

Abstract
The structures and reactivities of pseudoephedrine-derived dianionic Myers enolates are examined. A combination of NMR and IR spectroscopic, crystallographic, and computational data reveal that the homoaggregated dianions form octalithiated tetramers displaying S4-symmetric Li8O8 cores and overall C2 symmetry. Computational and isotopic labeling studies reveal strong N–Li contacts in the carboxamide enolate moiety. The method of continuous variations proves deceptive, as octalithiated tetrameric homoaggregates afford hexalithiated trimeric heteroaggregates. A lithium diisopropylamide–lithium enolate mixed aggregate is found to be a C2-symmetric hexalithiated species incorporating two enolate dianions and two lithium diisopropylamide (LDA) subunits. Structural and rate studies show that lithium chloride has little effect on the dynamics of the enolate homoaggregates but forms adducts of unknown structure. Rate studies of alkylations indicate that the aging of the aggregates can have effects spanning orders of magnitude. The LiCl–enolate adduct dramatically accelerates the reaction but requires superstoichiometric quantities owing to putative autoinhibition. Efforts and progress toward eliminating the requisite large excess of LiCl are discussed.
Graphical Abstract

Introduction
In 1994, Myers and co-workers reported that dianions of type 2 derived from acylated pseudoephedrines (1) undergo functionalizations with a range of electrophiles in high yields and selectivities (eq 1).1 The products are deprotected by facile hydrolysis that appears to benefit from anchimeric assistance from the pendant hydroxy moiety noted previously by Evans using pyrrolidinol-derived amides.2 A key observation was that ≥5.0 equiv of LiCl was essential for optimal yields. Although no direct evidence attests to the role of LiCl, an artist’s rendition (3) was offered to illustrate the opposite selectivities observed for alkyl halide and epoxides. Despite the need for large quantities of LiCl, which has the effect of imposing concentration limits, the alkylations have found applications in a number of pharmaceutical-scale drug syntheses.3 They are now part of the pantheon of methods used for asymmetric synthesis, and they display special value in generating quaternary centers in exceptional yields and selectivities.1
 |
(1) |
Understanding reactivity and selectivity in solutions teeming with observable and fleeting species demands a sound understanding of solution structure. Challenges to determining the structures of Myers enolates promised to be especially daunting: (1) possible aggregate topologies illustrated in Chart 1 are representative but are by no means a comprehensive compilation of possible structural motifs4; (2) positional isomerism represented generically in eq 2 would amplify spectral complexity; and (3) the difficulty of characterizing dianions5 is exacerbated by the absence of observable scalar coupling of Li–O bonds.
 |
(2) |
Chart 1.

Selected possible topologies of Myers enolates.
We describe herein studies of the structure and reactivity of Myers enolates. We take unusual care to illustrate some of the trials and tribulations—the often unstated pain and suffering en route to the answer—while trying not to make the reader suffer proportionately. The story is one of tactics and strategies. Although we characterized several heteroaggregates and a lithium diisopropylamide (LDA)–enolate mixed aggregate spectroscopically (vide infra), our best efforts to understand the all-important homoaggregate of 2 left us with too many possible structures and too many niggling details that simply did not afford an unapologetic assignment. A low-resolution crystal structure that would be unpublishable in isolation provided us the answer key—the Rosetta stone. The crystallographic and spectroscopic data proved fully self-consistent. Which structure of the many under consideration was correct? In a word, none. We had many of the details right but missed the topology of the Li8O8 core that was, ironically, lurking in the Cambridge Structural Database.6
Dropping any pretense of presenting the data to the reader and then making the assignment, we provide the plot-spoiling solution structure of Myers enolate 2 as C2-symmetric octalithiated prism 4 constituted from four dianionic subunits manifesting an S4-symmetric core and overall C2 symmetry. The four N–Li contacts were foreshadowed computationally. Although four coordinated THF ligands are omitted for clarity, they were the subject of considerable experimental focus. As part of our efforts to use the method of continuous variations (MCV)—a favorite of ours for studying solution structures of enolates7—we generated and characterized heteroaggregates containing subunits of opposite absolute configuration (heterochiral) that afforded hexalithiated prisms of type 5. In short, MCV failed to provide heteroaggregates that attest to the structures of the homoaggregates. Control experiments to understand the consequences of excess LDA revealed C2-symmetric hexalithiated mixed aggregate 6 (comprising two LDA subunits and two dianionic subunits), which bears little relationship to the enolate homoaggregates but shares several unusual features with heteroaggregate 5. Rate studies of alkylations showed profound aging effects spanning orders of magnitude.8 As to the influence of LiCl, we observed nondescript lithium enolate–LiCl mixed aggregates.
Results
We confront the age-old problem of presenting data from a variety of sources that, taken in total, provide the assignments yet must be presented serially. We try to assist the reader by prefacing the results with a detailed survey of the methods, as well as the structural and stereochemical consequences of the polygonal prisms. The nonspecialist will find an adequate survey of the results in the Discussion.
Substrates.
Acylated pseudoephedrine-based enolates 2 and 7–22 (Chart 3) were prepared using standard protocols.1 [15N](R,R)-2 and [15N](S,S)-2 to probe N–Li contacts originated from [15N]NH4Cl, as shown in Scheme 1. [6Li]LDA and [6Li,15N]LDA were isolated as LiCl-free solids.9 Solvation was probed in several ways, including by using pyridine as a 6Li chemical shift reagent10 and [15N]isoquinoline (eq 3) as an inexpensive,11 relatively nonvolatile pyridine surrogate.
 |
(3) |
Chart 3.

Myers enolates.
Scheme 1. Synthesis of 15N-labeled enolate precursors.

(a) PhC(=O)H, MgCl2, TMSCl, NEt3, EtOAc, 24 h, rt; (b) TFA, MeOH, 10 min, rt; (c) TBDMSCl, imidazole, DMF, 18 h, rt; (d) LiOH, H2O2, THF, 6 h, 0 °C to rt; (e) (COCl)2, CH2Cl2, 2 h, rt; (f) 15NH4Cl, NaOH, 30 min, 0 °C to rt; (g) [hydroxyl(tosyloxy)iodo]benzene, CH3CN, 2 h, reflux; (h) acetic formic anhydride, CH2Cl2, 1 h, −30 °C; (i) LiAlH4, THF, overnight, reflux; (j) propionyl chloride, NEt3, THF, 10 min, 0 °C.
Myers enolates were generated at −78 °C with subsequent aging for 10 min at 25 °C owing to slow aggregate equilibration.8 Although we focused on aggregates 4–6 prepared from propionate enolate 2, we examined the behavior of 17 enolates that fall into several distinct categories: (1) enolates 8 and 11 bearing small aliphatic enolate substituents share common properties with enolate 2 that we comfortably assigned by analogy to homoaggregate 4; (2) 7, 9, 10, 12, 19, and 20, which manifest complex ensembles of otherwise sharp resonances suggesting isomerism is at play yet form well-defined heterochiral heteroaggregates sharing spectroscopic properties with 5; (3) arylated enolates 13–18, which are similar to one another but quite distinct from their aliphatic counterparts, yet again appear to form distinct type 5 heteroaggregates; and (4) enolate 21, which is insoluble, and enolate 22, which displays hopelessly broad resonances (possibly owing to instability mentioned by Myers).1
MCV.
We have developed general protocols for using MCV in conjunction with NMR spectroscopy to characterize lithium enolates.7 In its simplest form, MCV involves mixing binary pairs of lithium enolate homoaggregates, An and Bn, of unknown aggregation state, n, to generate an ensemble of homo- and heteroaggregates (eq 4). In principle, the homo- and heteroaggregates can be monitored using 1H, 6Li, 13C, 15N, or 19F NMR spectroscopies. In practice, only 6Li and 19F NMR spectroscopies played roles in this study. (Caveat: Large T1’s for 6Li mandated up to 50 s pulse delays to attain accurate 6Li integrations.) 15N NMR spectroscopy has been useful in MCV when labeled substrates are available.12 Despite the requisite resolution indicating that it would work well, [15N](R,R)-1 is simply too precious. Plotting the relative concentrations of different aggregates versus measured mole fraction of An (XA) or Bn (XB) affords a Job plot. (See Figure 7 for an example.)
| (4) |
Figure 7.

Job plot showing the relative integrations of octalithio homoaggregates (R4/S4) and hexalithio heteroaggregates (RS2/R2S) versus intended mole fractions21 of [6Li](R,R)-2 (XR) for 0.10 M mixtures of enantiomeric lithium enolates [6Li](R,R)-2 (R) and [6Li](S,S)-2 (S) in neat THF at −80 °C monitored with 6Li NMR spectroscopy. The curves result from a parametric fit to a tetramer–trimer model.20
Choosing the right pairing partners to ensure sufficient spectroscopic resolution of all homo- and heteroaggregates is critical and largely empirical and was particularly challenging in this case. The presumption is that the number and symmetries of the heteroaggregates attest to the aggregation number of homoaggregates An and Bn. For the first time in what is now hundreds of applications, the homo- and heteroaggregates did not share a common aggregation number. Therefore, we defer discussing MCV-based studies until after presenting a more conventional approach to assigning of the structure of homoaggregate 4.
Two-Dimensional NMR Spectroscopy.
Critical spatial relationships stem from a composite of NMR spectroscopies (COSY, TOCSY, HSQC, HMBC, and ROESY).13 Detailed discussions of the cross-correlations without a computer interface are poorly suited for print media. We only briefly allude to important correlations herein. In a pedagogically more tractable approach, a video tutorial in the supporting information walks the viewer through some of the assignments, as done in a previous paper.11 Pulsed field-gradient spin echo NMR spectroscopy (PFGSE)—or diffusion NMR spectroscopy—was used semi-quantitatively to determine relative aggregate sizes.14
Computations.
Density functional theory (DFT) calculations were carried out at the B3LYP/6–31G(d) level with energies from single-point calculations at the MP2 level of theory.15 They largely showed plausibility of structural motifs, provided support to the correlation spectroscopies, and allowed us to probe experimentally elusive three-dimensional details. The energies proved unhelpful in distinguishing structural and stereoisomers owing to resulting narrow energetic ranges. The computations were crucial, however, in predicting strong N–Li contacts that were eventually confirmed spectroscopically and crystallographically. They also played a central role in developing mechanistic and stereochemical models.
Isomerism.
Three general classes of isomerism alluded to throughout the case studies required constant attention because of their implications for symmetry and spectral complexity. We also occasionally saw evidence of minor, lower symmetry isomers. The three major forms of isomerism are as follows.
-
Helical isomerism is exemplified by mirror-image fragments 23a and 23b, which become diastereomers when stereogenic centers are included. Asymmetry within the prismatic structure of 4 rendered concerns about helical isomerism moot, but the possibility of such isomerism lingered within our models for months.

Isomerism resulting from N–Li bond scission and reassociation to form different N–Li contacts (Scheme 2) is represented by isomeric pairs 24/25 and 26/27. Chelates in 24 and 26 have phenyl moieties on the concave chelate face, whereas 25 and 27 have them on the convex face. Two-dimensional spectroscopic probes of 4 and 5 show both types (vide infra). Should only one of the multiple subunits reverse chelate orientation, however, any existing symmetry would be lost and the resonance counts would spike as each 6Li becomes magnetically distinct.
Positional isomerism such as that manifested in 24/26 or 25/27 pairs in Scheme 2 results from reversal of the enolate and alkoxide roles. Such a transposition would break the high symmetry.
Scheme 2.

Isomerism arising from reversal of chelate orientation or enolate and alkoxide positioning.
Case 1: Homoaggregate of (R,R)-2.
Structural studies of the homoaggregate of propionate enolate (R,R)-2 are the focus of this paper. Applications of MCV are deferred to subsequent case studies.
6Li NMR spectra of (R,R)-2 generated at −78 °C show complex mixtures of resonances (Figure 1A). Full aggregate equilibration required aging at 25 °C for 10 min, affording two groups of 6Li resonances at chemical shift extremes in carefully measured 1:1:1:1 proportions (Figure 1B). (The details of this aging process are delineated below.) Varying the THF concentration between 2.0 equiv per Li and 12.3 M (neat THF) and the enolate concentration from 0.020 M to 0.20 M resulted in no change in relative integrations, indicating that the resonances arose from either a single aggregate with four magnetically inequivalent lithiums or two isomeric aggregates in a precise 1:1 proportion. Failure to achieve coalescence at >40 °C owing to slow inter- and intraaggregate exchange,16 in conjunction with requisite aging at ambient temperature, attested to the remarkable robustness of the aggregate core.
Figure 1.

6Li NMR spectra of a solution of 0.10 M [6Li](R,R)-2 in neat (12.3 M) tetrahydrofuran (THF) recorded at −80 °C. (A) Unaged; (B) aged at 25 °C for 10 min. Inset in (B) shows the resolution at −40 °C. (C) and (D) NMR spectra of an aged solution of 0.10 M [6Li ,15N](R,R)-2. (C) 6Li NMR spectrum recorded at −20 °C (to improve resolution). (D) 15N NMR spectrum recorded at −80 °C.
Early in the study, DFT computations of dianion subunits in isolation or as part of larger aggregates evidenced strong N–Li contacts, prompting us to insert an 15N label as shown in Scheme 1. [15N](R,R)-2 showed 6Li–15N coupling in the downfield resonances at 2.32 and 2.35 ppm (JN–Li = 0.90 and 1.40 Hz, respectively), whereas the upfield resonances remained as sharp singlets (Figure 1C). The corresponding 15N NMR spectrum shows two 1:1:1 15N triplets correlated by single-frequency decoupling (Figure 1D).
Studies of solvation showed that only two of the four lithiums—logically those lacking N–Li contacts—contain one ligated THF each. For example, titrations of toluene solutions of 2 with THF showed that only 0.5 equiv THF per lithium was required to convert broad mounds to the four characteristic sharp resonances; additional THF caused minimal further change. More surprisingly, free and bound THF were in slow exchange on the NMR timescale at room temperature (Figure 2A). Such slow ethereal solvent exchange on lithium is rarely observed17 and is without precedent at ambient temperature. Single-frequency decoupling of the β protons on the bound THF revealed a single AB quartet for the α protons (Figure 2B). Given that two magnetically inequivalent THF ligands would show as many as four distinct AB quartets, we presumed that a combination of free rotation about the Li–O bond and near magnetic equivalence is at play. The decoupling was mimicked using 3,3,4,4-tetradeuteriotetrahydrofuran (THF-d4),18 affording the same AB quartet.
Figure 2.

Partial 1H NMR spectra of an aged solution of 0.10 M (R,R)-2 in 0.44 M THF and 9.1 M toluene-d8 at 20 °C. (A) Coupled; (B) single-frequency irradiated at 1.40 ppm.
Pyridine functions as a 6Li NMR chemical shift reagent, imparting large (>1 ppm) downfield shifts for pyridine-ligated 6Li nuclei and muted downfield shifts for more distal 6Li resonances.10 Indeed, serial additions of pyridine to 2 showed large (1.1–1.4 ppm) shifts of the formerly THF-bearing lithiums and small shifts (≈0.4 ppm) of the 6Li resonances coordinated by the carboxamide nitrogens. Readily available and relatively inexpensive [15N]isoquinoline11 caused analogous shifts and showed two of the four 6Li resonances as doublets (JN–Li = 3.9 and 3.6 Hz) and the remaining two as sharp singlets (Figure 3). 15N NMR spectra showed broad mounds rather than sharp resonances. Broad-band 15N decoupling caused the two 6Li doublets to collapse to singlets. The coupling constants are large compared with those of other 6Li-15N dative bonds.17a
Figure 3.

6Li NMR spectra of aged solutions of 0.10 M [6Li](R,R)-2 at −95 °C. (A) 0.20 M THF; (B) 0.40 M [15N]isoquinoline and 0.20 M THF. The asterisk (*) flags THF–isoquinoline mixed-solvated 4.
The resonance count of the homoaggregate of 2 excludes a single hexalithiated form by symmetry. The remaining options shown in Chart 4 include: (1) a 1:1 mixture of isomeric dilithiated forms, such as isomers 28 and 29, displaying different convexity, each with two magnetically inequivalent lithium nuclei; (2) a 1:1 mixture of two high-symmetry tetralithio isomers, such as 30 and 31; (3) a single low-symmetry tetralithio form, such as 32 or 33; (4) either 30 or 31 paired with its helical isomer (not drawn but akin to 23a,b); (5) two high-symmetry hexalithio aggregates 34 and 35 (see below) or analogous pairs of octalithio or higher polygonal prismatic helical isomers; and (6) any one of a host of octalithio derivatives with a symmetry element affording a reduced resonance count (vide infra).
Chart 4.

Possible di-, tetra-, and hexalithio aggregates of enolate (R,R)-2.
The full gamut of two-dimensional NMR spectroscopies led to the assignment of all protons and carbons within the two magnetically inequivalent subunits of (R,R)-2 (Figure 4). The critical spatial correlations are as follows:
Multiple correlations between the two magnetically distinct dilithio subunits excluded all models based on pairs of isomers such as dilithio isomers 28 and 29, high-symmetry tetralithio isomers 30 and 31, and helical isomers 34 and 35.
Strong inter-subunit correlations resulting from Ph–Ph contacts (a) and Me–H contacts (b) exclude tetralithio isomers 30 or 31, as well as unsymmetrical tetralithio isomers 32 and 33.
Apparent intra-subunit correlations suggest opposite convexities, as drawn in Figure 4. The Me–Ph correlations (c) and Me–H correlation (d) place the phenyl moieties on the convex and concave faces, respectively. We hasten to add, however, that deception by inter-subunit interactions between two magnetically equivalent subunits was a persistent concern. Moreover, the isomer with the phenyl on the convex face, as in subunit 1, is also calculated using simple (dilithio) models to be >2.0 kcal/mol more stable.
Correlations a and b seem to exclude a number of plausible aggregates by showing that both faces of both subunits are proximate.
Figure 4.

Critical resonance correlations of the (R,R)-2-derived homoaggregate.
By eliminating all di-, tetra-, and hexalithio homoaggregates, we were left with octalithio derivatives. Additional support came from two independent measurements. A diffusion NMR spectrum14 showed that the homoaggregate is larger than a demonstrably hexalithio heteroaggregate (vide infra). We tried using mass spectrometry (MS) on a DART MS, but the crude effort suffered from considerable protonation problems and left us leery of potential for user bias.19
Considering all connectivities of octagonal prisms and stacked cubes consistent with two inequivalent subunits and four magnetically inequivalent lithium nuclei left eight prisms and 18 stacked cubes as possible structures. Including the restriction that subunits 1 and 2 have opposite convexities (Figure 4), the number of possibilities reduces to six prisms and 12 stacked cubes (Chart 5). The DFT-computed relative energies showed plausibility but would not make the assignment for us. The nastier problem was that none accounted for the concurrent correlations without some overtly optimistic interpretation.
Chart 5.


Renditions of DFT-computed structures showing a representative stacked cube and octagonal prism of octalithio homoaggregates of (R,R)-2.
We needed an X-ray crystal structure. Early efforts failed, but we eventually obtained a low-resolution crystal structure that provided the needed insight (Figure 5). The structure of 4 shows an S4-symmetric core,6 which becomes a C2-symmetric octalithiated aggregate with the chiral appendages. The lithium counts, N–Li contacts, and most importantly, through-space interactions match the resonance correlations without exception. Differential 6Li–15N coupling noted above is reflected in markedly different N–Li bond lengths. Even with a graphic interface, one has to look carefully to see two pairs of subunits of opposite convexity. The pairs of magnetically inequivalent THF ligands reside in environments that appear so similar that the spectroscopic detection of a single observable THF subunit is not surprising.
Figure 5.

Partial X-ray crystal structure of octalithiated enolate 4 showing the core with one subunit and the total structure missing four THF ligands.
Case 2: (R,R)-2–(R,R)-8 Homochiral Heteroaggregates.
The use of MCV to characterize homoaggregated enolates is based on the notion that mixtures of two homoaggregates will produce a highly characteristic ensemble of heteroaggregates (eq 4). Taking a cue from previous studies of chiral β-amino ester enolates,20 we examined three pairing strategies. The first involved mixing combinations of enolates within a homochiral series—mixtures of (R,R)-2 and (R,R)-8, for example—to generate type-i heteroaggregates. Heteroaggregation was evidenced by intractable spectral complexity. This strategy was doomed from the start given that two structurally distinct Myers enolates can form seven distinct heteroaggregated analogs of homoaggregate 4 manifesting 48 6Li resonances.

Case 3: (S,S)-2–(R,R)-2 Heterochiral Heteroaggregates.
Mixing enantiomers of a single enolate—systematically generating scalemic mixtures—affords type-ii heteroaggregates with heterochiral subunits. A substantial advantage of this strategy is that x:y and y:x heteroaggregates are mirror images, which makes them appear spectroscopically as a single species, thereby markedly reducing the resonance count and minimizing resolution problems. By chance, heterochiral enolates also optimize resolution and favor heteroaggregates relative to homoaggregates nonstatistically. The fundamental presumption of MCV for determining aggregation state fails, however. Scalemic mixtures of octalithiated homoaggregates of enolates afforded hexalithiated heteroaggregates without exception (eq 5).

| (5) |
For example, scalemic mixtures formed from (R,R)-2 and (S,S)-2 with requisite aging at 25 °C display six new 6Li resonances in equal proportions (Figure 6A). The resonance count precludes an octalithiated aggregate. Resonances with mid-range chemical shifts (1.0–2.0 ppm) proved to be hallmarks of such heterochiral aggregates.
Figure 6.

6Li NMR spectra of aged solutions of enolate mixtures in neat THF at −80 °C: (A) 0.050 M [6Li](S,S)-2 and 0.050 M [6Li](R,R)-2; and (B) 0.050 M [6Li,15N](S,S)-2 and 0.050 M [6Li,15N](R,R)-2. The resonance labeled “alkoxides” corresponds to monolithiated substrate.
Monitoring the proportions of homo- and heteroaggregates as a function of mole fraction X(R,R)-2 afforded the Job plot in Figure 7. Although the data adequately fit to an all-hexalithio (trimer) model, the fit in Figure 7 corresponds to that derived for octalithio homoaggregates (tetramers) and hexalithio heteroaggregates (trimers).20 (Recall that only two spectroscopically discrete species exist.) The requisite fitting protocols are in the supporting information. The maximum at 0.50 derives from the superposition of enantiomeric 2:1 and 1:2 heteroaggregates. One could easily imagine confusing the maximum as evidence of a tetralithio heteroaggregate (dimer) if not for the resonance count and poor fit.
The relative and absolute aggregation state of heteroaggregate 5 was supported by diffusion NMR spectroscopy showing that the hexalithio appeared to be smaller than the octalithio (Figure 8).14 In fact, showing the reverse—the octalithio is larger than the more easily characterized hexalithio—was more important.
Figure 8.

Plot of diffusion coefficient (D) from diffusion NMR spectroscopy versus molecular weight of an aged solution of 0.040 M (S,S)-2 and 0.060 M (R,R)-2 in neat THF at −60 °C; y = a + bx, a = −4.622, b = 0.443. This experiment assumes four coordinated THF ligands per homotetramer and four per heterotrimer.
The gamut of two-dimensional NMR spectroscopies provided the key resonance correlations for the three discrete subunits of 5 shown in Figure 9. The convexities as drawn derive from the correlations labeled a, d, and e. Notably, subunit 3 is isolated from subunits 1 and 2 and displays no intersubunit correlation.
Figure 9.

Critical resonance correlations of subunits in the hexalithiated heterochiral aggregate formed from (R,R)-2 and (S,S)-2.
Labeling studies showed that the hexalithio heteroaggregates represented an aggregate that was altogether different from that of octalithio homoaggregate 4. In mixtures of [15N](R,R)-2 and [15N](S,S)-2, the resonance at 1.95 ppm appeared as a doublet (JN–Li = 1.25 Hz), whereas the resonance at 1.83 ppm appeared as a doublet of doublets (JN–Li = 1.30 and 2.17 Hz). This result indicates that two nitrogens are bound to a common lithium (Figure 6B). The remaining four upfield resonances are singlets. The 15N NMR spectra showed three 15N triplets with correlations confirmed by single-frequency decoupling. Mixtures of (S,S)-2 and [15N](R,R)-2 in which only one substrate is 15N labeled caused the lithium central to the N–Li–N fragment to appear as a doublet at all proportions of the two enolates. Thus, the subunits forming the N–Li–N contact—subunits 1 and 2 in Figure 9—are necessarily heterochiral as drawn. From connectivities alone, we could draw six isomeric cores with a central N–Li–N contact. Including additional restrictions imposed by the heterochiral core and established convexities, we were left with two pseudo-octahedral partial structures, 36 and 37, displaying cores that would be delta isomers.22 There are eight possible isomers obtained by the placement of either (R,R)-2 or (S,S)-2 as the third subunit (with positional isomerism considered). The gross structure of hexalithiated aggregate 5 relied on 1H and 13C correlation spectroscopy (supporting information). From the eight possible isomers, DFT computations suggested that the isomer shown as aggregate 5 with the 36 core is the most stable by >4 kcal/mol (Figure 10).
Figure 10.

DFT-computed structure of the most stable hexalithiated enolate aggregate, 5, showing the Li6O6 core (left) and distorted octahedral core described by 36 (right).
Case 4: (R,R)-8–(S,S)-11 Heterochiral Heteroaggregates.
Pairing enolates of opposite absolute configuration and containing different enolate substituents affords type-iii heteroaggregates displaying many of the properties of the heterochirality observed in case 3 but with magnetically distinct R′S2 and R′2S heteroaggregates (eq 6). Heterochiral binary combinations of 2, 8, and 11 were all tractable. Emblematically, mixtures of (S,S)-8 and (R,R)-11 displayed fully desymmetrized 2:1 and 1:2 heteroaggregates each with affiliated six equal-intensity resonances (Figure 11). Unlabeled debris in the spectra of both homo- and heteroaggregates is reproducible, implicating minor stereoisomers. Although detailed correlating spectroscopic analyses would offer marginal gain at great effort, monitoring the aggregates versus mole fraction of (R,R)-11, X(R,R)-11, affords the Job plot shown in Figure 12. The use of measured rather than intended mole fraction21 is especially suited for such mixtures.
Figure 11.

6Li NMR spectra of 0.10 M solutions of [6Li](S,S)-8 (S) and [6Li](R,R)-11 (R′) in neat THF at −80 °C. The measured mole fractions of [6Li](R,R)-11 (XR′) are 0.00, 0.40, 0.57, and 1.00 for A–D, respectively.
Figure 12.

Job plot showing the relative integrations of octalithio homoaggregates and hexalithio heteroaggregates versus measured mole fractions of [6Li](R,R)-11 (XR′) for 0.10 M mixtures of lithium enolates [6Li](S,S)-8 (S) and [6Li](R,R)-11 (R′) in neat THF at −80 °C monitored with 6Li NMR spectroscopy (Figure 11). The curves result from a parametric fit to a tetramer–trimer model.20

| (6) |
Case 5: (R,R)-14–(S,S)-16 Heterochiral Heteroaggregates.
Myers enolates with sterically demanding yet anion-stabilizing aryl substituents show behaviors that are distinctly different from those of their aliphatic counterparts (see Figure 1A). Each displays a major upfield resonance accompanied by a minor resonance in ratios that vary from enolate to enolate (Figure 13). The intensities are invariant to changes in enolate concentration, suggesting that they are isomers.23 The absence of downfield resonances shows that the aryls cause deep-seated structural changes. High steric demands and anion stabilization of the phenyl moiety would be expected to promote lower aggregates.24 Diffusion NMR spectroscopy confirmed that the homoaggregate is smaller than the hexalithio heteroaggregate (vide infra). Attempts to obtain X-ray-quality crystals of the homoaggregates were unsuccessful.
Figure 13.

6Li NMR spectra of 0.10 M solutions of [6Li](S,S)-16 (S) and [6Li](R,R)-14 (R′) in neat THF at −80 °C at the ratios indicated on the spectra.
Heterochiral mixtures of arylated enolates (S,S)-14 and (R,R)-16 showed clear evidence of hexalithiated heteroaggregates, but spectral overlap precluded a Job plot using 6Li NMR spectroscopy. The single fluorine nucleus per subunit (as opposed to two 6Li nuclei) enables 19F NMR spectroscopy while cutting the resonance counts in half. The 19F NMR spectrum shows a single resonance (Figure 14) despite normally high-resolution of 19F NMR spectroscopy. Mixtures showed that the 1:2 and 2:1 19F pairs are consistent with a hexalithiated species. Minor isomers of the 2:1 and 1:2 heteroaggregates also were observable. Monitoring the homo- and heteroaggregates versus X(R,R)-14 and arbitrarily assigning the homoaggregates as hexalithio, tetralithio, or even dilithio afforded reasonable Job plots. The plot in Figure 15 arbitrarily assumes a tetralithiated homoaggregate.
Figure 14.

19F NMR spectra of 0.10 M solutions of [6Li](S,S)-16 (S) and [6Li](R,R)-14 (R′) in neat THF at −80 °C. The measured mole fractions of [6Li](R,R)-14 (XR′) are 0.00, 0.34, 0.67, and 1.00 for A–D, respectively.
Figure 15.

Job plot showing the relative integrations of tetralithio homo- and hexalithio heteroaggregates versus measured mole fractions of [6Li](R,R)-14 (XR′) for 0.10 M mixtures of lithium enolates [6Li](S,S)-16 (S) and [6Li](R,R)-14 (R′) in neat THF at −80 °C monitored with 19F NMR spectroscopy (see Figure 14). The curves result from a parametric fit to a dimer–trimer model.20
Case 6: (R,R)-2–LDA Mixed Aggregate.
As a standard control experiment, lithium enolates were generated from [6Li,15N]LDA9 to identify any resonances attributable to LDA–enolate mixed aggregates. Although resulting LDA-derived mixed aggregate 6 bore little relationship to homoaggregate 4, it foreshadowed the unusual N–Li–N connectivity in heteroaggregate 5.
Mixtures of enolate (R,R)-2 with varying amounts of excess [6Li,15N]LDA included LDA homodimer, homoaggregate 4, and mixed dimer 6. The key characteristics of 6 were as follows: (1) four 6Li resonances appeared in a 1:2:2:1 ratio (Figure 16A), indicating a hexalithiated species with a symmetry element; (2) the two major 6Li resonances appeared as doublets (J = 5.45 and 5.45 Hz) owing to splitting by a single LDA-derived 15N nucleus (Figure 16B); (3) the mixed aggregate prepared from [15N](R,R)-2 and [6Li]LDA caused one of the minor resonances not connected to the LDA fragment to appear as a triplet (J = 2.4 Hz; Figure 16C), which correlated with one 15N triplet (1:1:1). The gamut of 1H and 13C NMR spectroscopies showed a single subunit with the phenyl moiety in the convex face, as drawn for 6. The stoichiometry, symmetry, and coupling demanded a hexalithio mixed aggregate with a C2 symmetry axis. The symmetry demanded that the dianions reside on opposite faces of 6. Although pyridine caused marked chemical shifts, attempts to probe solvation using [15N]-isoquinoline failed owing to decomposition. DFT computations showed that 6 is viable, revealed favorable N–Li contacts, and supported tetrasolvation, as displayed in Figure 17.
Figure 16.

6Li NMR spectra of 0.10 M solutions of [6Li](R,R)-2 and 0.20 M LDA in neat THF at −80 °C using (A) [6Li]LDA; (B) [6Li,15N]LDA; and (C) [6Li,15N]LDA and [15N](R,R)-2.
Figure 17.

Structure of enolate–LDA mixed aggregate 6. The four solvents supported by DFT computations have been omitted in the computed rendition for clarity.
Lithium Chloride: Influence on Structure.
Addition of LiCl to a pre-aged solution of aggregate 4 had no measurable effect (Figure 18A): aggregate 4 was observed along with free LiCl at the anticipated relative proportions. Warming the probe in 20 °C increments caused no broadening of the resonances of 4 or appearance of new resonances. Careful integration of the resonances, however, showed that the broad mound corresponding to LiCl increased at the expense of 4 (Figure 18, B–F). The mound appeared to include free LiCl in rapid exchange with a LiCl–enolate adduct(s) denoted nondescriptly as 2LiCl. Assuming conservation of mass, the proportions of homoaggregate 4 and 2LiCl could be teased out. Plotting the percent of enolate 2 that existed as homoaggregate 4 versus temperature (Figure 19) showed limited adduct formation at low temperatures and a marked change (a discontinuity) at ≈0 °C. Reversing the protocol—cooling the probe in 20 °C increments—did not reverse the changes; 2LiCl remained dominant.25 The spectrum re-recorded at −80 °C may include additional debris that corresponds to species in slow exchange at −80 °C, but it appears insignificant.1 Adduct formation was confirmed by LiCl-dependent chemical shifts observed in the 1H and 13C NMR spectra.26 The time-averaged resonances may be concealing multiple LiCl adducts of varying LiCl stoichiometries as discussed below.1 Although adduct 2LiCl cannot be characterized, it is decidedly important (vide infra).
Figure 18.

6Li NMR spectra of a pre-aged 0.10 M solution of [6Li](R,R)-2 with subsequent addition of 0.20 M LiCl in neat THF-d8 recorded at incrementally rising temperatures.
Figure 19.

Plot of percent enolate 4 in solutions of a pre-aged 0.10 M solution of [6Li](R,R)-2 with subsequent addition of 0.20 M LiCl in neat THF-d8 versus temperature (Figure 18). Incremental temperature increases (black) were followed by incremental temperature decreases (blue).
Lithium Chloride: Influence on Subunit Exchange.
In a fairly crude experiment, kinetically formed (pre-aged) ensembles of 2 (see Figure 1A) were warmed in 20 °C increments with cooling to −80 °C at each temperature level to record the 6Li spectra. The spectra indicate that aging to form 4 becomes consequential at >0 °C. Repeating the experiment with added LiCl showed little or no effect on the rate of aging.
In a cleaner experiment, samples of (S,S)-2 and (R,R)-2 were separately and fully aged before mixing to form a racemic mixture of aggregate 4. Warming in 20 °C increments revealed hexalithiated heteroaggregate 5, evidencing that interaggregate subunit exchange on laboratory timescales emerges only at 40 °C. (Subunit exchange in the octalithiated homoaggregate is slower than exchange in unaged ensembles.) The same experiment with added LiCl showed that rate of conversion of the two antipodes of 4 to give 5 occurs at >0 °C. Thus, LiCl influences the aging rate on laboratory timescales but only marginally and, as we discuss below, much more slowly than the alkylation.
Aging Effects on Enolate Reactivity.
We now present some curious observations on how aggregate aging and the stabilities of the aggregates influence their reactivities. Alkylations using allyl bromide (eq 7) were monitored using in situ IR spectroscopy following the formation of the product alkoxide at 1610 cm−1. (The enolate absorbance at 1640 cm−1 was poorly resolved from an absorbance of the product alkoxide.) The apparent percent conversions were confirmed with workup and 1H NMR spectroscopic analysis of the crude products. Fully aged homoaggregate 4 was marginally reactive toward allyl bromide at 0 °C for 1.0 h (Figure 20). Myers also noted such low conversions (10–13%). Varying the THF molarity offered no improvement. Elevated temperatures promoted O-alkylation byproducts. Reasoning that unaged samples contain structures that are less stable and, thus, more reactive, we found that addition of allyl bromide 1.0 min after enolization at −20 °C improved the conversion to 40%. Completely unaged samples kinetically formed at −78 °C (see Figure 1A) alkylated at −78 °C to 67% conversion before stalling. Surmising that the ensemble of unstable aggregates generated at −78 °C might depend on the source, we showed that metalation with lithium 2,2,6,6-tetramethylpiperidide increased the conversion to 78%, which proved to be the upper limit. Attempts to push the conversion of unaged samples through gentle warming failed, presumably because of competing aging to less reactive forms.
 |
(7) |
Figure 20.

Rates and percent conversions of alkylation of lithium enolate (R,R)-2 (0.040 M) in neat THF at −60 °C by allyl bromide (0.092 M) with various amounts of LiCl.
Influence of LiCl on Reactivity.
It is not surprising that homoaggregate 4 is inert; it looks inert. Addition of ≥5.0 equiv of LiCl as part of Myers’s optimized protocol is imperative. A more granular view obtained by adding various concentrations of LiCl to the allylation in eq 7 reveals a burst of reactivity followed by stalling to the rate that approximates the LiCl-free rate (Figure 20). Both the initial rates and the percent conversions were promoted by elevated LiCl concentrations. We attribute the acceleration to the nondescript adduct(s) denoted as 2LiCl and the stalling to an equally nondescript autoinhibition resulting from a lithium alkoxide-–LiCl mixed aggregate that occludes the LiCl.28 Monitoring the alkylation with 6Li NMR spectroscopy revealed only homoaggregate 4. The 6Li resonances of the product alkoxide were absorbed into the broad LiCl-derived mound in rapid time-averaged exchange.
Monitoring the initial alkylation rates proved instructive. The initial rates indicate a clean first-order dependence on allyl bromide (Figure 21) attesting to the general quality of the data and a rate-limiting alkylation.29 Curvatures in the LiCl dependence (Figure 22) and enolate dependence (Figure 23) could be construed as fractional-order dependencies emblematic of dimer–monomer and tetramer–monomer dissociations.28 Detection and quantitation of 2LiCl, however, clearly supported the curvatures in Figures 22 and 23 as saturation kinetics.
Figure 21.
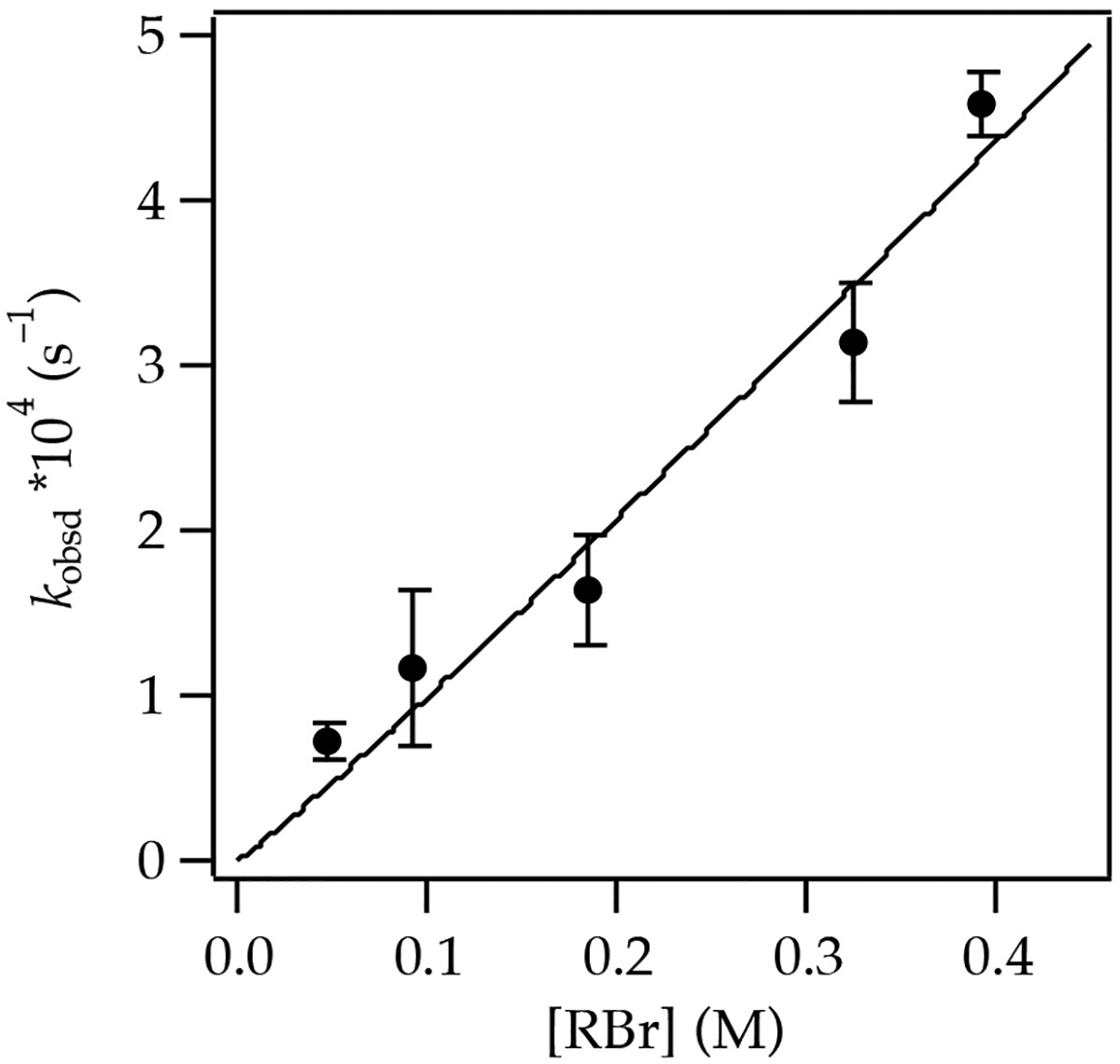
Plot of initial rate constants (kobsd) versus allyl bromide concentration for alkylation of 0.040 M (R,R)-2 and 0.040 M LiCl in neat THF at −60 °C. The curve depicts an unweighted least-squares fit to the function f(x) = axn such that a = (1.2 ± 0.26) × 10−2 and n = 1.1 ± 0.19.
Figure 22.

Plot of initial rate constants (kobsd) versus LiCl equivalence (per enolate) for alkylation of 0.040 M (R,R)-2 and 0.092 M allyl bromide in neat THF at −60 °C. The red curve depicts an unweighted least-squares fit to the function f(x) = axn/(bxn + 1) such that a = (1.6 ± 0.34) × 10−4, b = 0.57 ± 0.25, and n = 1 (set value). The blue curve depicts an unweighted least-squares fit with n as an adjustable parameter such that a = (2.3 ± 0.86) × 10−4, b = 1.2 ± 0.52, and n = 2.1 ± 0.77.
Figure 23.

Plot of initial rate constants (kobsd) versus the concentration of (R,R)-2 for allylation in eq 7 with 0.080 M LiCl and 0.18 M allyl bromide in neat THF at −60 °C. The asterisk (*) denotes a point at 0.010 M enolate; the absorbance at 0.010 M enolate is too low to be considered fully reliable. The curve depicts an unweighted least-squares fit to the function f(x) = axm/(bxm + 1) such that a = 0.014 ± 0.0043, b = 48 ± 18, and m = 1 (set value).
The red curve in Figure 22 represents first-order saturation behavior required if the equilibrium in eq 8 affords 2LiCl as a monomer-based tetralithiated mixed aggregate, (enolate)(LiCl)2 (eq 8, 1/n = 1 and 1/m = 1/4). The blue curve shows a fit with n as an adjustable parameter and fits to a second-order LiCl saturation (1/n = 2 and 1/m = 1) consistent with 2LiCl as a tetramer-based mixed aggregate, (enolate)4(LiCl)4. We hasten to add that the quality of the fits distinguishes the various possible structures for 2LiCl in principle but, in our opinion, not in practice. The enolate dependence in Figure 23 is fit to a first-order saturation behavior (eq 8, 1/m = 1), but a fit to a fractional-order saturation behavior (1/m = 1/4) is, practically speaking, indistinguishable. Plotting the initial rates versus THF concentration in hexane (Figure 24) reveals an upward curvature suggesting intermediate first or second order.
Figure 24.

Plot of initial rate constants (kobsd) versus THF concentration for alkylation of 0.040 M (R,R)-2 with 0.040 M LiCl by 0.092 M allyl bromide in hexanes cosolvent at −60 °C. The curve depicts an unweighted least-squares fit to the function f(x) = axn such that a = (0.042 ± 0.020) × 10−4 and n = 1.5 ± 0.21.
The saturation behaviors are consistent with the generic rate law in eq 9. Two critical pieces of information remained outstanding: (1) the structure of 2LiCl as described in eq 8 is unknown, leaving the values of m and n in eq 9 undefined; and (2) the reaction order in 2LiCl could not be measured owing to solubility constraints; knowing this would provide an important clue about the structure of 2LiCl. We consider three mechanisms in the Discussion.
| (8) |
| (9) |
Discussion
Myers’s central insight, which made pseudoephedrine-derived enolates popular reagents for asymmetric alkylations, was that excess LiCl is critical to obtaining high yields. From a structural and mechanistic perspective, the dianionic enolates present a host of challenges outlined in the introduction that are unlike any we have confronted to date.
Structural Studies: Protocols.
The aggregate structures are summarized in Scheme 3, but they tell only a small part of the story. The number of analytical methods and tactics is unusual. These include a bevy of two-dimensional correlation spectroscopies, 6Li–15N double-labeling studies, diffusion NMR spectroscopy, mass spectrometry, IR spectroscopy, MCV, X-ray crystallography, and DFT computations. This breadth of effort was demanded by the elusiveness of the answers rather than by some predilection toward thoroughness.
Scheme 3.

Structural summary
Standard 1H and 13C NMR two-dimensional correlation spectroscopies provided critical information but did not lead us to a definitive structural assignment. Attempts to use MS to determine aggregation19 presented potentially resolvable technical challenges that rendered the data from our first effort at risk of user-dependent interpretation. DFT computations often help resolve structural issues, but there were simply too many possible isomers in a narrow energetic range. After several years of study, a low-quality crystal structure provided the answer key: the solution structural studies came together into a fully coherent structural model. While we had obsessed over octagonal prisms and stacked cubes, the octalithiated aggregate proved to be 4, the core of which was known.6
MCV consumed considerable attention. By intent, mixtures of structurally similar homoaggregates afford ensembles of heteroaggregates with numbers and symmetries that attest to the structures of the homoaggregates. However, mixtures of octalithiated homoaggregates—dozens of combinations in total—afforded hexalithiated heteroaggregates. To the best of our knowledge, this failure of MCV is a first for us. A detailed study of two antipodes of enolate 2 revealed the heterochiral heteroaggregate of type 5. Aside from the different aggregation numbers of 4 and 5, the hexalithiated heteroaggregates include strikingly different structural details.
Structures.
Let us take a granular peek at structural details of the homo- and heteroaggregates and mixed aggregates30 summarized in Scheme 3.
Aging Effects. Implicitly embedded in Myers’s alkylation protocols are aging effects—requisite warming cycles—that prove to have a firm structural basis and became a major focus of our studies. Enolization at −78 °C affords a complex mixture that converges to form octalithiated aggregate 4 only on warming to ambient temperatures. The same aging effects are observed during formation of heteroaggregates of type 5. Although aging to form 4 could be construed as the structural change that Myers addressed by warming the samples, this conclusion is incorrect. The aging was needed to generate LiCl adducts (below).
N–Li contacts. The N–Li contacts of carboxamide-derived enolates appear to have gone undocumented until now. We discovered them computationally and then took them seriously enough only to confirm them using 15N-labeled substrate. Contemporaneously, Williard and co-workers crystallographically detected a number of such N–Li contacts in structurally simple carboxamide enolates.31 As to how general they are in solution remains unknown; a simple 15N-labeled propionamide enolate affords no N–Li coupling.32 From the outset, the importance of the N–Li contacts and the resulting convexity in the Myers enolates seemed certain to underlie the stereocontrol.
Slow THF exchange. Slow exchange of lithium-bound THF on 4 with free THF at ambient temperatures is surprising. It is rare that THF can be observed in the slow exchange limit even at low temperatures.17 We suspect that the slow exchange on aggregate 4 derives, at least in part, from suppression of the associative pathway.17a Although slow alkylations of 4 probably render rates of THF exchange irrelevant, highly reactive electrophiles could suffer consequentially (stereochemically) from restricted access to coordination sites.
- Stereochemically sensitive aggregation. The formation of octalithiated homochiral aggregates of type 4 and hexalithiated heterochiral heteroaggregates of type 5 are, in one sense, an idiosyncratic artifact of using MCV to study aggregation. (We are likely the only group to intentionally study racemates of chiral lithium enolates used in asymmetric synthesis.)20 Nonetheless, the hexalithiated heteroaggregates display a fundamentally different doubly chelated N–Li–N core (see 36 and 37). This structural feature is shared by LDA mixed aggregate 6 (Figure 17). The hexalithiated aggregates are also formed nonstatistically. The good news is that DFT studies suggest that the homochiral hexalithio derivatives prefer heterochirality (eq 10) and the octalithiated forms prefer homochirality (eq 11). The dubious result is that, when combined, DFT computations completely fail to predict hexalithiated aggregates of type 5 to be competitive with homoaggregate 4 (eq 12).

(10) 
(11) 
(12)
Aging Effects on Reactivity.
If one supposes that all aggregated forms converge on a fleeting dilithiated (monomeric) intermediate, then aggregate stability and reactivity should correlate inversely (Scheme 4). Fully aged 4 reacts slowly at 0 °C and stalls at <10% conversion, whereas unaged aggregates—the complex ensemble of species formed kinetically at −78 °C—are orders of magnitude more reactive, eventually stalling at 67–78% conversion. Presumably, the kinetically formed ensemble contains aggregates of varying reactivities. One cannot help but worry that the depiction of 5 may be susceptible to correction in its detail.
Scheme 4.

Reactivity of Myers enolate 2 without added LiCl as measured by percent conversion after 1 h.
The Role of LiCl: Enolate Structure and Reactivity.
Adding excess LiCl is imperative, as noted by Myers,1 and it serves the dual function of accelerating the alkylation and allowing the reaction to proceed to full conversion (Figure 20). We considered several possible roles for LiCl.
Catalyzed Deaggregation. One could imagine that LiCl catalyzes tetramer–monomer exchange, shifting the rate-limiting step from deaggregation to alkylation of the dissociated monomer.28,34 LiCl-catalyzed deaggregation33 was studied and documented in excruciating detail for LiCl-catalyzed deaggregation of LDA.28 Presuming that a catalyzed deaggregation would affiliate with a catalyzed exchange of subunits, we probed the influence of LiCl on rates of subunit exchange from a number of angles. Minor accelerations of subunit exchanges occurring on laboratory timescales near ambient temperatures were observed, but they could not explain LiCl-mediated alkylations at −60 °C.
- Mixed Aggregation. Lithium chloride forms a nondescript mixed aggregate denoted simply as 2LiCl (eq 8). Formation of 2LiCl is slow at −80 °C and occurs in earnest only near ambient temperatures on laboratory timescales (eq 13). It is the slow formation of 2LiCl that Myers seems to have overcome by warming the enolate–LiCl mixtures before alkylating the enolates. An odd temperature dependence (Figure 19) shows that the formation of 2LiCl is slow and irreversible. It also suggests, however, that other partial adducts at intermediate temperatures may exhibit limited reactivity. Once 2LiCl forms, the time averaging of free and bound LiCl in the 6Li NMR spectra (eq 14) made it difficult to detect—we almost missed it—and difficult to characterize to date.35 The potentially critical observation is that exchange of free and bound LiCl, as well as intraaggregate exchanges of the 6Li nuclei within 2LiCl, are exceedingly facile on NMR timescales at −80 °C.

(13) 
(14) - Autoinhibition. Alkylation of enolate 2 with allyl bromide stalls at percent conversions that qualitatively correlate with the added LiCl. In theory, this result could stem from incomplete formation of 2LiCl, but we cannot push the percent conversions effectively even under conditions in which LiCl could be recycled. We surmise that the product of the reaction—an alkoxide (R′OLi)—occludes LiCl in a mixed aggregate (eq 15). Once again, strikingly fast LiCl exchange did not refute the existence of such an adduct but did preclude structural studies.

(15)
It is instructive to ponder the structure of 2LiCl (Chart 6). The deep-seated structural changes in LiCl adducts 38 and 39 are appealing in light of remarkably low rates of LiCl incorporation, although 39 contains one or two LiCl fragments depending on whether the counterion is a simple or a cationic triple ion.36 Chloride-derived “ate” complexes analogous to 39 and 40, in which one could think of chloride as a hexamethylphosphoramide surrogate, have been observed37 and are computationally viable (supporting information). Retention of the intact octalithio framework, as depicted in 40, cannot be rigorously excluded—it is a computational minimum—but the spectroscopic properties of 2LiCl seem to argue against the intact Li8O8 core.
Chart 6.

Possible structures of enolate-LiCl adduct 2LiCl.
Rate Studies and Mechanism.
Detailed rate studies reveal curvatures consistent with saturation kinetics reflecting the lithium enolate- and LiCl-dependent formation of 2LiCl (Figures 22 and 23). The accompanying THF dependence (Figure 24) is compatible with almost any model with some imagination. We have considered a number of mechanisms for the alkylation that hinge on the structure of 2LiCl with a focus on mixed aggregates 38 or 39. We present a working hypothesis based on 38 as described by eq 16 and delineated in Scheme 5.
Scheme 5.

Mechanism of alkylation of cubic mixed aggregates.
Computational studies suggest a preference for 38a relative to 38b, displaying the opposite convexity in the resting state that superficially follows through to the most stable transition structures 41a and 41b. The mechanism also includes no provisions for an additional THF suggested by the rate studies, but that does not trouble us too much. The more systemic flaw is that the preference for transition structure 41b predicts the wrong (minor) isomer compared with that observed experimentally.
| (16) |
An alternative mechanism that could account for THF-dependent rates (maybe too much so depending on the resting state of 2LiCl) is based on LiCl ate complexes such as 39. Such ate complexes have been observed37 as have simple +Li(THF)n and more complex triple-ion-based counterions.36 The model reveals a strong (2.5 kcal/mol) preference for transition structure 42 relative to the second most stable of three other isomeric transition (supporting information). This model is consistent with the experimental results.

The third and simplest of the three mechanisms is based on direct alkylation via dilithiated monomer-based transition structure 43 stripped free of any LiCl influence. Although the model could readily account for the THF dependence, it also predicts a net inverse LiCl dependence of some form owing to requisite dissociation of LiCl from 2LiCl. We can easily imagine such details evading our detection by being embedded in the saturation kinetics required to form 2LiCl (Figure 22). This monomer-based transition structure 43 is computationally preferred relative to the three other choices (supporting information) and is consistent with experiment.
On first inspection, the notion that forming a LiCl adduct to simply dissociate it before alkylation seems to defy the Principle of Detailed Balance, which suggests that in such an ensemble the LiCl should impart a positive influence on rates only if it is included in the rate-limiting transition state.38 Yet the Principle of Detailed Balance applies only to ensembles of species in equilibrium. The Myers enolate and its LiCl adduct (eq 17) are not at equilibrium; the reluctant formation of 2LiCl requires ambient temperatures, affording 2LiCl, which seems to be in rapid exchange with monomer but slow exchange with homoaggregate 4. The role of LiCl is to overcome the otherwise insurmountable barrier to dissociating octalithiated prismatic tetramer 4. An analogous example of aggregate ensembles in which not all species are at equilibrium was recently observed for Evans enolates in which facile dimer–monomer equilibration occurs concurrently with slow formation of the more stable and unreactive tetramer.8d
 |
(17) |
Inspection of the transition structures (12 in total) with the aid of a graphical interface reveals no self-evident insight into the stereochemical preferences. There is no visibly discernible preference for a specific convexity of the chelated dianion or for an approach of the allyl bromide to the enolate that can be generalized. Notably, however, one can see a lengthening of the N–Li bond at the transition structure as the loss of the enolate character at the transition states that attenuates the Lewis basicity of the nitrogen ligand. IRC calculations indicate that the alkylation causes complete scission of the N–Li bond during the alkylation.
Omitting Lithium Chloride?
A potential impediment to using Myers enolates on pilot-plant or plant scales stems from the relatively high dilutions (0.10 M) imposed by the limited solubility (and cost) of the ≥5-fold LiCl excess. Evidence that precluding enolate aging markedly enhanced reactivities and percent conversions in halide free solutions offered reason for optimism, but we could not push past 78% conversion. Investigators at Merck were able to exclude LiCl in the alkylation of a highly stabilized enolate bearing an aryl group akin to enolates 13–18 using THF/TMEDA,3e but we showed those are structurally quite different than the more standard Myers enolates. It would be tempting to add ligands to selectively extract LiCl from the putative product mixed aggregate by forming +Li(ligand)//Cl− (eq 18), but according to the Principle of Detailed Balance, such a side equilibrium would decrease the concentration of 2LiCl as well. Nonetheless, we tried a few chelating ligands to no avail. We have, however, been making progress using a radically different strategy, but that story will have to wait.
 |
(18) |
Conclusions
Studies of dianionic Myers enolates certainly presented challenges and surprises. The influence of aging on aggregate structure and reactivity is profound and reinforces the notion that enolates age remarkably slowly compared with other organolithiums. The pseudoephedrine-derived Myers enolates are slower to equilibrate than most, but they are by no means unique.8 Indeed, limited solution structural studies of enolates to date suggest that most enolates—particularly the structurally complex variants—undergo aggregate exchanges so slowly that the unsuspecting user may be getting whipsawed by slow aggregate equilibrations without realizing it. Functionalizing an enolate under conditions in which aggregates have not equilibrated may be constructive or destructive. Worse, however, is when functionalizations of partially equilibrated aggregates wreak havoc on attempts to develop rational models to control and understand reactivity and selectivity.
In some ways, the study of Myers enolates is about refining protocols for characterizing complex organolithiums in solution. We are positioning to move our gaze to Myers’s second-generation enolates, derived from the 1,2-diphenyl-substituted amino ethanols. The new perspective might fill in some details that eluded us in the current investigation. Moreover, there are many dianionic enolates in which ancillary lithium alkoxides, lithiated sulfonamides, or lithiated Boc-amines are used in lieu of standard protecting groups. We are optimistic that we now have the tools to probe structure–reactivity relationships.
Experimental
Reagents and Solvents.
THF and toluene were distilled from solutions containing sodium benzophenone ketyl. Pyridine was distilled from solutions sodium benzophenone ketyl. LDA, [6Li]LDA, and [6Li,15N]LDA were prepared as described previously.9 Air- and moisture-sensitive materials were manipulated under argon using standard glove box, vacuum line, and syringe techniques. Myers enolate precursors were either purchased or prepared as described previously.1 Several previously unreported precursors were prepared by acylating the pseudoephedrine as described in supporting information.
NMR Spectroscopy.
Individual stock solutions of substrates and LDA were prepared at room temperature. An NMR tube under vacuum was flame-dried on a Schlenk line and allowed to return to room temperature. It was then backfilled with argon and placed in a dry ice/acetone bath at −78 °C. The appropriate amounts of acylated pseudoephedrine and LDA (1.1 equiv) were added sequentially via syringe. The tube was sealed under partial vacuum and vortexed three times on a vortex mixer for 5 s. Samples could be stored indefinitely in a freezer −86 °C. Each sample routinely contained 0.10 M total enolate with a 0.005 M excess of LDA. Standard 1H, 6Li, 13C, 15N, and 19F NMR spectra were recorded on a 500 MHz spectrometer at 499.92, 73.57, 125.72, 50.67, and 470.33 MHz, respectively. Quantitated 6Li integrations required the relaxation delay set to seven times the decay half-life. Integration of NMR signal was carried out using the peak area generated by a line-fitting method. The 6Li and 13C resonances are referenced to 0.30 M [6Li]LiCl/MeOH at −90 °C (0.0 ppm) and the CH2O resonance of THF at −80 °C (67.57 ppm). Two-dimensional NMR spectroscopies (COSY, TOCSY, HSQC, HMBC, and ROESY) were recorded using standard pulse sequences.
IR Spectroscopic Analyses.
IR spectra were recorded using an in situ IR spectrometer fitted with a 30-bounce, silicon-tipped probe. The spectra were acquired in 16 scans at a gain of 1 and a resolution of 4 cm−1. A representative reaction was carried out as follows: The IR probe was inserted through a nylon adapter and O-ring seal into an oven-dried, cylindrical flask fitted with a magnetic stir bar and a T-joint. The T-joint was capped with a septum for injections and a nitrogen line. After evacuation under full vacuum, heating, and flushing with nitrogen, the flask was charged with a stock solution of enolate 2 in THF or THF/hexane and LiCl in THF. The vessel was cooled to −60 °C in a constant-temperature bath controlled with a cold finger and prepared with fresh acetone. After a background spectrum was recorded, neat allyl bromide was added with stirring. IR spectra were recorded every 6 s with monitoring of the absorbance at 1610 cm−1 over the course of the reaction.
MCV.
The mathematical treatment for creating Job plots using Mathematica has been described previously.20 The specific case in which octalithiated homoaggregates (formally tetramers) afford hexalithiated heteroaggregates (trimers) required additional coding as described in the supporting information.
Supplementary Material
Chart 2.

Structurally characterized homo- and heteroaggregates and mixed aggregates (4–6) of Myers enolates.
Acknowledgments.
We thank the National Institutes of Health (GM077167) for support.
Footnotes
Supporting Information: Spectroscopic, crystallographic, computational, and rate data as well as additional Job plots. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
References and Footnotes
- 1.(a) Yang BH; Chen H; Gleason JL; Myers AG Use of Pseudoephedrine as a Practical Chiral Auxiliary for Asymmetric Synthesis. J. Am. Chem. Soc 1994, 116, 9361. [Google Scholar]; (b) McKinstry L; Myers AG Practical Syntheses of Enantiomerically Enriched γ-Lactones and γ-Hydroxy Ketones by the Alkylation of Pseudoephedrine Amides with Epoxides and their Derivatives. J. Org. Chem 1996, 61, 2428. [Google Scholar]; (c) Yang BH; Chen H; McKinstry L; Kopecky DJ; Gleason JL; Myers AG Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones. J. Am. Chem. Soc 1997, 119, 6496. [Google Scholar]
- 2.Evans DA; Takacs JM Enantioselective Alkylation of Chiral Enolates. Tetrahedron Lett. 1980, 21, 4233. [Google Scholar]
- 3.(a) Sandham DA; Taylor RJ; Carey JS; Fässler A A convergent synthesis of the renin inhibitor CGP60536B. Tetrahedron Letters, 2000, 41, 10091. [Google Scholar]; (b) Dragovich PS; Prins TJ; Zhou R Formal, stereoselective synthesis of hydroxyethylene dipeptide isosteres utilizing pseudoephedrine amides. J. Org. Chem 1997, 62, 7872. [Google Scholar]; (c) Sinz C; Bittner A; Brady E; Candelore M; Dallas-Yang Q; Ding V; Jiang G; Lin Z; Qureshi S; Salituro G; Saperstein R Discovery of N-Aryl-2-acylindole human glucagon receptor antagonists. Bioorganic & medicinal chemistry letters 2011, 21, 7124. [DOI] [PubMed] [Google Scholar]; (d) Narasimhulu CP; Das P Stereoselective Preparation of C1–C10 and C11-O14 Fragments of Narbonolide: Exploiting the Versatility of Thiazolidinethione Chiral Auxiliary. Synthesis 2009, 474. [Google Scholar]; (e) Smitrovich JH; DiMichele L; Qu C; Boice GN; Nelson TD; Huffman MA; Murry J Michael Reactions of Pseudoephedrine Amide Enolates: Effect of LiCl on Syn/Anti Selectivity. J. Org. Chem 2004, 69, 1903. [DOI] [PubMed] [Google Scholar]
- 4.(a) Seebach D Structure and Reactivity of Lithium Enolates. From Pinacolone to Selective C-Alkylations of Peptides. Difficulties and Opportunities Afforded by Complex Structures. Angew. Chem., Int. Ed. Engl 1988, 27, 1624. [Google Scholar]; (b) Braun M Lithium Enolates: ‘Capricious’ Structures - Reliable Reagents for Synthesis. Helv. Chim. Acta 2015, 98, 1. [Google Scholar]
- 5.(a) Gruver JM; West SP; Collum DB; Sarpong R Experimental Characterization and Computational Study of Unique C,N-Chelated Lithium Dianions. J. Am. Chem. Soc 2010, 132, 13212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li D; Keresztes I; Hopson R; Williard PG Characterization of Reactive Intermediates by Multinuclear Diffusion-Ordered NMR Spectroscopy (DOSY). Acc. Chem. Res 2009, 42, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gareyev R; Ciula JC; Streitwieser A Lithium and Cesium Ion-Pair Acidities of Dibenzyl Ketone. Aggregation of Lithium and Cesium Ion Pairs of the Enolate Ion and Dianion. J. Org. Chem 1996, 61, 4589. [DOI] [PubMed] [Google Scholar]; (d) Brand H; Capriotti JA; Arnold J New Lithium Porphyrin Derivatives: Synthesis of Li2(P)(Et2O)2 (P = TTP, TBPP) and Solution Structure of Li2(TTP)(Et2O)2 by 7Li and 15N NMR Inorg. Chem 1994, 33, 4334. [Google Scholar]; (e) Günther H Selected Topics from Recent NMR Studies of Organolithium Compounds. J. Braz. Chem. Soc 1999, 10, 241. [Google Scholar]; (f) Jacobson MA; Keresztes I; Williard PG On the Mechanism of THF Catalyzed Vinylic Lithiation of Allylamine Derivatives: Structural Studies Using 2-D and Diffusion-Ordered NMR Spectroscopy. J. Am. Chem. Soc 2005, 127, 4965. [DOI] [PubMed] [Google Scholar]; (g) Cohen Y; Roelofs NH; Reinhardt G; Scott LT; Rabinovitz M Novel carbocyclic dianions: NMR study of charge delocalization, paratropicity, and structure in the dianions of acephenanthrylene and aceanthrylene. J. Org. Chem 1987, 52, 4207. [Google Scholar]; (h) Matsuo T; Mizue T; Sekiguchi A Synthesis and Molecular Structure of a Dilithium Salt of the cis-Diphenylcyclobutadiene Dianion. Chem. Lett 2000, 896. [Google Scholar]; (i) Henderson KW; Dorigo AE; MacEwan GJ; Williard PG Structure of an unsymmetrical heptalithium cage complex containing aldolate and enolized aldolate dianion. Tetrahedron 2011, 67, 10291. [Google Scholar]
- 6.(a) MacLellan JG; Mulvey RE; Nichols PJ; Andrews PC New homo- and hetero-alkali metal alkoxide cages; crystal structures of [Me2N(CH2)2OLi]8 and [{Me2N(CH2)2O}12Li8K6]O. J. Chem. Soc., Dalton Trans, 2002, 1651. [Google Scholar]; (b) Allan JF; Nassar R; Specht E; Beatty A; Calin N; Henderson KW Characterization of a kinetically stable, highly ordered, octameric form of lithium tert-butoxide and its implications regarding aggregate formation. J. Am. Chem. Soc 2004, 126, 484. [DOI] [PubMed] [Google Scholar]; (c) Boyle TJ; Alam TM; Peters KP; Rodriguez MA Structural diversity of lithium neopentoxide compounds. Inorg. Chem 2001, 40, 6281. [DOI] [PubMed] [Google Scholar]
- 7.Renny JS; Tomasevich LL; Tallmadge EH; Collum DB Method of Continuous Variations: Applications of Job Plots to the Study of Molecular Associations in Organometallic Chemistry. Angew. Chem., Int. Ed 2013, 52, 11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Casy BM; Flowers RA On the Nature of the Oxidative Heterocoupling of Lithium Enolates. J. Am. Chem. Soc 2011, 133, 11492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jin KJ; Collum DB Solid-State and Solution Structures of Glycinimine-Derived Lithium Enolates J. Am. Chem. Soc 2015, 137, 14446. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) For a marked influence of aggregate aging on the reactivity and selectivity of organolithiums, see:; Xu F; Reamer RA; Tillyer R; Cummins JM; Grabowski EJJ; Reider PJ; Collum DB; Huffman JC Highly Enantioselective 1,2-Addition of Lithium Acetylide-Ephedrate Complexes: Spectroscopic Evidence for Reaction Proceeding via a 2:2 Tetramer, and X-ray Characterization of Related Complexes. J. Am. Chem. Soc 2000, 122, 11212. [Google Scholar]; (d) Tallmadge EH; Jermaks J; Collum DB Structure–Reactivity Relationships in Lithiated Evans Enolates: Influence of Aggregation and Solvation on the Stereochemistry and Mechanism of Aldol Additions. J. Am. Chem. Soc 2016, 138, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Y; Hoepker AC; Gupta L; Faggin MF; Collum DB 1,4-Addition of Lithium Diisopropylamide to Unsaturated Esters: Role of Rate-Limiting Deaggregation, Autocatalysis, Lithium Chloride Catalysis and Other Mixed Aggregation Effects. J. Am. Chem. Soc 2010, 132, 15610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jermaks J; Tallmadge EH; Keresztes I; Collum DB Lithium Amino Alkoxide–Evans Enolate Mixed Aggregates: Aldol Addition with Matched and Mismatched Stereocontrol. J. Am. Chem. Soc 2018, 140, 3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Garratt PJ; Vollhardt KPC Homophthalaldehyde. Synthesis, 1971, 8, 423. [Google Scholar]; (b) Huang X; Tanaka KS; Bennet AJ Glucosidase-catalyzed hydrolysis of α-D-glucopyranosyl pyridinium salts: kinetic evidence for nucleophilic involvement at the glucosidation transition state. J. Am. Chem. Soc 1997, 119, 11147. [Google Scholar]
- 12.Algera RF; Ma Y; Collum DB Sodium Diisopropylamide: Aggregation, Solvation, and Stability. J. Am. Chem. Soc 2017, 139, 7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a).Friebolin H Basic One- and Two-Dimensional NMR Spectroscopy; Wiley VCH: Weinheim, 2010. [Google Scholar]; (b) Claridge TDW High-Resolution NMR Techniques in Organic Chemistry, 2nd ed; Elsevier: Amsterdam, 2009. [Google Scholar]
- 14.(a).Sørland GH Dynamic Pulsed-Field-Gradient NMR, Springer: Berlin, 2014. [Google Scholar]; (b) Pöppler A-C; Granitzka M; Herbst-Irmer R; Chen Y-S; Iverseen BB; John M; Mata RA; Stalke D Characterization of oa Multicomponent Lithium Lithiate from a Combined X-Ray Diffraction, NMR Spectroscopy, and Computational Approach. Angew. Chem. Int. Ed 2014, 53, 13282. [DOI] [PubMed] [Google Scholar]; (c) Neufeld R; Teuteberg TL; Herbst-Irmer R; Mata RA; Stalke D Solution Structures of Hauser Base i-Pr2NMgCl and Turbo- Hauser Base i-Pr2NMgCl·LCl in THF and the Influence of LiCl on the Schlenk Equilibrium. J. Am. Chem. Soc 2016, 138, 4796. [DOI] [PubMed] [Google Scholar]
- 15.Gaussian 09, Revision A.02,; Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, and Fox DJ, Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]
- 16. It is often the case that exchange of magnetically inequivalent 6Li nuclei within aggregates can, on warming, undergo rapid intraaggregate exchange, causing each aggregate to display a single 6Li resonance. See reference 20.
- 17.(a) Lucht BL; Collum DB Lithium Hexamethyldisilazide: A View of Lithium Ion Solvation Through a Glass-Bottom Boat. Acc. Chem. Res 1999, 32, 1035. [Google Scholar]; (b) Boche G; Fraenkel G; Cabral J; Harms K; van Eikema Hommes NJR; Lohrenz J; Marsch M; Schleyer P. v. R. exo,exo-[1,3-Bis(trimethylsilyl)allyl]lithium-N,N,N’,N’-tetramethylethylenediamine Complex: Crystal Structure and Dynamics in Solution. J. Am. Chem. Soc 1992, 114, 1562. [Google Scholar]; (c) Hilmersson G; Davidsson OA Multinuclear NMR Study of a Chiral Lithium Amide with an Intramolecular Chelating Methoxy Group in Coordinating Solvents at the Slow Ligand Exchange Limit. J. Org. Chem 1995, 60, 7660. [Google Scholar]; (d) Reich HJ; Kulicke KJ Dynamics of Solvent Exchange in Organolithium Reagents. Lithium as a Center of Chirality. J. Am. Chem. Soc 1996, 118, 273. [Google Scholar]
- 18.(a) Bissell ER; Finger M Organic Deuterium Compounds. II. Some Deuterated Tetrahydrofurans. J. Org. Chem 1959, 24, 1259. [Google Scholar]; (b) Yang YK; Bergman RG Synthesis, molecular structure, and thermal chemistry of (.eta.5-cyclopentadienyl) dicarbonylrhenacyclopentane. Organometallics 1985, 4, 129. [Google Scholar]
- 19.(a) Putau A; Koszinowski K Probing Cyanocuprates by Electrospray Ionization Mass Spectrometry. Organometallics 2010, 29, 3593. [Google Scholar]; (b) Cvačka J; Svatoš A Matrix-assisted laser desorption/ionization analysis of lipids and high molecular weight hydrocarbons with lithium 2,5-dihydroxybenzoate matrix. Rapid Commun. Mass Spectrom 2003, 17, 2203. [DOI] [PubMed] [Google Scholar]
- 20.Liou LR; McNeil AJ; Toombes GES; Collum DB Structures of ß-Amino Ester Enolates: New Strategies Using the Method of Continuous Variation. J. Am. Chem. Soc 2008, 130, 17334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The intended mole fraction refers to the mole fraction based on what was added to the samples. The measured mole fraction—the mole fraction within only the ensemble of interest—eliminates the distorting effects of impurities.
- 22.Haq I; Lincoln P; Suh D; Norden B; Chowdhry BZ; Chaires JB Interaction of .DELTA.- and .LAMBDA.-[Ru(phen)2DPPZ]2+ with DNA: A Calorimetric and Equilibrium Binding Study. J. Am. Chem. Soc 1995, 117, 4788. [Google Scholar]
- 23.Additional resonances appear at low THF concentrations.
- 24.Reich HJ Role of Organolithium Aggregates and Mixed Aggregates in Organolithium Mechanisms. Chem. Rev 2013, 113, 7130. [DOI] [PubMed] [Google Scholar]
- 25.Allowing samples to stand at low temperature for weeks causes no detectable increase in free enolate concentration, suggesting that the failure to observe appreciable 4 is not the result of a suppressed rate of exchange.
- 26.In some cases, adding LiCl to hexalithiated heteroaggregates causes considerable additional debris to appear, attesting to significant formation of structurally complicated LiCl-enolate mixed aggregates.
- 27.Monitoring the influence of LiCl on the IR absorbance of enolate 4 at 1640 cm−1 revealed broadening of the peak with no chemical shift changes.
- 28.Algera RF; Gupta L; Hoepker AC; Liang J; Ma Y; Singh KJ; Collum DB Lithium Diisopropylamide: Non-Equilibrium Kinetics and Lessons Learned about Rate Limitation. J. Org. Chem 2017, 82, 4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LiBr is approximately 15-fold less effective at mediating the alkylation. The low concentrations generated during the alkylation are inconsequential.
- 30.We refer to (LiX)n and (LiX)m(LiX′)n as a “homoaggregate” and “heteroaggregate,” respectively, and reserve the term “mixed aggregate” for (LiX)m(LiY)n.
- 31.Williard PG, unpublished.
- 32.For a discussion of the relationship of Li-N coupling and Li-N bonding, see:; Engelhardt F; Maaß C; Andrada DM; Herbst-Irmer R; Stalke D Benchmarking lithium mide versus amine bonding by charge desnity and energy decomposition analysis arguments. Chem. Sci 2018, 9, 3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.In some literature reports, mixed aggregates containing a single subunit of substrate and inorganic salts are referred to as “deaggregated.” We find this usage to be unproductive.
- 34.Lithium sulfonate in catalytic quantities influenced the stereochemistry of acetylide additions without influencing the structure of the observable mixed aggregate:; Parsons RL Jr. Development of Commercially Viable, Enantioselective Processes for HIV-1 NNRTIs. Curr. Opin. Drug Discovery Dev 2000, 3, 783. [PubMed] [Google Scholar]
- 35.Attempts to grow crystals of 2LiCl afforded only homoaggregate 4.
- 36.Yakimansky AV; Müller AHE; Van Beylen M Density Functional Theory Study on the Aggregation and Dissociation Behavior of Lithium Chloride in THF and Its Interaction with the Active Centers of the Anionic Polymerization of Methyl Methacrylate and Styrene. Macromolecules 2000, 33, 5686. [Google Scholar]
- 37.Romesberg FE; Collum DB Lithium Dialkylamide Mixed Aggregation: An NMR Spectroscopic Study of the Influence of Hexamethylphosphoramide (HMPA). J. Am. Chem. Soc 1994, 116, 9198. [Google Scholar]
- 38.Alberty RA Principle of Detailed Balance in Kinetics. J. Chem. Educ 2004, 81, 1206 Hammes, G. G. Principles of Chemical Kinetics; Academic Press: New York, 1978; pp 14–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.