

Summary:
Small RNA-sequencing has been widely adopted to study the diversity of extracellular RNAs (exRNAs) in biofluids; however, exRNA samples can be challenging: they are vulnerable to contamination and artifacts from different isolation techniques, present in lower concentrations than cellular RNA, and are occasionally of exogenous origin. To address these challenges, we present exceRpt, the extracellular RNA processing toolkit of the NIH Extracellular RNA Communication Consortium (ERCC). exceRpt is structured as a cascade of filters and quantifications prioritized based on one’s confidence in a given set of annotated RNAs. It generates quality control reports and abundance estimates for RNA biotypes. It is also capable of characterizing mappings to exogenous genomes, which, in turn, can be used to generate phylogenetic trees. exceRpt has been used to uniformly process all ~3,500 exRNA-seq datasets in the public exRNA atlas and is available from genboree.org and github.gersteinlab.org/exceRpt.
Introduction:
Extracellular RNA (exRNA) have been found in the blood and other body fluids (Patton et al., 2015; Skog et al., 2008) and have been shown to be relatively stable within extracellular vesicles (EVs) or bound to proteins or lipids (Yanez-Mo et al., 2015). exRNA-based liquid biopsy is particularly attractive as a non-invasive mode for monitoring disease due to the significantly increased accessibility of biofluids over tissues, thereby allowing more frequent and longitudinal sampling (Byron et al., 2016). With better characterization of the differences between profiles secreted by diseased and healthy tissues, the diagnostic and prognostic utility of exRNA-based profiling is increasingly becoming a reality (Akat et al., 2014; McKiernan et al., 2016; Yuan et al., 2016).
Despite rapid technological and methodological progress in isolating EVs and exRNA, the analysis and interpretation of exRNA data is uniquely challenging. Biochemical methods for extraction, purification, and sequencing of exRNAs are much more vulnerable to contamination and artifacts than cellular RNA preparations, in large part due to relative low abundance (Danielson et al., 2017). Quality control prior to sequencing of samples derived from EV or exosome preparations is difficult due to the lack of reliable ‘housekeeping’ markers, such as the ratio of 18S and 28S ribosomal RNAs (Tataruch-Weinert et al., 2016). The variable presence of rRNA in mixtures of low- and high-density EVs (Lasser et al., 2017), deterministic cleavage of structured smallRNA (tRNAs and piRNAs) and longer RNA molecules, and imperfect annotation of miRNAs, piRNAs, and tRNAs all pose challenges for quantification and functional interpretation of exRNA. Furthermore, it has been suggested that exogenous exRNAs may be also be present at detectable levels in some biofluids (Freedman et al., 2016; Yeri et al., 2017), and careful analysis is required to differentiate these sequences from endogenous RNA molecules. For these reasons, existing computational tools capable of analyzing smallRNA-seq data (Barturen et al. 2014; Yuan et al. 2016; Wong et al. 2017) are not optimized for the new field of exRNA analysis.
To address these analytical challenges, we present the extracellular RNA processing tool (exceRpt). exceRpt is the primary smallRNA analysis pipeline used by the NIH Extracellular RNA Communication Consortium (ERCC). By providing an optimized and standardized bioinformatics platform, exceRpt reduces technical bias and allows for cross-study analyses to potentiate meaningful insights into exRNA biology.
Results:
Filtering and Quantification Cascade:
The exceRpt pipeline is comprises a serial cascade of computational filters and alignments, the order of which is designed to reflect an appropriate level of confidence in the various endogenous and exogenous sequences that may exist in the extracellular fraction (Figure 1A). To combat potential contamination in an RNA-seq library, reads aligned to known contaminants are removed before mapping to the host genome and to the exogenous sequences. The pipeline is highly modular (constructed as a makefile file containing shell, Java, and R modules), allowing the user to define the order of which smallRNA annotations are used during read-mapping. It includes support for random-barcoded libraries and spike-in sequences for calibration. The general workflow comprises steps for preprocessing, endogenous alignment, and exogenous alignment (Figure 1A and B).
Figure 1.
