

Abstract
We describe a strategy for the enantio- and diastereoselective synthesis of homoallylic α-trifluoromethyl amines by the catalytic hydroalkylation of terminal dienes. Trifluoromethyl-substituted isatin-derived azadienolate nucleophiles undergo γ-selective alkylation with a Pd–DTBM-SEGPHOS catalyst, which additionally promotes regioselective addition to the diene and delivers products in up to 86% yield, 10:1 dr, and 97.5:2.5 er.
Graphical Abstract

Chiral amines bearing an α-trifluoromethyl group hold a significant place among classes of medicinally important N-containing molecules. The CF3 group modulates a number of pharmacological parameters, alters the amine basicity and polarity significantly, and stands in as a non-hydrolyzable amide surrogate.1 Despite the beneficial properties this motif might impart upon drug-like molecules, methods for the catalytic enantioselective synthesis of α-trifluoromethyl amines are fairly uncommon, with most approaches to these enantioenriched compounds relying on chiral auxiliary chemistry.2
A valuable subset of these compounds are α-trifluoromethyl homoallylic amines. Recently, a number of groups have investigated allylic substitution approaches to these molecules utilizing azaallyl anion building blocks.3 With an isatin4 activating group for the nucleophile (Scheme 1), an Ircatalyzed procedure gives rise to branch-selective coupling of the allylic carbonate exclusively at the azadienolate’s α-position; however, this product spontaneously undergoes aza-Cope rearrangement to deliver a net γ-allylation. As a result, the unsaturated amines bear only one stereogenic center.4,5 The same class of nucleophiles have been utilized in a γ-selective allylation with Morita–Baylis–Hillman-type allylic carbonates by employing a chiral nucleophilic catalyst (Scheme 1).6 These coupling processes can yield homoallylic amines with vicinal syn stereogenic centers;6a however, the product scope is limited to aryl-substituted allylic centers and carbonyl-containing alkenes.7,8
Scheme 1.

Prior Catalytic Enantioselective α-CF 3 Homoallylic Amine Synthesis and Proposed Strategy
Given our longstanding interests in both umpolung synthesis of amines9 and diene hydrofunctionalization reactions,10 we envisioned that the merger of these strategies would allow for the enantio-, diastereo-, and regioselective preparation of α-CF3 homoallylic amines that bear vicinal stereogenic centers and internal alkenes,10a potentially enabling the anti diastereomer to be accessed (Scheme 1). Several challenges had to be met and overcome for the successful realization of this idea. Primary among these was regioselectivity. In contrast to previous metal-catalyzed allylic substitution methods, our goal was to develop a kinetically γ-selective allylation of isatin azadienolates, obviating the aza-Cope rearrangement and thus giving rise to a fundamentally different product connectivity than would otherwise be available. Could a catalyst be found that is γ-selective, and would this process still be efficient? Would the same catalyst also provide regioselectivity with respect to the diene? Finally, would such a catalyst allow for control of the relative and absolute stereochemistry of the homoallylic amine products? Only recently has a diene hydroalkylation that sets two stereogenic centers in one bond-forming event been reported.11 Herein, we illustrate that Pd–DTBM-SEGPHOS promotes the γ-selective coupling of azadienolates with the terminal olefin of dienes, generating the anti diastereomer of homoallylic α-CF3 amines with excellent levels of stereocontrol.12
We initiated our investigations by exploring the coupling of N-trifluoroethyl imine 1 and phenylbutadiene 2a (Table 1).13 Whereas most ligands favor azadienolate α-alkylation product 3a′ under Pd catalysis, bis(phosphines) comprised of 3,5-di-tert-butyl-4-methoxy (DTBM) aryl groups at phosphorus exclusively deliver the desired γ-alkylation product 3a (compare entries 3 and 6 with entries 1–2 and 4–5).14 Notably, the diastereomeric ratio for 3a is also considerably higher with the more γ-selective catalysts, affording the anti diastereomer as the major isomer (entries 3 and 6). Use of DTBM-SEGPHOS provides the greatest conversion to 3a, which is isolated in 9:1 dr and 91.5:8.5 er (entry 6).15 We discovered that the enantioselectivity could be improved by the addition of 10 mol % NaBArF4 (93:7 er, entry 7).16 By switching the solvent from diethyl ether to 1,4-dioxane, the γ-alkylation product 3a is isolated in 80% yield, 8:1 dr, and 95:5 er (entry 9).
Table 1.
