

Abstract
Middle East respiratory syndrome (MERS) is a viral respiratory illness first reported in Saudi Arabia in September 2012 caused by the human coronavirus (CoV), MERS-CoV. Using full-genome sequencing and phylogenetic analysis, scientists have identified three clades and multiple lineages of MERS-CoV in humans and the zoonotic host, dromedary camels. In this study, we have characterized eight MERS-CoV isolates collected from patients in Saudi Arabia in 2015. We have performed full-genome sequencing on the viral isolates, and compared them to the corresponding clinical specimens. All isolates were clade B, lineages 4 and 5. Three of the isolates carry deletions located on three independent regions of the genome in the 5′UTR, ORF1a and ORF3. All novel MERS-CoV strains replicated efficiently in Vero and Huh7 cells. Viruses with deletions in the 5′UTR and ORF1a exhibited impaired viral release in Vero cells. These data emphasize the plasticity of the MERS-CoV genome during human infection.
Keywords: Middle East respiratory syndrome human coronavirus, MERS-CoV, growth kinetics, clinical isolates, genomic sequences, coronavirus, phylogeny
Introduction
Middle East respiratory syndrome coronavirus (MERS-CoV) is a Beta coronavirus that can cause respiratory illnesses that range in severity from asymptomatic to severe. Since the virus was identified in the Middle East in 2012, there have been over 2000 laboratory-confirmed cases with a mortality rate of approximately 35 % of reported cases (WHO). Over 80 % of cases have been reported in Saudi Arabia with all cases epidemiologically linked to the Arabian Peninsula. Dromedary camels have been identified as a primary animal reservoir, and human outbreaks have been postulated to be caused by spillover events [1]. As of September 2019, MERS-CoV continues to circulate.
MERS-CoV is an enveloped positive-strand RNA virus with a 30 kb genome that encodes 16 non-structural proteins within ORF1ab at the 5′ end and multiple structural and accessory proteins encoded at the 3′ end. To date, multiple recombinations and non-synonymous mutations have been identified in clinical specimens with the majority in Spike, Matrix, ORF1ab and ORF4b [2–5], but only eight deletion variants have been identified in viruses isolated from humans. All of the deletion variants have been in accessory and structural proteins. The identified deletions have been present in spike, ORF3a, ORF4, ORF5 and between ORF5 and E generating an ORF5E fusion protein [6–10]. Of note, the accessory proteins ORF4 and ORF5 are unique to Group 2C Beta coronaviruses and thus are absent in the other highly pathogenic Beta coronaviruses. None of the deletion variants identified in humans have been characterized in vitro, but the 414 nt deletion variant in ORF5 and E was identified in the index patient in China that had close contact with the index patient in South Korea in 2015 who was later deemed as a ‘super-spreader’ upon starting an outbreak that infected 186 people [7]. A 20 nt ORF5 deletion variant was identified from a fatal case occurred in 2015, linked to nosocomial transmission in Riyadh, Saudi Arabia [8]. Notably, deletion variants, with deletions in ORF3 and/or ORF4b have also been identified in specimens collected from dromedary camels in West and North Africa, which are phylogenetically distinct from viruses isolated from the Arabian Peninsula [11].
In this study, we have isolated eight MERS-CoV viruses from clinical specimens collected during 2015 in Saudi Arabia [2]. We sequenced the full genomes from all eight isolates and compared them to the full genome or spike gene from the matched clinic samples. Using these sequenced viral isolates, we also performed phylogenetic analysis and characterized the growth and viability of four viral isolates with both complete genomes and novel deletions in the highly conserved ORF1ab (nsp2), ORF3 and the 5′ UTR regions.
Methods
Clinical specimens
Specimens were collected as part of an epidemiologic investigation between January–June 2015 [2]. Respiratory specimens were collected and shipped to the Centers for Disease Control and Prevention. Portions of the specimens were used for RNA extraction and sequencing, which have been previously published [2]. Remaining respiratory specimens were used for virus isolation.
Ethical approval
The use of human specimens to generate viral isolates was determined to be non-human subjects research by the National Center for Immunizations and Respiratory Diseases Human Research Protections Reviewer with the project ID P_2018_DVD_Thornburg_466.
