

Abstract
Bioflavonoids have a similar chemical structure to etoposide, the well-characterized topoisomerase II (Top2) poison, and evidence shows that they also induce DNA double-strand breaks (DSBs) and promote genome rearrangements. The purpose of this study was to determine the kinetics of bioflavonoid-induced DSB appearance and repair, and their dependence on Top2. Cells were exposed to bioflavonoids individually or in combination in the presence or absence of the Top2 catalytic inhibitor dexrazoxane. The kinetics of appearance and repair of γH2AX foci were measured. In addition, the frequency of resultant MLL-AF9 breakpoint cluster region translocations was determined. Bioflavonoids readily induced the appearance of γH2AX foci, but bioflavonoid combinations did not act additively or synergistically to promote DSBs. Myricetin-induced DSBs were mostly reduced by dexrazoxane, while genistein and quercetin-induced DSBs were only partially, but significantly, reduced. By contrast, luteolin and kaempferol-induced DSBs increased with dexrazoxane pre-treatment. Sensitivity to Top2 inhibition correlated with a significant reduction of bioflavonoid-induced MLL-AF9 translocations. These data demonstrate that myricetin, genistein, and quercetin act most similar to etoposide although with varying Top2-dependence. By contrast, luteolin and kaempferol have distinct kinetics that are mostly Top2-independent. These findings have implications for understanding the mechanisms of bioflavonoid activity and the potential of individual bioflavonoids to promote chromosomal translocations. Further, they provide direct evidence that specific Top2 inhibitors or targeted drugs could be developed that possess less leukemic potential or suppress chromosomal translocations associated with therapy-related and infant leukemias.
Keywords: double-strand break, topoisomerase II, etoposide, DNA damage, genome instability, environmental mutagenesis
1. Introduction
Infant acute leukemias are characterized by early onset after birth (<one year of age), rapid progression, and aggressive invasion. Approximately 85% of infant acute lymphoblastic leukemia (ALL) and 50% of infant acute myeloblastic leukemia (AML) cases harbor chromosomal translocations involving the MLL gene at chromosome position 11q23[1,2] almost exclusively concentrated in an 8.3-kb region named the breakpoint cluster region (BCR)[3,4]. Chromosomal rearrangements involving the MLL gene with more than 80 partner genes have been found though a majority of rearrangements occur with 9 partner genes[2,5,6]. MLL rearrangements can be detected in neonatal blood spots of infants who later develop leukemia, including in identical twins, supporting the theory that formation of the initiating rearrangement occurs in utero[7–9]. Sequenced translocation breakpoint junctions of infant leukemia patient samples cluster together in the MLL BCR with those of therapy-related leukemia (tAML) samples that are tightly associated with previous exposure to the potent topoisomerase II (Top2) poison etoposide[3,8,10,11]. Thus, it has been hypothesized that exposure to compounds biochemically similar to etoposide may contribute to infant leukemia from in utero exposure.
Top2 is an essential enzyme involved in relaxation of DNA for transcription and replication, and interference with normal Top2 function has been shown to cause DNA double-strand breaks (DSBs)[2,9–12]. Top2α is primarily active in proliferating cells and has roles in DNA replication, chromosome condensation and separation. Top2β is expressed in all cells and is primarily active in regions of active transcription[2,12]. Both Top2 isoforms have multistep cleavage and religation reactions involving initial binding to two dsDNA molecules, transient DSB in the first DNA molecule creating a cleavage complex, ATP hydrolysis passage of the second DNA helix through the DSB, and religation of the DSB[2,13]. While both isoforms act in this manner, recent studies have suggested interference with Top2β function is responsible for carcinogenic translocations[14,15]. Top2 poisons, including etoposide and doxorubicin, act by binding to the cleavage complex and preventing religation of the DSB from occurring[2,10]. By contrast, Top2 catalytic inhibitors, including dexrazoxane, act by binding to Top2 to prevent its initial interaction with dsDNA[16–18].
Bioflavonoids are found in fruits, vegetables, legumes, tea, coffee and wine[19–21]. Bioflavonoids are antioxidants and have anti-inflammatory, anti-viral, anti-carcinogenic, cardioprotective and cytoprotective properties[19,20,22]. Given the range of desirable health effects, bioflavonoids can also be found in over-the-counter supplements. However, similar to the Top2 poison etoposide, bioflavonoids contain pendant rings that feature a 4’-OH group essential for activity[23–25]. Additionally, bioflavonoids inhibit Top2α and Top2β activity[23,25], induce cleavage in the MLL gene locus, and can result in MLL rearrangements detectable by inverse PCR and karyotype analysis[11,26]. Along with the ability to poison Top2, bioflavonoids can impact DNA repair processes, alter epigenetic markers, and activate signal transduction pathways leading to altered protein expression in multiple pathways, such as cell cycle regulation, cell survival and cytokine expression[27–31]. Thus, it remains unclear which of these pleiotropic activities in cells are responsible for the potential mutagenic and carcinogenic impacts of bioflavonoids[32]. Average adult intake of soy that contains the bioflavonoid genistein is approximately 0.15–3.0 mg/day, but can reach up to 8.6 mg/day in women and 7.5 mg/day in men[33,34]. Additionally, bioflavonoids are bio-accumulative which can increase plasma concentrations. Natural and synthetic bioflavonoids cross the placental barrier [35], and exposure in utero is likely more damaging due to differences in metabolic and excretion rates of mother and fetus[36] as well as due to rapid proliferation and developmental programs of fetal cells that make them more sensitive to topoII inhibiting agents [37].
In this study, we examined a panel of bioflavonoids to quantify dose-dependent and time-dependent appearance, persistence, and repair of DSBs. We further determined the extent of bioflavonoid activity is due to inhibition of Top2 by treatment with dexrazoxane. Finally we examined the effect of Top2 inhibition on the formation of MLL translocations induced by bioflavonoid treatment. For these studies we used our recently established a stem cell reporter system that can systematically examine, quantitate, and stratify the potential for bioflavonoids and a larger panel of compounds to interfere with Top2 activity, induce DNA DSBs and promote chromosomal translocations involving the MLL BCR, analogous to those observed in infant leukemia[38]. This study demonstrates the range of potency of bioflavonoids to induce DNA DSBs and which bioflavonoids act primarily through Top2-dependent or Top2-independent mechanisms to promote chromosomal translocations. These findings have implications for understanding the biochemical activity of bioflavonoids and other dietary supplements with regard to their direct interaction with DNA, and they provide evidence to support a mode of action similar to that of etoposide in promoting the types of genome instability and chromosomal translocations that are associated with therapy-related and infant leukemias.
2. Materials and Methods
2.1. Compounds.
Etoposide and bioflavonoids were obtained from LKT Laboratories (St Paul, MN). Dipyrone and dexrazoxane were obtained from Sigma (St. Louis, MO). Compounds were diluted in dimethyl sulfoxide (DMSO; Sigma) and stored as 20 mM stock solutions. Stock solutions were diluted in 1X phosphate buffered saline pH 7 (PBS; Fisher, Hampton, NH) prior to each experiment.
2.2. Cell lines.
Mouse embryonic stem (ES) cells (all obtained from ATCC, Old Town Manassas, VA) D3[39], EtG2a[40] and MAG[38] cell lines were maintained at 37°C 5% CO2 on tissue culture dishes pre-treated with 0.2% gelatin (Sigma). Cells were authenticated by ATCC. Mycoplasma testing occurred yearly using InvivoGen Mycoplasma Detection kit according to manufacturer instructions. Cells were passaged and expanded 3–8 times, and then frozen as stocks. Stocks were thawed for each replicate experiment. Cells were maintained in non-selective medium consisting of Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Waltham, MA), 15% ES qualified STASIS fetal bovine serum (FBS; Gemini, Sacramento, CA), 100U/mL penicillin/streptomycin (Gemini), 2mM L-glutamine (Gemini), 0.1 mM non-essential amino acids (Gibco), 1000U/ml ESGRO® leukemia inhibitory factor (LIF; Gemini), 100 μM β-mercaptoethanol (Sigma).