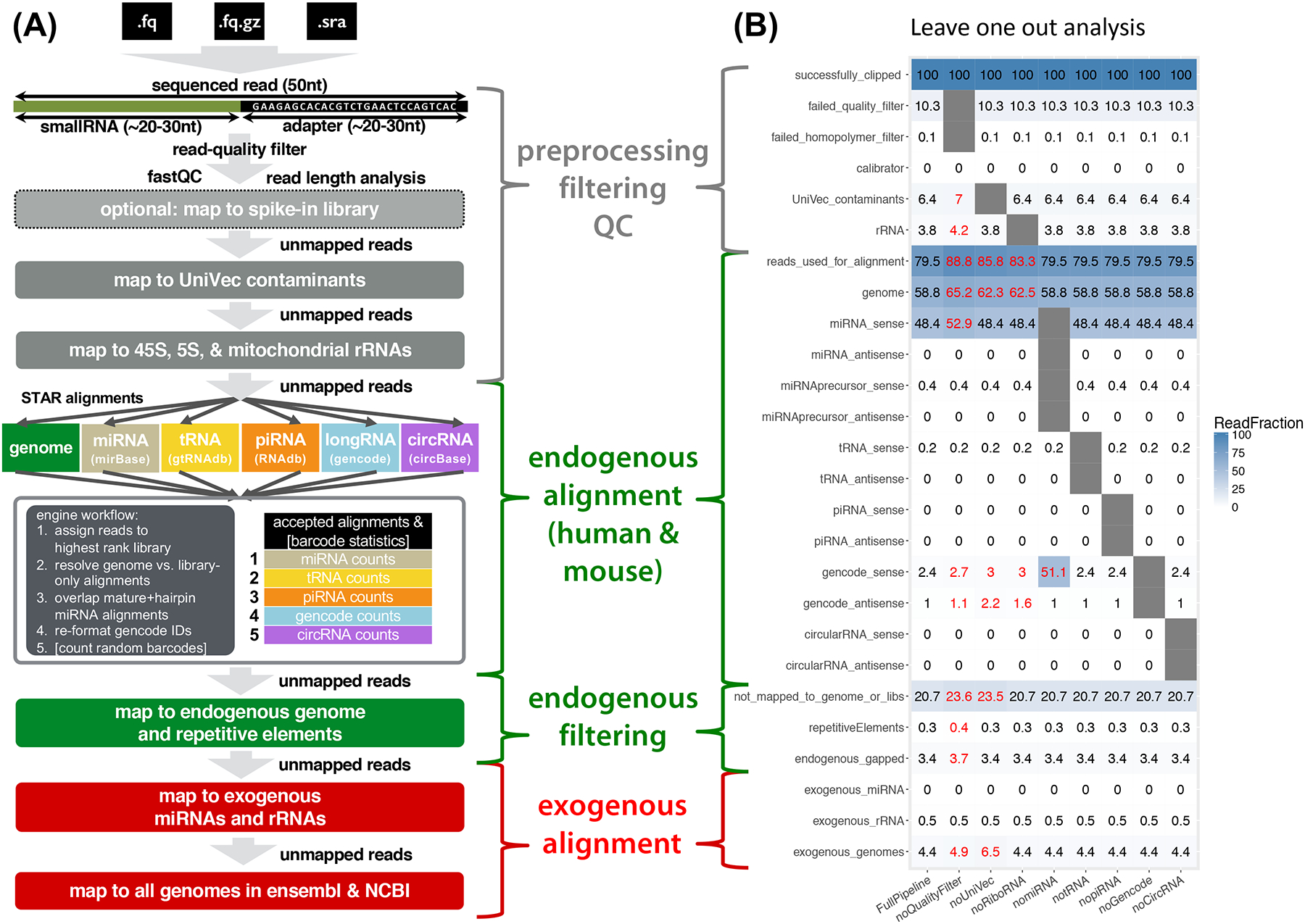
(A): exceRpt schema: Samples in FASTA, FASTQ or SRA file formats are used as inputs to exceRpt. Adapter and random barcode sequences are removed, followed by a read-quality filter, optional spike-in quantification and removal, and UniVec contaminant removal. High-quality filtered reads then enter the endogenous quantification engine, with RNA library prioritization defined by the user. After a second-pass endogenous genome and repetitive elements filter, reads are mapped to the exogenous miRNA, rRNA, and genomic libraries.
(B): Leave-one-out analysis: Running the pipeline multiple times with individual steps removed shows the effect of those steps on subsequent alignments. The sample used for this analysis was SRR822433, a plasma exRNA plasma sample. Low-quality and low-complexity reads and reads that align to UniVec or rRNA sequences account for a sizeable fraction of the total number sequenced. Removing the UniVec alignment step significantly increases the number of reads that, likely incorrectly, map to the exogenous genomes.
Preprocessing:
First, exceRpt automatically identifies and removes 3’ adapter sequences. If specified, it will also remove and store information from random-barcoded insert sequences, which are increasingly being used in smallRNA sequencing in an attempt to identify and compensate for ligation and/or amplification artifacts that have the potential to affect downstream quantification (Fu et al., 2014). If exogenous spike-in sequences are added to the library, exceRpt aligns adapter- and barcode-clipped reads against either standard NIST/ERCC sequences or sets of user-specified sequences. In order to filter out common laboratory contaminants, low-quality RNA-seq reads and reads aligned to sequences in the NCBI’s UniVec/Vecscreen database are removed. Finally, reads are aligned to all primary endogenous ribosomal RNAs (5S, 5.8S, 18S, 28S, and 45S), many of which are highly variable in abundance in EV preparations.