Cell culture, virus isolation, growth and titration
Vero and Huh-7 cells were obtained from ATCC and maintained in DMEM (Life Technologies, Grand Island, NY, USA) supplemented with 10 % heat-inactivated FBS (Hyclone, USA) and penicillin-streptomycin (PS) (Sigma-Aldrich, USA). Huh-7 cells were also supplemented with non-essential amino acids. MERS-CoV isolations were performed in sterile flat-bottom 96-well plates using Vero cells in a biosafety level 3 laboratory. Briefly, 50–100 µl respiratory specimens were serially diluted tenfold with DMEM and 100 ul of 3×105 Vero cells ml− 1 in suspension in DMEM/5 % FBS/PS. The specimens and cells were incubated at 37 °C/ 5 % CO2 and were monitored daily for signs of cytopathic effects (CPE). If wells showed CPE, half the well was used for passage, and half was used for RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR) to confirm the presence of MERS-CoV. Isolates that were confirmed to be MERS-CoV were expanded and titred by TCID50 using crystal violet.
To characterize MERS-CoV growth kinetics, Vero or Huh-7 cells were plated in 24-well tissue-culture plates (Corning Costar), and cells were inoculated at an m.o.i. of 0.1 in 500 µl DMEM. After 30 min adsorption at 37 ˚C/5 % CO2, the monolayers were washed twice with PBS, and replaced with 1 ml of DMEM/5 % FBS/PS. The supernatants and cells were harvested every 12 h for 120 h. The titres of both supernatant and cell-associated viruses were determined by TCID50 in triplicate.
Viral isolate sequencing and phylogenetic analysis
Overlapping primer pairs that span the entire MERS-CoV genome were developed and used in conjunction with the Fluidigm Access Array system [12]. The Fluidigm Access Array was used to set up and perform a throughput of 48 wells (15 samples with triplicate per sample) × 48 RT-PCRs according to the manufacturer’s protocol. Resulting amplicon pools from each sample were sheared from 800 to 1200 bp to 400–500 bp using a Covaris M220 sonicator. Sheared cDNA of each isolate was used to generate barcoded libraries for multiplexed sequencing using the NEB Next Ultra II DNA library prep kit (New England Biolabs). Sequencing was performed using an Illumina MiSeq instrument. Next-generation sequencing (NGS) data were analysed using a custom workflow in CLC Genomics Workbench 8.5. Trimmed reads were aligned to the reference Jordan-N3/2012 (KC776174.1). Consensus sequence was called based on regions that had 10× or greater coverage. The genome gaps after NGS sequence assembly were then filled by PCR followed by Sanger sequencing as described previously [12].
The genome nucleotide sequences from this study were first aligned in MAFFT v7.013 with published MERS-CoV sequences retrieved from GenBank. Phylogenetic trees were then inferred using the maximum-likelihood (ML) method using the available version 3.0 PhyML software assuming a general time-reversible (GTR) model with a discrete gamma distributed rate variation among sites (Gamma4) and a SPR tree swapping algorithm and visualized using mega version 6.
Results
Clinical specimens collected during an outbreak investigation had been previously characterized by qPCR and sequenced [2]. We used these clinical specimens for virus isolation. Several viruses were isolated from human respiratory clinical specimens including sputum, nasal swabs, nasopharyngeal or bronchoalveolar lavage in Vero cells. All viruses were isolated from specimens with Cts at or below 26. The full genome or spike gene of the novel isolates and the matched clinic samples were sequenced, phylogenetic analysis was performed, and sequences were compared to the sequences from the corresponding clinical specimens to identify potential tissue-culture adaptations. All viruses from this study were from clade B, lineages 4 and 5 (Table 1 and Fig. 1). The initial description of the sequences from these clinical specimens described these viral sequences as novel recombinant clade, however we have since adopted lineage language for clarity [2]. Of the eight isolates, five had full-length genomes, and three had genomic deletions. Four isolates with full-length genomes were isolated from clinical specimens in which the full genomes were sequenced. Six isolates had identical sequences in comparison to the available sequences (full genome or spike gene) of their matched clinical samples (Table 1), indicating no tissue-culture adaptations. The full-length isolate Hu/Khobar_KSA-6736_2015 had 1 nt non-synonymous change (F2242I) in ORF1ab, nsp3 in comparison to its source clinic specimen.
Table 1.