2.3. Exposure to bioflavonoids and detection of γ-H2AX foci.
1 × 106 stem cells were plated on 15 mm gelatin-coated coverslips (Neuvitro Corporation, Vancouver, WA) with 4 mL non-selective medium and incubated 12 h at 37°C 5% CO2. Cells were treated with each compound at 0, 25, 50, 75, 100, 150, or 200 μM doses by addition to culture medium and incubated 1 h at 37°C 5% CO2. In preliminary trials no difference was observed between untreated and vehicle only treated cells. Thus in all experiments in this report, 0 μM dose indicated untreated cells. All dose ranges were selected based around approximate LD50 values as previously reported by our lab [38]. Eto & M: 50 μM,G & Q: 75 μM, K: 100 μM, & L: 175 μM [38]. Medium was aspirated and cells washed with 1X PBS, then cells were incubated in media for 0, 1, 4, or 8 h at 37°C 5% CO2. Culture medium was aspirated and cells washed with 1X PBS, then cells were fixed with 3% paraformaldehyde (Sigma) for 15 min at room temperature with rocking. Then coverslips were incubated with 0.2% Triton-X (Sigma) for 15 min at room temperature. Next, coverslips were blocked with 5% non-fat milk resuspended in 1x Tris-Buffered Saline-Tween 20 (TBS-T, Fisher, Waltham, MA) for 30 min at room temperature with rocking. Anti-phospho-Histone H2A.X antibody conjugated to Alexa Fluor 488 (Millipore, Burlington, MA) was diluted 1:100 in 5% non-fat milk/TBS-T and applied to coverslips for 1 h at room temperature in the dark. Coverslips were washed thrice with 1X PBS and mounted to slides (VWR, Radnor, PA) with ProLong Gold Antifade Mountant with DAPI (Life Technologies, Carlsbad, CA). Slides were stored at −20°C until confocal images were recorded with an Olympus FV1000 microscope. One-way ANOVA with Sidak’s multiple comparisons test was used for statistical analysis.
2.4. Pre-treatment with dexrazoxane, exposure to bioflavonoids, and detection of γ-H2AX Foci.
1 × 106 stem cells were plated on 15 mm gelatin-coated coverslips with 4 mL of culture medium, and incubated 12 h at 37°C 5% CO2. Cells were untreated (0 μM) or treated with 200 μM dexrazoxane for 1 or 5 h at 37°C 5% CO2. After dexrazoxane treatment, cells were treated with compounds for 1 h at 37°C 5% CO2 [38]. Medium was aspirated and cells were washed with 1X PBS. Then cells were stained for γ-H2AX foci and mounted to slides as described above. Slides were stored at −20°C until confocal images were recorded with an Olympus FV1000 microscope.
2.5. Quantification of chromosomal translocations.
MAG ES translocation Reporter cells [38] were plated at a density of 2 × 107 cells per 10 cm tissue culture plate. Cells were treated with 200 μM dexrazoxane for 1 or 5 h, then compounds at doses of 0, 50, 75, or 100 μM were added to the culture medium and incubated 1 h 37°C 5% CO2. Following exposure, cells were washed with 1X PBS before trypsinization. Cells were washed with 1X PBS again following trypsinization and plated on 96 well culture dishes with ES media. All treatment cohorts were washed with 1X PBS on the first 2 d after treatment, then incubated in ES media 37°C 5% CO2. For each experiment, all cells on all plates in all treatment groups were screened at days 5–7 post-treatment for GFP+ fluorescence at 400X magnification on an inverted Zeiss Axiovert25 microscope with images recorded by Zeiss AxioCam MRc digital camera. GFP+ cell colonies represent a translocation between two engineered GFP exons within huMLL and huAF9 bcr transgene inserts (Supplemental Fig. 1)[38]. Each complete experiment including controls was repeated a minimum of three independent times (n = 3). One-way ANOVA using Bonferroni post-hoc tests were used for statistical analysis.
3. Results
3.1. Bioflavonoids stimulate the appearance of DNA DSBs in a dose-dependent manner
We tested the potential of bioflavonoids to directly induce DNA DSBs as determined by the appearance of γH2AX foci. Untreated asynchronous cultures of ES cells contained few, if any, spontaneous γH2AX foci across all experiments and timepoints. As a control, cells were exposed to the potent Top2 poison etoposide. An increasing number of γH2AX foci were scored immediately following exposure to etoposide (Fig. 1A). All averages and p-values are listed in Supplemental Table 1. A similar pattern of persistence and kinetics of DSB repair was observed for all doses of etoposide tested; the average number of γH2AX foci persisted or slightly increased 1h post-exposure and then significantly declined over the total 8 h period (Fig. 1A).
Figure 1. Bioflavonoids Stimulate DNA DSBs and Kinetics of DNA Damage and Repair.
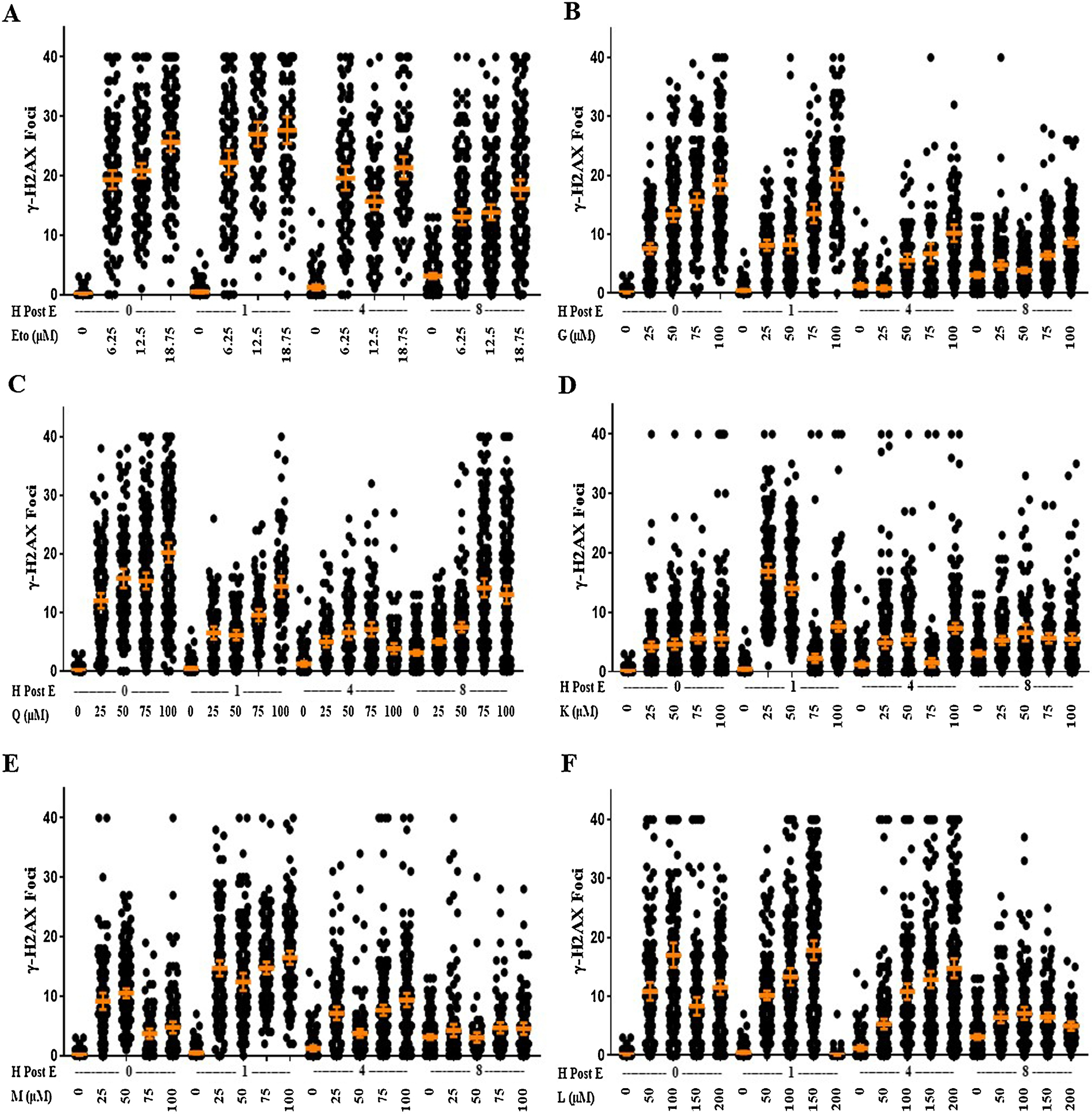
Cells were treated with A, etoposide (Eto), B, genistein (G), C, quercetin (Q), D, kaempferol (K), E, myricetin (M), or F, luteolin (L) at a range of doses before being stained for γH2AX foci either immediately after treatment or after 1, 4, or 8 h post-exposure. Doses for etoposide include 6.25, 12.5, and 18.75 μM. Doses for genistein, quercetin, kaempferol, and myricetin include 25, 50, 75, and 100 μM. Doses for luteolin doses include 50, 100, 150, and 200 μM. Each point represents the number of foci in one cell, a minimum of 100 cells were examined in each group. The mean is displayed with the 95% confidence interval as error bars. All p-values are provided in Supplemental Table 1.