Endogenous Alignment:
RNA-seq reads are aligned to the host genome and transcriptome of human or mouse, and transcript abundances are calculated (RNAs are quantified using both raw read counts and normalized reads per million (RPM)). Because of the variety of RNA preparations available (totalRNA, smallRNA, miRNA), exceRpt allows the user to prioritize the order in which annotations (miRBase, tRNAscan, piRNA, GENCODE, circRNA) are used for quantification based on our confidence in the presence of a given annotation in a given sample. For example, reads from a miRNA-seq prep can be assigned to miRBase miRNA annotations before piRNA annotations. Likewise, reads from long- or total-RNA preparations can be assigned to longer GENCODE transcripts before (or instead of) the other smallRNA libraries. This feature is particularly relevant for lower-confidence annotations; piRNAs, for example, are generally given lower priority than tRNAs to ensure correct read assignments.
Exogenous Alignment:
exceRpt has been designed to enable confident assessment of non-human sequences in biofluids after careful, explicit removal of as many known or likely contaminants as possible. Before interrogating non-contaminant and non-endogenous reads for potential exogenous sequences, exceRpt includes a second-pass alignment against the host genome and known repetitive sequences using significantly relaxed mapping criteria. This step serves to remove additional sequences that most likely derive from the host genome and makes exceRpt more conservative in the identification of exogenous sequences. Reads are then aligned to curated libraries of annotated exogenous miRNAs in miRBase and exogenous rRNA sequences in the Ribosomal Database Project (RDP), followed by alignment to the full genomes of all sequenced bacteria, viruses, plants, fungi, protists, metazoa, and selected vertebrates that are potentially part of the host diet.
Existing approaches for exogenous sequence alignment remove degenerate sequences (i.e. those that co-occur across multiple species), which results in a loss of potentially valuable data as reads frequently align across multiple species/strains. By characterizing exogenous genome alignments generated by exceRpt (in terms of the NCBI taxonomy tree) and assigning reads to the most specific possible node in the phylogenetic tree (many reads can only be assigned to nodes higher up in the phylogenetic tree due to non-unique mapping to a specific sub-species’ genome), users may obtain valuable information regarding the contribution of flora and fauna to various exRNA samples, and they can generate phylogenies for cross-sample comparison. For example, in Figures 2A, 2B and C, we present the reads that we assign to bacterial ribosomal and genome sequences for a specific saliva sample as phylogenetic trees. In both trees, we find an abundance of reads assigned to the node corresponding to the genus Streptococcus. Given that the sets of short reads used for constructing these two trees are disjoint, we have a high degree of confidence in these results.
Figure 2.
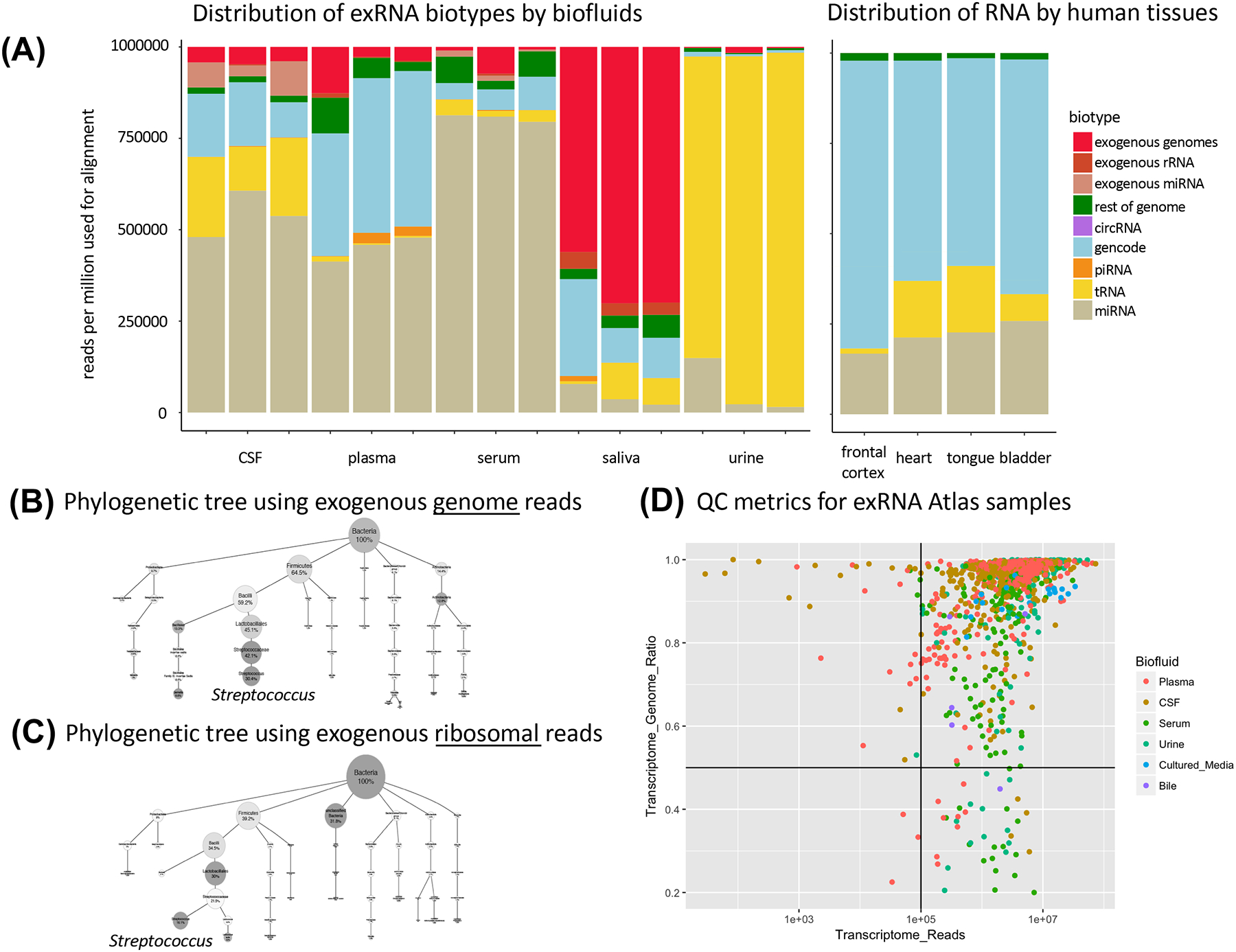