Genomic characteristics MERS-CoV isolates
|
Isolate |
Strain name |
Clinical specimen accession |
Isolate accession |
Clade |
Lineage |
Genomic deletions |
|---|---|---|---|---|---|---|
|
Hu/Khobar_KSA_6736_2015 |
KT806048 (Full genome) |
B |
5 |
Full length – 1 nt difference between clinical specimen and tissue-culture isolate |
||
|
Hu/Hofuf_KSA_11442_2015 |
KT806025 (Spike) |
5 |
Full length |
|||
|
Hu/Hofuf_KSA_11767_2015 |
KT806031 (Spike) |
5 |
Full length |
|||
|
Hu/Hofuf_KSA_11002_2015 |
KT806046 (Full genome) |
5 |
Full length |
|||
|
Hu/Hofuf_KSA_11401_2015 |
KT806024 (Spike) |
5 |
Full length |
|||
|
Hu/Riyadh_KSA_2959_2015 |
KT026453 (Full genome) |
5 |
Δ9 nt 5’ UTR, detected in clinical specimen and tissue-culture isolate |
|||
|
Hu/Riyadh_KSA-4050_2015 |
KT026454 (Full genome) |
5 |
Δ3 nt ORF1a, Δ1AA NSP2 |
|||
|
Hu/Aseer_KSA_RS924_2015 |
KT805992 (Spike) |
4 |
Δ41 nt ORF3 |
|||
|
Previously described |
Hu/Florida/USA-2_Saudi Arabia_2014 |
n/a |
1 |
Full length |
||
|
Hu/England-Qatar_2012 |
n/a |
4 |
Full length |
|||
|
Hu/Jordan_N3_2012 |
n/a |
A |
A |
Full length |
Fig. 1.
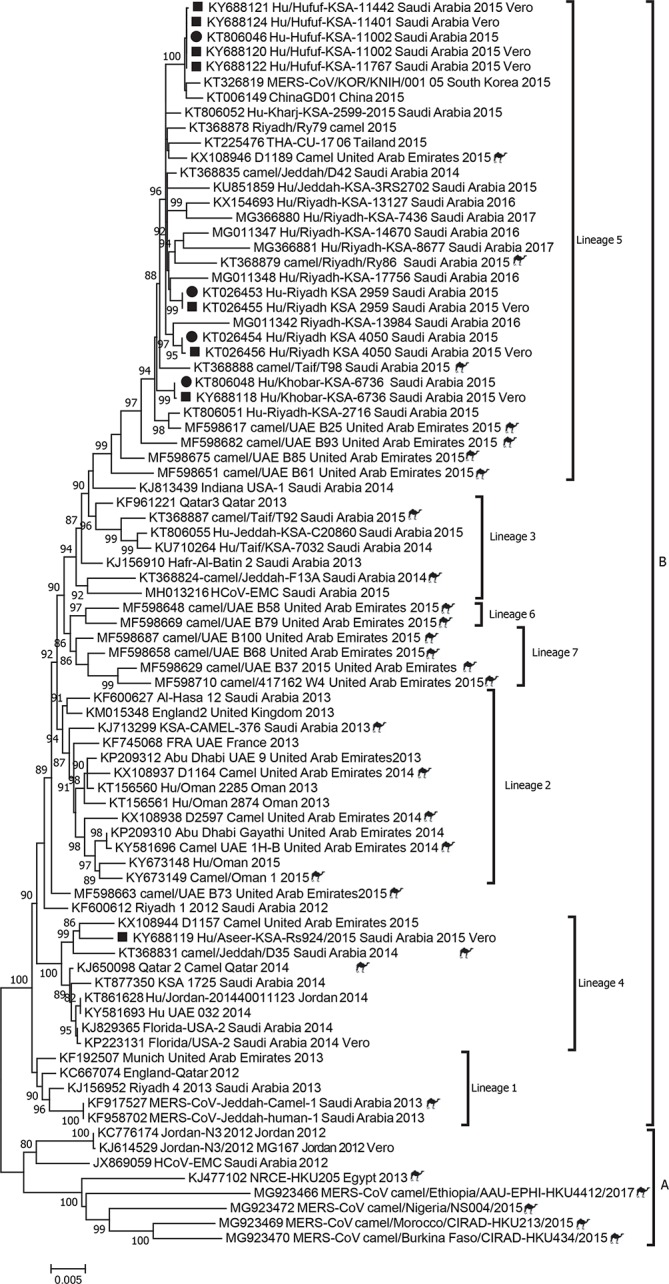
Phylogenetic analysis of MERS-CoV full genomes previously sequenced from humans and camels in comparison to novel clinical isolates (highlighted in grey).