We then exposed ES cells to increasing doses of a panel of bioflavonoids. Consistent with previous studies, genistein and quercetin robustly induced the appearance of γH2AX foci in a dose-dependent manner (Fig. 1B&C). Kaempferol only weakly induced γH2AX foci (Fig.1D). Myricetin and luteolin induced γH2AX foci but the number of observed foci peaked at 50 μM or 100 μM respectively. The number of γH2AX foci at higher doses of myricetin and luteolin, while still significantly higher than untreated cells, were quenched in comparison to the lower doses (Fig. 1E&F).
3.2. Persistence and kinetics of repair of bioflavonoid-induced DNA DSBs
Since bioflavonoids have structural similarity to etoposide and induced DSBs across the genome with no distinct pattern of distribution, we wanted to determine if the DSBs induced by bioflavonoids are sensed and repaired with kinetics similar to those induced by etoposide. To quantify the persistence and repair of bioflavonoid-induced DNA damage, cells were exposed to increasing doses of bioflavonoids, and γH2AX foci scored at 1, 4, and 8 h post-exposure (Fig. 1).
The overall cellular response, persistence, and repair kinetics of bioflavonoid-induced DSBs were similar to those observed following etoposide exposure. Agents induced a rapid appearance and significant average number of γH2AX foci/cell immediately post-exposure Consistent with the kinetics of DDR signaling and spread of γH2AX marks across chromatin away from each DSB, the appearance of γH2AX foci continued over the first hour post-exposure. DSBs persisted then disappeared, indicating repair of DSBs, with slower kinetics. As expected, since lower concentrations of bioflavonoids produced an absolute lower number of foci/cell, their disappearance and repair led to a return to baseline levels at earlier timepoints post-exposure than higher concentrations. Several bioflavonoids induced a second wave of γH2AX foci at later timepoints indicating a secondary indirect impact on chromosome stability.
In cells exposed to genistein, the average number of γH2AX foci persisted at 1 h post-exposure as compared to the 0 h timepoint. The presence of γH2AX foci decreased at 4 h and 8 h post-exposure following all doses. By 4 h post-exposure, the average number of foci/cell with 25 μM treatment decreased to baseline, and by 8 h post-exposure, 50 μM treatment decreased to baseline as well (Fig. 1B). Overall, when accounting for all four concentrations of genistein, the average number of DNA DSBs decreased slightly 1 h post-exposure, before drastically decreasing at 4 h and remaining low or undetectable by 8 h post-exposure.
Cells exposed to quercetin showed significant number of γH2AX foci immediately following exposure at all doses, that rapidly and significantly decreased within 1 h at all concentrations. At 1 h post-exposure with 25 μM, the average number foci/cell decreased by approximately 50%. The average foci/cell remained constant or further decreased by 4 h post-exposure. Interestingly, by 8 h post-exposure of higher concentrations, we observed a significant increase in the average number of foci/cell compared to the 4 h (Fig. 1C). When all quercetin concentration treatments are considered, there was a significant decline in DSBs by 1 and 4 h post-exposure, with a significant increase 8 h post-exposure.
Increasing concentrations of kaempferol led to an immediate small, but statistically significant increase, in γH2AX foci. All doses of kaempferol caused a similar number of γH2AX foci. By 4 h and 8 h post-exposure, γH2AX foci decreased down to approximately the same amount observed immediately following treatment at the 0 h timepoint (Fig. 1D).
Cells treated with increasing doses of myricetin or luteolin led to an overall significant increase in the average number foci/cell, although not with dose dependent increases observed with other bioflavonoids tested. However, the appearance of γH2AX persisted or continued to significantly increase by 1 h post-exposure, before significantly decreasing at 4 and 8 h post-exposure (Fig. 1E&F). Myricetin treatment showed the most similar trend to etoposide treatment with an increase in γH2AX foci after 1 h, followed by decreases by 4 and 8 h, with all doses returning to baseline by 8 h post-exposure (Fig. 1E).
We wanted to determine if combinatorial exposure to bioflavonoids would promote an additive or synergistic impact on the appearance, persistence, and repair of γH2AX foci that would better mimic dietary exposure to these compounds that are often contained in the same dietary sources. Cells were exposed to equal concentrations of both genistein and quercetin (G/Q) at the same concentrations used for single exposures to each. Unexpectedly, cells exposed to increasing concentrations of G/Q produced a similar average number of foci/cell compared to genistein or quercetin individually (Fig. 2A compared to Fig. 1B&C). By 1 h post-exposure, the observed range of average foci/cell in the G/Q cells mirrored exposure to genistein alone and consistent with the 0 h timepoint.
Figure 2. Combinations of Bioflavonoid Exposures Display Similar DNA DSBs and Kinetics to Singular Bioflavonoid Exposures.