(A): Read distributions: exceRpt outputs endogenous alignment quantifications which can be used to compare RNA biotype distributions in exRNA samples. Here, saliva has a higher proportion of exogenous sequences than other samples, and urine has a higher proportion of tRNA sequences. Quantifications can also be performed for cellular datasets, such as ENCODE samples, where the majority of reads align to long coding and non-coding RNAs in GENCODE.
(B+C): Exogenous alignment phylogeny with genome reads: Exogenous sequence quantifications based on exogenous genome reads and rRNA reads can be represented using phylogenetic trees. The tree in (B) was constructed using 1.74M genome reads from a saliva exRNA-seq sample, and the tree in (C) was constructed from 1,127K ribosomal reads in the same saliva sample. Saliva biofluids are distinguished from other biofluids by their exposure to a robust and complex bacterial community in the oral cavity (Hasan et al., 2014), which causes a greater contribution of reads of bacterial origin (and not human genome) to the sample. In both the phylogenetic trees constructed using either bacterial genome mapped reads (B) or ribosomal mapped reads(C), we find an abundance of reads assigned to the node corresponding to the genus Streptococcus.
(D): Quality control metrics: ERCC QC metrics are based on number of transcriptome reads and ratio of RNA-annotated reads to the genome reads. The horizontal and vertical lines define QC threshold minima. Most exRNA Atlas samples meet the standards and fall in the upper right quadrant.
Pipeline Output and Analysis:
The pipeline generates bulk statistics for abundance of the various RNA biotypes in addition to sample-level quality control (QC) and processing reports. Due to the heterogeneity of the various exRNA isolation techniques, we developed QC metrics in collaboration with the ERCC for identification of clear experimental outliers (Figure 2D). These metrics are based on transcriptome-mapped read count and the proportion of genomic reads that map to annotated RNA transcripts, which can be used to distinguish exRNA samples that have significant cellular DNA contaminants (see STAR Methods). Descriptions of the post-processing output files and diagnostic plots generated by exceRpt are listed in Table 1. As a performance evaluation, we found that the endogenous miRNA abundance estimates produced by exceRpt are in close agreement with existing tools. Comparing exceRpt-filtered read counts for miRBase miRNAs for representative datasets from different biofluids, we obtain Pearson correlations of between 0.93 and 0.99 with the read counts produced by miRDeep2 (Friedländer et al. 2012), see Figure S1. As another performance evaluation, running the same sample through the pipeline with individual steps excluded shows the effect of the filters and alignments on downstream quantifications (Figure 1B). This analysis shows that the pre-filtering of low-quality and low-complexity reads and reads that align to UniVec or rRNA sequences account for a sizeable fraction of the total number sequenced and, without explicit removal, can align to the human genome leading to potential confounding and added quantification variability. UniVec-based contaminant filtering has the largest effect on the fraction of reads aligning to exogenous genomes, and its omission substantially increases the number of reads that appear to be, but are very likely not, exogenous in origin.
Table 1:
Description of Output Files
| File Name | Description of File |
|---|---|
| QC Data | |
| exceRpt_DiagnosticPlots.pdf | All diagnostic plots automatically generated by the tool |
| exceRpt_readMappingSummary.txt | Read-alignment summary including total counts for each library |
| exceRpt_ReadLengths.txt | Read-lengths (after 3’ adapters/barcodes are removed) |
| Raw Transcriptome Quantifications | |
| exceRpt_miRNA_ReadCounts.txt | miRNA read-counts quantifications |
| exceRpt_tRNA_ReadCounts.txt | tRNA read-counts quantifications |
| exceRpt_piRNA_ReadCounts.txt | piRNA read-counts quantifications |
| exceRpt_gencode_ReadCounts.txt | gencode read-counts quantifications |
| exceRpt_circularRNA_ReadCounts.txt | circularRNA read-count quantifications |
| Normalized Transcriptome Quantifications | |
| exceRpt_miRNA_ReadsPerMillion.txt | miRNA RPM quantifications |
| exceRpt_tRNA_ReadsPerMillion.txt | tRNA RPM quantifications |
| exceRpt_piRNA_ReadsPerMillion.txt | piRNA RPM quantifications |
| exceRpt_gencode_ReadsPerMillion.txt | gencode RPM quantifications |
| exceRpt_circularRNA_ReadsPerMillion.txt | circularRNA RPM quantifications |
| R Objects | |
| exceRpt_smallRNAQuants_ReadCounts.RData | All raw data (binary R object) |
| exceRpt_smallRNAQuants_ReadsPerMillion.RData | All normalized data (binary R object) |
These bulk statistics can be used to discriminate between biofluids (or tissues, if exceRpt is run on cellular samples) based on their RNA distribution. For example, results from samples selected from the exRNA Atlas (Figure 2A) show that, relative to other biofluids, saliva samples tend to have more reads that are unmapped or that map to exogenous genomes (this is true for the majority of saliva samples in the exRNA Atlas), which is consistent with saliva’s high potential for bacterial contamination and exposure to the external environment. Moreover, abundance quantifications for specific RNA biotypes can show which miRNAs (or other RNA biotype) are most highly represented in a particular sample (Figure S2). This information is critical for understanding the composition of exRNA profiles and for interrogating their biological significance.
Discussion:
The exceRpt pipeline was built to address the need for a standardized bioinformatics processing platform in extracellular RNA research, and is structured as a principled, biologically-driven series of alignment, filtering, and quantification steps in which unmapped reads are used as inputs to the next step. The prioritization of steps is biased in favor of conservative estimates for RNA quantifications, with higher-confidence RNA reference sets (as measured by degree of expectation or annotation quality) having higher priority. To date, exceRpt has uniformly processed and applied QC standards to all of the datasets in the ERCC exRNA Atlas; http://exrna-atlas.org/) (Subramanian et al., 2015). The exceRpt pipeline enables a variety of user-specified customizations, including RNA reference prioritization, random-barcoding, spike-in support, and detailed quantification reports. The pipeline is available as source code or wrapped in a user-friendly, browser-based interface available at genboree.org.
STAR Methods:
PREPROCESSING
Input files of sequenced reads can be in FASTA, FASTQ, or SRA formats. exceRpt begins the preprocessing step by automatically identifying and removing 3’ adapter sequences. Randomly barcoded 5’ and/or 3’ adapter sequences are increasingly being used in smallRNA sequencing in an attempt to identify and compensate for ligation and/or amplification artifacts. exceRpt is capable of removing and quantifying these biases at both the insert level, which reveals ligation/amplification bias, and the transcript level, which provides an opportunity to compensate for the bias by counting unique N-mer barcodes rather than counting the number of inserts. Random-barcode/UMI information from the raw reads is preserved throughout the alignment and quantification steps and may be used as an alternative to read-counting in the quantitation of smallRNAs. The pipeline then aligns against an input set of known spike-in sequences (if used in the library construction), followed by a filter to remove low-quality reads and reads with large homopolymer repeats using the FASTX toolkit (QFILTER_MIN_READ_FRAC = 80, QFILTER_MIN_QUAL = 20). As the final preprocessing step, exceRpt aligns reads to annotated sequences in the UniVec database (designed for filtration of common laboratory contaminants) and then to endogenous ribosomal RNAs, both of which are highly variable in abundance in EV preparations. The mapping to calibrators (spike-ins) step uses Bowtie2. All other preprocessing mapping steps use STAR.