Strain Hu/Riyadh_KSA_2959_2015 had a 9 nt deletion in the 5′ untranslated region (UTR), which was also present in the clinic sample, presumably resulting in no changes in viral protein sequences (Table 1). The isolate Hu/Riyadh_KSA-4050_2015 had a 3 nt deletion leading to a single amino acid deletion in NSP2 (I, at position 811) (Table 1) and a 1 nt synonymous change in the spike gene. The full genome of the matched clinical sequence was obtained, in which these mutations were absent, indicating potential tissue-culture adaptation (Table 1). Strain Hu/Aseer_KSA_RS924_2015 had a 41 nt deletion in ORF3 resulting in a frame shift (Table 1 and Fig. S1, available in the online version of this article). This deletion justified the distance of KY688119 by phylogenetic analysis (Fig. 1). Unfortunately, only the spike gene was sequenced from the clinical specimen due to very limited volume, so it is unclear if the deletion was generated during culturing. It remains a limitation of our comparison.
We performed growth-curve analysis on the selected virus isolates (three isolates with full genome and three isolates with deletions) from this study and three previously published viral isolates, novel full length and deletion mutant viruses in Vero cells to determine if genomic deletions corresponded with growth delays or inhibitions. Vero cells were inoculated with equivalent virus titres, and supernatants were collected every 12 h for 120 h and titrated by TCID50. Peak viral titres were observed between 48 and 96 h post inoculation, and did not differ between full-length isolates and deletion mutants (Fig. 2a). Peak titre of full-length viruses ranged from 105 to 106 TCID50 ml−1, whereas deletion mutants ranged from 5×104−1×105 TCID50 ml−1 (Fig. 2a), suggesting that these deletions may confer some growth inhibition, but not abrogate it.
Fig. 2.

Multistep growth curve analysis in Vero cells. Vero-cell monolayers were inoculated at an m.o.i. of 0.1 with previously described, novel full length or novel deletion mutant MERS-CoV isolates. Cell and supernatants combined (a) or cell and supernatant separately (b) were collected every 12 h for 120 h. Timepoints were titred in Vero cells.
In order to determine if the lower titres observed in the supernatants of Vero cells inoculated with deletion mutants represented lower viral replication or a budding deficiency, viruses were titred from Vero-cell lysates and supernatants after inoculation with full-length and deletion mutant viral isolates. In previously described and novel full-length viruses, the peak titre of each virus is approximately equal in cell-associated and supernatant fractions with a maximum of half a log difference (Fig. 2b). However, two of the three deletion mutant viruses exhibited higher titres in cell-associated fractions than supernatants (Fig. 2b). Hu/Riyadh_KSA_2959_2015 cell-associated titre peaked at 5×106 TCID50 ml−1 with supernatant peaking at 5×104 TCID50 ml−1. Similarly, Hu/Riyadh_KSA-4050_2015 cell-associated titre peaked at 1×106 TCID50 ml−1 with supernatant peaking at 5×104 TCID50 ml−1. The third deletion mutant virus Hu/Aseer_KSA_R2924_2015 exhibited similar titres in cell and supernatant fractions.
Genetically engineered viruses deleted in ORF 3, 4a, 4b and 5 exhibit efficient replication in Vero cells, but reduced replication in Calu3 cells [13]. Vero cells are known to be deficient in type I IFN responses, and MERS-CoV accessory proteins, ORF4a, ORF4b and ORF 5 have some IFN antagonist functions [14]. While none of our deletion mutants have deletions in ORF4a, ORF4b or ORF5, we wanted to test their abilities to replicate in cells with intact IFN systems. We tested the growth of full-length Hu/Florida/USA-2_KSA_2014, Hu/Riyadh_KSA_2959_2015 and Hu/Riyadh_KSA_4050_2015 in Huh7 cells, which have an intact IFN system. Neither deletion mutant exhibited delayed growth or reduced titres (Fig. 3), indicating they have an intact ability to antagonize the IFN system.
Fig. 3.

Multistep growth-curve analysis in HUH7 cells. HUH7 cell monolayers were inoculated at an m.o.i. of 0.1, with previously described Hu/Florida/USA-2_KSA_2014 or novel deletion mutants Hu/Riyadh_KSA_2014 or Hu/Riyadh_KSA_4050_2015, and cell and supernatants were collected separately every 12 h for 84 h. Cell and supernatant fractions were titred on Vero cells.
Discussion
In this manuscript, we describe eight MERS-CoV isolates collected from patients in 2015 in Saudi Arabia. Of those isolates, all were identified to be in clade B, five had full-length genomes, and three exhibited genomic deletions. This work is the first example of in vitro characterization of human clinical isolates with genomic deletions. These deletions were located in the 5′ UTR, ORF1a in NSP2, and ORF3. Growth-curve analysis indicated the viruses still grew efficiently in cell culture, however Hu/Riyadh_KSA_2959_2015 and Hu/Riyadh_KSA-4050_2015 exhibited higher cell-associated viral titres than titres in supernatant, indicating they may have a deficiency in viral budding or release.
Other MER-CoVs have been isolated from specimens collected from dromedary camels in North and West Africa with deletions in ORF3 and/or ORF4b [11]. The location and size of the deletions varied, however many resulted in frame shifts or premature stop-codons. Notably, these viruses were of a distinct clade (clade C1), but still had deletions in a similar region to our isolates from clade B human viruses. The clade C camel MERS viruses replicated less efficiently in Calu-3 cells than the human MERS-CoV isolate, EMC. These deletion mutants exhibited increased type I and III IFN responses with increased mRNA expression of IFN-β and IFN-γ1. When the deletion was engineered into a human strain of MERS-CoV (EMC), the viruses also exhibited higher type I and III, but did not exhibit growth inhibition suggesting the deletion is not solely responsible for growth inhibition. These data are consistent with the growth characteristics of the viruses described in our study.
It is clear from the ability of these novel MERS isolates with deletions, that none of the deletions dramatically affected the ability of the viruses to replicate in cell culture. This is not surprising given that two of the deletions are located in accessory proteins, and one is outside of a coding region. There may be compensating mutations elsewhere in the genome, however we have not yet been able to identify any, despite full-genome analysis.
Hu/Aseer_KSA_RS924_2015 exhibited a 41 nt deletion in ORF3 (Fig. S1). The function of ORF3 is unknown, but the protein is conserved across coronaviruses. Other groups have generated coronavirus mutants lacking ORF3, 4a, 4b and 5 that also replicate efficiently in cell culture [13, 15, 16]. Replication of an engineered quadruple deletion mutant was attenuated Calu3 cells but not in IFN-defective Vero cells, suggesting host response may affect the ability of these deletion mutants to grow [13, 15, 16]. This deletion mutant also induced elevated IFNβ and IFNγ in cell culture, and exhibited reduced replication in mice suggesting a potential role in pathogenesis [13]. Hu/Aseer_KSA_RS924_2015 was isolated from an ill hospitalized patient without comorbidities who was eventually discharged to home. Despite the large deletion and frame shift in ORF3, Hu/Aseer_KSA_RS924_2015 still retained the ability to cause illness. Unlike the engineered mutant lacking ORF3, 4a, 4b and 5 generated by reverse genetics, Hu/Aseer_KSA_RS924_2015 still retained most of ORFs 4a, 4b and 5, which may be stronger contributors to pathogenesis than ORF3.
The deletion in ORF1ab in Hu/Riyadh_KSA-4050_2015 maps to nsp2. SARS-CoV nsp2 is dispensable for replication, however it has been found to bind host proteins, prohibitin 1 and prohibitin 2 [17]. These direct protein–protein interactions have been hypothesized to alter the host environment. We have not yet examined similar functions with Hu/Riyadh_KSA-4050_2015, but because this deletion is absent in the matched clinical specimen, it may indicate a tissue-culture adaptation.
The deletion mutant, Hu/Riyadh_KSA_2959_2015, exhibited a 9 nt deletion in the 5′ UTR. The 5′ UTR is thought to play a role in the discontinuous synthesis of subgenomic RNAs as well as bind several host proteins [18]. Limited research has been done on the roles of group-specific accessory proteins or regions such as the 5′ UTR in replication, virulence and pathogenicity.
Efficient replication of viruses with genomic deletions in cell culture do not eliminate the possibility that the gene products may play important roles in vivo. All of these viruses were isolated from ill patients. Hu/Riyadh_KSA_2959_2015 (Δ9 nt 5′ UTR) and Hu/Aseer_KSA_RS924_2015 (Δ41 nt ORF3) were isolated from patients who eventually died, and Hu/Riyadh_KSA-4050_2015 (Δ3nt ORF1a) was isolated from a hospitalized patient who was discharged to home. We confirmed the deletion of the 5′ UTR in the matched clinical specimen. The patient infected with Hu/Riyadh_KSA-4050_2015 (Δ3nt ORF1a) died during the course of illness confirming that the virus mutant retained its pathogenicity for humans. The 3 nt deletion in ORF1a in Hu/Riyadh_KSA-4050_2015 was absent in the matched clinical specimen suggesting that only full-genome viruses in the sample were able to grow in tissue culture. Additionally, we were unable to confirm that the ORF3 deletion in Hu/Aseer_KSA_RS924_2015 is also present in the clinical specimen because we were only able to recover the spike sequence from the clinical specimen due to limited volume. It is a limitation of the study. The patient from which Hu/Aseer_KSA_RS924_2015 (ΔORF3) was isolated died during the course of the illnesses. By isolating and characterizing MERS-CoV deletion variants detected in ill patients, we have confirmed that these viruses are viable and are not simply an artifact of directly sequencing clinical specimens. We have not yet determined if these deletions render less pathogenic viruses, however pathogenesis studies in animal models of these isolates are ongoing.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Names of specific vendors, manufacturers or products are included for public health and informational purposes; inclusion does not imply endorsement of the vendors, manufacturers or products by the Centers for Disease Control and Prevention or the US Department of Health and Human Services.
Supplementary Data
Funding information
This work received no specific grant from any funding agency.
Author contributions
Azaibi Tamin – conceptualization, methodology, investigative and writing – review and editing Krista Queen – investigative, formal analysis, writing – review and editing Clinton R. Paden – investigative, formal analysis, writing – review and editing Xiaoyan Lu – methodology, investigative, writing – review and editing Erica Andres – writing – original draft reparation, formal analysis Senthilkumar K. Sakthivel – investigative, writing – review and editing Yan Li - investigative, formal analysis, writing – review and editing Ying Tao – investigative, formal analysis, writing – review and editing Jing Zhang – investigative, formal analysis, writing – review and editing Shifaq Kamili – investigative, writing – review and editing Abdullah M. Assiri – supervision, resources, writing – review and editing Ali Alshareef – supervision, resources, writing – review and editing Taghreed A. Alaifan – supervision, resources, writing – review and editing Asmaa M. Altamimi – supervision, resources, writing – review and editing Hani Jokhdar – supervision, resources, writing – review and editing John T. Watson – supervision, resources, writing – review and editing Susan I. Gerber – supervision, resources, writing – review and editing Suxiang Tong- supervision, resources, writing – review and editing Natalie J. Thornburg – conceptualization, supervision, formal analysis, visualization, writing – original draft preparation
Conflicts of interest
Azaibi Tamin is listed as an inventor for licensed Hu/Florida/USA-2_Saudi Arabia_2014 with Zeptometrix. No other authors have any conflicts of interest.
Footnotes
Abbreviations: CoV, Coronavirus; CPE, Cytopathic effect; DMEM, Dulbecco’s modified eagle media; FBS, fetal bovine serum; GTR, general time-reversible; IFN, interferon; MERS, Middle East respiratory syndrome; ML, maximum likelihood; NGS, next generation sequencing; ORF, open reading from; PBS, phosphate buffered saline; PCR, polymerase chain reaction; PS, Pennicillin-Streptomycin; qPCR, quantitative PCR; RT-PCR, reverse transcription PCR; TCID50, tissue culture infectious dose 50%; UTR, untranslated region; WHO, World Health Organization.
One supplementary figure is available with the online version of this article.
References
- 1.Mohd HA, Al-Tawfiq JA, Memish ZA. Middle East respiratory syndrome coronavirus (MERS-CoV) origin and animal reservoir. Virol J. 2016;13:87. doi: 10.1186/s12985-016-0544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assiri AM, Midgley CM, Abedi GR, Bin Saeed A, Almasri MM, et al. Epidemiology of a novel recombinant Middle East respiratory syndrome coronavirus in humans in saudi arabia. J Infect Dis. 2016;214:712–721. doi: 10.1093/infdis/jiw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cotten M, Watson SJ, Zumla AI, Makhdoom HQ, Palser AL, et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio. 2014;5:e01062-13. doi: 10.1128/mBio.01062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu R, Wang Y, Wang W, Nie K, Zhao Y, et al. Complete genome sequence of Middle East respiratory syndrome coronavirus (mers-cov) from the first imported mers-cov case in china. Genome Announc. 2015;3 doi: 10.1128/genomeA.00818-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Liu D, Shi W, Lu R, Wang W, et al. Origin and possible genetic recombination of the Middle East respiratory syndrome coronavirus from the first imported case in china: phylogenetics and coalescence analysis. mBio. 2015;6:e01280–15. doi: 10.1128/mBio.01280-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu X, Rowe LA, Frace M, Stevens J, Abedi GR, et al. Spike gene deletion quasispecies in serum of patient with acute MERS-CoV infection. J Med Virol. 2017;89:542–545. doi: 10.1002/jmv.24652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie Q, et al. Two deletion variants of Middle East respiratory syndrome coronavirus found in a patient with characteristic symptoms. Arch Virol. 2017 doi: 10.1007/s00705-017-3361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Assiri AM, Biggs HM, Abedi GR, Lu X, Bin Saeed A, et al. Increase in Middle East respiratory syndrome-coronavirus cases in saudi arabia linked to hospital outbreak with continued circulation of recombinant virus, July 1–August 31, 2015. Open Forum Infect Dis. 2016;3:p. ofw165. doi: 10.1093/ofid/ofw165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamers MM, Raj VS, Shafei Mah'd, Ali SS, Abdallh SM, et al. Deletion variants of Middle East respiratory syndrome coronavirus from humans, jordan, 2015. Emerg Infect Dis. 2016;22:716–719. doi: 10.3201/eid2204.152065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Payne DC, Biggs HM, Al-Abdallat MM, Alqasrawi S, Lu X, et al. Multihospital outbreak of a Middle East respiratory syndrome coronavirus deletion variant, jordan: a molecular, serologic, and epidemiologic investigation. Open Forum Infect Dis. 2018;5:p. ofy095. doi: 10.1093/ofid/ofy095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu DKW, Hui KPY, Perera RAPM, Miguel E, Niemeyer D, et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc Natl Acad Sci USA. 2018;115:3144–3149. doi: 10.1073/pnas.1718769115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yusof MF, Queen K, Eltahir YM, Paden CR, Al Hammadi ZMAH, et al. Diversity of Middle East respiratory syndrome coronaviruses in 109 dromedary camels based on full-genome sequencing, abu dhabi, united arab emirates. Emerg Microbes Infect. 2017;6:1–10.:e101. doi: 10.1038/emi.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menachery VD, Mitchell HD, Cockrell AS, Gralinski LE, Yount BL, et al. MERS-CoV accessory orfs play key role for infection and pathogenesis. mBio. 2017;8 doi: 10.1128/mBio.00665-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y, Zhang L, Geng H, Deng Y, Huang B, et al. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell. 2013;4:951–961. doi: 10.1007/s13238-013-3096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scobey T, Yount BL, Sims AC, Donaldson EF, Agnihothram SS, et al. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc Natl Acad Sci USA. 2013;110:16157–16162. doi: 10.1073/pnas.1311542110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almazán F, DeDiego ML, Sola I, Zuñiga S, Nieto-Torres JL, et al. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio. 2013;4:e00650–13. doi: 10.1128/mBio.00650-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cornillez-Ty CT, Liao L, Yates JR, Kuhn P, Buchmeier MJ, et al. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol. 2009;83:10314–10318. doi: 10.1128/JVI.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang D, Leibowitz JL. The structure and functions of coronavirus genomic 3' and 5' ends. Virus Res. 2015;206:120–133. doi: 10.1016/j.virusres.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.