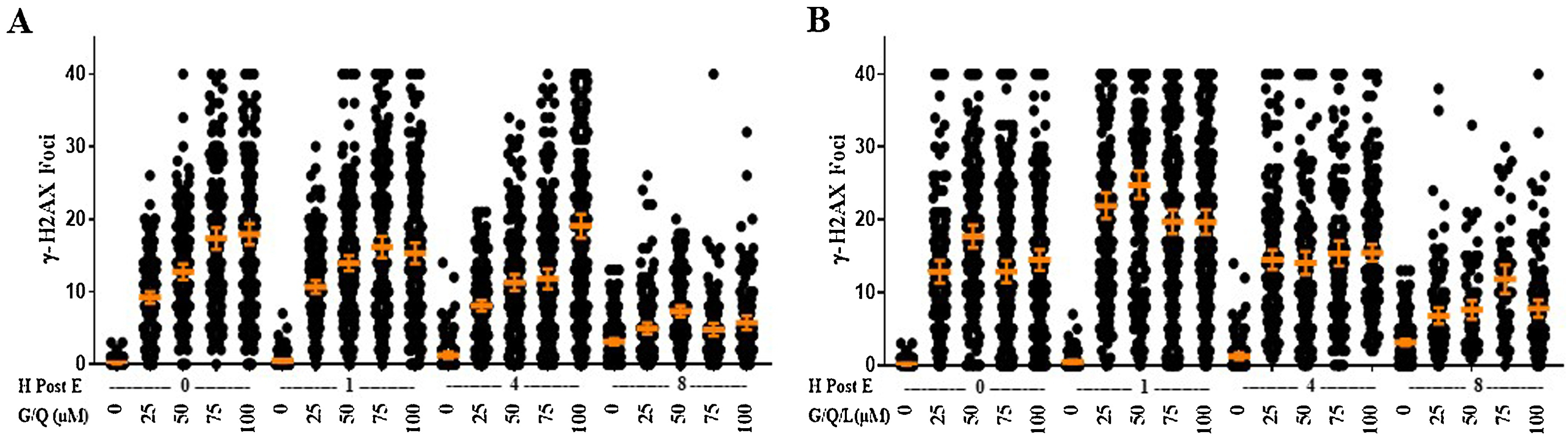
Cells were treated with A, genistein/quercetin (G/Q), or B, genistein/quercetin/luteolin (G/Q/L) at a range of doses before being stained for γH2AX foci either immediately after treatment or after 1, 4, or 8 h post-exposure. Doses for genistein/quercetin include 25/25, 50/50, 75/75, and 100/100 μM and for genistein/quercetin/luteolin doses include 25/25/50, 50/50/100, 75/75/150, and 100/100/100/200 μM. Each point represents the number of foci in one cell, a minimum of 100 cells were examined in each group. The mean is displayed with the 95% confidence interval as error bars. All p-values are provided in Supplemental Table 1.
Overall, the amount of DNA DSBs in cells exposed to G/Q decreased slightly at 4 h, then drastically decreased by 8 h post-exposure, except in the 100/100 μM group which showed a significant increase in average foci/cell from 1 to 4 h post-exposure, before decreasing by 8 h post-exposure (Fig. 2A).
Cells were exposed to the same concentrations of genistein, quercetin and luteolin (G/Q/L) as used for individual exposures. Increasing concentrations of G/Q/L induced an immediate increase in the incidence of γH2AX foci to levels similar to what was observed in cells exposed to each individual bioflavonoid at 0 h post-exposure (Fig. 2B compared to Fig. 1B,C,&F). γH2AX foci continued to appear over the first hour post-exposure to G/Q/L. These observations suggest an additive, rather than synergistic, impact of exposure to luteolin to the combination of G/Q. The kinetics of repair of DNA DSBs mirrored single exposures and significantly decreased by 4 h post-exposure compared to 1 h, but at elevated levels similar to the 0 h timepoint. The average number of foci/cell continued to decrease by 8 h post-exposure. The kinetics with all four doses is similar to the trend observed with etoposide exposure, with an increase in DNA DSBs by 1 h post-exposure followed by repair of DSBs by 4 and 8 h post-exposure (Fig. 2B).
3.3. Bioflavonoid-induced DNA DSBs through Top2-dependent and -independent mechanisms
Some bioflavonoids such as kaempferol, quercetin and myricetin have been shown to directly inhibit Top2 in cell free systems, but bioflavonoids also have pleiotropic effects. To understand what portion of the bioflavonoid-induced DNA DSBs are a direct effect of Top2 inhibition, cells were pre-treated with 200 μM dexrazoxane (+DEX) for 1 or 5 h, followed by 1 h exposure to each of the compounds, and immediately scored for the presence of γH2AX foci. As expected, asynchronous cultures +DEX contained few, if any, spontaneous γH2AX foci across all experiments and timepoints (Fig. 3). As expected, pre-treatment with DEX was sufficient to produce a significant reduction in the average number of foci/cell following etoposide exposure. Exposure to etoposide alone induced an average 20.1 foci/cell that was significantly reduced by 51% with 1 h/+DEX and reduced by almost 75% with 5 h/+DEX (Fig. 3A).
Figure 3. Dexrazoxane Pre-treatment Reduces Bioflavonoid-induced DNA DSBs Through Top2-dependent Mechanisms.
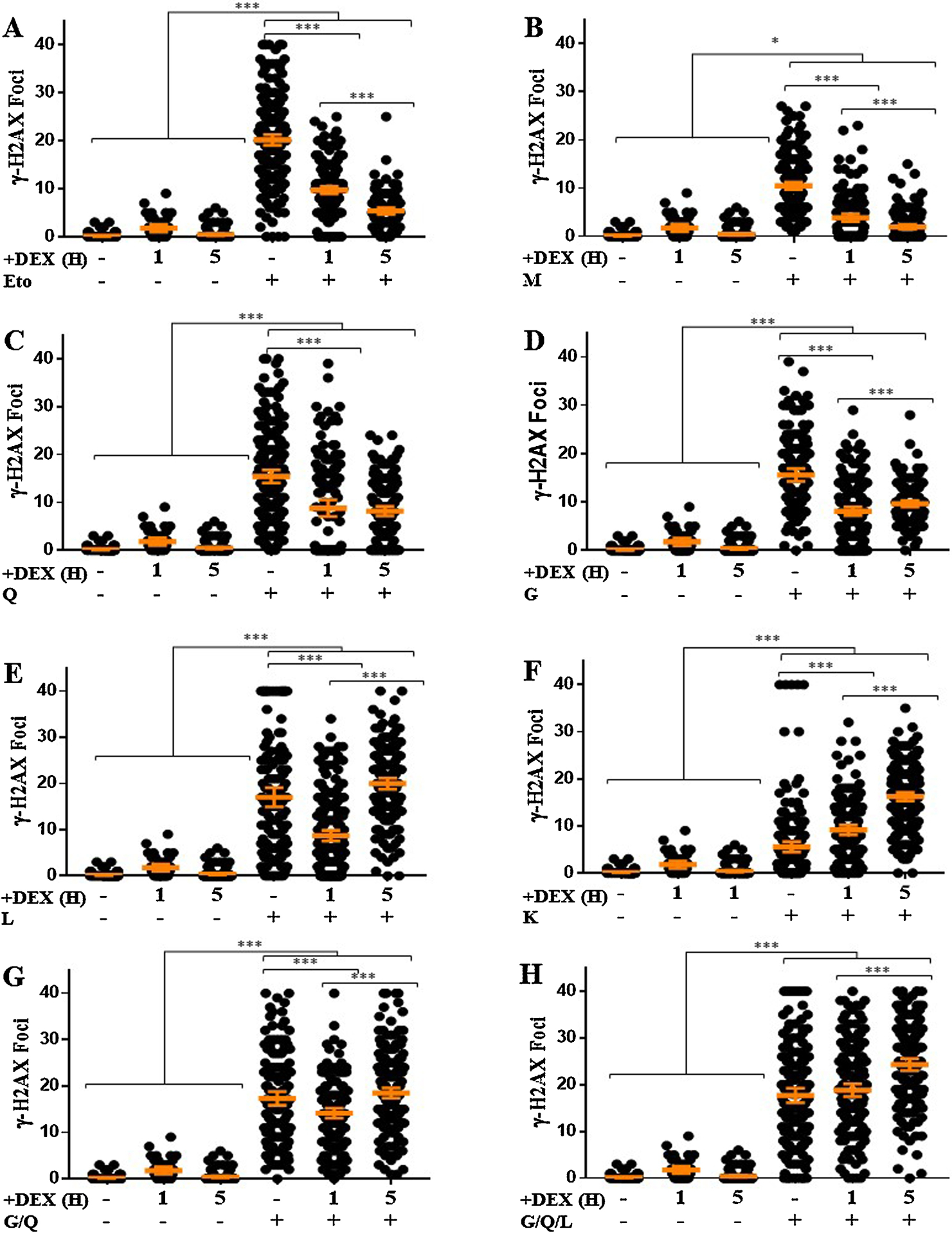
Cells were pre-treated with dexrazoxane at 200 μM for either 1 or 5 h before 1 h treatment with A, etoposide (Eto) 6.25 μM, B, myricetin (M) 50 μM, C, quercetin (Q) 75 μM, D, genistein (G) 75 μM, E, luteolin (L) 100 μM, F, kaempferol (K) 100 μM, G, genistein/quercetin (G/Q) 75/75 μM, or H, genistein/quercetin/luteolin (G/Q/L) 50/50/100 μM. Immediately after treatment, cells were stained for γH2AX foci. Each point represents the number of foci in one cell, a minimum of 100 cells were examined in each group. The mean is displayed with the 95% confidence interval as error bars. * p<0.05, *** p<0.0001
A significant portion of bioflavonoid-induced DSBs was inhibited by the pre-treatment of cells with dexrazoxane consistent with a Top2-dependent mechanism of bioflavonoid activity. Myricetin exposure alone induced an average 10.5 foci/cell that was significantly reduced with 1 h/+DEX and inhibited by approximately 80% or to baseline levels with 5 h/+DEX indicating that the significant majority of induced DSBs were created via a Top2-dependent mechanism (Fig. 3B). Quercetin exposure induced an average 15.4 foci/cell that was significantly reduced by 57% with 1 h/+DEX and remained consistent with 5 h/+DEX (Fig. 3C). Exposure to genistein induced an average 15.6 foci/cell that was significantly reduced by 48% with 1 h/+DEX and slightly but significantly increased with 5 h/+DEX, although still significantly lower than genistein only (Fig. 3D).
Luteolin exposure induced an average 17.0 foci/cell. 1 h/+DEX treatment resulted in a 51% decrease in the average foci/cell, but 5 h/+DEX led to a small but significant increase in the average foci/cell (Fig. 3E). This increase was greater than what was observed for luteolin alone. Although 1 h/+DEX significantly inhibited DNA DSBs induced by quercetin, genistein, or luteolin, increased pre-treatment time to 5 h was not sufficient for any further reduction in the average number of foci/cell, and in fact increased after luteolin exposure, supporting a Top2-independent mechanism for the remaining observed DSBs. Uniquely, dexrazoxane pre-treatment did not inhibit the appearance or persistence of γH2AX foci after kaempferol exposure. 1 hr/+DEX produced a significant 165% increase in γH2AX foci, and 5 h/+DEX showed a 300% increase compared to exposure to kaempferol alone (Fig. 3F).
We wanted to determine if dexrazoxane pre-treatment would inhibit the appearance of γH2AX foci induced by combinatorial exposure to bioflavonoids. Cells exposed to G/Q induced an average 17.3 foci/cell that significantly decreased with 1 h/+DEX. However, the average number foci/cell significantly increased with 5 h/+DEX, compared to the 1 h/+DEX, though the 5 h/+DEX group to levels statistically consistent with G/Q alone (Fig. 3G). Cells exposed to G/Q/L alone induced an average 17.8 foci/cell that remained constant after 1 h/+DEX and significantly increased after 5 h/+DEX compared to both G/Q/L alone and 1 h/+DEX (Fig. 3H).
3.4. Bioflavonoid-induced chromosome translocations through Top2-dependent and -independent mechanisms
We wanted to understand the consequences of bioflavonoid-induced DSBs and determine what portion of, bioflavonoid-induced chromosomal translocations are Top2-dependent and sensitive to dexrazoxane pre-treatment. We used our previously developed MAG Reporter cell line that only expresses green fluorescent protein (GFP) if DSB repair leads to a chromosomal translocation [38] (Sup. Fig. 1). In the MAG Reporter cell line, two engineered GFP exons within huMLL and huAF9 bcr transgene inserts are upstream (GFPe1) or downstream (GFPe2) of mapped breakpoints identified in infant- and t-AML. DNA DSBs in each of chromosome transgene inserts may be repaired to ligate the GFPe1 exon and the GFPe2 exon onto the same DNA duplex, generate a chromosomal translocation, and generate a functional GFP gene (Sup. Fig. 1). The Reporter cell line previously demonstrated that exposure to etoposide, a panel of bioflavonoids, or ROS was sufficient to promote the formation of DSB-induced chromosomal translocations[38,41].
As expected, the translocation frequency observed after etoposide exposure alone significantly decreased by 71% following dexrazoxane pre-treatment (Table 1). Similarly, the translocation frequency observed after myricetin exposure significantly decreased following dexrazoxane pre-treatment (Table 1). By contrast, the formation of translocations induced by genistein or kaempferol exposure was not sensitive to Top2-inhibition. The translocation frequency observed after genistein exposure did not decrease but increased slightly although not significantly, and the translocation frequency observed after kaempferol exposure had a significant increase by more than 400% (Table 1). The impact of dexrazoxane and Top2 inhibition of GFP+ chromosomal translocations was overall consistent with the observed γH2AX scoring.
Table 1.
Inhibition of Top2 with Pre-treatment with Dexrazoxane and Frequency of Bioflavonoid-induced Translocations.
| Scored GFP+ Events | Average Frequency | ||||
|---|---|---|---|---|---|
| Compound | Dose | −DEX | +DEX | −DEX | +DEX |
| Untreated | n/a | 0 | 0, 2, 4 | <0.007 × 10−6 | 0.20 × 10−6 |
| Etoposide | 50 μM | 80, 92, 68, 97 | 23, 25, 26 | 8.43 × 10−6 | 2.47 × 10−6 |
| Genistein | 75 μM | 15, 8, 14 | 11, 17, 19 | 1.23 × 10−6 | 1.57 × 10−6 |
| Kaempferol | 100 μM | 6, 2, 3, 4 | 10, 18, 19 | 0.38 × 10−6 | 1.57 × 10−6 |
| Myricetin | 100 μM | 1, 1, 2 | 1, 0, 0 | 0.13 × 10−6 | 0.03 × 10−6 |
The MAG translocation reporter cell line was treated with 200 μM dexrazoxane for 5 h before 1 h treatment with etoposide (50 μM), genistein (75 μM), kaempferol (100 μM) or myricetin (50 μM). Number of GFP+ colonies were counted after 5–7 days. Experiment was repeated in triplicate and compared to our previous reported data without dexrazoxane pre-treatment. The translocation frequency decreased with dexrazoxane pre-treatment for etoposide (p= 0.0006), increased for kaempferol (p= 0.0058) and remained statistically unchanged for untreated, genistein, and myricetin (p= 0.272, 0.363, and 0.101, respectively).
4. Discussion
Overall, this study demonstrated the potential for individual bioflavonoids to lead to DNA DSBs in a dose-dependent manner through both Top2-dependent and Top2-independent mechanisms. Consistent with its known major activity as a Top2 inhibitor in the cleavage-ligation reaction, this study supported etoposide’s potential to rapidly induce multiple and persistent DNA DSBs and translocations that were significantly reduced with Top2 inhibition. The bioflavonoids examined here all demonstrated the similar ability to directly cause DSBs immediately post-exposure, although at lower absolute levels than induced by etoposide. Exposure to etoposide and myricetin showed the most similar overall kinetics of DNA damage and repair, as well as dependence on Top2. Etoposide and its metabolites act as both an interfacial (traditional) and as a covalent Top2 poison[10,42]. Bandele et al demonstrated that myricetin also works through both mechanisms to poison Top2 in cell free systems suggesting that the biochemical mechanism of Top2 poisoning is more predictive of a bioflavonoid’s potential to induce DNA DSBs, than its subclass[10]. Bandele et al also showed myricetin causes more DNA cleavage than traditional poisons but at a slower rate, which could in part explain the significantly greater inhibition of DSB formation in cells exposed to myricetin as compared to cells exposed to genistein or quercetin that act exclusively as traditional poisons[10].