ENDOGENOUS QUANTIFICATION
Reads that were not filtered out in the preprocessing steps are aligned to the endogenous genome and transcriptome of either human or mouse, using STAR. STAR uses the following parameters for the endogenous alignments by default, MIN_READ_LENGTH = 18, MAX_MISMISMATCH = 1, MISMATCH_OVER_L_MAX = 0.3, MATCH_N_MIN_OVER_L_READ = 0.9. Transcript abundances are calculated (RNAs are quantified using both raw read counts and normalized reads per million (RPM)). The user can prioritize the order in which annotations (miRBase, tRNAscan, piRNA, GENCODE, circRNA) are used for quantification based on confidence in the presence of a given RNA biotype in a given sample. By default the order for quantification is miRNAs, tRNAs, piRNAs, GENCODE transcripts (snRNAs, snoRNAs, miscRNAs, protein coding genes and lncRNAs) followed by circular RNAs based on our confidence of the likelihood of these annotations to be present in a small exRNA-Seq sample. The exceRpt pipeline can be used with the human host endogenous genome (either hg19 or GRCh38) or the mouse host endogenous genome (mm10) with corresponding annotations. By default, the exceRpt pipeline can be run on human and mouse RNA-Seq datasets (due to the high-quality annotations of these two genomes). The annotation files may be substituted for other species - the results however would be contingent on the quality of the annotations in the specific species.
We have benchmarked extracellular RNA-Seq datasets and for typical datasets with ~10 million reads the endogenous portion of the pipeline completes on a typical compute node in about 10 mins (some bigger datasets take up to 1 hour).
EXOGENOUS QUANTIFICATION
Other tools exist for the metagenomics analysis of sequenced data, such as KRAKEN (Wood et al. 2014), CLARK (Ounit et al. 2015) and MG-RAST (Wilke et al. 2015). However most of these tools are primarily focused on genomic sequencing data that is targeted specifically at microbial communities. Thus, these do not have the same issues with short reads from small RNA-Seq data in the context of extracellular RNA, in which case one would first need to carefully filter reads from the host genome before the exogenous analysis.
Before analyzing the remaining reads for potential exogenous sequences, exceRpt performs a second pass alignment using STAR, first against the host genome (allowing for novel gapped alignments) with more relaxed mapping criteria, and then against a database of known repetitive sequences. Reads are then aligned to curated libraries of annotated exogenous miRNAs in miRBase and exogenous rRNA sequences in the Ribosomal Database Project (RDP), followed by alignment to the full genomes of all sequenced bacteria, viruses, plants, fungi, protist, metazoa, and the following 12 vertebrate genomes: chicken, cod, cow, dog, duck, frog, horse, rabbit, pig, sheep, tilapia, and turkey. Multiple STAR indexes were constructed provided in the exceRpt database for the bacterial genomes, the plant genomes, the metazoan genomes, fungi, protest and virus genomes and the vertebrate genomes so that exogenous genome alignment, which is the most time-consuming step of the exceRpt pipeline, can be parallelized. exceRpt then uses STAR to align the remaining reads to these genomes. By default, exceRpt allows for no mismatches during this step (in order to be as conservative as possible in identifying possible exogenous sequences). In the Github repository for the exceRpt pipeline (github.gersteinlab.org/exceRpt) we have included the directory “scriptsToMakeExogenousGenomes”. This directory contains the code as well as the complete list of URLs for all the exogenous genomes that are indexed by STAR for the exogenous processing of the pipeline. Indices are created separately for all bacteria (split into 10), viruses, plants, fungi, protists, metazoa, and the 12 vertebrate genomes.
Since many exogenous genomes have a high degree of sequence similarity based on evolution, we find that many reads that align to an exogenous genome align to multiple genomes. We assign reads that align to exogenous genomes to the position in the phylogenetic taxonomy tree based on the node is most parsimonious with the different genomes that the read aligns (Figure S3). Reads that align to a unique genome are aligned to the corresponding leaf node in the phylogenetic tree while reads that align to multiple genomes are assigned to nodes higher up in the tree. exceRpt also independently performs this assignment for the reads that align to exogenous rRNA sequences in the context of the phylogenetic taxonomy tree constructed using rRNA sequences. One can compare the structures of the exogenous genomic and rRNA phylogenetic taxonomy trees for consistency, as the trees are constructed independently using the disjoint sets of exogenous genome and rRNA reads.