Optimization for γ-Selective Addition of an Isatin-Derived Azadienolate to Phenylbutadienea
 | ||||||
|---|---|---|---|---|---|---|
| entry | L | NaBArF4 (Y/N) | 3a:3a′ b | conv to 3a/3a′ b (%) | dr of 3ab | er of 3ac |
| 1 | BINAP | N | 1:2.5 | 48 | 1:1 | nd |
| 2 | MeO-BIPHEP | N | 1:2.5 | 66 | 1.5:1 | nd |
| 3 | DTBM-MeO-BIPHEP | N | >20:1 | 49 | 7:1 | nd |
| 4 | SEGPHOS | N | 1:1.1 | 30 | 3:1 | nd |
| 5 | DM-SEGPHOS | N | 1:1.5 | 38 | 3:1 | nd |
| 6 | DTBM-SEGPHOS | N | >20:1 | >98 | 9:1 | 91.5:8.5 |
| 7 | DTBM-SEGPHOS | Y | >20:1 | >98 | 9:1 | 93:7 |
| 8d | DTBM-SEGPHOS | N | >20:1 | >98 | 8:1 | 94:6 |
| 9d,e | DTBM-SEGPHOS | Y | >20:1 | 97 (80)f | 8:1 | 95:5 |
Reactions run under N2 with 0.15 mmol of diene 2a (0.75 M).
Determined by 376 MHz 19F NMR spectroscopy of the unpurified mixture.
Determined by HPLC analysis of purified 3a.
1,4-Dioxane as solvent.
Reaction for 12 h.
Isolated yield of purified 3a. nd = not determined.
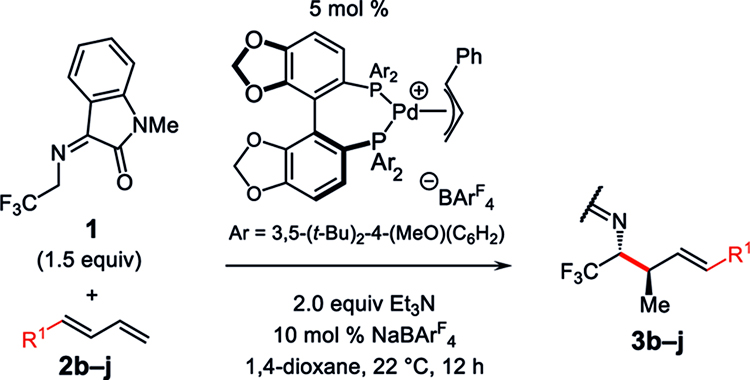
Taking the conditions in Table 1, entry 9, as optimal, we next explored the scope of aryl-substituted terminal dienes for coupling with 1 (Table 2). Both electron-rich (e.g., entries 1 and 6–7) and electron-poor (entries 2–3) dienes generate the homoallylic amine products with ≥87% conversion, ≥7:1 dr, and moderate to good enantioselectivity. Heterocycles are tolerated, with furyl-substituted 3i (entry 8) and pyridyl-containing 3j (entry 9) isolated in good yields and stereo-selectivities. The majority of dienes lead exclusively to the γ-alkylation product; however, an ortho substituent on the arene results in a more sluggish reaction (44% conversion in 12 h) that furnishes approximately 10% α-coupling of the azadienolate (entry 5). Improving reaction efficiency required the omission of NaBArF4, and while diastereoselectivity was lower, enantioselectivity remained high (92.5:7.5 er). In all, aryl-substituted dienes readily participate in couplings with 1 at room temperature, affording homoallylic α-trifluoromethyl amines 3b−j in 59–86% yield.
Table 2.
Aryl Diene Scope in Azadienolate Couplinga
 | ||||
|---|---|---|---|---|
| entry | product (3); R1 | conv to 3ab (%); yield of 3c (%) | dr of 3b | er of 3d |
| 1 | 3b; 4-(MeO)(C6H4) | 87; 68 | 10:1 | 94:6 |
| 2 | 3c; 4-CI(C6H4) | 98; 75 | 7:1 | 90.5:9.5 |
| 3 | 3d; 4-(F3C)(C6H4) | 94; 74 | 8:1 | 90:10 |
| 4 | 3e; 3-Me(C6H4) | 89; 72 | 6:1 | 93:7 |
| 5e | 3f; 2-Me(C6H4) | 63; 59 | 2:1 | 92.5:7.5 |
| 6 | 3g; 2-naphthyl | 94; 62 | 8:1 | 94:6 |
| 7 | 3h; 3,4-dioxolato(C6H3) | 94; 86 | 10:1 | 91.5:8.5 |
| 8 | 3i; 2-furyl | 69; 59 | 8:1 | 93:7 |
| 9 | 3j; 3-pyridyl | 83; 82 | 7:1 | 94:6 |
Reactions run under N2 with 0.15 mmol of diene 2 (0.75 M).
Determined by 376 MHz 19F NMR or 400 MHz 1H NMR spectroscopy of the unpurified mixture.
Isolated yield of purified 3.
Determined by HPLC analysis of purified 3.
Reaction run without NaBArF4; 9:1 3f:3f′.
Alkyl-substituted terminal dienes are also effective coupling partners; however, the Pd–DTBM-SEGPHOS-catalyzed processes with imine 1 require elevated temperature (50 °C) to proceed effectively (Table 3). Consequently, we also omitted the NaBArF4 additive to enable the reaction to proceed at a higher rate. Both linear (entries 1–3) and α-branched (entries 4–5) dienes participate in the reactions, affording homoallylic amines 3k−o in 43–83% yield in up to 4:1 dr and 91:9 er. Products derived from isomerization of the diene along the alkyl chain (“chain walking”) prior to enolate addition could not be detected, including with phenethyl 2k or heptadienoate 2m. Alkyl dienes largely or solely lead to γ-alkylation of the azadienolate, although it is notable that piperidine 2o affords ca. 9% α product 3o′. We also observed roughly 8% of an additional γ-alkylation product 3k″ with phenethyl diene 2k. Homoallylic amine 3k″ bears a different connectivity with respect to the diene-derived fragment, and a series of experiments suggest that 3k″ is formed from the aza-Cope rearrangement of α-alkylation product 3k′.17
Table 3.
Azadienolate–Alkyl Diene Coupling Scopea
 | ||||
|---|---|---|---|---|
| entry | product (3); R1 | conv to 3 (%);b yield of 3 (%)c | dr of 3b | er of 3d |
| 1e | 3k; Ph(CH2)2 | 82; 69 | 4:1 | 88:12 |
| 2 | 31; n-hexyl | 65; 60 | 4:1 | 81.5:18.5 |
| 3 | 3m; EtO2C(CH2)2 | 84; 83 | 3:1 | 91:9 |
| 4 | 3n; Cy | 61; 43 | 3:1 | 90:10 |
| 5f |  |
83; 83 | 3:1 | 86.5:13.5 |
Reactions run under N2 with 0.15 mmol of diene 2 (0.75 M).
Determined by 376 MHz 19F NMR or 400 MHz 1H NMR spectroscopy of the unpurified mixture.
Isolated yield of purified 3.
Determined by HPLC analysis of purified 3.
A 10:1 mixture of γ-alkylation products 3k and 3k″ was formed; see ref 17.
11:1 3o:3o′.
Intriguingly, in the course of our alkyl diene studies, we discovered that hexadienoate 2p (Scheme 2) undergoes diene isomerization into conjugation with the ester prior to its coupling with 1, furnishing the ethyl-substituted stereogenic center of homoallylic amine 3p. The process is reasonably efficient, with the α-CF3 amine obtained in 48% yield, 12:1 dr, and 97.5:2.5 er. Comparatively, its internal diene analogue 4 also delivers acrylate 3p with similar levels of stereoselectivity but lower conversion.
Scheme 2.

Isomerization/Alkylation of 3,5-Hexadienoate and Comparison to Its Internal Diene Isomer
We have explored a number of additional reaction partners to expand the scope of the hydrofunctionalization (Figure 1). Imine 5 was tested in a coupling with diene 2a in order to access α-difluoromethyl amines, but unfortunately, the nucleophile undergoes complete decomposition without alkylation. Substituted imines, such as 6, would form products bearing tetrasubstituted stereogenic centers5,6b,8 but were unreactive. Other dienes were also investigated. Alcohol- and silyl ether-containing alkyl dienes 2q and 2r lead to a complex mixture of products, which we surmise to be a combination of the desired γ-alkylation 3, the regiomeric α-alkylation 3′, and the aza-Cope rearrangement products 3″, all as a mixture of diastereomers, rather than products attributable to chain walking.
Figure 1.
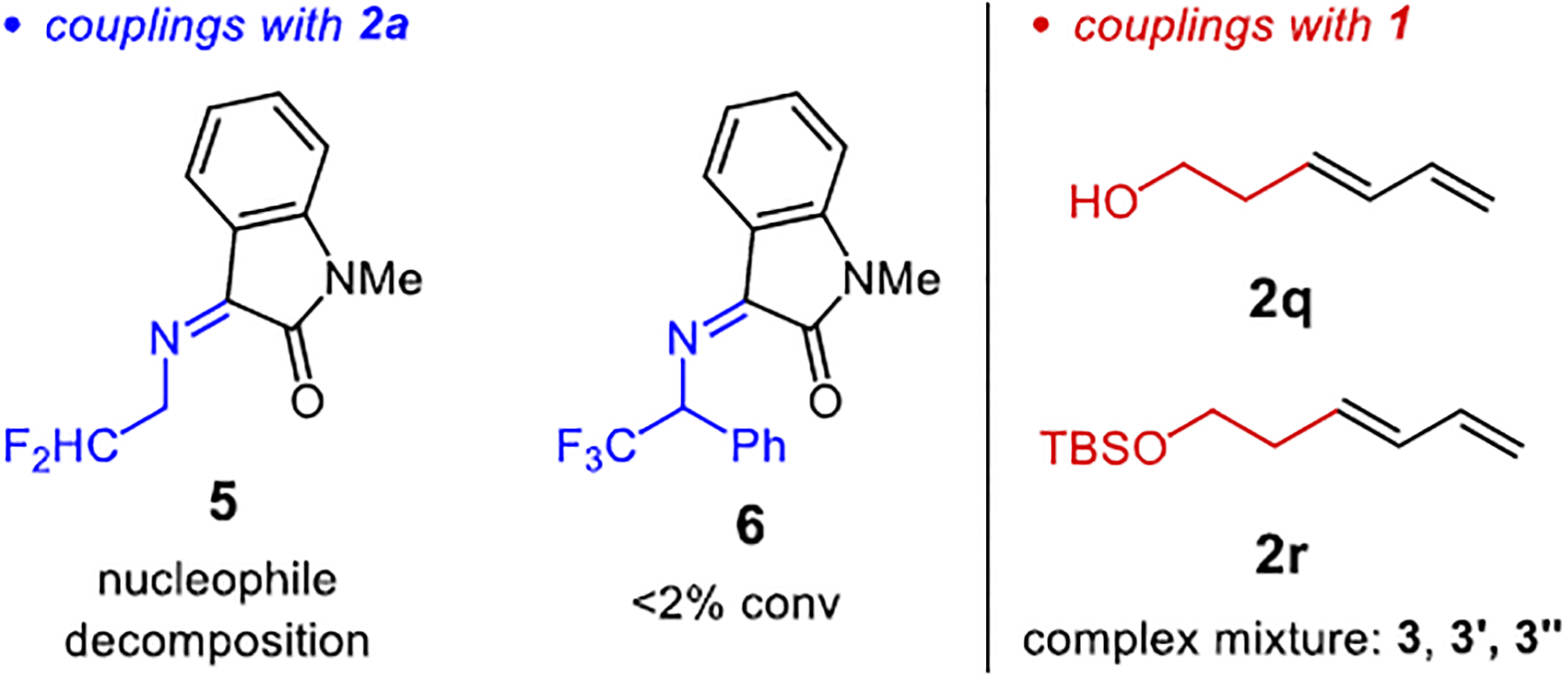
Limitations in Reaction Partners.
The diene hydroalkylation with imine 1 can be performed on a 1.0 mmol scale to furnish the α-trifluoromethyl isatin-protected homoallylic amine 3a in 82% yield (Scheme 3). Additionally, hydrolysis of the isatin moiety under mildly acidic conditions delivers the free amine 7 in 77% yield.
Scheme 3.
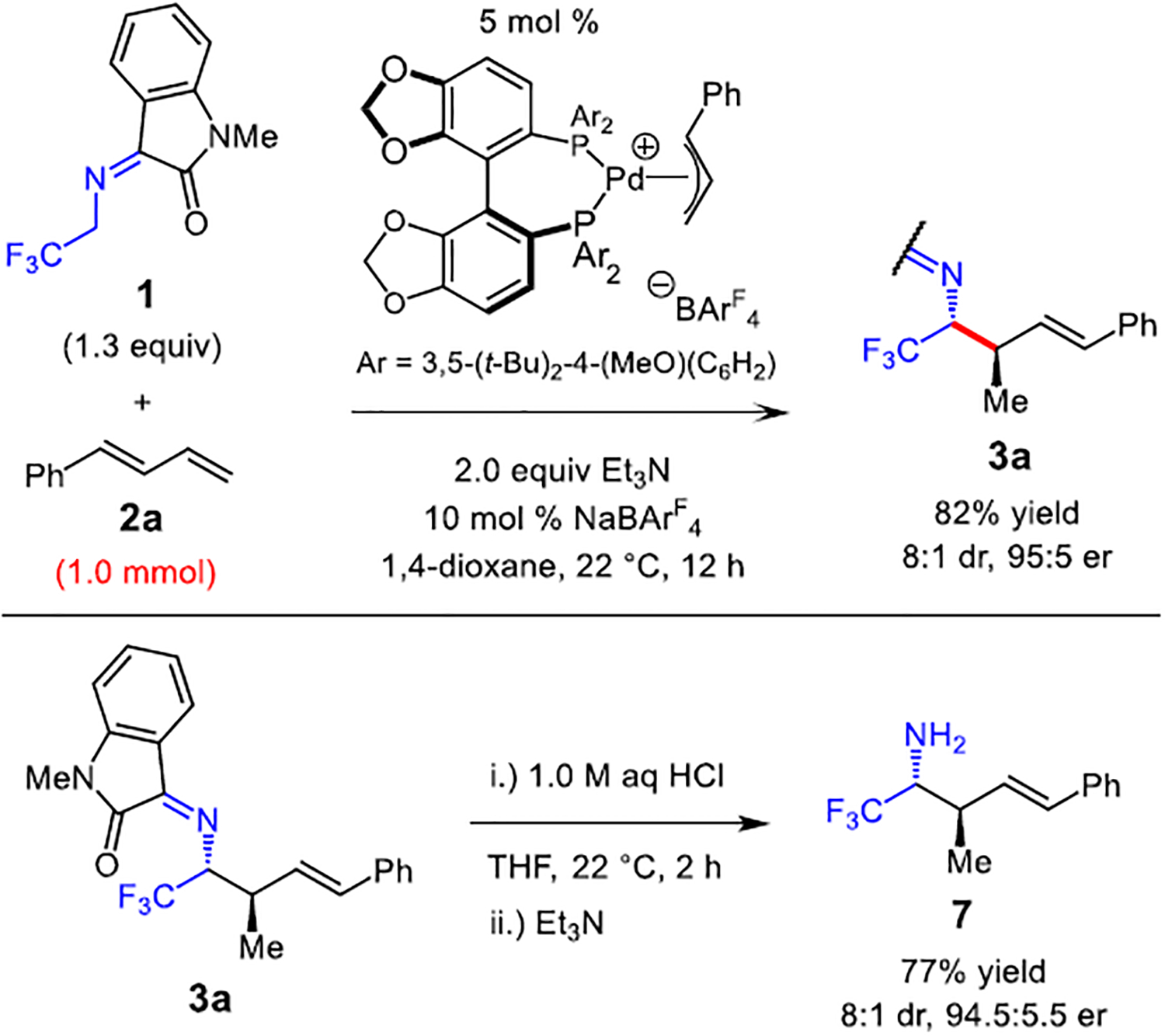
Scale Up of Azadienolate Hydroalkylation and Product Imine Hydrolysis
Catalytic enantioselective diene hydrofunctionalization provides an enabling route toward highly valuable chemical building blocks that are not readily prepared by other methods. Here, in combination with imine umpolung,3 we have shown that important homoallylic α-trifluoromethyl amines bearing contiguous stereogenic centers and an internal olefin can be accessed for the first time. Utilizing an isatin auxiliary, we have discovered that, in contrast to other transition-metal-catalyzed processes, palladium ligated with DTBM-SEGPHOS allows for regioselective γ-alkylation of the derived azadienolate, generating the anti diastereomer of the homoallylic α-CF3 amines with high levels of stereocontrol. This catalytic process should open up new chemical space for drug discovery.
Supplementary Material
ACKNOWLEDGMENTS
This work was generously supported by the NIH (GM124286) and the NSF (CHE-1800012). C.I.O. is grateful to the Duke Chemistry Department for a Burroughs-Wellcome fellowship. All X-ray crystallographic measurements were made in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University; we thank Dr. Roger Sommer (NC State) for assistance with analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.0c00342.
Experimental procedures, analytical data for new compounds, X-ray crystallographic data, and NMR spectra (PDF)
Accession Codes
CCDC 1978720 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61, 5822. [DOI] [PubMed] [Google Scholar]; (b) Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315. [DOI] [PubMed] [Google Scholar]
- (2).For reviews, see:; (a) Fioravanti S Trifluoromethyl Aldimines: An Overview in the Last Ten Years. Tetrahedron 2016, 72, 4449. [Google Scholar]; (b) Nie J; Guo H-C; Cahard D; Ma J-A Asymmetric Construction of Stereogenic Carbon Centers Featuring a Trifluoromethyl Group from Prochiral Trifluoromethylated Substrates. Chem. Rev 2011, 111, 455. [DOI] [PubMed] [Google Scholar]; For additional examples not highlighted in these reviews, see:; (c) Grellepois F; Ben Jamaa A; Saraiva Rosa N α-Trifluoromethylated Tertiary Homoallylic Amines: Diastereoselective Synthesis and Conversion into β-Aminoesters, γ- and δ-Amino-alcohols, Azetidines and Pyrrolidines. Org. Biomol. Chem 2017, 15, 9696. [DOI] [PubMed] [Google Scholar]; (d) Grellepois F; Ben Jamaa A; Gassama A Diastereoselective Addition of Organomagnesium and Organolithium Reagents to Chiral Trifluoromethyl N-tert-Butanesulfinyl Hemiaminals. Eur. J. Org. Chem 2013, 2013, 6694. [Google Scholar]; (e) Guo T; Song R; Yuan B-H; Chen X-Y; Sun X-W; Lin G-Q Highly Efficient Asymmetric Construction of Quaternary Carbon-Containing Homoallylic and Homopropargylic Amines. Chem. Commun 2013, 49, 5402. [DOI] [PubMed] [Google Scholar]
- (3).For a review on azaallyl anions as imine umpolung reagents, see:; (a) Tang S; Zhang X; Sun J; Niu D; Chruma JJ 2-Azaallyl Anions, 2-Azaallyl Cations, 2-Azaallyl Radicals, and Azomethine Ylides. Chem. Rev 2018, 118, 10393. [DOI] [PubMed] [Google Scholar]; For other reviews on umpolung chemistry, see:; (b) Mahatthananchai J; Bode JW On the Mechanism of N-Heterocyclic Carbene-Catalyzed Reactions Involving Acyl Azoliums. Acc. Chem. Res 2014, 47, 696. [DOI] [PubMed] [Google Scholar]; (c) Boyce GR; Greszler SN; Johnson JS; Linghu X; Malinowski JT; Nicewicz DA; Satterfield AD; Schmitt DC; Steward KM Silyl Glyoxylates. Conception and Realization of Flexible Conjunctive Reagents for Multicomponent Coupling. J. Org. Chem 2012, 77, 4503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Izquierdo J; Hutson GE; Cohen DT; Scheidt KA A Continuum of Progress: Applications of N-Heterocyclic Carbene Catalysis in Total Synthesis. Angew. Chem., Int. Ed 2012, 51, 11686. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smith AB III; Wuest WM Evolution of Multi-Component Anion Relay Chemistry (ARC): Construction of Architecturally Complex Natural and Unnatural Products. Chem. Commun 2008, 44, 5883. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Seebach D Methods of Reactivity Umpolung. Angew. Chem., Int. Ed. Engl 1979, 18, 239. [Google Scholar]
- (4).Shi L-M; Sun X-S; Shen C; Wang Z-F; Tao H-Y; Wang C-J Catalytic Asymmetric Synthesis of α-Trifluoromethyl Homoallylic Amines via Umpolung Allylation/2-Aza-Cope Rearrangement: Stereoselectivity and Mechanistic Insight. Org. Lett 2019, 21, 4842. [DOI] [PubMed] [Google Scholar]
- (5).Azaallyl anions formed from fluorenyl imines have likewise been shown to undergo enantioselective allylation followed by 2-aza-Cope rearrangement; see:; (a) Wang Y; Deng L-F; Zhang X; Niu D Catalytic Asymmetric Synthesis of α-Tetrasubstituted α-Trifluoromethyl Homoallylic Amines by Ir-Catalyzed Umpolung Allylation of Imines. Org. Lett 2019, 21, 6951. [DOI] [PubMed] [Google Scholar]; (b) Shen C; Wang R-Q; Wei L; Wang Z-F; Tao H-Y; Wang C-J Catalytic Asymmetric Umpolung Allylation/2-Aza-Cope Rearrangement for the Construction of α-Tetrasubstituted α-Trifluoromethyl Homoallylic Amines. Org. Lett 2019, 21, 6940. [DOI] [PubMed] [Google Scholar]
- (6).(a) Li X; Su J; Liu Z; Zhu Y; Dong Z; Qiu S; Wang J; Lin L; Shen Z; Yan W; Wang K; Wang R Synthesis of Chiral α-Trifluoromethylamines with 2,2,2-Trifluoroethylamine as a “Building Block. Org. Lett 2016, 18, 956. [DOI] [PubMed] [Google Scholar]; For a similar reaction, see:; (b) Chen P; Yue Z; Zhang J; Lv X; Wang L; Zhang J Phosphine Catalyzed Asymmetric Umpolung Addition of Trifluoromethyl Ketimines to Morita–Baylis–Hillman Carbonates. Angew. Chem., Int. Ed 2016, 55, 13316. [DOI] [PubMed] [Google Scholar]; For a related method with allenoate electrophiles, see:; (c) Gao Y-N; Shi M Enantioselective Synthesis of Isatin-Derived α-(Trifluoromethyl)imine Derivatives: Phosphine-Catalyzed γ-Addition of α-(Trifluoromethyl)imines and Allenoates. Eur. J. Org. Chem 2017, 2017, 1552. [Google Scholar]
- (7).For additional examples of enantioselective homoallylic amine synthesis involving stereoselective sigmatropic rearrangement, see:; (a) Wei L; Zhu Q; Xiao L; Tao H-Y; Wang C-J Synergistic Catalysis for Cascade Allylation and 2-aza-Cope Rearrangement of Azomethine Ylides. Nat. Commun 2019, 10, 1594. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huo X; Zhang J; Fu J; He R; Zhang W Ir/Cu Dual Catalysis: Enantioand Diastereodivergent Acess to α,α-Disubstituted α-Amino Acids Bearing Vicinal Stereocenters. J. Am. Chem. Soc 2018, 140, 2080. [DOI] [PubMed] [Google Scholar]; (c) Wei L; Zhu Q; Xu S-M; Chang X; Wang C-J Stereodivergent Synthesis of α,α-Disubstituted α-Amino Acids via Synergistic Cu/Ir Catalysis. J. Am. Chem. Soc 2018, 140, 1508. [DOI] [PubMed] [Google Scholar]
- (8).For additional examples of catalytic enantioselective synthesis of α-trifluoromethyl amines not covered in refs 2 and 4−6, see:; (a) Hu B; Deng L Direct Catalytic Asymmetric Synthesis of Trilfuoromethylated γ-Amino Esters/Lactones via Umpolung Strategy. J. Org. Chem 2019, 84, 994. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu J; Huang L; Dong W; Li N; Yu X; Deng W-P; Tang W Enantioselective Rhodium-Catalyzed Addition of Arylboroxines to N-Unprotected Ketimines: Efficient Synthesis of Cipargamin. Angew. Chem., Int. Ed 2019, 58, 16119. [DOI] [PubMed] [Google Scholar]; (c) Hu B; Bezpalko MW; Fei C; Dickie DA; Foxman BM; Deng L Origin of and a Solution for Uneven Efficiency by Cinchona Alkaloid-Derived, Pseudoenantiomeric Catalysts for Asymmetric Reactions. J. Am. Chem. Soc 2018, 140, 13913. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li Z; Hu B; Wu Y; Fei C; Deng L Control of Chemoselectivity in Asymmetric Tandem Reactions: Direct Synthesis of Chiral Amines Bearing Nonadjacent Stereocenters. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 1730. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hu B; Deng L Catalytic Asymmetric Synthesis of Trifluoromethylated γ-Amino Acids through the Umpolung Addition of Trifluoromethyl Imines to Carboxylic Acid Derivatives. Angew. Chem., Int. Ed 2018, 57, 2233. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hu L; Wu Y; Li Z; Deng L Catalytic Asymmetric Synthesis of Chiral γ-Amino Ketones via Umpolung Reactions of Imines. J. Am. Chem. Soc 2016, 138, 15817. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kawai H; Kusuda A; Nakamura S; Shiro M; Shibata N Catalytic Enantioselective Trifluoromethylation of Azomethine Imines with Trimethyl-(trifluoromethyl)silane. Angew. Chem., Int. Ed 2009, 48, 6324. [DOI] [PubMed] [Google Scholar]
