into an “OFF” state; subsequently, the system was taken over by enzyme‐mediated ATP depletion, which turned the motors into an “ON” state owing to the increase of the hydrophilicity of the polymers, and the subsequent increased access of the substrate to the nanocavity of the stomatocytes.
Figure 6.
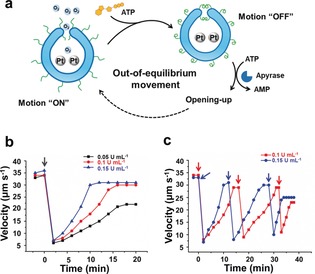
a) Schematic of the ATP‐mediated adaptive stomatocyte nanomotors. b) Velocity of the nanomotors as a function of time on the addition of ATP (62.5 μm) to a PtNP‐loaded PLL‐stomatocyte solution in the presence of different concentrations of apyrase. c) Three cycles of an adaptive nanomotor system upon the repeated addition of ATP (62.5 μm) showing the out‐of‐equilibrium movement of the system. The arrows in (b) and (c) indicate the addition of ATP.
To demonstrate control over this coupling of transient motor function, we studied the dynamic regulation of the motors under different apyrase levels (Figure 6 c). Notably, in all situations, the motors attained Brownian motion upon addition of ATP, followed by a spontaneous increase in velocity until it leveled off to almost the starting value. At the highest concentration of apyrase (0.15 U mL−1), the velocity returned to the initial value after just 10 min. When a lower enzyme concentration (0.1 U mL−1) was applied, the system reached its approximately original state of motion after a longer time (around 15 min), which shows that increasing the enzyme concentration leads to a faster recovery and a shorter lifetime of the temporal OFF state. At the lowest apyrase level (0.05 U mL−1), the motors also rapidly switched off movement, followed by a gradual speed increase but the final velocity (22 μm s−1) of the motors was significantly lower than the initial value, which was caused by the lower ATP hydrolytic rate. These observations demonstrate that the biocatalytic pathway that dissipates energy plays a crucial role in controlling the transient states of the motors.
Next, we investigated the fatigue behavior of our system to see whether this adaptive process can be re‐activated by consecutive ATP addition. When the system returned to its initial state, fresh ATP (62.5 μm) was re‐added into the motor solutions, leading to a new cycle (Figure S22, Supporting Information). To tune the periodicity of this transient behavior, we varied the level of apyrase and kept the ATP concentration constant. As expected, a significant enhancement of movement was observed when the enzyme level was increased from 0.1 to 0.15 U mL−1. In a control experiment, in which motion was recorded in the absence of ATP, the velocity of the motors was maintained for a sufficiently long time (40 min), which confirms that the amount of H2O2 is not limiting in the study (Figure S23, Supporting Information). These results indicate that our nanomotor system could be used to power cellular machines and act as transmitter module in ATP‐dependent physiological feedback systems.18
Conclusion
We have developed a transient stomatocyte nanosystem, activity of which can be regulated by a molecule‐mediated dynamic process, which captures the physicochemical complexity of a biological system in a synthetic system. The competition between ATP and PLL complexation on the surface of the stomatocytes, with the decomposition of ATP by apyrase, enables the precise regulation of the activity/movement in a catalytically and time‐programmed fashion. The ATP/PLL complexes have a considerable effect on the access of substrate into the cavity of the stomatocyte through the collapsed hydrophobic domains, leading to the diminished activity of the nanoreactors and speed of the nanomotors. Consumption of ATP switches the system back to the initial condition. Moreover, the temporal control over movement relies on the amount of ATP input and the rate of ATP consumption, endowing the system with life‐like properties. This design is distinct from traditional responsive motors that operate at a thermodynamic ground state. We expect that this concept could promote a novel class of artificial adaptive nanosystems, guided by biological design principles.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Acknowledgements
The authors acknowledge the Dutch Science Foundation (VICI grant), the ERC Advanced grant Artisym 694120, the Dutch Min‐istry of Education, Culture and Science (Gravitation program 024.001.035) and the European Union's Horizon 2020 research and innovation programme Marie Sklodowska Curie Innovative Training Networks Nanomed, (No. 676137) for funding.
H. Che, J. Zhu, S. Song, A. F. Mason, S. Cao, I. A. B. Pijpers, L. K. E. A. Abdelmohsen, J. C. M. van Hest, Angew. Chem. Int. Ed. 2019, 58, 13113.
The copyright line for this article was changed on 4 March 2020 after original online publication.
Contributor Information
Dr. Loai K. E. A. Abdelmohsen, Email: l.k.e.a.abdelmohsen@tue.nl.
Prof. Jan C. M. van Hest, Email: j.c.m.v.hest@tue.nl.
References
- 1.
- 1a. Guix M., Mayorga-Martinez C. C., Merkoçi A., Chem. Rev. 2014, 114, 6285–6322; [DOI] [PubMed] [Google Scholar]
- 1b. Ren L., Wang W., Mallouk T. E., Acc. Chem. Res. 2018, 51, 1948–1956; [DOI] [PubMed] [Google Scholar]
- 1c. Wang J., Gao W., ACS Nano 2012, 6, 5745–5751; [DOI] [PubMed] [Google Scholar]
- 1d. Sengupta S., Patra D., Ortiz-Rivera I., Agrawal A., Shklyaev S., Dey K. K., Córdova-Figueroa U., Mallouk T. E., Sen A., Nat. Chem. 2014, 6, 415–422; [DOI] [PubMed] [Google Scholar]
- 1e. Parmar J., Vilela D., Villa K., Wang J., Sánchez S., J. Am. Chem. Soc. 2018, 140, 9317–9331. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Badjić J. D., Balzani V., Credi A., Silvi S., Stoddart J. F., Science 2004, 303, 1845–1849; [DOI] [PubMed] [Google Scholar]
- 2b. Spudich J. A., Science 2011, 331, 1143–1144; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Koumura N., Zijlstra R. W., van Delden R. A., Harada N., Feringa B. L., Nature 1999, 401, 152–155; [DOI] [PubMed] [Google Scholar]
- 2d. Ismagilov R. F., Schwartz A., Bowden N., Whitesides G. M., Angew. Chem. Int. Ed. 2002, 41, 652–654; [Google Scholar]; Angew. Chem. 2002, 114, 674–676; [Google Scholar]
- 2e. Lewandowski B., De Bo G., Ward J. W., Papmeyer M., Kuschel S., Aldegunde M. J., Gramlich P. M., Heckmann D., Goldup S. M., D'Souza D. M., Science 2013, 339, 189–193. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Mei Y., Huang G., Solovev A. A., Ureña E. B., Mönch I., Ding F., Reindl T., Fu R. K., Chu P. K., Schmidt O. G., Adv. Mater. 2008, 20, 4085–4090; [Google Scholar]
- 3b. Gao W., Sattayasamitsathit S., Orozco J., Wang J., J. Am. Chem. Soc. 2011, 133, 11862–11864; [DOI] [PubMed] [Google Scholar]
- 3c. Ma X., Wang X., Hahn K., Sánchez S., ACS Nano 2016, 10, 3597–3605. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Wilson D. A., Nolte R. J., Van Hest J. C. M., Nat. Chem. 2012, 4, 268–274; [DOI] [PubMed] [Google Scholar]
- 4b. Wu Y., Wu Z., Lin X., He Q., Li J., ACS Nano 2012, 6, 10910–10916; [DOI] [PubMed] [Google Scholar]
- 4c. Peng F., Tu Y., van Hest J. C. M., Wilson D. A., Angew. Chem. Int. Ed. 2015, 54, 11662–11665; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11828–11831. [Google Scholar]
- 5.
- 5a. Pavlick R. A., Sengupta S., McFadden T., Zhang H., Sen A., Angew. Chem. Int. Ed. 2011, 50, 9374–9377; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9546–9549; [Google Scholar]
- 5b. Paxton W. F., Kistler K. C., Olmeda C. C., Sen A., Angelo S. K. St., Cao Y., Mallouk T. E., Lammert P. E., Crespi V. H., J. Am. Chem. Soc. 2004, 126, 13424–13431; [DOI] [PubMed] [Google Scholar]
- 5c. Wu Y., Si T., Gao C., Yang M., He Q., J. Am. Chem. Soc. 2018, 140, 11902–11905. [DOI] [PubMed] [Google Scholar]
- 6. Fletcher D. A., Mullins R. D., Nature 2010, 463, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Merindol R., Walther A., Chem. Soc. Rev. 2017, 46, 5588–5619; [DOI] [PubMed] [Google Scholar]
- 7b. della Sala F., Neri S., Maiti S., Chen J. L., Prins L. J., Curr. Opin. Biotechnol. 2017, 46, 27–33; [DOI] [PubMed] [Google Scholar]
- 7c. van Roekel H. W., Rosier B. J., Meijer L. H., Hilbers P. A., Markvoort A. J., Huck W. T., de Greef T. F., Chem. Soc. Rev. 2015, 44, 7465–7483. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Te Brinke E., Groen J., Herrmann A., Heus H. A., Rivas G., Spruijt E., Huck W. T., Nat. Nanotechnol. 2018, 13, 849; [DOI] [PubMed] [Google Scholar]
- 8b. Boekhoven J., Hendriksen W. E., Koper G. J., Eelkema R., van Esch J. H., Science 2015, 349, 1075–1079. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Hao X., Chen L., Sang W., Yan Q., Adv. Sci. 2018, 5, 1700591; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Maiti S., Fortunati I., Ferrante C., Scrimin P., Prins L. J., Nat. Chem. 2016, 8, 725–731; [DOI] [PubMed] [Google Scholar]
- 9c. Heuser T., Weyandt E., Walther A., Angew. Chem. Int. Ed. 2015, 54, 13258–13262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13456–13460. [Google Scholar]
- 10.
- 10a. Che H., Buddingh’ B. C., van Hest J. C. M., Angew. Chem. Int. Ed. 2017, 56, 12581–12585; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12755–12759; [Google Scholar]
- 10b. Che H., Cao S., van Hest J. C. M., J. Am. Chem. Soc. 2018, 140, 5356–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. della Sala F., Maiti S., Bonanni A., Scrimin P., Prins L. J., Angew. Chem. Int. Ed. 2018, 57, 1611–1615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1627–1631; [Google Scholar]
- 11b. Dhiman S., Jain A., Kumar M., George S. J., J. Am. Chem. Soc. 2017, 139, 16568–16575; [DOI] [PubMed] [Google Scholar]
- 11c. Van Ravensteijn B. G., Hendriksen W. E., Eelkema R., Van Esch J. H., Kegel W. K., J. Am. Chem. Soc. 2017, 139, 9763–9766. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Yehl K., Mugler A., Vivek S., Liu Y., Zhang Y., Fan M., Weeks E. R., Salaita K., Nat. Nanotechnol. 2016, 11, 184; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Cha T.-G., Pan J., Chen H., Salgado J., Li X., Mao C., Choi J. H., Nat. Nanotechnol. 2014, 9, 39–43; [DOI] [PubMed] [Google Scholar]
- 12c. Mao C., Sun W., Shen Z., Seeman N. C., Nature 1999, 397, 144–146. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Pijpers I. A., Abdelmohsen L. K., Williams D. S., van Hest J. C. M., ACS Macro Lett. 2017, 6, 1217–1222; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Meeuwissen S. A., Kim K. T., Chen Y., Pochan D. J., van Hest J. C. M., Angew. Chem. Int. Ed. 2011, 50, 7070–7073; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7208–7211; [Google Scholar]
- 13c. Kim K. T., Zhu J., Meeuwissen S. A., Cornelissen J. J., Pochan D. J., Nolte R. J., van Hest J. C. M., J. Am. Chem. Soc. 2010, 132, 12522–12524. [DOI] [PubMed] [Google Scholar]
- 14. Molnar J., Lorand L., Arch. Biochem. Biophys. 1961, 93, 353–363. [DOI] [PubMed] [Google Scholar]
- 15. Abdelmohsen L. K., Nijemeisland M., Pawar G. M., Janssen G.-J. A., Nolte R. J., van Hest J. C. M., Wilson D. A., ACS Nano 2016, 10, 2652–2660. [DOI] [PubMed] [Google Scholar]
- 16. Tu Y., Peng F., Sui X., Men Y., White P. B., van Hest J. C. M., Wilson D. A., Nat. Chem. 2017, 9, 480–486. [DOI] [PubMed] [Google Scholar]
- 17. Nijemeisland M., Abdelmohsen L. K., Huck W. T., Wilson D. A., van Hest J. C. M., ACS Cent. Sci. 2016, 2, 843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Dahl G., Philos. Trans. R. Soc. London Ser. B 2015, 370, 20140191; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Burnstock G., Nat. Rev. Drug Discovery 2008, 7, 575–599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary