

Abstract
We recently reported the discovery of phenylacetate decarboxylase (PhdB), representing one of only ten glycyl‐radical‐enzyme reaction types known, and a promising biotechnological tool for first‐time biochemical synthesis of toluene from renewable resources. Here, we used experimental and computational data to evaluate the plausibility of three candidate PhdB mechanisms, involving either attack at the phenylacetate methylene carbon or carboxyl group [via H‐atom abstraction from COOH or single‐electron oxidation of COO− (Kolbe‐type decarboxylation)]. In vitro experimental data included assays with F‐labeled phenylacetate, kinetic studies, and tests with site‐directed PhdB mutants; computational data involved estimation of reaction energetics using density functional theory (DFT). The DFT results indicated that all three mechanisms are thermodynamically challenging (beyond the range of many known enzymes in terms of endergonicity or activation energy barrier), reflecting the formidable demands on PhdB for catalysis of this reaction. Evidence that PhdB was able to bind α,α‐difluorophenylacetate but was unable to catalyze its decarboxylation supported the enzyme's abstraction of a methylene H atom. Diminished activity of H327A and Y691F mutants was consistent with proposed proton donor roles for His327 and Tyr691. Collectively, these and other data most strongly support PhdB attack at the methylene carbon.
Keywords: glycyl radical enzyme, Kolbe decarboxylation, phenylacetate decarboxylase, radical reactions, reaction mechanisms
Three candidate mechanisms for the novel glycyl radical enzyme (GRE) phenylacetate decarboxylase (PhdB) were evaluated using a combination of in vitro experimental data and computational data involving estimation of reaction energetics. The data best support a methylene carbon attack (green arrows) that differs substantially from the reported mechanism for p‐hydroxyphenylacetate decarboxylase.

Introduction
Phenylacetate decarboxylase (PhdB), which was recently discovered via activity‐guided metaproteomic studies of toluene‐producing microbial communities,1 has promise as a catalyst for first‐time biochemical synthesis of toluene from renewable resources. PhdB represents one of ten glycyl radical enzyme (GRE) reaction types that have been discovered to date, namely, pyruvate formate‐lyase (EC 2.3.1.542), anaerobic ribonucleotide reductase (EC 1.17.4.13), benzylsuccinate synthase (EC 4.1.99.114, 5, 6), p‐hydroxyphenylacetate decarboxylase (EC 4.1.1.837, 8, 9), B12‐independent glycerol (and propane‐1,2‐diol) dehydratase (EC 4.2.1.3010), choline trimethylamine‐lyase (EC 4.3.99.411, 12), and the very recently discovered (since 2017) trans‐4‐hydroxy‐l‐proline dehydratase,13 phenylacetate decarboxylase,1 indoleacetate decarboxylase,14 and isethionate sulfite‐lyase.15, 16
GREs share certain features, including 1) a conserved glycyl radical motif (RVxG[FWY]x6‐8[IL]x4Qx2[IV]x2R1, 17) near the C terminus of the protein, 2) a mechanism involving abstraction of a hydrogen atom from the substrate (p‐hydroxyphenylacetate decarboxylase is a variation on this, and catalyzes de facto hydrogen‐atom abstraction as the concerted abstraction of an electron from the carboxyl group and a proton from the phydroxy group9, 18), 3) a conserved cysteine residue near the middle of the protein sequence (the location of the thiyl radical in the active site that initiates hydrogen‐atom abstraction from the substrate), 4) a cognate activase, which is a radical S‐adenosylmethionine (SAM) enzyme, and 5) operation under strictly anaerobic conditions due to irreversible inactivation upon exposure to molecular oxygen.
There are three known GRE decarboxylases: p‐hydroxyphenylacetate decarboxylase (aka CsdBC or HpdBC), phenylacetate decarboxylase (PhdB), and indoleacetate decarboxylase (IAD). Noteworthy similarities and differences exist among these three decarboxylases: 1) All three act on aryl acetates derived from aromatic amino acids (the aryl acetates are p‐hydroxyphenylacetate, phenylacetate, and indoleacetate derived, respectively, from tyrosine, phenylalanine, and tryptophan), but only PhdB acts on an aromatic moiety that is not substituted with the electronegative elements O or N; 2) There is a single subunit type for PhdB1 and IAD (for Clostridium scatologenes 14), but large and small subunits for CSD/HPD; and 3) The three decarboxylases are each very specific to their cognate substrates, and do not use each other's substrates. For example, IAD does not use phenylacetate or p‐hydroxyphenylacetate,14 PhdB does not use p‐hydroxyphenylacetate,1 and CsdBC does not use phenylacetate.8, 19
Several factors suggest that PhdB has a different mechanism than other known GRE decarboxylases, including that the aryl acetate substrates of p‐hydroxyphenylacetate decarboxylase and indoleacetate decarboxylase enable H‐bonding to the aromatic moiety or its substituents, whereas this is not possible with the unsubstituted ring of phenylacetate. Here, we used experimental and computational data to evaluate the plausibility of three candidate PhdB mechanisms, involving either attack at the phenylacetate methylene carbon or carboxyl group (protonated versus deprotonated). In vitro experimental data included assays with F‐ and D‐labeled phenylacetate, kinetic studies, and tests with site‐directed PhdB mutants; computational data involved estimation of reaction energetics using density functional theory (DFT). Collectively, the data most strongly support PhdB attack of the phenylacetate methylene carbon via a decarboxylation mechanism that differs substantially from that reported for p‐hydroxyphenylacetate decarboxylase.
Results and Discussion
Considering hydrogen‐atom abstraction from the methylene carbon of phenylacetate by PhdB
Characterized GREs initiate transformation of their substrate by abstracting a hydrogen atom.17, 20 We were interested in the position of the hydrogen atom abstracted by PhdB, specifically, whether hydrogen‐atom abstraction is from the methylene carbon or carboxyl group of phenylacetic acid. To examine whether the hydrogen atom was abstracted from the methylene carbon, we carried out in vitro experiments with α,α‐difluorophenylacetate as a substrate for PhdB. The rationale for these experiments was as follows: because C−F bonds are very strong (ca. 500 kJ mol−1 bond dissociation energy in difluoromethane21), their presence in α,α‐difluorophenylacetate would preclude PhdB attack at the methylene carbon if this were the site of hydrogen‐atom abstraction. However, these methylene C−F bonds would not be expected to interfere with formation of the expected product, [methyl‐F2]difluorotoluene, if the hydrogen‐atom abstraction site were from the carboxyl group.
As shown in Figure 1, there was no [methyl‐F2]toluene produced in assays with radical‐activated PhdB and α,α‐difluorophenylacetate. PhdB activity for the positive control (13C‐labeled phenylacetate) was robust (Figure 1), and GC/MS analysis of [methyl‐F2]toluene standards demonstrated that this product should have been easily detectable at a concentration range relevant to PhdB activity in this experiment. We also searched for production of [methyl‐F1]toluene, which would have resulted from the abstraction of an F atom from the methylene carbon of α,α‐difluorophenylacetate if this reaction were possible; no signal was detectable for this product. Overall, the observation of no detectable activity with α,α‐difluorophenylacetate is consistent with the deduction that PhdB abstracts a hydrogen atom from the methylene carbon of phenylacetate.
Figure 1.

In vitro production of substituted toluenes in an anaerobic assay containing purified, recombinant PhdB and PhdA, and SAM. The substituted toluenes are [methyl‐13C]toluene from phenylacetate‐2–13C (positive control, n=2) and [methyl‐F2]toluene from α,α‐difluorophenylacetate (n=3).
An alternative explanation for the results in Figure 1 could be that α,α‐difluorophenylacetate does not actually bind to the enzyme, in which case negative results would not be informative about the site of hydrogen‐atom abstraction. However, if it were shown that α,α‐difluorophenylacetate was binding to PhdB and was a competitive inhibitor of phenylacetate, this would support the deduction offered above, that α,α‐difluorophenylacetate is not acted upon by PhdB because hydrogen‐atom abstraction is not possible at the F‐substituted methylene site. We investigated whether α,α‐difluorophenylacetate was binding to PhdB and acting as a competitive inhibitor for phenylacetate, as described below.
α,α‐Difluorophenylacetate as a competitive inhibitor of PhdB and the implications for hydrogen‐atom abstraction from the methylene carbon
To test whether α,α‐difluorophenylacetate was actually binding to PhdB, we carried out kinetic studies to determine whether α,α‐difluorophenylacetate was a competitive inhibitor of phenylacetate for PhdB. Kinetic studies involving phenylacetate‐2‐13C as the substrate, α,α‐difluorophenylacetate as the potential competitive inhibitor, and radical‐activated PhdB provided evidence that α,α‐difluorophenylacetate is indeed a competitive inhibitor of phenylacetate (Figure 2 A,B, Table S1 in the Supporting Information). These results are consistent with the above deduction about hydrogen‐atom abstraction from the methylene carbon.
Figure 2.

Kinetic studies of PhdB in the presence of a substrate, phenylacetate‐2‐13C, and competitive inhibitor, α,α‐difluorophenylacetate. A) Michaelis–Menten plots of velocity [μU mg protein−1] vs. phenylacetate concentration [mm]. B) Lineweaver–Burk plots for the same data set shown in A. For biological replicates, symbols represent averages and error bars represent one standard deviation (15 of 20 data points represent duplicates from independent experiments, but error bars are not visible in all cases; see Table S1 for more details on statistics).
The kinetic data for different concentrations of α,α‐difluorophenylacetate (0, 0.5, 1, and 2 mm) showed a strong fit (R 2=0.9745) to a competitive inhibition model using nonlinear regression (Table S1; Prism, GraphPad Software, Inc.). Figure 2 B clearly reveals trends characteristic of competitive inhibition, that is, assays with different inhibitor concentrations that show the same V max values (the y intercept in Lineweaver–Burk plots represents 1/V max) but different apparent K M values (the x intercept in Lineweaver–Burk plots represents −1/K M). The K i value determined for α,α‐difluorophenylacetate was 0.37±0.03 mm, whereas the K M value for phenylacetate was 2.54±0.38 mm (Table S1). The K M value for PhdB was similar to that determined for another GRE decarboxylase, HpdBC (2.8 mm 7), although a lower K M value of 0.65 mm has also been reported for the same enzyme.8 A Ki value for α,α‐difluorophenylacetate that is lower than the K M value for phenylacetate is suggestive of tighter binding at the active site by the fluorinated analogue. Fluorine‐substituted analogues have been reported to have an enhanced binding affinity toward their target proteins for multiple possible reasons;22 in the case of PhdB, electron withdrawal by the highly electronegative fluorine atoms on methylene CF2 would likely decrease the pK a of the substrate and potentially lead to stronger binding. As the van der Waals radius of fluorine (1.47 Å) is only slightly larger than that of hydrogen (1.20 Å), its size should not contribute to a significant change in the mode of substrate binding to the protein.22
Computed energetics for candidate PhdB decarboxylation mechanisms
An important consideration in assessing the site of attack by PhdB is the energy barriers associated with candidate mechanisms. Three candidate mechanisms (Figure 3) were subjected to scrutiny using DFT computations. Structures were optimized and characterized as minima or transition‐state structures using the SMD(chloroform)‐ωB97X‐D/6‐311+G(d,p) method;23 details can be found in the Supporting Information, along with results from other levels of theory (although gas‐phase and aqueous calculations were explored, chloroform results are shown here, as the dielectric of chloroform is often suggested to be similar to that of an enzyme active site24).
Figure 3.
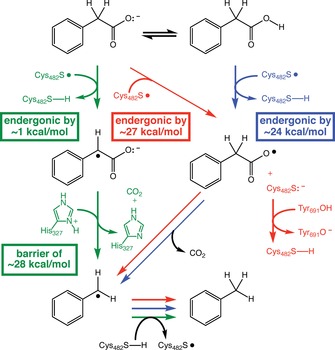
Three candidate PhdB decarboxylation mechanisms considered in this study (highlighted with green, red, and blue arrows; see text). Residues indicated include Cys482, the highly conserved site of the thiyl radical, and putative proton donors His327 and Tyr691. Energies shown are predicted free energies [SMD(chloroform)‐ωB97X‐D/6‐311+G(d,p)]. Energies predicted for the gas phase [ωB97X‐D/6‐311+G(d,p)] are ≈7 kcal mol−1 (green “barrier” box) and ≈22 kcal mol−1 (red and blue boxes).