- (9).(a) Malcolmson SJ; Li K; Shao X 2-Azadienes as Enamine Umpolung Synthons for the Preparation of Chiral Amines. Synlett 2019, 30, 1253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shao X; Malcolmson SJ Catalytic Enantioand Diastereoselective Cyclopropanation of 2-Azadienes for the Synthesis of Aminocyclopropanes Bearing Quaternary Carbon Stereogenic Centers. Org. Lett 2019, 21, 7380. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Daniel PE; Onyeagusi CI; Ribeiro AA; Li K; Malcolmson SJ Palladium-Catalyzed Synthesis of α-Trifluoromethyl Benzylic Amines via Fluoroarylation of gem-Difluoro-2-azadienes Enabled by Phosphine-Catalyzed Formation of an Azaallyl–Silver Intermediate. ACS Catal. 2019, 9, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shao X; Li K; Malcolmson SJ Enantioselective Synthesis of anti-1,2-Diamines by Cu-Catalyzed Reductive Couplings of Azadienes with Aldimines and Ketimines. J. Am. Chem. Soc 2018, 140, 7083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li K; Shao X; Tseng L; Malcolmson SJ 2-Azadienes as Reagents for Preparing Chiral Amines: Synthesis of 1,2-Amino Tertiary Alcohols by Cu-Catalyzed Enantioselective Reductive Couplings with Ketones. J. Am. Chem. Soc 2018, 140, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Li K; Weber AE; Tseng L; Malcolmson SJ Diastereoselective and Enantiospecific Synthesis of 1,3-Diamines via 2-Azaallyl Anion Benzylic Ring-Opening of Aziridines. Org. Lett 2017, 19, 4239. [DOI] [PubMed] [Google Scholar]; (g) Daniel PE; Weber AE; Malcolmson SJ Umpolung Synthesis of 1,3-Amino Alcohols: Stereoselective Addition of 2-Azaallyl Anions to Epoxides. Org. Lett 2017, 19, 3490. [DOI] [PubMed] [Google Scholar]
- (10).(a) Adamson NJ; Malcolmson SJ Catalytic Enantio- and Regioselective Addition of Nucleophiles in the Intermolecular Hydrofunctionalization of 1,3-Dienes. ACS Catal. 2020, 10, 1060. [Google Scholar]; (b) Park S; Adamson NJ; Malcolmson SJ Brønsted Acid and Pd–PHOX Dual-Catalysed Enantioselective Addition of Activated CPronucleophiles to Internal Dienes. Chem. Sci 2019, 10, 5176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Park S; Malcolmson SJ Development and Mechanistic Investigations of Enantioselective Pd-Catalyzed Intermolecular Hydroaminations of Internal Dienes. ACS Catal. 2018, 8, 8468. [Google Scholar]; (d) Adamson NJ; Wilbur KCE; Malcolmson SJ Enantioselective Intermolecular Pd-Catalyzed Hydroalkylation of Acyclic 1,3-Dienes with Activated Pronucleophiles. J. Am. Chem. Soc 2018, 140, 2761. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Adamson NJ; Hull E; Malcolmson SJ Enantioselective Intermolecular Addition of Aliphatic Amines to Acyclic Dienes with a Pd–PHOX Catalyst. J. Am. Chem. Soc 2017, 139, 7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).This report utilizes a dual metal-catalyzed strategy; see:; (a) Zhang Q; Yu H; Shen L; Tang T; Dong D; Chai W; Zi W Stereodivergent Coupling of 1,3-Dienes with Aldimine Esters Enabled by Synergistic Pd and Cu Catalysis. J. Am. Chem. Soc 2019, 141, 14554. [DOI] [PubMed] [Google Scholar]; For transformations with allenes/alkynes, see:; (b) Bora PP; Sun G-J; Zheng W-F; Kang Q Rh/Lewis Acid Catalyzed Regio-, Diastereo- and Enantioselective Addition of 2-Acyl Imidazoles with Allenes. Chin. J. Chem 2018, 36, 20. [Google Scholar]; (c) Cruz FA; Dong VM Stereodivergent Coupling of Aldehydes and Alkynes via Synergistic Catalysis Using Rh and Jacobsen’s Amine. J. Am. Chem. Soc 2017, 139, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhou H; Wei Z; Zhang J; Yang H; Xia C; Jiang G From Palladium to Brønsted Acid Catalysis: Highly Enantioselective Regiodivergent Addition of Alkoxyallenes to Pyrazolones. Angew. Chem., Int. Ed 2017, 56, 1077. [DOI] [PubMed] [Google Scholar]; (e) Trost BM; Xie J; Sieber JD The Palladium Catalyzed Asymmetric Addition of Oxindoles and Allenes: An Atom-Economical Versatile Method for the Construction of Chiral Indole Alkaloids. J. Am. Chem. Soc 2011, 133, 20611. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Trost BM; Ja kel C; Plietker B Palladium-Catalyzed Asymmetric Addition of Pronucleophiles to Allenes. J. Am. Chem. Soc 2003, 125, 4438. [DOI] [PubMed] [Google Scholar]
- (12).A preprint of this manuscript was published on January 20, 2020; see:; Onyeagusi CI; Shao X; Malcolmson SJ Enantio- and Diastereoselective Synthesis of Homoallylic α-Trifluoromethyl Amines by Catalytic Hydroalkylation of Dienes. 2020. (ChemRxiv). 10.26434/chemrxiv.11653143.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13). For additional screening data, see the Supporting Information.
- (14). The transformations in Table 1 utilize a preformed Pd–bis(phosphine) complex derived from [Pd(η3-cinnamyl)Cl]2, bis(phosphine), and NaBArF4. When conducting reactions directly with in situ-generated catalyst, we observe a significant induction period in the reaction that is avoided by employing an isolated complex. See the Supporting Information for further details.
- (15).Pd–DTBM-SEGPHOS has been shown to be the optimal catalyst in a handful of enantioselective diene hydrophosphinylation reactions; see:; Nie S-Z; Davison RT; Dong VM Enantioselective Coupling of Dienes and Phosphine Oxides. J. Am. Chem. Soc 2018, 140, 16450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). Although NaBArF4 improves enantioselectivity, it greatly reduces the reaction rate. Under the conditions shown in Table 1, entry 6 without NaBArF4, the reaction is complete within 6 h, but longer reaction times are needed the more NaBArF4 is added. The data in Table 1 are all shown for 16 h reaction time for comparative purposes. See the Supporting Information for additional details.
-
(17). See the Supporting Information for a detailed discussion.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.