Top2 has two isoforms, α and β, with distinct patterns of expression and functions. Top2α is highly expressed in highly proliferative cell types including stem cells. Top2α associates with replication forks and remains bound to chromosomes in mitotic cells. Top2β is expressed in most cell types, including stem cells, regardless of proliferation status and is required for viability of some post-mitotic cells such as neurons[1,2]. Unrelated to its direct activity to release torsional strain in DNA through DSBs, Top2β binding is enriched at promoters and associated with cohesion, TAD boundaries, CTCF binding sites, and H3K4me2, a mark of active chromatin[43,44]. Dexrazoxane inhibits both Top2 isoforms by stabilizing the closed clamp Top2 conformation preventing DNA binding, however with longer DEX treatment the drug induces the proteasomal degradation of Top2β [18]. Therefore, any persistent DNA damage after DEX treatment is likely due to Top2-independent mechanisms.
Only a portion of DNA DSBs induced by genistein and quercetin were inhibited by dexrazoxane pre-treatment, and formation of DSBs induced by luteolin and kaempferol were resistant to dexrazoxane pre-treatment and increased. These data indicate these compounds promote at least some DNA damage and chromosomal translocations through Top2-indepependent mechanisms. Bioflavonoids may lead to replication fork collapse, transcription machinery collision, or early apoptotic triggers leading to DSBs[45].
Repair of DNA DSBs can occur by one of several competing pathways including homologous recombination, classical nonhomologous end-joining (c-NHEJ), or alternative end-joining (alt-EJ)[46]. Recent studies suggest that alt-EJ preferentially leads to translocations, and inhibition of proteins involved in alt-EJ suppress translocation formation[47]. Exposure to bioflavonoids in our Reporter cell system both decreases expression of c-NHEJ proteins and increases expression of alt-EJ proteins[48]. Thus, bioflavonoid activity could direct DSBs to more mutagenic repair associated with chromosomal translocations.
Unexpectedly, DSBs induced by quercetin and luteolin were repaired rapidly post-exposure, to be followed by a second wave of DSBs appearing at later timepoints. When all quercetin concentration treatments are considered, there is a significant drop in the number of DSBs at 1 and 4 h post-exposure, with a significant increase 8 h post-exposure. The delayed appearance of new γH2AX foci suggests an indirect secondary effect of quercetin.
Oxidative stress is implicated in multiple chronic diseases and cancers[49,50]. While bioflavonoids have been shown to have antioxidant capabilities, some antioxidants can induce oxidative stress at higher doses[40,41,50]. The kinetics of γH2AX following quercetin and luteolin treatment would be consistent with a role of inducing oxidative stress through increased reactive oxygen/nitrogen species (ROS/RNS), that in turn lead to DSBs at later times post-exposure[40,50]. In support of this, ROS can cause DNA damage, and we previously showed that ROS are sufficient to promote translocations between MLL and AF9 bcrs in our Reporter cell line[41].
Exposure to combinations of bioflavonoids did not additively nor synergistically induce DNA DSBs. Exposure to multiple bioflavonoids led to the appearance and persistence of γH2AX foci with kinetics of the individual bioflavonoid that led to the greatest level of persistent damage at a single timepoint. This result suggests some redundancy or overlap of the chromosomal loci susceptible to damage by bioflavonoids even if individual bioflavonoids induce a greater or lesser average number foci/cell. If susceptible breakpoints overlap then exposure to multiple compounds at a given time would not present on a global scale with breaks at new loci. In addition, if luteolin indirectly induces damage at a later secondary timepoint after repair of any immediate G/Q/L breakpoints, then these would be expected to appear as newly formed foci.
Recently, END-seq has been used for genome mapping of etoposide-induced chromosomal breakpoints and the correlation of these with Top2 binding sites[43]. Direct mapping of bioflavonoid-induced breakpoints by END-seq could provide insight into specific loci of the genome are susceptible to bioflavonoid damage, the overlap of susceptible loci to individual bioflavonoids and Top2 binding sites. We would predict that genome mapping will reveal that quercetin promotes cleavage at a smaller number of hotspots that largely overlap with a subset of genistein-induced cleavage hotspots. Further, genome mapping studies may reveal which bioflavonoids promote DNA damage with unique patterns to suggest possible synergy in their dietary consumption. Identification of overlap between bioflavonoid-induced and etoposide-induced cleavage hotspots may be the strongest predictors of potential oncogenesis events, particularly if they occur in known leukemic BCRs or proto-oncogene loci.
Top2β activity is required for etoposide-induced DNA DSBs and chromosomal rearrangements initiating within the MLL locus in cell lines[51]. Similarly, inhibition, but not complete loss, of Top2 activity by dexrazoxane, led to a reduction of etoposide-induced translocations in this study. We were also able to stratify the potential for individual bioflavonoids to promote translocations and show which ones specifically were also sensitive to Top2 inhibition. Overall, these findings have implications for understanding the biochemical activity of bioflavonoids and other dietary supplements to interact directly with DNA to promote DNA DSBs and genome instability, and provide direct evidence that specific Top2 inhibitors or targeted drugs could be developed that possess less leukemic potential or suppress chromosomal translocations associated with therapy-related and infant leukemias. This work further highlights the need for in vivo models to understand physiological consequences of bioflavonoids and their contribution to infant and t- AML.
Supplementary Material
Supplemental Figure 1. Structure of Transgene Constructs in MLL-AF9-GFP (MAG) Translocation Reporter Cell Line. A, Schematic diagram showing GFPe1 and GFPe2 constructs in huMLL and huAF9 breakpoint cluster regions on heterologous chromosomes. GFPe1 includes promoter and upstream region, engineered exon 1, and splice donor. GFPe2 includes adenovirus branch sequence, splice acceptor, engineered exon 2, and polyA sequence. Schematic of DNA DSBs and resultant translocation between the huMLL and huAF9 bcrs to result in GFPe1 and GFPe2 on the same DNA duplex to allow for GFP expression. B, GFP+ colonies were scored by inverted fluorescent microscopy. Representative phase contrast (left panel) and fluorescent (right panel) microscopy images of GFP+ colonies are shown side by side.
Highlights:
Bioflavonoids induce persistent DNA damage similar to etoposide
Bioflavonoid-induced DNA damage is partially dependent on Top2 activity
Bioflavonoid-induced translocations can be reduced by inhibition of Top2
Bioflavonoid structure impacts mechanism of DNA damage and chromosomal translocations
Funding:
This work was supported by funds provided by the University of North Carolina at Charlotte and the National Institutes of Health R15GM128571.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest.
References
- [1].Felix CA, Kolaris CP, Osheroff N, Topoisomerase II and the etiology of chromosomal translocations, DNA Repair (Amst). 5 (2006) 1093–1108. 10.1016/j.dnarep.2006.05.031. [DOI] [PubMed] [Google Scholar]
- [2].Pendleton M, Lindsey RH, Felix CA, Grimwade D, Osheroff N, Topoisomerase II and leukemia, Ann. N. Y. Acad. Sci 1310 (2014) 98–110. 10.1111/nyas.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Broeker PLS, Super HG, Thirman MJ, Pomykala H, Yonebayashi Y, Tanabe S, Zeleznik-Le N, Rowley JD, Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: Correlation with scaffold attachment regions and topoisomerase II consensus binding sites, Blood. 87 (1996) 1912–1922. [PubMed] [Google Scholar]
- [4].Gu Y, Alder H, Nakamura T, Schichman SA, Prasad R, Canaani O, Saito H, Croce CM, Canaani E, Sequence analysis of the breakpoint cluster region in the ALL-1 gene involved in acute leukemia., Cancer Res. 54 (1994) 2327–30. http://www.ncbi.nlm.nih.gov/pubmed/8162575. [PubMed] [Google Scholar]
- [5].Muntean AG, Hess JL, The Pathogenesis of Mixed-Lineage Leukemia, Annu. Rev. Pathol. Mech. Dis 7 (2012) 283–301. 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, Ben Abdelali R, Macintyre E, De Braekeleer E, De Braekeleer M, Delabesse E, de Oliveira MP, Cavé H, Clappier E, van Dongen JJM, Balgobind BV, van den Heuvel-Eibrink MM, Beverloo HB, Panzer-Grümayer R, Teigler-Schlegel A, Harbott J, Kjeldsen E, Schnittger S, Koehl U, Gruhn B, Heidenreich O, Chan LC, Yip SF, Krzywinski M, Eckert C, Möricke A, Schrappe M, Alonso CN, Schäfer BW, Krauter J, Lee DA, zur Stadt U, Te Kronnie G, Sutton R, Izraeli S, Trakhtenbrot L, Lo Nigro L, Tsaur G, Fechina L, Szczepanski T, Strehl S, Ilencikova D, Molkentin M, Burmeister T, Dingermann T, Klingebiel T, Marschalek R, New insights to the MLL recombinome of acute leukemias, Leukemia. 23 (2009) 1490–1499. 10.1038/leu.2009.33. [DOI] [PubMed] [Google Scholar]
- [7].Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, Greaves MF, Backtracking leukemia to birth: Identification of clonotypic gene fusion sequences in neonatal blood spots, Proc. Natl. Acad. Sci. U. S. A 94 (1997) 13950–13954. 10.1073/pnas.94.25.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gole B, Wiesmüller L, Leukemogenic rearrangements at the mixed lineage leukemia gene (MLL)-multiple rather than a single mechanism, Front. Cell Dev. Biol 3 (2015) 1–13. 10.3389/fcell.2015.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Spector LG, Xie Y, Robison LL, Heerema NA, Hilden JM, Lange B, Felix CA, Davies SM, Slavin J, Potter JD, Blair CK, Reaman GH, Ross JA, Maternal Diet and Infant Leukemia: The DNA Topoisomerase II Inhibitor Hypothesis: A Report from the Children’s Oncology Group, Cancer Epidemiol. Biomarkers Prev 14 (2005) 651–655. 10.1158/1055-9965.EPI-04-0602. [DOI] [PubMed] [Google Scholar]
- [10].Bandele OJ, Clawson SJ, Osheroff N, Dietary polyphenols as topoisomerase II poisons: B ring and C ring substituents determine the mechanism of enzyme-mediated DNA cleavage enhancement, Chem. Res. Toxicol 21 (2008) 1253–1260. 10.1021/tx8000785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Strick R, Strissel PL, Borgers S, Smith SL, Rowley JD, Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia, Proc. Natl. Acad. Sci 97 (2000) 4790–4795. 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gómez-Herreros F, Romero-Granados R, Zeng Z, Álvarez-Quilón A, Quintero C, Ju L, Umans L, Vermeire L, Huylebroeck D, Caldecott KW, Cortés-Ledesma F, TDP2-Dependent Non-Homologous End-Joining Protects against Topoisomerase II-Induced DNA Breaks and Genome Instability in Cells and In Vivo, PLoS Genet. 9 (2013). 10.1371/journal.pgen.1003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Deweese JE, Osheroff N, The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing, Nucleic Acids Res. 37 (2009) 738–748. 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, Meeker AK, Netto G, De Marzo AM, Nelson WG, Yegnasubramanian S, Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements., Nat. Genet 42 (2010) 668–75. 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cowell IG, Sondka Z, Smith K, Lee KC, Manville CM, Sidorczuk-Lesthuruge M, Rance HA, Padget K, Jackson GH, Adachi N, Austin CA, Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 8989–8994. 10.1073/pnas.1204406109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Andoh T, Bis(2,6-dioxopiperazines), catalytic inhibitors of DNA topoisomerase II, as molecular probes, cardioprotectors and antitumor drugs., Biochimie. 80 (1998) 235–46. 10.1016/s0300-9084(98)80006-0. [DOI] [PubMed] [Google Scholar]
- [17].Classen S, Olland S, Berger JM, Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187, Proc. Natl. Acad. Sci. U. S. A 100 (2003) 10629–10634. 10.1073/pnas.1832879100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yi LL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF, Topoisomerase IIβ-mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane, Cancer Res. 67 (2007) 8839–8846. 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- [19].Yao LH, Jiang YM, Shi J, Tomás-Barberán FA, Datta N, Singanusong R, Chen SS, Flavonoids in food and their health benefits., Plant Foods Hum. Nutr 59 (2004) 113–22. 10.1212/01.CON.0000389954.27724.00. [DOI] [PubMed] [Google Scholar]
- [20].Galati G, O’Brien PJ, Potential toxicity of flavonoids and other dietary phenolics: Significance for their chemopreventive and anticancer properties, Free Radic. Biol. Med 37 (2004) 287–303. 10.1016/j.freeradbiomed.2004.04.034. [DOI] [PubMed] [Google Scholar]
- [21].Scalbert A, Williamson G, Dietary intake and bioavailability of polyphenols., J. Nutr 130 (2000) 2073S–85S. 10.1093/jn/130.8.2073S. [DOI] [PubMed] [Google Scholar]
- [22].Turmagambetova AS, Sokolova NS, Bogoyavlenskiy AP, Berezin VE, Lila MA, Cheng DM, Dushenkov V, New functionally-enhanced soy proteins as food ingredients with anti-viral activity, VirusDisease. 26 (2015) 123–132. 10.1007/s13337-015-0268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Azarova AM, Lin R-K, Tsai Y-C, Liu LF, Lin C-P, Lyu YL, Genistein induces topoisomerase IIbeta- and proteasome-mediated DNA sequence rearrangements: Implications in infant leukemia., Biochem. Biophys. Res. Commun 399 (2010) 66–71. 10.1016/j.bbrc.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Long BH, Musial ST, Brattain MG, Comparison of Cytotoxicity and DNA Breakage Activity of Congeners of Podophyllotoxin Including VP16–213 and VM26: A Quantitative Structure-Activity Relationship, Biochemistry. 23 (1984) 1183–1188. 10.1021/bi00301a024. [DOI] [PubMed] [Google Scholar]
- [25].Bandele OJ, Osheroff N, Bioflavonoids as poisons of human topoisomerase IIα and IIβ, Biochemistry. 46 (2007) 6097–6108. 10.1021/bi7000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].van W. van Doorn-Khosrovani SB, Janssen J, Maas LM, Godschalk RWL, Nijhuis JG, van Schooten FJ, Dietary flavonoids induce MLL translocations in primary human CD34+ cells, Carcinogenesis. 28 (2007) 1703–1709. 10.1093/carcin/bgm102. [DOI] [PubMed] [Google Scholar]
- [27].Russo GL, Vastolo V, Ciccarelli M, Albano L, Macchia E, Ungaro P, Luigi G, Vastolo V, Ciccarelli M, Albano L, Dietary polyphenols and chromatin remodeling, Crit. Rev. Food Sci. Nutr 57 (2017) 2589–2599. 10.1080/10408398.2015.1062353. [DOI] [PubMed] [Google Scholar]
- [28].Phillips PA, Sangwan V, Borja-Cacho D, Dudeja V, Vickers SM, Saluja AK, Myricetin induces pancreatic cancer cell death via the induction of apoptosis and inhibition of the phosphatidylinositol 3-kinase (PI3K) signaling pathway, Cancer Lett. 308 (2011) 181–188. 10.1016/j.canlet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pommier Y, Sun Y, Huang SYN, Nitiss JL, Roles of eukaryotic topoisomerases in transcription, replication and genomic stability, Nat. Rev. Mol. Cell Biol 17 (2016) 703–721. 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aalinkeel R, Bindukumar B, Reynolds JL, Sykes DE, Mahajan SD, Chadha KC, Schwartz SA, The dietary bioflavonoid, quercetin, selectively induces apoptosis of prostate cancer cells by down-regulating the expression of heat shock protein 90, Prostate. 68 (2008) 1773–1789. 10.1002/pros.20845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cho S, Park S, Kwon M, Jeong T-S, Bok S, Choi W, Jeong W, Ryu S, Do S, Lee C, Song J, Jeong K, Quercetin suppresses proinflammatory cytokines production through MAP kinases and NF- κ B pathway in lipopolysaccharide-stimulated macrophage, Mol. Cell. Biochem 243 (2003) 153–160. [DOI] [PubMed] [Google Scholar]
- [32].Bariar B, Vestal CG, Richardson C, Long-Term Effects of Chromatin Remodeling and DNA Damage in Stem Cells Induced by Environmental and Dietary Agents, J. Environ. Pathol. Toxicol. Oncol 32 (2013) 307–327. 10.1615/JEnvironPatholToxicolOncol.2013007980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mulligan AA, Welch AA, McTaggart AA, Bhaniani A, Bingham SA, Intakes and sources of soya foods and isoflavones in a UK population cohort study (EPIC-Norfolk), Eur. J. Clin. Nutr 61 (2006) 248 10.1038/sj.ejcn.1602509. [DOI] [PubMed] [Google Scholar]
- [34].Ross JA, Kasum CM, Dietary flavonoids : Bioavailability, metabolic effects, and safety Reproduced with permission of the copyright owner. Further reproduction prohibited without permission., Annu. Rev. Nutr 22 (2002) 19–34. [DOI] [PubMed] [Google Scholar]
- [35].Vanhees K, Godschalk RW, Sanders A, Van SB Waalwijk van Doorn-Khosrovani, F.J. Van Schooten, Maternal quercetin intake during pregnancy results in an adapted iron homeostasis at adulthood, Toxicology. (2011). 10.1016/j.tox.2011.10.017. [DOI] [PubMed] [Google Scholar]
- [36].Todaka E, Sakurai K, Fukata H, Miyagawa H, Uzuki M, Omori M, Osada H, Ikezuki Y, Tsutsumi O, Iguchi T, Mori C, Fetal exposure to phytoestrogens—The difference in phytoestrogen status between mother and fetus, Environ. Res 99 (2005) 195–203. 10.1016/j.envres.2004.11.006. [DOI] [PubMed] [Google Scholar]
- [37].Zandvliet DWJ, Hanby AM, Austin CA, Marsh KL, Clark IBN, Wright NA, Poulsom R, Analysis of foetal expression sites of human type II DNA topoisomerase a and β mRNAs by in situ hybridisation, Biochim. Biophys. Acta - Gene Struct. Expr 1307 (1996) 239–247. 10.1016/0167-4781(96)00063-2. [DOI] [PubMed] [Google Scholar]
- [38].Bariar B, Vestal CG, Deem B, Goodenow D, Engledove RW, Sahyouni M, Richardson C, Bioflavonoids promote stable translocations between MLL - AF9 breakpoint cluster regions independent of normal chromosomal context : Model system to screen environmental risks, Environ. Mol. Mutagen (2018). https://doi.org/10.0.3.234/em.22245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Doetschman TC, Eistetter H, Katz M, The in vitro development of blastocyst-derived embryonic stem cell lines: Formation of visceral yolk sac, blood islands and myocardium, J. Embryol. Exp. Morphol VOL. 87 (1985) 27–45. [PubMed] [Google Scholar]
- [40].Hooper M, Hardy K, Handyside A, Hunter S, Monk M, HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells., Nature. 326 (n.d) 292–5. 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- [41].Richardson C, Yan S, Vestal CG, Oxidative stress, bone marrow failure, and genome instability in hematopoietic stem cells, Int. J. Mol. Sci 16 (2015) 2366–2385. 10.3390/ijms16022366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Smith NA, Byl JAW, Mercer SL, Deweese JE, Osheroff N, Etoposide quinone is a covalent poison of human topoisomerase IIβ, Biochemistry. 53 (2014) 3229–3236. 10.1021/bi500421q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Canela A, Maman Y, Huang SN, Wutz G, Tang W, Zagnoli-Vieira G, Callen E, Wong N, Day A, Peters J-M, Caldecott KW, Pommier Y, Nussenzweig A, Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity, Mol. Cell 75 (2019) 252–266.e8. 10.1016/j.molcel.2019.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tiwari VK, Burger L, Nikoletopoulou V, Deogracias R, Thakurela S, Wirbelauer C, Kaut J, Terranova R, Hoerner L, Mielke C, Boege F, Murr R, Peters AHFM, Barde YA, Schübeler D, Target genes of Topoisomerase IIβ regulate neuronal survival and are defined by their chromatin state, Proc. Natl. Acad. Sci. U. S. A 109 (2012). 10.1073/pnas.1119798109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Betti CJ, Villalobos MJ, Jiang Q, Cline E, Diaz MO, Loredo G, Vaughan ATM, Cleavage of the MLL gene by activators of apoptosis is independent of topoisomerase II activity, Leukemia. 19 (2005) 2289–2295. 10.1038/sj.leu.2403966. [DOI] [PubMed] [Google Scholar]
- [46].Ranjha L, Howard SM, Cejka P, Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes, Chromosoma. 127 (2018) 187–214. 10.1007/s00412-017-0658-1. [DOI] [PubMed] [Google Scholar]
- [47].Zhang Y, Jasin M, An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway., Nat. Struct. Mol. Biol 18 (2011) 80–4. 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ghosh A, Richardson CA, Abstract 2571: Role of XRCC4 downregulation in bioflavonoid-induced chromosomal translocations, in: Mol. Cell. Biol. / Genet, American Association for Cancer Research, 2019: pp. 2571–2571. 10.1158/1538-7445.AM2019-2571. [DOI] [Google Scholar]
- [49].Ito F, Sono Y, Ito T, Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: Oxidative stress in diabetes, atherosclerosis, and chronic inflammation, Antioxidants 8 (2019). 10.3390/antiox8030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lee JH, Shu L, Fuentes F, Su Z-Y, Kong A-NT, Cancer Chemoprevention by Traditional Chinese Herbal Medicine and Dietary Phytochemicals: Targeting Nrf2-Mediated Oxidative Stress/Anti-Inflammatory Responses, Epigenetics, and Cancer Stem Cells, J. Tradit. Complement. Med 3 (2013) 69–79. 10.4103/2225-4110.107700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau JYN, Wang JC, Liu LF, Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 11014–11019. 10.1073/pnas.0704002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Structure of Transgene Constructs in MLL-AF9-GFP (MAG) Translocation Reporter Cell Line. A, Schematic diagram showing GFPe1 and GFPe2 constructs in huMLL and huAF9 breakpoint cluster regions on heterologous chromosomes. GFPe1 includes promoter and upstream region, engineered exon 1, and splice donor. GFPe2 includes adenovirus branch sequence, splice acceptor, engineered exon 2, and polyA sequence. Schematic of DNA DSBs and resultant translocation between the huMLL and huAF9 bcrs to result in GFPe1 and GFPe2 on the same DNA duplex to allow for GFP expression. B, GFP+ colonies were scored by inverted fluorescent microscopy. Representative phase contrast (left panel) and fluorescent (right panel) microscopy images of GFP+ colonies are shown side by side.