As discussed above, one possible mechanism for PhdB‐catalyzed phenylacetate decarboxylation involves hydrogen‐atom abstraction from the methylene carbon (Figure 3, left, green); this step is predicted to be only slightly endergonic (+0.8 kcal mol−1; computed using HC(O)NHCH2CH2S. as a model of the radical form of Cys482). After hydrogen‐atom abstraction by the thiyl radical at Cys482, the mechanism involves decarboxylation and net protonation of the substrate radical by the imidazolium group of His327. Protonation is stoichiometrically required for the PhdB reaction when the substrate is bound as the anion,1 and His327 is the closest potential proton donor to phenylacetate identified in the PhdB homology model presented previously.1 According to this homology model, His327 (π‐N) is located approximately 5.2 Å from the phenylacetate methylene carbon and 3.1 Å from the carboxyl oxygen (note that interatomic distances calculated from the homology model should be viewed with caution, as they are not based on experimental protein structural data). This mechanism would require His327 to be in its cationic, conjugate acid form. Although the estimated pK a of the histidine side chain is 6.5,25 which would not favor the cationic form at a physiological pH of 7, this generic pK a value is not specific to this PhdB residue, and would be altered (likely increased) by the presence of the anionic substrate. Thus, we cannot rule out the cationic form. [Note that we have not attempted to refine pK a values in this study with available pK a prediction software (continuum electrostatic calculations) in conjunction with the PhdB homology model because of concerns about the propagation of inherent error in homology models as compared to experimentally determined crystal structures; an additional concern is that available pK a prediction methods have particular limitations with regard to accurate prediction of doubly protonated histidine,26 which is directly relevant to His327.] The calculated energy barrier for the concerted decarboxylation/protonation step to form a benzyl radical (computed using imidazolium as a model of protonated His327) is 28 kcal mol−1, which is considerably higher than the approximately 20 kcal mol−1 range of barriers considered characteristic of enzyme‐catalyzed reactions.27 In summary, this mechanism involves (Figure 3, left, green): 1) An H‐atom abstraction step between the Cys482 thiyl radical and the substrate to form a substrate radical, 2) a concerted decarboxylation/protonation step (note that the reaction coordinate for this reaction involves an asynchronous combination of initial H‐atom transfer from protonated His327 to the substrate and subsequent decarboxylation/electron transfer, rather than simple two‐electron decarboxylation and proton transfer events; see Supporting Information for additional details), and 3) an H‐atom transfer step between the resultant benzyl radical and the Cys482 thiol to regenerate the Cys482 thiyl radical.
In an alternative mechanism, hydrogen‐atom abstraction by the Cys482 thiyl radical could occur at the protonated carboxyl group (Figure 3, right, blue). Subsequent decarboxylation would result in formation of the benzyl radical (Figure 3). The initial hydrogen atom transfer [computed using HC(O)NHCH2CH2S. as a model of the Cys482 radical] is predicted to be uphill by approximately 24 kcal mol−1 (also above the upper end of the range considered to characterize enzymatic reactions), with the subsequent steps being more energetically favorable.
A third possibility involves electron transfer from the substrate carboxylate to the Cys482 thiyl radical (Figure 3, center, red). This mechanism is analogous to the Kolbe‐type decarboxylation proposed for p‐hydroxyphenylacetate decarboxylase,9, 18 with the important distinction that there is no concerted proton abstraction from the substrate proposed here for PhdB. Predicting the energy for electron transfer with DFT has proven difficult, with estimates varying according to the method used (see the Supporting Information for details). Still, we predict with SMD(chloroform)‐ωB97X‐D/6‐311+G(d,p) that electron transfer from phenylacetate to a model of Cys482S. [i.e., HC(O)NHCH2CH2S. with internal hydrogen‐bonding] is uphill by 27 kcal mol−1, again outside the upper end of the typical enzymatic range. Our computational results also suggest that this electron transfer process will be more difficult, by >10 kcal mol−1, for the fluorinated substrate analogue (α,α‐difluorophenylacetate), as expected for a structure bearing electron‐withdrawing groups on the putative electron donor. This result could provide an alternative explanation for the experimental results shown in Figure 1, namely, that α,α‐difluorophenylacetate decarboxylation by PhdB is thermodynamically infeasible. For the Kolbe‐type mechanism, a proton donor is required to protonate the Cys482 thiolate (Figure 3, red). The proposed proton donor for this mechanism is Tyr691, which, based on the PhdB homology model, is closer to the Cys482 S atom (5.8 Å) than is His327 (7.8 Å).
Based on our computed energies, there is no clear‐cut preferred mechanism. All three mechanisms examined are predicted to have barriers that are higher than expected for an enzymatic reaction, but not by much. These barriers can, of course, be modulated by the surrounding enzyme. For the mechanism involving H‐atom abstraction from the methylene carbon of phenylacetate (Figure 3, left, green), the enzyme would need to lower the barrier for concerted H‐transfer/decarboxylation, which could be accomplished by improving the electron‐accepting ability of His327. For the Kolbe‐type mechanism (Figure 3, center, red), the enzyme would need to promote initial electron transfer, for example, by H‐bonding to Cys482, so as to selectively stabilize the thiolate anion produced upon electron‐transfer. For the mechanism involving initial H‐atom transfer from CO2H (Figure 3, right, blue), the enzyme would need to modulate the substrate pK a to favor the conjugate acid form and would also need to lower the barrier for H‐atom transfer. Unfortunately, without an experimentally determined, high‐resolution PhdB structure, we are unable to assess the likelihood of each of these possibilities.
Considering PhdB attack of the substrate in protonated versus unprotonated form
In weighing the feasibility of the three candidate mechanisms, one consideration is whether the substrate is likely to be protonated (as it is in Figure 3, blue, but not in Figure 3, green or red). The following two factors weigh against attack (hydrogen‐atom abstraction) at the protonated carboxyl group (Figure 3, blue): 1) Protonation of the carboxyl group would be unlikely at a circumneutral physiological pH and 2) PhdB activity on the protonated substrate would be inconsistent with a proposed physiological role for the reaction (see below).
Regarding PhdB attack at the protonated carboxyl group, the pK a of phenylacetic acid is 4.31,28 so the acid would be >99.5 % deprotonated at a physiological pH of 7, for example. Unless active‐site residues modulate the acidity of the substrate to a pK a>7, the abstraction of a hydrogen‐atom from the carboxyl group seems unlikely, as the substrate would be predominantly ionized under likely physiological conditions. Notably, in the proposed mechanism for p‐hydroxyphenylacetate decarboxylase, the analogous substrate is presented as being bound to the enzyme in fully ionized form based on a continuum electrostatic model.9
A possible selective advantage of the PhdB reaction has been proposed1 that has relevance to the protonation state of the substrate: phenylacetate decarboxylation could provide an advantage through intracellular pH homeostasis and/or development of a proton motive force (pmf). To clarify, if the anion phenylacetate were imported into the cell, the PhdB‐catalyzed decarboxylation to toluene consumed a proton from the cytoplasm (consistent with the balanced reaction of C8H7O2 −+H+→C7H8+CO2), and the neutral reaction products toluene and CO2 exited the cell (e.g., by diffusion), the result would be alkalinization of the cytoplasm and indirect development of a pmf (by depletion of protons from the cytoplasm rather than the canonical pumping of protons across the cytoplasmic membrane). Clearly, this selective advantage would not be operative if the substrate were protonated.
The fate of the methylene hydrogen atoms of phenylacetate in the PhdB reaction
Depending on whether PhdB attack is at the methylene carbon or the carboxyl group, the fate of the methylene hydrogen atoms of phenylacetate in the PhdB reaction could be of interest for elucidating the PhdB mechanism. In the former case, a C−H methylene bond is broken, and the fate of the released hydrogen atom is non‐obvious. In the latter case, no C−H bonds on the methylene carbon are broken, so the fate of these hydrogen atoms is not in question.
Considering the possibility that hydrogen‐atom abstraction is from the methylene carbon, we investigated the fate of the hydrogen atoms from the methylene carbon during the PhdB reaction. In vitro studies of PhdB conducted with a range of deuterium‐labeled phenylacetates, including α,α‐D2‐phenylacetate (C6H5CD2CO2 −), 2,3,4,5,6‐D5‐phenylacetate (C6D5CH2CO2 −), and α,α‐2,3,4,5,6‐phenyl‐D7‐acetate (C6D5CD2CO2 −), are described in detail in the Supporting Information. In summary, the data provide strong evidence that, if a hydrogen atom is abstracted from the methylene carbon of phenylacetate, it is retained on the toluene product. Analogous results were reported for another GRE, benzylsuccinate synthase (BSS), where it was found that the hydrogen atom abstracted from the methyl carbon of toluene (the substrate of BSS) was retained in the succinyl moiety of the benzylsuccinate product.29, 30 By analogy to BSS, it is likely that a methylene hydrogen atom abstracted by the PhdB thiyl radical at Cys482 to form a thiol (Figure 3, green), would then be donated back from Cys482 to quench the substrate benzyl radical (Figure 3).
Site‐directed mutations of PhdB
A number of putative active‐site residues were targeted for mutagenesis to assess their criticality to PhdB function. The rationales for selection of these residues are given below along with the effects of the mutations on PhdB activity.
Highly conserved Gly and Cys residues play an essential role in GREs. All GREs share the same mechanism for radical generation that is initiated by their cognate activating enzyme.17, 20 Radical activation involves reductive cleavage of a molecule of SAM bound to the [4Fe−4S]+ cluster of the activating enzyme. Cleavage of SAM results in the formation of a 5′‐deoxyadenosyl (5′‐dAdo) radical and methionine. The 5′‐dAdo radical in turn stereospecifically abstracts a hydrogen atom from the glycine residue of the highly conserved glycyl radical motif (Gly815 in PhdB; Figure 4 A). During catalysis, the glycyl radical comes into close proximity to a conserved active‐site cysteine residue (Cys482 in PhdB; Figure 4 A), generating a thiyl radical upon hydrogen‐atom abstraction, which triggers catalytic attack of the GRE′s substrate to form a substrate radical. We constructed and assayed loss‐of‐function mutations of the active‐site, radical‐propagating glycine and cysteine residues in PhdB, whereby these residues were substituted with alanine. As expected, these mutations resulted in total loss of activity for Cys482 (C482A; Figure 4 B) and Gly815 (G815A1). Comparable results have been reported for all other GREs for which such mutations have been tested (for example, C489A and G821A in CutC;11 C438S and G817A in HypD13).
Figure 4.

A) Multiple‐sequence alignment of selected active‐site regions of GREs including the two known copies of PhdB. Sequences used in this alignment are indicated in parenthesis [as Protein Data Bank (PDB), GenBank, or UniProt accession number]. The “s” and “l” suffixes for PhdB stand for sewage and lake cultures, respectively. CsIAD, C. scatologenes indoleacetate decarboxylase; OsIAD, Olsenella scatoligenes IAD; PflB, pyruvate formate‐lyase; BssA, benzylsuccinate synthase α subunit; CutC, choline trimethylamine‐lyase; Gdh, glycerol dehydratase; HypD, trans‐4‐hydroxy‐l‐proline dehydratase; and DvIseG, Desulfovibrio vulgaris isethionate sulfite‐lyase. Numbering is based on PhdB. B) Relative PhdB activity of key active‐site mutants relative to the wild‐type (WT) version (WT=100 % by definition). Error bars represent one standard deviation. The E132A mutant was used as a positive control; note that residue 132 is an E in the sewage culture version of PhdB but is an A in the lake sediment version of PhdB, so this E132A mutation was not expected to disrupt activity. The G815A results were reported previously.1 C) Predicted positioning of residues targeted for mutations, based on a homology model of PhdB.1 The bound phenylacetate substrate is shown in gray.
As discussed above, homology modeling of the PhdB active site with bound substrate1 indicates a Tyr691 residue in the vicinity of Cys482 and a His327 residue in the vicinity of the phenylacetate carboxyl group (Figure 4 C). These residues were proposed as proton donors for certain PhdB mechanisms: Tyr691 for the Kolbe‐type decarboxylation mechanism (Figure 3, red) and His 327 for the methylene carbon attack (Figure 3, green). The roles of these residues were examined by creating and assaying Y691F and H327A PhdB mutants. The Y691F mutant showed no activity (Figure 4 B), supporting, but not proving, that it could have a role in protonating the Cys482 thiolate in the Kolbe‐type decarboxylation mechanism; an additional potential role for this residue is discussed in the following paragraph. Although the proton‐donating role of Tyr691 may appear unlikely based on the estimated pK a of a generic tyrosine side chain (10.025), the actual pK a may be modulated due to hydrogen‐bonding to nearby Ser689 (Figure 4 C; 4.2 Å separates the O atoms in the side chains of Ser689 and Tyr691) and electrostatic interactions with the Cys482 thiolate (Figure 3, red). The H327A mutant showed an 88 % decrease in activity (Figure 4 B), strongly suggestive of a role in PhdB catalysis. However, one might expect complete loss of activity if His327 were fully responsible for proton donation in the methylene attack mechanism (Figure 3, green). It is conceivable that Tyr691, which is located somewhat further from the phenylacetate carboxyl O atom than His327 (5.0 vs. 3.1 Å) but is a similar distance from the phenylacetate methylene carbon (5.2 to 5.3 Å), could have partially compensated for the loss of protonation from His327 in the H327A mutant.
The PhdB homology model also indicates a hydrophobic binding pocket comprising residues Trp495, Tyr691, and Val693 (Figure 4 C), which appear to be positioned to sterically orient the aromatic ring of phenylacetate within the active site. Similarly, a hydrophobic region has been described in the GRE benzylsuccinate synthase that helps position the aromatic ring of the toluene substrate.20 In this study, the Y691F mutant showed a complete loss of activity (Figure 4 B), despite the preservation of the aromatic ring in the mutant as phenylalanine. This result suggests a role for Tyr691 beyond substrate binding. Following from the above discussion about Tyr691 as a potential proton donor, it is possible that this residue has a primary role as a proton donor and a secondary role in substrate binding. The W495F mutant also showed a complete loss of activity, despite the preservation of the aromatic ring in the mutant as phenylalanine; the PhdB homology model suggests the possibility for aromatic (π) stacking between the W495 residue and the phenylacetate substrate (Figure 4 C).
Superimposition of the substrate‐bound active site of modeled PhdB1 with that of p‐hydroxyphenylacetate decarboxylase, CsdB (PDB ID: https://www.rcsb.org/structure/2YAJ), and multiple‐sequence alignment of PhdB, CsdB, and other GREs (Figure 4 A), reveal additional conserved residues that could be involved in substrate binding and catalysis. Notably, the Cys‐loop Glu residue (‐CXE‐), conserved across most known GREs, is substituted with a Gln (‐CXQ‐) residue in both known PhdB versions,1 while in PflB and BssA it is substituted by a Ser residue (Figure 4 A). In some GREs, the Cys‐loop Glu residue has been described as being involved in acid‐base catalysis, such as deprotonation of a hydroxy group in trans‐4‐hydroxy‐l‐proline dehydratase, choline trimethylamine‐lyase, and B12‐independent glycerol (and propane‐1,2‐diol) dehydratase,13, 20 or protonation of the thiolate group on the highly conserved active‐site Cys in p‐hydroxyphenylacetate decarboxylase.9, 18 In this study, we used sitedirected mutagenesis to replace the Cys‐loop Gln residue in PhdB with a Glu residue (Q484E), which resulted in complete loss of activity; similarly, the Q484A and Q484L mutants showed negligible activity (Figure 4 B). The result for Q484E provides a stark contrast with the other two known GRE decarboxylases, p‐hydroxyphenylacetate decarboxylase (CsdBC/HpdBC) and indoleacetate decarboxylase (IAD), both of which contain a Glu (rather than Gln) residue in this Cys‐loop position14, 18 (Figure 4 A). This is an indication that PhdB uses a different catalytic mechanism than these other GRE decarboxylases. Another indication of a different catalytic mechanism is that CsdBC has an unusual proposed mechanism for hydrogen atom abstraction that involves abstraction of a proton from the p‐hydroxy group of p‐hydroxyphenylacetate,9, 18 which is clearly not possible with the unsubstituted phenylacetate substrate of PhdB. The Q484E mutant result for PhdB (Figure 4 B) also stands in contrast to other GREs, for which substitution of a conserved Cys‐loop Glu with Gln resulted in abolished activity; this was observed for the E440Q mutation in trans‐4‐hydroxy‐l‐proline dehydratase13 as well as for the E491Q mutation in choline trimethylamine‐lyase.31 In the latter enzyme, an E491A mutation also abolished activity.31 While the role of the Cys‐loop Gln residue in PhdB is not yet clear, the mutant results and comparisons with other GREs provide some basis for speculation. The Q484E mutant results for PhdB suggest that, unlike proposed roles for a conserved Cys‐loop Glu in other GREs, the role of the conserved Cys‐loop Gln in PhdB (Q484) is probably not direct involvement in acid‐base catalysis, since the proton is considerably more exchangeable for Glu than Gln, and the Q484E mutant was inactive.
Mutant versions of PhdB were subjected to size‐exclusion chromatography (SEC) to verify that they behaved similarly to wild‐type PhdB in solution, which would be consistent with proper folding. The tested PhdB mutants eluted at the same retention volume as the wild‐type protein (Figure S1), indicating no perceptible changes in folding or oligomerization state of the mutant proteins relative to the wild‐type. These results suggest that observed changes in activity of the PhdB mutants did not result from changes in folding, but rather from the targeted biochemical change in the active site. Notably, the SEC results indicate that wild‐type PhdB occurs as a functional homodimer in solution, as the molecular weight estimated by SEC was approximately 200 kDa and the calculated, sequence‐based value is 95.6 kDa (accounting for the N‐terminal GH scar; see Experimental Section). Other single‐subunit GREs that have been characterized also occur as homodimers.14, 16, 17, 20
Conclusion
Collectively, the data presented here most strongly support PhdB attack of the phenylacetate methylene carbon (Figure 3, green) rather than attack of the carboxylate group via a Kolbe‐type decarboxylation (single‐electron oxidation; Figure 3, red) or hydrogen‐atom abstraction from the protonated carboxyl group (Figure 3, blue). Evidence that PhdB was able to bind α,α‐difluorophenylacetate (Figure 2) but was unable to catalyze its decarboxylation (Figure 1) supported the enzyme's abstraction of a methylene H atom. Further, the methylene attack on the ionized substrate is consistent with the pK a of phenylacetic acid and with a hypothesis about the selective physiological advantage of PhdB acting on phenylacetate rather than phenylacetic acid (discussed above). DFT calculations of reaction energetics do not clearly favor any of the three candidate mechanisms, although they may provide an alternative explanation for the α,α‐difluorophenylacetate results (that is, the decarboxylation reaction is much less favorable with the fluorinated substrate for the Kolbe‐type mechanism). Diminished activity of H327A and Y691F mutants was consistent with proposed proton donor roles for His327 and Tyr691, although these results more strongly support the role of Tyr691, which could play a role in the Kolbe‐type mechanism or the methylene attack mechanism (discussed above): 0 % activity for the Y691F mutant versus 12 % residual activity for the H327A mutant (Figure 4 C).
If true, the methylene attack mechanism (Figure 3, green) would differ dramatically from the Kolbe‐type decarboxylation that has been proposed for p‐hydroxyphenylacetate decarboxylase,9, 18 another GRE decarboxylase with a similar substrate. Indeed, even the Kolbe‐type decarboxylation mechanism described here for PhdB (Figure 3, red), if true, would differ from that for p‐hydroxyphenylacetate decarboxylase and be unusual among all other known GREs, as it only entails electron abstraction from the substrate, whereas other characterized GREs catalyze hydrogen‐atom (electron plus proton) abstraction (or de facto hydrogen‐atom abstraction, in the case of p‐hydroxyphenylacetate decarboxylase, as discussed above). Experimental characterization of the PhdB structure will be needed to better assess the mechanism of this intriguing enzyme.
Experimental Section
Unless noted otherwise, all protein preparations and assays were carried out under strictly anaerobic conditions1 in an anaerobic glove box (Type B, Coy Laboratory Products, Inc., Grass Lake, MI, USA) filled with a nominal gas composition of 85 % N2/10 % CO2/5 % H2 (ultra‐high purity, anaerobic mixture) and maintained at ambient temperature (≈22 °C). All aqueous solutions were prepared in high‐purity water (18 MΩ resistance; Barnstead Nanopure system, Thermo Scientific, Waltham, MA) that was autoclaved and nitrogen‐purged before use.
Protein expression and purification: Expression and anaerobic purification of phenylacetate decarboxylase activating enzyme, PhdA (UniProt ID: A0A2P6WDZ2), and phenylacetate decarboxylase, PhdB (UniProt ID: A0A2P6WDZ5), were performed under strictly anaerobic conditions as described previously.1 Briefly, codon‐optimized phdA and phdB were cloned into a modified pET28b vector producing N‐terminal hexahistidine‐tagged proteins. In the case of PhdA, the construct yielded His6‐PhdA, and in the case of PhdB, the gene encoding Maltose‐Binding Protein (MBP) was inserted between the N‐terminal hexahistidine tag and phdB, yielding a His6‐MBP‐PhdB fusion protein. The His6‐MBP tag was cleavable using Tobacco Etch Virus (TEV) protease, yielding full‐length PhdB (with an N‐terminal scar containing G and H residues upstream of methionine). Bacterial strains and plasmids used in this study are listed in Table S2.
Strain and plasmid availability: Strains and plasmids listed in Table S2 along with their associated information (annotated GenBank‐format sequence files) have been deposited in the public version of the JBEI Registry (https://public-registry.jbei.org) and are physically available from the authors and/or addgene (http://www.addgene.org) upon request.
In vitro, GC‐MS‐based assays for labeled toluene production using recombinant PhdA and PhdB: Assays measuring the production of toluene from labeled phenylacetate in the presence of recombinant PhdA and PhdB were carried out under strictly anaerobic conditions, as described previously.1, 19 Briefly, assays contained approximately 0.2 mm PhdA, 2 mm dithionite, 2.5 μm PhdB, 2 mm SAM [S‐(5′‐adenosyl)‐l‐methionine chloride dihydrochloride; Sigma–Aldrich], and 2.5 mm substrate (either phenylacetate‐2–13C or D‐ or F‐labeled phenylacetate, as described in the following sections) in buffer containing 50 mm Tris (pH 7.5), 150 mm NaCl, 1 mm MgCl2, 5 mm (NH4)2SO4, and 5 mm DTT (dithiothreitol) in a final volume of 1 mL or 1.5 mL. Assays were performed in 4‐mL glass vials fitted with 13‐mm PTFE Mininert screw‐cap valves (Sigma–Aldrich), and the vials were shaken at low speed on a tabletop orbital shaker. Labeled toluene production was assayed by sampling 100 μL of headspace with a 500‐μL gastight syringe (Sample‐Lok series A‐2; Sigma–Aldrich) and immediately analyzing the gas sample by electron ionization GC–MS in selected ion monitoring (SIM) mode. Detection and quantification of [methyl‐13C]toluene (from phenylacetate‐2‐13C) has been described in detail elsewhere.1, 19 The detection and quantification of D‐ or F‐labeled toluene was confirmed with expected ions, as noted in the following sections. Quantitative standards, when available, were prepared with the same headspace/liquid ratios as the assays, and a dimensionless Henry's constant of 0.271 was used to calculate the aqueous concentration. Controls were run with each experiment, as described below.
In vitro PhdB studies with D‐ and F‐labeled phenylacetate: For various mechanistic studies, labeled analogues of phenylacetic acid were used wherein D or F atoms replaced H atoms on the methylene and/or ring carbons. Labeled phenylacetic acid analogues included: α,α‐D2‐phenylacetic acid (Sigma–Aldrich, 98 atom% D), 2,3,4,5,6‐D5‐phenylacetic acid (Sigma–Aldrich, 98 atom% D), α,α‐2,3,4,5,6‐phenyl‐D7‐acetic acid (Sigma–Aldrich, 98 atom% D), and α,α‐difluorophenylacetic acid (99 %, Alfa‐Aesar, Haverhill, MA). All substrates were prepared in 50 mm Tris (pH 7.5) at a stock concentration of 8 mm. All experiments involving D‐ or F‐labeled phenylacetic acid were performed alongside controls containing phenylacetic acid‐2–13C, as described previously.1 Contingent on commercial availability, production of D‐ or F‐labeled toluene was verified by comparison of mass spectra of products to those of authentic labeled toluene standards. Labeled toluene standards included α,α,α‐D3‐toluene (Sigma–Aldrich, 99 atom% D), 2,3,4,5,6‐D5‐toluene (Sigma–Aldrich, 98 atom% D), D8‐toluene (Sigma–Aldrich, 100 atom% D), and [methyl‐F2]toluene (Sigma–Aldrich, 97 %). Although α,α,α‐D3‐toluene and D8‐toluene were not expected products, they were used to determine fragmentation patterns of D‐labeled toluene products for which standards were not available (see the Supporting Information).
For assessing D‐ or F‐labeled toluene production, GC–MS data acquisition methods were tailored to the labeled product expected; when available, authentic labeled toluene standards were used to establish major ions for monitoring and confirmatory ion ratios. To confirm the proper selection of major ions for monitoring labeled toluenes for which no authentic standards were available, data were also acquired in the full‐scan mode for the assay products of each type of labeled phenylacetate. The following ions were monitored for labeled toluenes: for α,α‐D2‐toluene—m/z 91, 92, 93, and 94; for 2,3,4,5,6‐D5‐toluene—m/z 94, 95, 96, and 97 (standard available); for α,α‐2,3,4,5,6‐phenyl‐D7‐toluene—m/z 96, 97, 98, 99, and 100; for α‐fluorotoluene—m/z 109 and 110; for α,α‐difluorotoluene—m/z 127 and 128 (standard available). Additional standards run were α,α,α‐D3‐toluene (major ions at m/z 93, 94, and 95) and D8‐toluene (major ions at m/z 98, 99, and 100). Other than differences in specific ions monitored, GC and acquisition parameters (such as ion dwell time) were the same for all labeled toluenes as those described for the detection of [methyl‐13C]toluene.1
In vitro kinetic assays with PhdB: For analysis of kinetics associated with phenylacetate decarboxylation catalyzed by PhdB, kinetic assays were performed under strictly anaerobic conditions, as described previously.1 To determine K M and V max, reactions were performed with varying substrate concentrations in buffer containing 50 mm Tris (pH 7.5), 150 mm NaCl, 1 mm MgCl2, 5 mm (NH4)2SO4, 5 mm DTT, and 0.2 mm PhdA. This buffer was pre‐incubated with 2 mm dithionite for approximately 30 min, followed by the addition of ≈20 μm PhdB and 2 mm SAM. These reactions, in a final volume of 1.5 mL, were initiated by the addition of the substrate phenylacetate‐2‐13C at concentrations of 0.4, 0.6, 0.8, 1.0, 1.2, 2.0, 2.4, and 3.0 mm. Headspace samples (100 μL) were assayed for [methyl‐13C]toluene production by GC–MS hourly for a total of 5 h, beginning immediately upon the addition of substrate (defined as time zero). Linear initial velocity profiles were determined for each substrate condition, and kinetic parameters were obtained by fitting velocity versus substrate concentration data with the nonlinear least squares Michaelis–Menten rate Equation (1) using Prism 7.0d (GraphPad Software, Inc., San Diego, CA):
| (1) |
in which v is the measured velocity, V max is the maximal velocity, K M is the Michaelis constant for phenylacetate‐2‐13C, and [A] is the concentration of phenylacetate‐2‐13C.
Inhibition of PhdB‐catalyzed toluene production in the presence of α,α‐difluorophenylacetate was determined by varying concentrations of the substrate, phenylacetate‐2‐13C, while maintaining a fixed concentration of the inhibitor, in a manner described by Kakkar et al.32 Kinetic inhibitor assays were performed as described above, except that the PhdB concentration was ≈40 μm. Control and inhibition experiments consisted of individual assays with four different substrate concentrations: 0.7, 1.0, 2.0, and 3.0 mm. Inhibition experiments were performed with a single inhibitor concentration of 0.5, 1.0, or 2.0 mm (the controls contained no inhibitor). Data sets were analyzed using Lineweaver–Burk plots (Prism 7.0d) to identify the nature of inhibition by α,α‐difluorophenylacetate (for example, competitive, noncompetitive, uncompetitive). K M(obs) calculated at each inhibitor concentration was used to determine the K i of α,α‐difluorophenylacetate for PhdB using Equation (2). Velocity vs. substrate concentration data were fit with a competitive inhibition nonlinear least squares regression rate Equation (3) using Prism 7.0d,
| (2) |
| (3) |
in which K M(obs) is the K M value observed in the presence of the inhibitor, [I] is the inhibitor concentration, and K i is the inhibition constant.
Generation of PhdB mutants: Selected PhdB residues were targeted for mutagenesis based in part on a homology model of active‐site residues, as previously described. The homology model was visualized using PyMOL v.2.2.3 (Schrödinger, Inc., New York, NY, USA). PhdB point mutations (H327A, C482A, Q484A, Q484E, Q484L, W495F, and Y691F) were generated using a QuikChange Lightning site‐directed mutagenesis kit (Agilent) using the His6‐MBP‐PhdB construct. Primers used for site‐directed mutagenesis are listed in Table S3. Mutations were confirmed by plasmid sequencing (Illumina MiSeq platform). The resulting mutant proteins were expressed and purified in a manner identical to that described above for wild‐type PhdB.
PhdB oligomeric state as determined by size‐exclusion chromatography: To determine the oligomeric state of wild‐type and mutant PhdB, the His6‐MBP tag was cleaved by incubating His6‐MBP‐PhdB (purified by passage of cell lysates through a 5‐mL MBPTrap column; GE Healthcare) with lab‐purified TEV protease at a 5:1 concentration ratio overnight (≈18 h) at 4 °C. The protein mixture was then concentrated to 0.5 mL using a 50‐kDa molecular weight cutoff Amicon concentrator (EMD Millipore, Burlington, MA) and loaded onto a HiLoad 16/60 Superdex 200 gel filtration column (GE Healthcare) that was pre‐equilibrated with 50 mm Tris (pH 7.5), 50 mm NaCl, and 5 mm DTT run at a flow rate of 0.5 mL min−1. To establish a molecular weight calibration curve, a gel filtration standard consisting of a mixture of proteins of molecular weights 670 kDa, 158 kDa, 44 kDa, 17 kDa, and 1.35 kDa (Bio‐Rad, Hercules, CA, USA) was run on the same column under identical conditions. The identity of tag‐cleaved, purified PhdB was confirmed by shotgun proteomics.1 Protein amounts were determined by the Bradford assay using BSA as a standard (Bio‐Rad). The oligomeric state of all tested PhdB mutants (H327A, Q484E, Q484A, Q484L, Y691F, and W495F) was similarly verified by size‐exclusion chromatography, as just described for wild‐type PhdB.
Computations: Computations were carried out at the ωB97X‐D/6‐311+G(d,p) level of theory,23 unless otherwise noted. Tests with other levels of theory (for example, MP2) and comparisons with experimental data were carried out for some structures. Solvation calculations were carried out using SMD(chloroform). Structures were confirmed to be minima or transition state structures by frequency analysis. Coordinates and energies for all computed structures, along with additional details and discussion, are available in the Supporting Information.