We have benchmarked the exogenous component for the exceRpt pipeline for typical extracellular RNA-Seq datasets with approximately ~10 million reads. The runtime is strongly dependent on the fraction of endogenously unmapped reads. For a saliva sample with a high fraction of exogenous aligning reads, we find that the full pipeline takes 2–8 hours to run on a typical high-memory compute node.
QUALITY METRICS
To evaluate the quality of the samples and to identify outliers, we developed QC data standards in collaboration with the ERCC which exceRpt evaluates uniformly on all input samples: (1) datasets are required to have at least 100,000 reads that overlap with any annotated RNA transcript in the host genome, and (2) over 50% of the reads that map to the host genome also align to any RNA annotation. The first criterion ensures that enough reads are generated for quantification (the minimal read depth required for the minimal normalized expression of an annotated RNA should be greater than 1 RPM) and the second criterion ensures that the reads mostly align to RNA, as opposed to DNA contamination from cellular sources. We find that 95% of the ~2500 exRNA-Seq datasets that have been uniformly processed in the exRNA Atlas with exceRpt meet both criteria (Figure 2D), with most datasets well above both thresholds.
QUANTIFICATION AND STATISTICAL ANALYSIS:
All quantification analyses were performed in R and Java. Annotated RNAs are quantified using both raw read counts and are also normalized by sample across all RNA biotypes to reads per million mapped reads (RPM)). We chose to normalize the quantification in units of RPM, as the short nature of small RNAs annotations precludes the need to normalize with respect to the transcript length (such as RPKM or TPM). Diagnostic plots and output statistics are also automatically generated by exceRpt.
DATA AND SOFTWARE AVAILABILITY AND HARDWARE REQUIREMENTS:
The graphical, browser-based, user-friendly interface for uploading and processing of exRNA-seq datasets with exceRpt is available at the Genboree Workbench (Figure S4A): http://genboree.org/theCommons/projects/exrna-tools-may2014/wiki/Small%20RNA-seq%20Pipeline. The exceRpt source code may be downloaded and installed manually for the greatest degree of flexibility (github.gersteinlab.org/exceRpt). For this option, the dependencies and databases used by the pipeline must be downloaded and installed, and the makefile must be properly configured for use (instructions for installation and use are available from the GitHub page). Alternatively, the exceRpt Docker image (Figure S4B) with all required dependencies may be used for installation on the user’s own machine or cluster (github.gersteinlab.org/exceRpt). exceRpt also includes a script (mergePipelineRuns.R) to combine outputs from multiple samples for downstream comparative analysis. A small test dataset with matching output is available from the ‘ExampleData’ directory in the GitHub repository.
The exceRpt pipeline requires hardware with at least 16 GB of RAM and at least 4 CPU cores. Machines with sufficient memory will almost certainly be equipped with a sufficiently powerful CPU. However, machines with fewer than 4 CPU cores may struggle to run the alignments required by exceRpt. Also, each uncompressed reference index requires around 35GB of disk space.
The exceRpt pipeline makes use of a makefile which allows the code to be executed up to any intermediate step by specifying the intermediate filename during execution. This allows the user to choose between endogenous-only [the default], endogenous+exogenous(rRNA), or endogenous+exogenous(rRNA+genomes) as endpoints for the pipeline.
For example, to run only the read filtering/QC portion of exceRpt (and stop before any endogenous genome/transcriptome alignment) one might specify:
make -f exceRpt_smallRNA
$OUTPUT_DIR/$SAMPLE_ID.clipped.trimmed.filtered.fastq.gz
[…remaining options…]
where the environment variables:
- $OUTPUT_DIR is the base of the results directory [e.g. ~/exceRptTest/Results]
- $SAMPLE_ID [e.g. testData_human.fastq] specifies the name of the results subdirectory for this run. One can initially run the pipeline in endogenous mode and then later modify the makefile options to include exogenous processing will resume the pipeline post endogenous processing.
Supplementary Material
KEY RESOURCES TABLE:
Acknowledgements:
This publication is part of the NIH Extracellular RNA
Communication Consortium paper package and was supported by the NIH
Common Fund’s exRNA Communication Program. We acknowledge support from the NIH Common Fund (U54DA036134) and from the AL Williams Professorship funds. JJP was supported by the NIH Medical Scientist Training Program Training Grant (T32GM007205).
Footnotes
ADDITIONAL RESOURCES:
The ERCC exRNA Atlas can be found here: https://exrna-atlas.org/
The ERCC quality control standards can be found here: https://exrna.org/resources/data/data-quality-control-standards/
Supplemental Information:
Document S1. Figures S1–S4.
References:
- Akat KM, Moore-McGriff D, Morozov P, Brown M, Gogakos T, Correa Da Rosa J, Mihailovic A, Sauer M, Ji R, Ramarathnam A, et al. (2014). Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci U S A 111, 11151–11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barturen G, Rueda A, Hamberg M, Alganza A, Lebron R, Kotsyfakis M, et al. (2014). sRNAbench: profiling of small RNAs and its sequence variants in single or multi-species high-throughput experiments. Methods in Next Generation Sequencing: Methods in Next Generation Sequencing; 2014. [Google Scholar]
- Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, and Craig DW (2016). Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet 17, 257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2009). GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37, D93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, and Tiedje JM (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42, D633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson KM, Rubio R, Abderazzaq F, Das S, and Wang YE (2017). High Throughput Sequencing of Extracellular RNA from Human Plasma. PLoS One 12, e0164644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman JE, Gerstein M, Mick E, Rozowsky J, Levy D, Kitchen R, Das S, Shah R, Danielson K, Beaulieu L, et al. (2016). Diverse human extracellular RNAs are widely detected in human plasma. Nat Commun 7, 11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Wei Chen NL, and Rajewsky N (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012 January; 40(1): 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu GK, Xu W, Wilhelmy J, Mindrinos MN, Davis RW, Xiao W, and Fodor SP (2014). Molecular indexing enables quantitative targeted RNA sequencing and reveals poor efficiencies in standard library preparations. Proc Natl Acad Sci U S A 111, 1891–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazar P, Papavasileiou P, and Rajewsky N (2014). circBase: a database for circular RNAs. RNA 20, 1666–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S (2004). The microRNA Registry. Nucleic Acids Res 32, D109–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan NA, Young BA, Minard-Smith AT, Saeed K, Li H, Heizer EM, McMillan NJ, Isom R, Abdullah AS, Bornman DM, et al. (2014). Microbial community profiling of human saliva using shotgun metagenomic sequencing. PLoS One 9, e97699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser C, Shelke GV, Yeri A, Kim DK, Crescitelli R, Raimondo S, Sjostrand M, Gho YS, Van Keuren Jensen K, and Lotvall J (2017). Two distinct extracellular RNA signatures released by a single cell type identified by microarray and next-generation sequencing. RNA Biol 14, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majem B, Li F, Sun J, and Wong DTW (2017). RNA sequencing analysis of salivary extracellular RNA. Methods Mol Biol. 2017; 1537: 17–36. doi: 10.1007/978-1-4939-6685-1_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKiernan J, Donovan MJ, O’Neill V, Bentink S, Noerholm M, Belzer S, Skog J, Kattan MW, Partin A, Andriole G, et al. (2016). A Novel Urine Exosome Gene Expression Assay to Predict High-grade Prostate Cancer at Initial Biopsy. JAMA Oncol 2, 882–889. [DOI] [PubMed] [Google Scholar]
- Mudge JM, and Harrow J (2015). Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm Genome 26, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounit R, Wanamaker S,Close TJ, and Lonardi S (2015). CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics. 2015; 16(1): 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton JG, Franklin JL, Weaver AM, Vickers K, Zhang B, Coffey RJ, Ansel KM, Blelloch R, Goga A, Huang B, et al. (2015). Biogenesis, delivery, and function of extracellular RNA. J Extracell Vesicles 4, 27494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sai Lakshmi S, and Agrawal S (2008). piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res 36, D173–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr., Carter BS, Krichevsky AM, and Breakefield XO (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian SL, Kitchen RR, Alexander R, Carter BS, Cheung KH, Laurent LC, Pico A, Roberts LR, Roth ME, Rozowsky JS, et al. (2015). Integration of extracellular RNA profiling data using metadata, biomedical ontologies and Linked Data technologies. J Extracell Vesicles 4, 27497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke A, Bischof J, Gerlach W, Glass E, Harrison T, Keegan KP, Paczian T, Trimble WL, et al. (2015). The MG-RAST metagenomics database and portal in 2015. Nucleic Acids Res. 2016 January 4; 44(Database issue): D590–D594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DE, and Salzberg (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014; 15(3): R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, et al. (2015). Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeri A, Courtright A, Reiman R, Carlson E, Beecroft T, Janss A, Siniard A, Richholt R, Balak C, Rozowsky J, et al. (2017). Total Extracellular Small RNA Profiles from Plasma, Saliva, and Urine of Healthy Subjects. Sci Rep 7, 44061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T, Huang X, Woodcock M, Du M, Dittmar R, Wang Y, Tsai S, Kohli M, Boardman L, Patel T, et al. (2016). Plasma extracellular RNA profiles in healthy and cancer patients. Sci Rep 6, 19413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.