Conflict of interest
J.D.K. has a financial interest in Amyris, Lygos, Demetrix, Napigen, Maple Bio, and Apertor Labs.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Bo Pang (LBNL) for helpful discussions and Jose H. Pereira and Paul D. Adams (LBNL) for providing an electronic version of the PhdB homology model developed for ref. 1. This work was part of the DOE Joint BioEnergy Institute (https://www.jbei.org) supported by the US Department of Energy, Office of Science, Office of Biological and Environmental Research through contract DE‐AC02‐05CH11231 between Lawrence Berkeley National Laboratory and the US Department of Energy. Computational support from the NSF XSEDE program is gratefully acknowledged. The views and opinions of the authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a nonexclusive, paid‐up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
A. V. Rodrigues, D. J. Tantillo, A. Mukhopadhyay, J. D. Keasling, H. R. Beller, ChemBioChem 2020, 21, 663.
References
- 1. Beller H. R., Rodrigues A. V., Zargar K., Wu Y. W., Saini A. K., Saville R. M., Pereira J. H., Adams P. D., Tringe S. G., Petzold C. J., Keasling J. D., Nat. Chem. Biol. 2018, 14, 451–457. [DOI] [PubMed] [Google Scholar]
- 2. Becker A., Fritz-Wolf K., Kabsch W., Knappe J., Schultz S., Volker Wagner A. F., Nat. Struct. Biol. 1999, 6, 969–975. [DOI] [PubMed] [Google Scholar]
- 3. Larsson K. M., Andersson J., Sjoberg B. M., Nordlund P., Logan D. T., Structure 2001, 9, 739–750. [DOI] [PubMed] [Google Scholar]
- 4. Heider J., Spormann A. M., Beller H. R., Widdel F., FEMS Microbiol. Rev. 1998, 22, 459–473. [Google Scholar]
- 5. Leuthner B., Leutwein C., Schulz H., Horth P., Haehnel W., Schiltz E., Schagger H., Heider J., Mol. Microbiol. 1998, 28, 615–628. [DOI] [PubMed] [Google Scholar]
- 6. Beller H. R., Spormann A. M., FEMS Microbiol. Lett. 1999, 178, 147–153. [DOI] [PubMed] [Google Scholar]
- 7. Selmer T., Andrei P. I., Eur. J. Biochem. 2001, 268, 1363–1372. [DOI] [PubMed] [Google Scholar]
- 8. Yu L., Blaser M., Andrei P. I., Pierik A. J., Selmer T., Biochemistry 2006, 45, 9584–9592. [DOI] [PubMed] [Google Scholar]
- 9. Feliks M., Martins B. M., Ullmann G. M., J. Am. Chem. Soc. 2013, 135, 14574–14585. [DOI] [PubMed] [Google Scholar]
- 10. O′Brien J. R., Raynaud C., Croux C., Girbal L., Soucaille P., Lanzilotta W. N., Biochemistry 2004, 43, 4635–4645. [DOI] [PubMed] [Google Scholar]
- 11. Craciun S., Balskus E. P., Proc. Natl. Acad. Sci. USA 2012, 109, 21307–21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalnins G., Kuka J., Grinberga S., Makrecka-Kuka M., Liepinsh E., Dambrova M., Tars K., J. Biol. Chem. 2015, 290, 21732–21740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levin B. J., Huang Y. Y., Peck S. C., Wei Y., Martinez-Del Campo A., Marks J. A., Franzosa E. A., Huttenhower C., Balskus E. P., Science 2017, 355, eaai8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu D., Wei Y., Liu X., Zhou Y., Jiang L., Yin J., Wang F., Hu Y., Nanjaraj Urs A. N., Liu Y., Ang E. L., Zhao S., Zhao H., Zhang Y., Nat. Commun. 2018, 9, 4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peck S. C., Denger K., Burrichter A., Irwin S. M., Balskus E. P., Schleheck D., Proc. Natl. Acad. Sci. USA 2019, 116, 3171–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xing M., Wei Y., Zhou Y., Zhang J., Lin L., Hu Y., Hua G., Nanjaraj Urs A. N., Liu D., Wang F., Guo C., Tong Y., Li M., Liu Y., Ang E. L., Zhao H., Yuchi Z., Zhang Y., Nat. Commun. 2019, 10, 1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Selmer T., Pierik A. J., Heider J., Biol. Chem. 2005, 386, 981–988. [DOI] [PubMed] [Google Scholar]
- 18. Martins B. M., Blaser M., Feliks M., Ullmann G. M., Buckel W., Selmer T., J. Am. Chem. Soc. 2011, 133, 14666–14674. [DOI] [PubMed] [Google Scholar]
- 19. Zargar K., Saville R., Phelan R. M., Tringe S. G., Petzold C. J., Keasling J. D., Beller H. R., Sci. Rep. 2016, 6, 31362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Backman L. R. F., Funk M. A., Dawson C. D., Drennan C. L., Crit. Rev. Biochem. Mol. Biol. 2017, 52, 674–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lemal D. M., J. Org. Chem. 2004, 69, 1–11. [DOI] [PubMed] [Google Scholar]
- 22. Shah P., Westwell A. D., J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]
- 23. Sengupta A., Raghavachari K., Org. Lett. 2017, 19, 2576–2579. [DOI] [PubMed] [Google Scholar]
- 24. Simonson T., Brooks C. L., J. Am. Chem. Soc. 1996, 118, 8452–8458. [Google Scholar]
- 25. Stryer L., Biochemistry, Freeman, New York, 1988. [Google Scholar]
- 26. Fisher S. J., Wilkinson J., Henchman R. H., Helliwell J. R., Crystallogr. Rev. 2009, 15, 231–259. [Google Scholar]
- 27. Wang S. C., Tantillo D. J., J. Org. Chem. 2008, 73, 1516–1523. [DOI] [PubMed] [Google Scholar]
- 28. Duer W. C., Robinson R. A., J. Chem. Soc. B 1971, 0, 2375. [Google Scholar]
- 29. Beller H. R., Spormann A. M., J. Bacteriol. 1997, 179, 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beller H. R., Spormann A. M., J. Bacteriol. 1998, 180, 5454–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bodea S., Funk M. A., Balskus E. P., Drennan C. L., Cell Chem. Biol. 2016, 23, 1206–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kakkar T., Pak Y., Mayersohn M., J. Pharmacol. Exp. Ther. 2000, 293, 861–869. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary