

Abstract
Alphavirus budding is driven by interactions between nucleocapsids assembled in the cytoplasm and envelope proteins present at the plasma membrane. So far, the expression of capsid and envelope proteins in infected cells has been considered an absolute requirement for alphavirus budding and propagation. In the present study, we show that Semliki Forest virus and Sindbis virus lacking the capsid gene can propagate in mammalian and insect cells. This propagation is mediated by the release of infectious microvesicles (iMVs), which are pleomorphic and have a larger size and density than wild-type virus. iMVs, which contain viral RNA inside and viral envelope proteins on their surface, are released at the plasma membrane and infect cells using the endocytic pathway in a similar way to wild-type virus. iMVs are not pathogenic in immunocompetent mice when injected intravenously, but can infect different organs like lungs and heart. Finally, we also show that alphavirus genomes without capsid can mediate the propagation of heterologous genes, making these vectors potentially interesting for gene therapy or vaccination studies. The minimalist infectious system described in this study shows that a self-replicating RNA able to express membrane proteins with binding and fusion properties is able to propagate, providing some insights into virus evolution.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-016-2230-1) contains supplementary material, which is available to authorized users.
Keywords: Alphavirus, Semliki Forest virus, Sindbis virus, Capsid deficiency, Infectious microvesicles
Introduction
For most viruses, the capsid is an essential protein structure that encloses and protects the viral genome. Many viruses also have an envelope wrapping the viral capsid, which is usually derived from host cell membranes and incorporates some viral glycoproteins. These proteins are important for virus attachment and penetration into host cells.
Alphaviruses have been extensively studied because they represent a good model for simple envelope RNA viruses. Among alphaviruses, Semliki Forest virus (SFV), Sindbis virus (SIN), and Venezuelan Equine Encephalitis virus (VEE) have been characterized as the most representative members of this genus. Although SFV and SIN are able to infect and produce disease in humans, VEE and other closely related alphaviruses can cause lethal encephalitis and are considered a significant public health threat in many areas of South, Central and North America. However, massive outbreaks of the SFV-related alphavirus chikungunya (CHIKV) in Indian Ocean islands and Asia, and more recently in the Americas, has produced a high morbidity, drawing worldwide attention to this type of viruses [1, 2].
Alphaviruses contain a single-stranded positive-sense RNA genome of 11–12 kb that is packaged into a nucleocapsid (NC) of icosahedral symmetry (T = 4) formed by 240 subunits of the capsid protein [3]. The NC is enveloped by a lipid bilayer containing 240 heterotrimers of glycoproteins E1 and E2, which mediate virus entry into target cells [4]. Viral entry takes place through endocytosis followed by low pH-mediated fusion of viral envelope and endosome membranes, disassembly of NC, and release of viral RNA into the cytoplasm (for a review see [5]). Upon cell entry, viral RNA is translated to produce the replicase (Rep), formed by four nonstructural protein subunits (nsP1–4), which is able to synthesize a complementary minus-sense genomic RNA (−RNA) during the early phase of infection. Then, Rep uses this −RNA as template to amplify viral genomes and to synthesize a subgenomic RNA (sgRNA), which is translated to produce a large structural polyprotein (capsid–p62–6K–E1). The capsid protein contains a serine protease activity, which is responsible for its auto-proteolytical cleavage from the nascent chain. The rest of the polyprotein is processed in the ER into three envelope proteins (p62, 6K, and E1), which are then translocated to the plasma membrane. p62 and E1 associate to form heterotrimers and at a late stage of the transport process p62 is cleaved by the host enzyme furin, thus generating the E2 form and a small glycoprotein called E3. E2 and E1 constitute the mature viral spikes [5]. E3 is lost in SIN, but remains on the virion surface in SFV. In both SFV and SIN, the initial 100–200 nucleotides of the capsid gene function as a translation enhancer, leading to very high levels of viral structural proteins in infected cells [6, 7]. The generation of new viral particles (VPs) involves two processes that have been studied in great detail: (1) packaging of viral RNAs into NCs and (2) budding of NCs at the plasma membrane. The capsid protein can assemble into NCs through specific interactions between capsid monomers and between these monomers and genomic viral RNA [8–11]. Neither the −RNA nor the sgRNA are usually packaged into VPs, because they lack a specific packaging sequence located within the Rep gene.
The budding process is driven by specific interactions between preformed NCs and spike glycoproteins E1 and E2, leading to the release of virus into the extracellular medium [12–15]. The expression of both capsid and envelope proteins in infected cells has been so far considered a requirement for alphavirus budding and propagation [16, 17]. Expression vectors based on several alphaviruses have been developed by replacing the region coding for the viral structural proteins with a heterologous gene of interest [18–20]. Vector RNAs can be packaged into VPs by co-transfecting them into cells together with helper RNAs coding for the structural proteins [19, 21, 22].
In the present study, we show that SFV and SIN lacking the capsid gene can propagate in mammalian and mosquito cells. This propagation is mediated by the envelope proteins and seems to occur through the production of infectious microvesicles (iMVs). These iMVs, which display envelope proteins on their surface and contain viral RNA, are produced at the plasma membrane and are able to infect cells in a similar way as wild-type (wt) alphavirus.
Materials and methods
Cells
BHK-21 cells (ATCC: CCL-10) were cultured in BHK Glasgow MEM (Gibco BRL, UK) supplemented with 5 % FCS, 10 % tryptose phosphate broth, 2 mM glutamine, and 20 mM HEPES (BHK complete medium). HuH-7 (Japanese Collection of Research Bioresources Cell Bank: 0403), Hep3B (ATCC: HB-8064), and Vero (ATCC: CCL-81) cells were grown in DMEM (Gibco BRL) supplemented with 10 % FCS and 2 mM glutamine. Aedes albopictus cell line C6/36 (ATCC: CRL-1660) was grown in Eagle’s minimum essential medium, containing 10 % FCS. All media contained 100 μg/ml streptomycin and 100 IU/ml penicillin.
Plasmids
Plasmids pSFV-1, pSFV-LacZ, pSFV-helperS2, and pSFV-helper-C-S219A have already been described [22, 23]. To construct pSFV-spike and pSFV-enh-spike, a fragment of 3.9 or 4.2 kb containing the sequence including “subgenomic promoter + p62–6K–E1 + 3′UTR” was obtained from pSFV-helper-E [22] or pSFV-helper-S2, respectively, by digestion with Xba I + Spe I and inserted into pSFV-1 digested with the same enzymes. pSFV-spike has the same structure as the one previously described [17]. In pSFV-enh-spike, the ORF p62–6K–E1 is fused to the sequence coding for the first 34 amino acids of the capsid (translation enhancer b1) using the foot and mouth disease virus 2A autoprotease as a linker [22, 24]. To construct pSFV-enh-spikeΔ6K, plasmid pSFV-enh-spike was digested with NdeI and the 2.1 kb fragment spanning from position 708 in p62 to position 1171 in E1 genes was substituted by a synthetic fragment (Genescript, Piscataway, NJ) of 1.9 kb covering the same area, but in which the 6K sequence was completely deleted, as previously described [23]. To construct pSFV-enh-spike-GFP, we first generated an intermediate plasmid containing a multiple cloning site (mcs) between the end of the E1 gene and the 3′UTR (pSFV-enh-spike-mcs) (information available upon request). A PCR fragment spanning the sequence from the subgenomic promoter to the end of 3′UTR was obtained from pSFV-GFP (kindly provided by Prof. P. Liljeström, Karolinska Institute, Sweden) and cloned into pSFV-enh-spike-mcs digested with Avr II (present in the mcs) and Spe I. To construct pSIN-spike, a PCR fragment containing SIN p62–6K–E1 sequence was obtained using primers 5′-TCTAGA CCACC ATGTCCGCAGCACCACTGGTCAC-3′ (forward) and 5′-GGGCCCTCATCTTCGTGTGCTAGTC-3′ (reverse), using plasmid DHBB(5′SIN) as template [21]. In the forward primer, an ATG (underlined) and a ribosome-binding site (bold) were included, since they are not present in the p62 sequence. The 2.9 kb PCR fragment was digested with Xba I and Apa I (in italics in the forward and reverse primers, respectively) and cloned into the same sites in pSINrep5 [21]. A pSinRep5 vector expressing GFP was used for in vitro transcription of SIN-GFP RNA (kindly provided by T. Furuta, Kyoto University, Japan).
Transfection of cells and production of viral particles (VPs)
RNA synthesis and transfection into BHK, Hep3B, and Vero cells by electroporation was performed as described previously [18]. Electroporation of HuH-7 cells was performed as described by Robinson et al. [25]. C6/36 cells were transfected using Cellfectin® Reagent (Life Technologies). Co-culture studies were performed by mixing electroporated and non-electroporated cells at 1:20 ratio and plating 106 cells/9.5 cm2 dish, resulting in confluent monolayers after cell attachment.
Packaging of recombinant SFV-LacZ or SFV-GFP RNAs into SFV VPs was performed as described [22]. Briefly, BHK cells were co-electroporated with each recombinant RNA and both SFV-helper-C-S219A and SFV-helper-S2 RNAs, providing in trans the capsid and envelope proteins, respectively. Packaging of SFV-spike, SFV-enh-spike, and SFV-enh-spike-GFP into VPs was performed by co-electroporating BHK cells with each of these RNAs and helper-C-S219A RNA (Supplementary Fig. 1). SFV VPs were harvested and purified by ultracentrifugation through a 20 % sucrose cushion as described previously [26]. For UV-inactivation, the total amount of SFV-LacZ VPs to be used was exposed to a 254 nm UV light (two cycles of 15 min) by using a uvgl-25 compact UV 4 W lamp (UVP, Upland, CA). After inactivation, the viral titre had decreased by a factor of >107.
Purification of infectious microvesicles (iMVs) and control exosomes
Approximately, 107 HuH-7 cells were electroporated with 50 μg of SFV-enh-spike in vitro-transcribed RNA by applying a pulse of 270 V and 950 μF using a Gene Pulser II electroporator (BioRad, Hercules, CA). These conditions resulted in approximately 50 % transfection efficiency. This process was repeated ten times to electroporate a total of 108 cells. Cells were incubated during 48 h at 33 °C, and the medium was clarified by centrifugation at 800g during 15 min to eliminate cell debris. This procedure yielded a total amount of 1.5 × 107 ± 1.6 × 106 IU (mean ± SD of eight independent iMV stocks). To purify iMVs, the supernatant was filtered using a 0.22 μm filter, centrifuged at 10,000g during 2 h in a SW28 rotor (Beckman), and the pellet was resuspended in a total volume of 200 μl of TN buffer (50 mM Tris–HCl, pH 7.4, 100 mM NaCl) for further use. The iMV recovery yield after the centrifugation step was of 50.1 ± 22.9 % (n = 8). To determine the density of purified iMVs or VPs, corresponding samples were centrifuged through a 5–60 % sucrose gradient at 100,000g during 90 min. After centrifugation, fractions of 1 ml were collected and analysed for infectivity and density.
For purification of control exosomes, we used standard protocols previously described [27]. Briefly, untransfected HuH-7 cells were grown in medium containing 2 % FBS during 48 h. Then, the medium was collected and clarified by centrifugation at 1500g for 15 min, yielding supernatants that were further clarified by centrifugation at 10,000g for 1 h. Exosomes in the clarified supernatants were pelleted at 100,000g through a 30 % sucrose cushion for 16 h.
Infection of cells
Cells were infected with SFV VPs or iMVs as described earlier [18]. For titrations, cells infected with serial dilutions of VPs or iMVs were fixed with cold methanol at 24 or 6 h post-infection (h p.i.), respectively. Indirect immunofluorescence was performed to determine the titres as described [28]. A rabbit antiserum specific for the nsP2 subunit of SFV Rep [29] or a murine polyclonal antiserum specific for the SFV envelope proteins was used as primary antibodies. The latter was obtained by immunizing C57BL/6 mice intravenously with four doses of 107 VPs of SFV-enh-spike given every 2 weeks. For neutralization studies, VP or iMV samples were incubated with 5 μg/ml RNAse during 20 min at 25 °C, 0.5 mg/ml proteinase K during 45 min at 37 °C, or with 1 % Triton X-100 during 1 h at 25 °C, respectively. For neutralization of endosomal pH, cells were either incubated with 20 mM NH4Cl during adsorption (1 h) and/or a 4 h period after adsorption. To determine the number of PFUs present in VP stocks, confluent monolayers of BHK cells were infected with different dilutions of the virus, the medium was removed after 1 h of adsorption, and overlay medium was added (BHK complete medium–10 % FCS and Glasgow MEM–0.2 % agarose, [1:1]). Cells were incubated for 72 h at 37 °C, fixed with 0.5 % glutaraldehyde in PBS, and stained with 0.1 % crystal violet in methanol–H2O (20:80).
Antibody labelling and electron microscopy
Immunogold labelling of SFV-LacZ VPs and iMVs was performed using the protocol described by Gowen and co-workers [30], but omitting the use of detergent. Murine polyclonal antiserum specific for SFV envelope proteins was used as primary antibody, which was then detected by exposure to anti-mouse immunoglobulin G (IgG) conjugated to 10 nm colloidal gold (Sigma). The labelled samples were then stained using Nanovan (Nanoprobes, Yaphank, NY) prior to visualization at 30,000× magnification by conventional electron microscopy using a JEOL JEM-1230 (Tokyo, Japan) microscope operating at 80 kV. Images were recorded on a digital camera SC1000 ORIUS (Gatan, Germany). Estimation of particle size was carried out using ImageJ [31].
RT-PCR
Samples containing purified VPs or iMVs were treated with 5 μg/ml RNAse, or mock incubated, during 40 min at 25 °C, and RNA was extracted first with phenol and then with chloroform in the presence of 10 μg of Poly-A Carrier RNA (Qiagen). RNA was precipitated with ethanol and 0.1 M sodium acetate, washed with 70 % ethanol, and resuspended in 20 μl of H20. Two μg of RNA obtained in this way was mixed with 5 ng of in vitro-transcribed SFV-ΔRep-GFP RNA. This latter RNA does not contain the replicase sequence and was used as an internal control for the RT-PCR. After DNAse treatment, RNAs were transcribed into cDNA with GoScript™ Reverse Transcriptase (Promega) using a mix of random primers. cDNAs were quantified by real-time PCR with the GoTaq® 2-Step RT-qPCR System kit (Promega) using oligonucleotides 5′-CTGTTCTCGACGCGTCGTC-3′ (forward) and 5′-GAGGTGTTTCCACGACCC-3′ (reverse) to amplify a 202 nt fragment of nsP2 subunit of SFV Rep. GAPDH was amplified using primers 5′-CCAAGGTCATCCATGACAAC-3′ (forward) and 5′-TGTCATACCAGGAAATGAGC-3′ (reverse), and human β-actin with primers 5′-AGAGCTACGAGCTGCCTGAC-3′ (forward) and 5′-AGCACTGTGTTGGCGTACAG-3′ (reverse). GFP, used as internal control, was amplified with primers: 5′-ACGGCAAGCTGACCCTGAA-3′ (forward) and 5′-GGGTGCTCAGGTAGTGGTT-3′ (reverse). For analysis of RNA present in cells, monolayers were collected at the indicated times after electroporation and RNA was extracted with the MaxWell 16 research system (Promega). 1.25 μg of RNA from each sample was analysed by quantitative RT-PCR as described above for iMVs. To determine the relative levels of SFV plus- and minus-RNA strands, RNAs extracted from iMVs, VPs, or cells, were transcribed into cDNA using primers 5′-GACGCGTCGTCAGCCAGGG-3′ and 5′-CCACGACCCCTGCACCTGC-3′, which hybridize to the SFV nsP2 region and are complementary to the minus- and plus-RNA strands, respectively. cDNAs were quantified by real-time PCR using these last two oligonuleotides as described above. Data were analysed using CFX Manager software (Bio-Rad).
Analysis of protein expression by Western blot
Whole cell lysates from SFV-transfected BHK or HuH-7 cells were obtained by incubation in a buffer containing 1 % IGEPAL (Sigma, St. Louis, MO), 50 mM Tris HCl pH 7.6, 150 mM NaCl, 2 mM EDTA, and 1 μg/ml PMSF (Sigma), cleared by centrifugation for 6 min at 6000 rpm in a refrigerated microcentrifuge, and quantified by Bradford assay. iMVs were obtained from the supernatant of electroporated HuH-7 cells, which was clarified by low speed centrifugation, filtered, and purified as described above. Purified VPs, iMVs, or cell lysates were analysed by SDS-PAGE in 10 % polyacrylamide gels, transferred to a nitrocellulose membrane, and incubated with the following primary antibodies: murine polyclonal antiserum against SFV envelope proteins (in this case, samples were run under non-reducing conditions to allow a good separation of E1 and E2 bands), murine monoclonal antibody against SFV capsid protein (clone C42 kindly provided by Dr. Irene Greiser-Wilke, Institute of Virology, Hannover, Germany) [32], rabbit polyclonal antisera specific for SFV nsP1 and nsP4 (kindly provided by Dr. Tero Ahola, University of Helsinki, Finland) or for nsP2 and nsP3 Rep subunits (generated in our laboratory), rabbit polyclonal antiserum specific for β-actin (Sigma), and murine monoclonal antibodies specific for human CD63 (Developmental Studies Hybridoma Bank, IA) and TSG101 (Abcam, Cambridge, UK). Peroxidase-conjugated goat anti-mouse (Thermo Fisher Scientific, Waltham, MA) and sheep anti-rabbit (Cell Signalling, Danvers, MA) antisera were used as secondary antibodies. Proteins were visualized using the Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer Life Sciences, Waltham, MA).
Animal manipulations
Five weeks old C57BL/6 female mice were purchased from Harlan (Barcelona, Spain) and maintained under standard pathogen-free conditions according to the ethical institution’s guidelines. Vectors were injected intravenously at the indicated doses diluted in 100–300 μl of saline (Fig. 7). At the indicated times, blood samples were obtained by orbital sinus bleeding, cells were removed by low-speed centrifugation, and the supernatant was frozen at −80 °C for subsequent virus assay. At 48 h, a brain fraction was homogenized and suspended in EMEM with 2 % FBS to obtain a 10 % suspension (wt/vol), which was then centrifuged at 733×g for 10 min. The supernatant was decanted, and the suspension was frozen at −80 °C for subsequent virus assay. The number of infectious units present in the serum and brain homogenates was determined as described above. For RT-PCR analysis, mice were killed 48 h after treatment and 20 mg of the indicated tissues was homogenized in 200 μl of chilled 1-thioglycerol/homogenization solution using Fisherbrand™ Disposable Pestle System (Fisher Scientific). Samples were then processed on a MaxWell 16 research system (Promega) and analysed by quantitative RT-PCR as described above.
Fig. 7.

Analysis of iMV pathogenicity and infectivity in mice. a, b For pathogenicity studies, C57BL/6 mice were intravenously injected with 106 IU of iMVs, 105 PFU of wtSFV or saline (n = 5). Animal weight was determined at the indicated times. a The weight of wt-SFV-inoculated mice is only shown until day 4, since only two mice in this group survived after this time point. b Kaplan–Meier plots of mouse survival. c–e For infectivity studies, C57BL/6 mice were intravenously injected with 107 IU of iMVs (n = 3), 107 PFU of wtSFV (wt, n = 6), 107 VPs of SFV-LacZ (Z, n = 9), the same dose of UV-inactivated SFV-LacZ (UV-Z, n = 4), or saline (sal., n = 6). Sera collected at the indicated times (c) and brain homogenates prepared at 48 h p.i. (d) were titrated in BHK monolayers. The titre expressed as IU/ml serum or IU/g tissue was determined by either X-gal staining (Z and UV-Z), IF against SFV envelope proteins (iMV) or plaque forming assays (wt). No infectious material was detected at any time in mice that received SFV-LacZ, UV-SFV-LacZ, and saline (not shown), or at >6 h in mice that received iMVs (not detected, n.d. detection limit: ≥40 IU/ml in serum; ≥100 IU/g in brain). e The indicated organs were homogenized at 48 h p.i. and analysed by quantitative RT-PCR using oligonucleotides specific for SFV replicase or mouse β-actin as described in “Materials and methods”. Data are shown as 2Δ(Ct β-actin-Ct Rep) × 104. In all graphs, values correspond to the mean + SD. *p < 0.05; **p < 0.01; ***p < 0.001 (asterisks correspond to comparisons with the saline group)
Statistical analysis
All error terms are expressed as the standard deviation (SD). Prism software (GraphPad Software Inc., San Diego, CA) was employed for statistical analysis. Mann–Whitney U test was used to compare two experimental groups. Paired t test was used in those experiments where the same samples were analysed for different parameters or under different treatments (Figs. 4c, 5b–d). Two-way ANOVA followed by Tukey’s post-tests was used for comparison among groups in Fig. 8b. p values <0.05 were considered to be statistically significant.
Fig. 4.

Cells transfected with SFV RNA devoid of capsid release infectious material. a The indicated cell lines were electroporated with SFV-enh-spike RNA as described in “Materials and methods”, supernatants were collected at 24 h and titrated by infecting BHK cell monolayers that were analysed by IF with α-spike serum. b Propagation of infectious material released by SFV-enh-spike-transfected HuH-7 cells was assessed by infecting BHK cells monolayers at MOI 0.05 and analysing spike expression at the indicated times by IF (magnification: ×100; scale bars 50 μm). c Effect of different agents on the infectivity of infectious microvesicles (iMVs) or wtSFV used as control (MOI 0.1 in both cases). For NH4Cl treatment, cells were incubated with this agent at a concentration of 20 mM during the 60 min. virus adsorption period (ad), during adsorption and a 4-h post-infection period (ad + inf), or only during the 4 h post-infection period (inf). The percentage of infectivity in each case was calculated considering the infectivity of non-treated samples as 100 % (non-treat). Prot K, proteinase K; Trit. X100, Triton X-100. In a and c data are the mean + SD of three experiments. ***p < 0.001
Fig. 5.

Characterization of iMVs. a Microvesicles purified from HuH-7 cells electroporated with the indicated RNAs were analysed by Western blot with a mixture of α-spike and anti-capsid sera. The gel was loaded with 105 IU of SFV-enh-spike iMVs (e-spike) or an equivalent volume of material purified from SFV-LacZ (LacZ) or mock-electroporated cells. A sample containing 5 × 107 purified SFV-LacZ VPs was used as control (VP). b Samples containing 106 IU of purified SFV-enh-spike iMVs (e-spike), or equivalent volume of material purified from SFV-LacZ (Z) or mock-electroporated cells (mock), were analysed by quantitative RT-PCR, with or without previous RNAse treatment, using oligonucleotides specific for SFV Rep or GAPDH as described in “Materials and methods”. A sample containing 106 purified SFV-LacZ VPs was used as positive control (VP). When indicated, iMV samples were treated with 0.1 % Triton X-100 for 15 min at RT previous to RNAse treatment (Triton). Data is shown as 2−Δ(Ct mock−Ct iMV or VP) for each gene. c HuH-7 cells electroporated with SFV-enh-spike (e-spike), SFV-LacZ (LacZ) RNAs or mock-electroporated were analysed at 24 h post-electroporation by quantitative RT-PCR as described in b. d The relative amounts of positive- and negative-strand SFV RNA (RNA+ and RNA−, black and grey columns, respectively) present in iMVs (5 × 105 IU) and cells electroporated with SFV-enh-spike RNA (at 24 h postelectroporation) was analysed by RT using oligonucleotides specific for each SFV RNA strand, followed by quantitative PCR as described in “Materials and methods”. 107 VPs of SFV-LacZ (VP) and material purified from mock-electroporated cells (mock) were used as positive and negative controls, respectively. In b–d data represent the mean + SD of at least three independent experiments analysed in triplicates. **p < 0.01, *p < 0.05. e The density of purified iMVs or VPs was determined by equilibrium centrifugation on a 5–60 % sucrose gradient from which 11 fractions were collected. The infectivity of each fraction was determined as described in “Materials and methods” and is represented as the relative titre (left Y-axis scale). The density of each fraction was calculated by measuring its refractive index (dashed line, right Y-axis scale). The iMV peak corresponds to 5 × 107 IU/ml and the VP peak to 8 × 107 VPs/ml. ns not significant, nd not detected
Fig. 8.

Propagation of SFV-enh-spike-GFP. BHK cell monolayers were infected with the indicated vectors at MOI 0.5 (a) or MOI 0.05 (b). a Cell monolayers were fixed at the indicated times and GFP expression was visualized with the appropriate filter. Spike expression was analysed in the same preparations by indirect immunofluorescence with an antiserum specific for SFV envelope proteins. Magnification of images: ×200; scale bars 50 μm. b Cells were trypsinized, fixed with 2 % paraformaldehyde and analysed by flow cytometry (FACS-Canto II, BD-Biosciences) to determine the percentage of GFP-positive cells. Data analysis was carried out using FlowJo software (Tree Star Inc., Ashland, OR). Results correspond to one of two representative experiments, each performed in triplicate that gave very similar results. Error bars represent the mean + SD. *p < 0.05; ***p < 0.001; ns not significant
Ethics statement
Mice were maintained under standard pathogen-free conditions according to our ethical institution’s guidelines. Animals were anesthetized with an intraperitoneal injection of a ketamine/xylazine mixture and killed by cervical dislocation. The protocol to perform the animal experiments described in this work was reviewed and approved by our local Institutional Animal Care and Use Committee (IACUC): Comité de Ética para la Experimentacion Animal de la Universidad de Navarra (protocol identification number: 105-14). Our local IAUCC adheres to the Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes.
Results
SFV and SIN devoid of capsid gene can propagate in cell culture
An SFV vector containing the sequence of SFV envelope proteins (p62–6K–E1) fused to the minimal SFV capsid translation enhancer (SFV-enh-spike) was initially constructed as a tool to generate a polyclonal antiserum against SFV envelope proteins (Supplementary Fig. 1). This vector was packaged into VPs by co-transfecting BHK-21 (BHK) cells with SFV-enh-spike and SFV-helper-C-S219A RNAs, the latter lacking the SFV packaging signal and providing the capsid gene in trans [22] (Supplementary Fig. 1). In this last RNA, the S219A mutation in the capsid abolishes its proteolytic activity, preventing the processing of the structural polyprotein if a full-length genome is generated by recombination [22]. Unexpectedly, SFV-enh-spike VPs were able to mediate the spreading of viral protein expression in monolayers of different mammalian cell lines including BHK, Hep3B, HuH-7, and the mosquito cell line C6/36 (Fig. 1). This effect was not due to the generation of replication-proficient virus (RPV), since no RPVs were detected by a plaque-forming unit (PFU) assay. In addition, propagation of this vector was not as efficient as that of wtSFV (Fig. 1b). However, to rule out that recombinant genomes containing all or part of the capsid gene could have been generated during the packaging of SFV-enh-spike, BHK cells were electroporated only with SFV-enh-spike RNA, and co-cultured with non-electroporated cells at a 1:20 ratio (Fig. 2a). As shown in Fig. 2b, SFV-enh-spike was able to propagate from transfected cells to neighbouring cells, reaching most cells in the monolayer at 36 h. p.i. This effect was not observed in a co-culture of cells electroporated with SFV-LacZ RNA (Supplementary Fig. 1), which gives a pattern of individual cells at all analysed time points (Fig. 2b). As observed with infected cells, the propagation of wtSFV from transfected cells was also more efficient than that of SFV-enh-spike (Fig. 2c).
Fig. 1.

Propagation of SFV devoid of capsid in different cell lines. a SFV RNA genomes devoid of capsid (SFV-enh-spike or SFV-spike, abbreviated in the diagram as SFV-ΔC) were packaged into VPs by co-electroporation of BHK cells with a helper RNA able to provide the SFV capsid in trans (SFV-helper-C-S219A, abbreviated as SFV-helper-C). SFV-LacZ VPs were generated by co-electroporation of BHK cells with SFV-LacZ RNA and two helper RNAs providing the SFV capsid and envelope proteins in trans, respectively [22] (not shown in the diagram). VPs were collected at 24 h post-electroporation and used to infect monolayers of BHK cells (b) or the indicated cell lines (c) at MOI 0.05. Cells were fixed at the indicated times and analysed by immunofluorescence (IF) with a polyclonal antiserum specific for the nsP2 subunit of SFV Rep (α-nsp2) (b), or with a polyclonal antiserum specific for SFV envelope proteins (α-spikes) (c). Images correspond to a representative experiment from at least two independent experiments performed in triplicate with similar results. Magnification of images: ×200 (except for images of C6/36 cells that were taken at ×400). Scale bar in all images: 50 μm
Fig. 2.

Propagation of alphavirus genomes without capsid upon cell transfection. a BHK cells were electroporated with the indicated alphavirus vector RNAs and co-cultured with non-electroporated cells at a ratio of 1:20. Cells were fixed at the indicated times and analysed by IF with α-nsp2 serum (b), α-spikes serum (c and upper images in e), or a monoclonal antibody specific for SIN E2 protein (lower images in d). GFP expression was visualized with the appropriate filter (upper images in d). Lower images in e show DAPI staining of nuclei. All panels correspond to a representative experiment from at least two independent experiments performed in triplicate with similar results. Magnification of images is ×200 (b–d) or ×100 (e). Scale bar in all images: 50 μm
SFV-enh-spike expresses the first 34 amino acids of the capsid, which according to many studies are insufficient to form functional NCs [8]. However, to discard any possible implication of capsid fragments in propagation, we constructed an SFV vector that expresses the envelope proteins without any capsid sequence (SFV-spike, Supplementary Fig. 1). The absence of capsid expression from this vector, as well as from SFV-enh-spike, was confirmed by Western blot analysis of cells electroporated with their respective RNAs (Supplementary Fig. 2a). SFV-spike was able to propagate after electroporation of vector RNA into BHK cells co-cultured with non-electroporated cells, demonstrating that propagation of SFV can take place in the complete absence of capsid sequences (Fig. 2b). This process was also observed in cells infected with SFV-spike VPs packaged as described for SFV-enh-spike (Fig. 1b). Propagation appeared to be more efficient when using the SFV-enh-spike RNA, indicating that overexpression of envelope proteins favoured this process (Figs. 1b, 2b; see also Supplementary Fig. 2 for a comparison of protein expression between vectors). To assess whether this phenomenon takes place in other alphaviruses, a SIN vector expressing its envelope proteins and with the whole capsid sequence deleted was also constructed (SIN-spike, Supplementary Fig. 1). SIN-spike was also able to propagate very efficiently in co-cultures of vector RNA-electroporated and non-electroporated BHK cells (Fig. 2d). In contrast, a SIN vector in which the structural genes were replaced by GFP, used as control (SIN-GFP, Supplementary Fig. 1), was unable to propagate.
The small 6K envelope protein was also dispensable for propagation, since an SFV-enh-spike vector RNA in which the 6K sequence had been deleted (SFV-enh-spikeΔ6K, Supplementary Fig. 1) was also able to propagate in co-cultures of vector RNA-electroporated and non-electroporated BHK cells (Fig. 2e). This vector was able to express levels of spike proteins that were similar to those of SFV-spike (Supplementary Fig. 2b), but did not express 6K (data not shown). These results show that propagation of alphavirus can take place even if only Rep and spike glycoproteins p62 and E1 genes are present in the viral genome.
Propagation is mediated by the envelope proteins and does not require cell fusion
The fact that SFV and SIN vectors expressing Rep and a reporter gene (SFV-LacZ and SIN-GFP) were unable to propagate indicates that propagation of alphavirus genomes devoid of capsid should be mediated by the expression of the envelope proteins. The role of envelope proteins was supported by the fact that transmission of the SFV-enh-spike vector was inhibited by incubating infected cells with a murine polyclonal antiserum against the SFV envelope proteins, a phenomenon that was also observed in cells infected with wtSFV, used as positive control (Fig. 3a). In contrast, in both cases, propagation was not inhibited when cells were incubated with preimmune serum, used as negative control. Since the spike proteins are able to mediate the fusion of membranes, one possible mechanism for the propagation of SFV without capsid could be the fusion of cells expressing the envelope proteins with neighbouring cells. However, the fusogenic activity of SFV-spike proteins requires a pH < 6 [33], which was not detected in the extracellular medium of infected or transfected cultures (data not shown). Additionally, syncytia were not observed in cell monolayers infected with SFV-enh-spike in which the vector had propagated (Fig. 3b). This result was confirmed by labelling cellular plasma membranes with wheat germ agglutinin conjugated with a fluorescent dye (Supplementary Fig. 3). Nevertheless, when these monolayers were incubated at pH 5, during a few minutes syncytia were induced very efficiently, suggesting that fusion does not take place under normal cell culture conditions (Fig. 3b, Supplementary Fig. 3). A similar outcome was observed in cells infected with wtSFV, although in this case viral propagation to most cells led to a general fusion of the monolayer at pH 5. As expected, syncytia were not observed in cell monolayers infected with SFV-LacZ when incubated at pH 5.
Fig. 3.

Propagation of SFV without capsid is mediated by the envelope proteins and does not require cell fusion. a BHK cell monolayers were infected with SFV-enh-spike VPs at MOI 0.1, or with wtSFV at MOI 0.025, and incubated for 24 h (SFV-enh-spike) or 10 h (wtSFV) in the presence of α-spikes serum, or with preimmune serum (in both cases diluted 1:50). b BHK cells were infected with SFV-enh-spike and SFV-LacZ VPs at MOI 0.05, or with wtSFV at MOI 0.025, and 16 h later were incubated during 3 min with PBS at the indicated pH. Cells were washed and incubated for 3 h with BHK complete medium. In both a and b, cells were fixed and analysed by IF with α-spikes (a) or α-nsp2 sera (left images for each pH in b). Nuclei in (b) were visualized with DAPI (right images for each pH). Magnification: ×100 (a) or ×200 (b); scale bars 50 μm. Panels correspond to a representative experiment from at least two independent experiments performed in triplicate with similar results. See also Supplementary Fig. 3 for higher magnification images in which cell membranes have also been labelled
Cells transfected with SFV genomic RNA devoid of the capsid sequence release infectious material to the supernatant
Propagation of SFV devoid of capsid could result from the release of infectious material from infected or transfected cells to the medium. We analysed the presence of infectious material in the supernatant of mammalian or mosquito cell lines transfected with an SFV RNA devoid of the capsid. As shown in Fig. 4a, all cell lines electroporated with SFV-enh-spike RNA were able to produce infectious material at a level of 103–105 infection units (IU)/ml/24 h, with the highest titres being produced by HuH-7 cells. Interestingly, the infectious material produced by transfected cells was also able to propagate after infection of new cell monolayers, as shown by a dramatic increase in the number of cells expressing the spike proteins between 6 and 40 h p.i. (Fig. 4b). No infectious material was detected in cells electroporated with SFV-LacZ. Furthermore, the infection material was able to transfer SFV RNA into infected cells, with cellular viral RNA levels increasing approximately 15-fold between 6 and 24 h p.i. (Supplementary Fig. 4), while viral RNA levels in cells infected with a non-propagative SFV vector expressing GFP (SFV-GFP, Supplementary Fig. 1) did not show a significant increase. As observed earlier, propagation was more efficient in cells infected with wtSFV, in which cellular viral RNA levels increased >3000 fold during a similar time interval. These data conclusively demonstrate that SFV devoid of capsid can propagate in a way similar to an infectious virus.
Purification of infectious material and neutralization of infectivity
Next, we characterized in detail the infectious material produced by SFV without capsid. For that purpose, we first generated a suitable amount of infectious material to be analysed. Approximately, 108 HuH-7 cells were electroporated with SFV-enh-spike, SFV-LacZ RNAs or mock electroporated as described in “Materials and methods”. Briefly, supernatants were collected at 48 h p.i. and, after clarification at low speed, centrifuged at 10,000g for 2 h. These conditions allowed to efficiently pellet the infectious material. After centrifugation, an average titre of >1.9 × 107 IU/ml was obtained from cells transfected with SFV-enh-spike with an average recovery yield of >50 %. No IUs were detected in the case of supernatants purified from SFV-LacZ RNA- and mock-electroporated cells.
To characterize the role of the different components present in infectious material produced by SFV-enh-spike-transfected cells, purified IUs were treated with ribonuclease A (RNAse), proteinase K, or Triton X-100 before infection. While RNAse treatment had no effect on their infectivity, both proteinase K and Triton X-100 were able to almost completely block infectivity (Fig. 4c). These data suggest that the infectious material contains viral RNA within an envelope that has the spike glycoproteins on its surface. For this reason from now on, we will refer to this material as infectious microvesicles (iMVs). Furthermore, the infectivity of iMVs was strongly inhibited in the presence of a lysosomotropic agent (20 mM NH4Cl), which is able to accumulate in lysosomes (Fig. 4c). If this agent was present only during a 60 min viral adsorption period, iMV infection was inhibited by 88.2 %, which was comparable to wtSFV infectivity inhibition (96.8 %). This inhibition was even higher if NH4Cl was present during the adsorption and a 4-h post-infection period, reaching >99 % for both iMVs and wtSFV. However, if NH4Cl was added after the adsorption period, the infectivity was only modestly inhibited (23.7 and 26.2 % inhibition for iMVs and wtSFV, respectively). These data suggest that iMVs use a mechanism of entry that requires acidification of endosomes in a similar way to wtSFV [34].
Analysis of protein and RNA content of iMVs
The presence of SFV envelope proteins in iMVs was confirmed by Western blot (Fig. 5a). Both E1 and E2 were detected in iMVs purified from cells electroporated with SFV-enh-spike RNA, while these proteins were not present in material purified from SFV-LacZ- or mock-electroporated cells. As expected, the capsid protein was only detected in SFV VPs used as control. Interestingly, iMVs produced a much stronger signal for E1 and E2 than SFV VPs (notice that in Fig. 5a, the gel was loaded with 5 × 107 VPs of SFV-LacZ and 105 IU of iMVs to obtain a comparable signal). This suggests that the particle-to-infectivity ratio of iMVs is higher than that of SFV VPs, although differences in size and envelope proteins density between VPs and iMVs could also result in different amount of spike proteins per infection unit.
To determine the origin of iMVs, we first analysed whether they contained markers from different subcellular compartments. This analysis showed that SFV-enh-spike iMVs only contained trace amounts of lysosomal (Lamp2, Supplementary Fig. 5a) and exosome markers (TSG101 and CD63 [35], Supplementary Fig. 5b), indicating that most likely these vesicles are not from lysosomal or exosomal origin. In contrast, enrichment in iMVs of Na+K+ATPase, a ubiquitously expressed oligomeric plasma membrane complex, indicates that they are likely generated at the cytoplasmic membrane (Supplementary Fig. 5a). To confirm this possibility, we transfected HuH-7 cells with SFV-enh-spike RNA, labelled them with [35S] methionine-cysteine, and performed cell surface biotinylation as described [36]. We then allowed the release of iMVs from cells during 18 h and incubated the supernatant with streptavidin-coated magnetic beads. As shown in Supplementary Fig. 6, we were able to visualize SFV-spike proteins only in streptavidin-retrieved supernatants derived from cells in which we had performed cell surface biotinylation, strongly suggesting that iMVs are generated at the plasma membrane. To obtain additional evidence of iMVs budding at the plasma membrane, we performed thin-section transmission electron microscopy (TEM) of cells transduced with SFV-spike. These cells showed some 100- to 150-nm spherical structures that sometimes appeared to be attached to the cell surface by a small “neck” and that could represent iMVs budding from the plasma membrane (Supplementary Fig. 7). wtSFV-infected cells used as control showed numerous VPs of 60–70 nm on the cell surface.
We also assessed whether iMVs could be derived from virus replication complexes (RCs), which originate at the plasma membrane, being later transported to the surface of large acidic vacuoles by different components of the cytoskeleton [37, 38]. RCs function in the synthesis of double-stranded RNA replicative intermediates and contain all viral nsPs [39, 40]. NsP1, nsP3, and nsP4 subunits of SFV Rep were not detectable in iMVs by Western blot analysis (Supplementary Fig. 5c). The observation that other cell proteins, like β-actin, were detected at low levels in iMVs (and trace amounts of nsP2; see Supplementary Fig. 5a–c, e-spike lane) could indicate that some proteins are unspecifically incorporated into iMVs. The absence or the lack of enrichment of nsPs in iMVs suggests that they are not derived from RCs.
To quantify the viral RNA packaged inside iMVs, we isolated RNA from purified iMVs and analysed it by qRT-PCR using oligonucleotides specific for the SFV Rep sequence. As shown in Fig. 5b, purified iMVs contained SFV RNA that was relatively protected from RNase digestion. Pretreatment of iMV samples with Triton X-100 resulted in almost complete elimination of SFV RNA upon RNAse treatment, confirming that the viral RNA is included in a lipid vesicle (Fig. 5b). The relative amount of protected SFV RNA in iMVs was about 60-fold higher than that observed in SFV VPs used as control, probably due to a higher particle-to-infectivity ratio in iMVs (for this analysis, we used the same amount of infectious units for both types of materials). To our surprise, the protected SFV RNA was also detected in the material purified from cells electroporated with SFV-LacZ, although at lower levels. This suggests that microvesicles containing viral RNA could be produced in the absence of spikes, although in this case they would not be infectious, as spike glycoproteins are required for propagation. iMVs mostly contained plus-strand SFV RNA, as determined by strand-specific RT-qPCR (Fig. 5d) although minus-strand RNA was also detected in iMVs. However, the relative levels of minus-strand RNA were low and comparable to those present in transfected cells. In fact, the plus-strand/minus-strand RNA ratios in cells and iMVs were not significantly different (p = 0.057), suggesting that the minus-strand RNA is not enriched in iMVs. Moreover, cellular mRNAs corresponding to GAPDH and β-actin were detected in purified iMVs before RNAse treatment, but only the latter one, which seemed to be more abundant in cells (Fig. 5c), was detected in RNAse-treated iMVs, suggesting that the cellular level of an mRNA could determine its inclusion into iMVs.
Size and density
To determine the density of iMVs, they were sedimented to equilibrium in a 5–60 % sucrose gradient and each fraction was assessed for infectivity. As shown in Fig. 5e, the majority of iMVs were localized in a fraction with a density of 1.19 g/cm3. This value was slightly higher than the one observed for SFV VPs analysed in a parallel gradient, localized at an average density of 1.17 g/cm3, which is very similar to the one previously described for this virus [41].
TEM analysis showed the presence of pleomorphic vesicles with an estimated size ranging from ~50 to ~150 nm in SFV-enh-spike samples that were labelled specifically with anti-spike antibodies (Fig. 6a–g). No labelling was observed in the material purified from SFV-LacZ- or mock-electroporated cells or when iMVs were directly incubated with colloidal gold-conjugated secondary antibody (data not shown). Virions of about 70 nm in diameter were clearly detected in a sample of pelleted SFV VPs used as positive control (Fig. 6h).
Fig. 6.
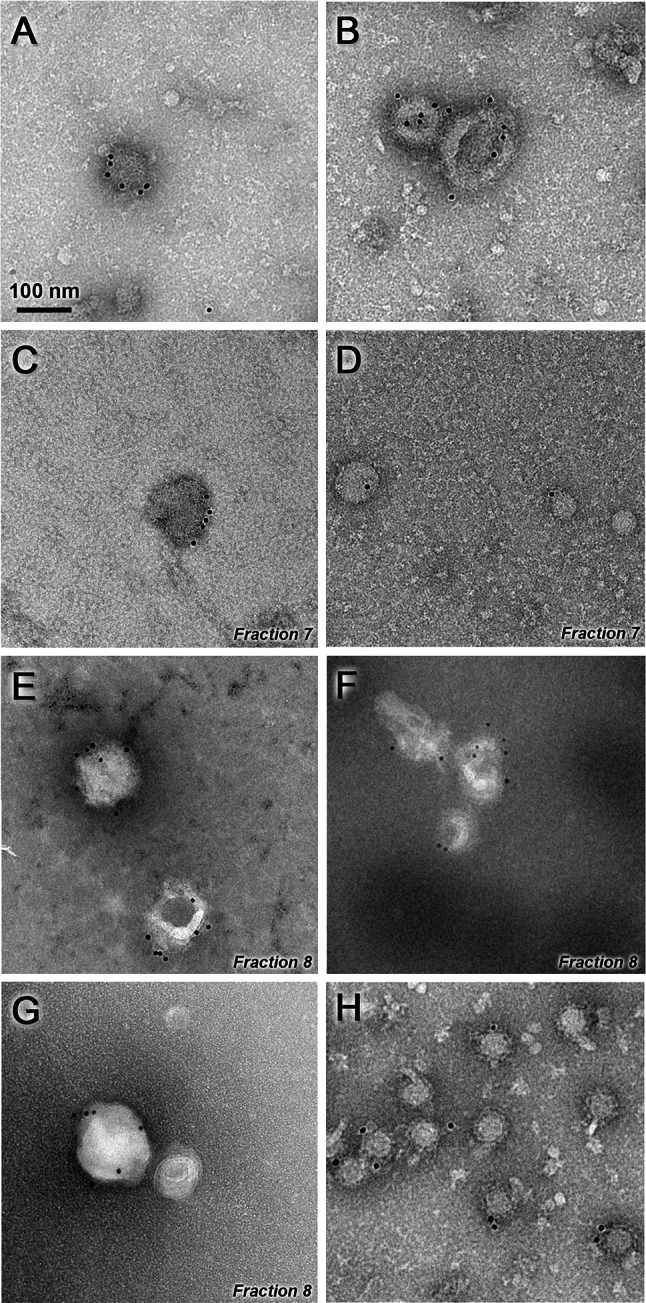
Electron microscopy of iMVs. Purified iMVs were incubated with an antiserum specific for SFV envelope proteins, followed by incubation with a gold-labelled secondary antibody and visualized by negative staining (a–g). Representative images of iMVs present before sucrose gradient ultracentrifugation (a, b) or in the indicated fractions after gradient purification (c–g) are shown. SFV-LacZ VPs isolated as described in “Materials and methods” were used as positive control (h). Magnification in all pictures is ×30,000. Scale bar 100 nm
Analysis of iMVs infectivity in mice
Given the propagative nature of iMVs we tested their infectivity in mice, since these animals are readily infected by wtSFV. C57BL/6 mice intravenously injected with 106 IU of iMVs remained healthy, showing no pathological signs, no weight loss, and long-term survival similar to control mice injected with saline (Fig. 7a. b). In contrast, 60 % of mice inoculated with 105 PFU of wtSFV succumbed at 6 days p.i.
In a parallel experiment, we analysed whether iMVs could be produced in vivo. Mice inoculated with 107 IU showed very low titres of iMVs in blood at 6 h p.i. (Fig. 7c), which were probably derived from the inoculum. In contrast, mice injected with 107 PFU of wtSFV showed sustained viremia during at least 48 h, with the highest values at 24 h p.i., as described previously [42]. In addition, wtSFV was also detected in brain at 48 h p.i. (Fig. 7d). However, neither iMVs nor control SFV-LacZ vector (injected at a dose of 107 VPs) was detected in the brain tissue.
Finally, we studied the infectivity of iMVs in different organs by measuring the amount of SFV RNA present in tissue homogenates by specific RT-qPCR. This analysis showed that iMVs were able to infect the heart with similar efficacy as SFV-LacZ and wtSFV. Interestingly, the ability of iMVs to infect the lung was significantly higher than that of the other two viruses (Fig. 7e). In contrast, only wtSFV genomes were detected in brain. No SFV RNA was detected in any organ from mice that received UV-inactivated SFV-LacZ, used as an additional control, or saline.
These data indicate that although iMVs do not seem to be able to give rise to a productive infection in immune-competent mice, they can infect different organs, without producing pathogenicity.
SFV without capsid is able to mediate transmission of a heterologous gene
Alphavirus vectors have been extensively used to express heterologous proteins for recombinant protein production, as well as for vaccination and gene therapy applications. To test if the SFV genome devoid of capsid was able to allow the expression and transmission of foreign genes, the ORF coding for GFP was cloned into the SFV-enh-spike vector downstream of a second subgenomic promoter (SFV-enh-spike-GFP, Supplementary Fig. 1). SFV-enh-spike-GFP RNA was efficiently packaged into VPs when co-transfected into BHK cells together with SFV-helper-C-S219A RNA. When these VPs were used to infect cell monolayers at low MOI, the number of GFP-expressing cells increased along time leading to both spike and GFP expression in a high proportion of cells at 30 h p.i. (Fig. 8a). In contrast, a non-propagative SFV-GFP vector used as control resulted in a pattern of individual GFP-expressing cells at all times tested. Accordingly, flow cytometry analysis showed a considerable increase (>5 fold at 24 h p.i.) in the percentage of GFP-positive cells when they were infected with SFV-enh-spike-GFP (Fig. 8b), indicating that this vector was able to transmit the GFP gene in cell monolayers. A similar result was observed in a co-culture experiment performed with cells electroporated with SFV-enh-spike-GFP RNA mixed with non-electroporated cells (data not shown). This result indicates that alphavirus genomes without capsid can incorporate foreign genes and propagate their expression together with viral genes.
Discussion
The role of the nucleocapsid revisited
It has been previously shown that propagation of alphaviruses requires expression of envelope and capsid proteins in the same cells [17]. It was later demonstrated that specific interactions between NCs and the cytoplasmic tail of E2 protein were necessary for budding of VPs and propagation [43]. In the present study, we have demonstrated that alphaviruses devoid of capsid sequences can also propagate in cell monolayers. For this purpose, we engineered alphavirus genomes in which the whole capsid gene was deleted (SFV-spike and SIN-spike), or in which the first 34 amino acids of the capsid (translation enhancer) were fused in frame with the sequence coding for the envelope polyprotein (SFV-enh-spike). This last strategy, which was used to increase the expression of envelope proteins, led to a more pronounced propagation of the vector. Although SFV-enh-spike is able to express a small capsid fragment, we can rule out that this peptide could mediate the formation of functional NCs, since it lacks all domains necessary for NC assembly and budding. These missing domains include most of the amino-terminal domain (residues 1–118), which mediates binding of capsid to viral RNA, and the carboxy-terminal domain (residues 119–267) needed for NC assembly and budding [14]. Although the amino-terminal domain can tolerate large deletions [8] the carboxy-terminal domain is essential for NC assembly and budding [43]. Furthermore, the fact that both SFV-spike and SIN-spike were able of propagating in cell monolayers demonstrates that no capsid sequences are involved in this phenomenon. Interestingly, the propagation of SFV-spike had not been observed previously, even though this vector had been used in early studies to show that both envelope and capsid proteins are necessary for budding and production of VPs [17]. A possible explanation of these apparent antithetic results lays in the fact that in those studies, cells infected with SFV-spike VPs were analysed only at early times post-infection (8–10 h), when propagation was still not evident (Fig. 1b).
Spike glycoproteins involvement in propagation
The propagation of alphavirus genomes without capsid sequences seems to be mediated by the expression of envelope proteins, since the vector was unable to propagate in the presence of an antiserum raised against these proteins. The envelope proteins include the E1 and E2 mature proteins that form the viral spikes and the small 6K protein, which is not necessary for spike formation but is involved in the correct assembly and release of VPs [23]. A possible contribution of 6K to the propagation of SFV genomes without capsid sequences was discarded when we observed that an SFV genome lacking both the capsid and 6K proteins (SFV-enh-spikeΔ6K) was also able to spread in cell monolayers. This vector, which was engineered by fusing the p62 and E1 genes as previously described [44], was able to express levels of spike proteins comparable to those of SFV-spike (Supplementary Fig. 2), indicating that the mutated polyprotein was correctly processed.
A formal possibility for the propagation of capsid-deleted alphavirus could be the fusion of cells expressing functional spikes with neighbouring cells. However, we did not observe fusion events under normal cell culture conditions (Fig. 3b, Supplementary Fig. 3). It has been described that SFV-infected cells can fuse to each other, but only when they are exposed to an acidic pH [45]. We were able to reproduce this phenomenon by incubating infected or transfected cell monolayers at pH 5. This result confirmed that these cells were expressing functional spikes on their surface, since only correctly folded spikes can undergo the conformational changes induced by low pH needed for the E1-mediated fusion.
Thus, propagation of alphavirus genomes devoid of capsid can be explained either by some sort of cell-to-cell transmission process, or by the release of infectious material to the extracellular medium. Regarding the first possibility, it has been recently shown that cell-to-cell transfer of SIN VPs can be mediated by filopodia-like extensions induced in infected cells [46]. However, this kind of transmission required interactions between spike and capsid proteins, making this process unlikely in the absence of the latter protein. Furthermore, in our study we demonstrate that cells transfected with SFV devoid of capsid are able to produce infectious material in different cell types. This material is formed by heterogeneous microvesicles that contain protected viral RNA and display spike proteins on their surface. Interestingly, these iMVs infect cells using the endocytic pathway, in a similar way to wt alphavirus (Fig. 4c) and are able to propagate from initially infected cells to neighbouring cells (Fig. 4b). However, the production of iMVs is not very efficient, reaching a maximum titre of 105 IU/ml in HuH-7 cells, which is about three–four orders of magnitude lower than the reported production of wtSFV VPs in mammalian cells [47]. This low titre could explain why propagation of vectors without capsid is much less efficient than that of the wt virus (Figs. 1b, 2b, c, Supplementary Fig. 4). Despite the low production of iMVs, both quantitative RT-PCR and Western blot experiments indicate that the amount of viral RNA and envelope proteins per infection unit is higher in iMVs compared to SFV VPs (Fig. 5a, b). The higher RNA level/infection unit in iMVs could indicate that each iMV incorporates several copies of viral RNA, but alternatively it could also indicate that the “particle-to-infectivity ratio” is higher for iMVs. The higher envelope proteins level/infection unit in iMVs might be a reflection of the larger size of iMVs compared to VPs, but it could also be due to a higher “particle-to-infectivity ratio” for iMVs. Although further studies are needed to clarify these possibilities, we cannot discard that a proportion of iMVs produced by transfected cells might be defective in infection.
Previous studies had shown that an SFV vector devoid of all viral structural proteins, but expressing vesicular stomatitis virus (VSV) G glycoprotein could also propagate in cell culture [48]. This chimeric SFV–VSV vector was also able to release infectious vesicles containing viral RNA and expressing VSV-G protein on their surface. Recently, using an elegant in vitro evolution assay, it has been found that the appearance of several mutations in the SFV Rep increases the production of infectious SFV–VSV-G vesicles [49]. Interestingly, vesicles produced by these mutants are derived from viral RC spherules that get trapped on the cell surface. Whether these mutations could also increase the production of iMVs derived from SFV genomes devoid of capsid deserves further investigation.
Viral RNA in iMVs
Our experiments with cells electroporated with SFV-LacZ suggest that microvesicles containing viral RNA could also be produced in the absence of spike proteins, albeit at lower levels (Fig. 5b). However, these microvesicles would be unable to infect cells, since they lack the glycoproteins necessary for binding and fusion to cell membranes. SFV packaging of viral RNA into VPs requires specific interactions between the capsid and a packaging signal present in viral RNA, and between the capsid and the E2 cytoplasmic tail. Since none of these interactions are possible in the absence of capsid, the inclusion of RNAs into iMVs should be therefore mediated by a different mechanism. The observation that β-actin RNA was detected in iMVs at low levels (Fig. 5b) suggests that packaging might be non-specific and only highly abundant RNAs are efficiently incorporated into iMVs. The fact that envelope proteins from a different virus, like VSV-G [48], can also mediate this incorporation excludes specific interactions between RNA and SFV-spike glycoproteins.
To our knowledge, the present study demonstrates for the first time that an enveloped virus can propagate in the absence of its capsid. In principle, this alternative way of viral propagation could also take place in the context of a wt virus infection. However, to validate this hypothesis, further studies are required. These have to overcome the challenges of the relatively small amounts of iMVs versus wtSFV and of the large production of wtSFV capsid protein likely to efficiently sequester the viral genomic RNA and thus limit its incorporation inside iMVs during an infection scenario.
Morphological features and possible origins of iMVs
We investigated the possibility that iMVs observed in this study could derive from exosomes, since it has been described that different viruses, including HCV, can use this type of vesicles to facilitate viral transmission [50–52]. However, several features observed in SFV-derived iMVs point to a different origin: (1) while exosomes appear as spherical vesicles with a diameter of 50–100 nm [53, 54], SFV-derived iMVs possess a less regular shape and in some cases a larger size (~150 nm in diameter), as shown by TEM studies (Fig. 6); (2) while exosomes are usually sedimented at 100,000g, iMVs were efficiently sedimented at 10,000g [55], and (3) we could not detect an enrichment of exosome markers in iMVs (Supplementary Fig. 5b). We also evaluated the possibility that iMVs could derive from RCs. However, this hypothesis was not supported by several observations: (1) we were unable to significantly detect nsPs 1, 2, 3, and 4 in iMVs (Supplementary Fig. 5c), suggesting that the whole Rep complex, which is an important component of RCs [40], is not present within iMVs; (2) iMVs contain mostly plus-strand RNA, with very low relative minus-strand RNA levels (Fig. 5d), suggesting that double-stranded RNA, an intermediate product of RNA replication that is generated in RCs, is not abundant in iMVs. These data strongly support that the novel infectious vesicles described in this study, unlike those produced by SFV–VSV [49], are not derived from RCs, suggesting that alphavirus genomes could use different mechanisms of propagation in the absence of capsid.
Considering that viral spike proteins can be normally transported to the plasma membrane in the absence of capsid [56] and that these proteins are also present on the surface of iMVs, it is more likely that they originate at that location by shedding and/or budding. This origin is further strengthened by the fact that iMVs were enriched in the oligomeric plasma membrane complex Na+/K+-ATPase (Supplementary Fig. 5a), as well as by cell surface biotinylation and thin-section TEM studies (Supplementary Figs. 6 and 7). It has been reported that SFV glycoproteins possess budding activity mediated by their own interactions [57]. Furthermore, structural studies have shown that SFV E1 protein displays lateral interactions that induce membrane curvature and direct budding of closed particles [58]. For other enveloped viruses, such as coronaviruses and flaviviruses, it has also been described that membrane glycoproteins assembled at the plasma membrane can create a “pulling” force that drives membrane curvature and bud formation even in the absence of capsid proteins [59].
A non-icosahedral enveloped virus naturally lacking an NC has been recently described [60]. This virus, infecting an archaeal host, is solely composed of a membrane vesicle randomly fenestrated by glycoproteins. This membrane encloses a circular single-strand DNA genome devoid of nucleic acid-binding nucleoproteins. It is enticing that the architectural comparison between this minimalist archaeal enveloped viruses and the iMVs produced by alphaviruses without capsid is described here. In this view, the appearance of the NC in this type of virus could represent a further architectural and functional element necessary for efficient packaging and delivering of viral genomes.
Infectivity of iMVs in mice and their potential use as transfer vectors
Animal studies indicated that, despite the capacity of iMVs to propagate in cell culture, they did not give rise to a productive infection in immunocompetent mice. Although further studies are needed to conclusively determine whether iMVs can propagate in vivo, our results demonstrated that iMVs can infect different organs in mice without producing any observable pathogenicity and without crossing the blood brain barrier, in contrast to wtSFV (Fig. 7). The higher infectivity of iMVs observed in lungs, compared to VPs, could be due to the larger size of iMVs, which might result in a longer retention time in the small lung capillaries, facilitating the infection of this organ.
Given the propagative nature of iMVs, we also studied whether SFV vectors without capsid could mediate the transfer of heterologous genes. We were able to show that this was the case by introducing the GFP reporter gene into SFV-enh-spike genome. These results, together with the fact that iMVs can infect different organs in vivo, suggest that this kind of new vectors could be useful for vaccination or gene therapy. In particular, alphavirus-derived iMVs could be interesting for vaccination against pathogenic members of this group of viruses, like CHIKV, given the fact that they can express very high levels of their own envelope proteins. In fact, it has been recently reported that a CHIKV vector based on a viral genome lacking the capsid gene (a construct very similar to our SFV-spike construct) was able to induce anti-CHIKV antibody and T cell responses similar to those obtained by single immunizations with wtCHIKV [61, 62]. It is enticing to consider that the potency of this type of vaccine vector could also lie in the production of iMVs, which would increase the number of infected cells and hence also antiviral immune responses.
Nevertheless, we are aware that the low efficiency of iMVs generation in vitro could limit their use for clinical applications. This low production of iMVs could, however, be circumvented by packaging SFV vectors carrying spike and heterologous genes into VPs, providing the capsid protein in trans (as described in “Materials and methods” for the production of SFV-enh-spike-GFP VPs). These VPs are produced at much higher levels than iMVs (we routinely obtained >108 VPs/ml for SFV-enh-spike-GFP), can infect and mediate the propagation of heterologous gene expression in cell culture (Fig. 8), and could be used to deliver propagation-competent alphavirus vectors in vivo in a more efficient way.
Conclusions
We have shown that alphaviruses can propagate in the complete absence of their capsids. This remarkable phenomenon allows spreading of a self-replicating RNA through the production of pleomorphic microvesicles that are assembled at the plasma membrane. These microvesicles contain spike glycoproteins on their surface and are capable of infecting neighbouring cells through endocytosis in a similar way to wt virus (Fig. 9). Although this new mechanism of viral transmission is less efficient than the one used by the parental virus, it could also contribute to its propagation. In addition, we speculate that this minimalist infectious system resembles a primitive alphavirus ancestor not yet complemented by an NC, providing some clues for virus evolution.
Fig. 9.

Schematic model of alphavirus propagation in the absence of capsid. Once the alphavirus RNA genome without capsid (alphavirus-ΔC) is delivered to the cytoplasm of a cell, it will replicate leading to the production of many genomic (gRNA) and subgenomic (sgRNA) copies. The envelope glycoproteins E1 and p62 will be translated from sgRNAs at the endoplasmic reticulum (ER) where they will form heterodimers that will be transported to the plasma membrane through Golgi. In late Golgi, p62 will mature to E2 by furin cleavage, rendering the spikes functional for fusion. iMVs will form when gRNAs get trapped inside pleomorphic microvesicles which are generated by budding at the plasma membrane. These iMVs, which have functional spikes on their surface, can interact with alphavirus receptors on neighbouring cells and enter them through endocytosis. The low pH of endosomes will trigger conformational changes on the spike glycoproteins, leading to the fusion of the iMV membrane and the endosome, resulting in the release of gRNA to the cytoplasm
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Sandra Delgado and Isaac Santos-Pérez (CIC bioGUNE) for skilful technical assistance with EM imaging of purified iMVs. We also acknowledge the personnel of the University of Navarra Histology Department for help with thin-section TEM studies and Rafael Aldabe (CIMA) for help with confocal microscopy. We are grateful to Juan Manuel Falcón (CIC bioGUNE) for advice on exosome isolation. We also thank Puri Fortes and Tomás Aragon (CIMA) for critical reading of the manuscript and suggestions. This work was supported by grants from the Spanish Health and Science Ministries (FIS PI11/02190 and PI14/01442 to CS, and SAF 2012-39578 to RH), European Regional Development Fund (FEDER), the Government of Navarra (GNE-VECTORES ALFAVIRUS to CS), the Center for Applied Medical Research-UTE Project (to CS), and the Spanish “Ministerio de Economia y Competitividad” BFU2012-33947 and BFU2015-64541-R (to NGAA).
Footnotes
M. Ruiz-Guillen and E. Gabev contributed equally to this work.
References
- 1.Her Z, Kam YW, Lin RT, Ng LF. Chikungunya: a bending reality. Microbes Infect. 2009;11:1165–1176. doi: 10.1016/j.micinf.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Johansson MA. Chikungunya on the move. Trends Parasitol. 2015;31:43–45. doi: 10.1016/j.pt.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paredes AM, Brown DT, Rothnagel R, Chiu W, Schoepp RJ, Johnston RE, Prasad BV. Three-dimensional structure of a membrane-containing virus. Proc Natl Acad Sci USA. 1993;90:9095–9099. doi: 10.1073/pnas.90.19.9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mancini EJ, Clarke M, Gowen BE, Rutten T, Fuller SD. Cryo-electron microscopy reveals the functional organization of an enveloped virus, Semliki Forest virus. Mol Cell. 2000;5:255–266. doi: 10.1016/S1097-2765(00)80421-9. [DOI] [PubMed] [Google Scholar]
- 5.Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frolov I, Schlesinger S. Translation of Sindbis virus mRNA: effects of sequences downstream of the initiating codon. J Virol. 1994;68:8111–8117. doi: 10.1128/jvi.68.12.8111-8117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sjoberg EM, Suomalainen M, Garoff H. A significantly improved Semliki Forest virus expression system based on translation enhancer segments from the viral capsid gene. Biotechnology (N Y) 1994;12:1127–1131. doi: 10.1038/nbt1194-1127. [DOI] [PubMed] [Google Scholar]
- 8.Forsell K, Suomalainen M, Garoff H. Structure-function relation of the NH2-terminal domain of the Semliki Forest virus capsid protein. J Virol. 1995;69:1556–1563. doi: 10.1128/jvi.69.3.1556-1563.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geigenmuller-Gnirke U, Nitschko H, Schlesinger S. Deletion analysis of the capsid protein of Sindbis virus: identification of the RNA binding region. J Virol. 1993;67:1620–1626. doi: 10.1128/jvi.67.3.1620-1626.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen KE, Kuhn RJ. Identification of a region in the Sindbis virus nucleocapsid protein that is involved in specificity of RNA encapsidation. J Virol. 1996;70:2757–2763. doi: 10.1128/jvi.70.5.2757-2763.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss B, Nitschko H, Ghattas I, Wright R, Schlesinger S. Evidence for specificity in the encapsidation of Sindbis virus RNAs. J Virol. 1989;63:5310–5318. doi: 10.1128/jvi.63.12.5310-5318.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S, Owen KE, Choi HK, Lee H, Lu G, Wengler G, Brown DT, Rossmann MG, Kuhn RJ. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure. 1996;4:531–541. doi: 10.1016/S0969-2126(96)00059-7. [DOI] [PubMed] [Google Scholar]
- 13.Owen KE, Kuhn RJ. Alphavirus budding is dependent on the interaction between the nucleocapsid and hydrophobic amino acids on the cytoplasmic domain of the E2 envelope glycoprotein. Virology. 1997;230:187–196. doi: 10.1006/viro.1997.8480. [DOI] [PubMed] [Google Scholar]
- 14.Skoging U, Vihinen M, Nilsson L, Liljestrom P. Aromatic interactions define the binding of the alphavirus spike to its nucleocapsid. Structure. 1996;4:519–529. doi: 10.1016/S0969-2126(96)00058-5. [DOI] [PubMed] [Google Scholar]
- 15.Zhao H, Lindqvist B, Garoff H, von Bonsdorff CH, Liljestrom P. A tyrosine-based motif in the cytoplasmic domain of the alphavirus envelope protein is essential for budding. EMBO J. 1994;13:4204–4211. doi: 10.1002/j.1460-2075.1994.tb06740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez S, Yao JS, Kuhn RJ, Strauss EG, Strauss JH. Nucleocapsid-glycoprotein interactions required for assembly of alphaviruses. J Virol. 1994;68:1316–1323. doi: 10.1128/jvi.68.3.1316-1323.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suomalainen M, Liljestrom P, Garoff H. Spike protein-nucleocapsid interactions drive the budding of alphaviruses. J Virol. 1992;66:4737–4747. doi: 10.1128/jvi.66.8.4737-4747.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liljestrom P, Garoff H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology (N Y) 1991;9:1356–1361. doi: 10.1038/nbt1291-1356. [DOI] [PubMed] [Google Scholar]
- 19.Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. Replicon-helper systems from attenuated Venezuelan Equine Encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology. 1997;239:389–401. doi: 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- 20.Xiong C, Levis R, Shen P, Schlesinger S, Rice CM, Huang HV. Sindbis virus: an efficient, broad host range vector for gene expression in animal cells. Science. 1989;243:1188–1191. doi: 10.1126/science.2922607. [DOI] [PubMed] [Google Scholar]
- 21.Bredenbeek PJ, Frolov I, Rice CM, Schlesinger S. Sindbis virus expression vectors: packaging of RNA replicons by using defective helper RNAs. J Virol. 1993;67:6439–6446. doi: 10.1128/jvi.67.11.6439-6446.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smerdou C, Liljestrom P. Two-helper RNA system for production of recombinant Semliki forest virus particles. J Virol. 1999;73:1092–1098. doi: 10.1128/jvi.73.2.1092-1098.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liljestrom P, Lusa S, Huylebroeck D, Garoff H. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol. 1991;65:4107–4113. doi: 10.1128/jvi.65.8.4107-4113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryan MD, Drew J. Foot-and-mouth disease virus 2A oligopeptide mediated cleavage of an artificial polyprotein. EMBO J. 1994;13:928–933. doi: 10.1002/j.1460-2075.1994.tb06337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson M, Yang H, Sun SC, Peng B, Tian Y, Pagratis N, Greenstein AE, Delaney WE. Novel hepatitis C virus reporter replicon cell lines enable efficient antiviral screening against genotype 1a. Antimicrob Agents Chemother. 2010;54:3099–3106. doi: 10.1128/AAC.00289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleeton MN, Sheahan BJ, Gould EA, Atkins GJ, Liljestrom P. Recombinant Semliki Forest virus particles encoding the prME or NS1 proteins of louping ill virus protect mice from lethal challenge. J Gen Virol. 1999;80:1189–1198. doi: 10.1099/0022-1317-80-5-1189. [DOI] [PubMed] [Google Scholar]
- 27.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protocols Cell Biol. 2006;30:3.22.1–3.22.29. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- 28.Salminen A, Wahlberg JM, Lobigs M, Liljestrom P, Garoff H. Membrane fusion process of Semliki Forest virus. II: cleavage-dependent reorganization of the spike protein complex controls virus entry. J Cell Biol. 1992;116:349–357. doi: 10.1083/jcb.116.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casales E, Rodriguez-Madoz JR, Ruiz-Guillen M, Razquin N, Cuevas Y, Prieto J, Smerdou C. Development of a new noncytopathic Semliki Forest virus vector providing high expression levels and stability. Virology. 2008;376:242–251. doi: 10.1016/j.virol.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 30.Gowen B, Bamford JK, Bamford DH, Fuller SD. The tailless icosahedral membrane virus PRD1 localizes the proteins involved in genome packaging and injection at a unique vertex. J Virol. 2003;77:7863–7871. doi: 10.1128/JVI.77.14.7863-7871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greiser-Wilke I, Moenning V, Kaaden OR, Figueiredo LT. Most alphaviruses share a conserved epitopic region on their nucleocapsid protein. J Gen Virol. 1989;70:743–748. doi: 10.1099/0022-1317-70-3-743. [DOI] [PubMed] [Google Scholar]
- 33.White J, Helenius A. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc Natl Acad Sci USA. 1980;77(6):3273–3277. doi: 10.1073/pnas.77.6.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kielian M, Jungerwirth S. Mechanisms of enveloped virus entry into cells. Mol Biol Med. 1990;7:17–31. [PubMed] [Google Scholar]
- 35.Urbanelli L, Magini A, Buratta S, Brozzi A, Sagini K, Polchi A, Tancini B, Emiliani C. Signaling pathways in exosomes biogenesis, secretion and fate. Genes (Basel) 2013;4:152–170. doi: 10.3390/genes4020152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu YE, Kielian M. Semliki forest virus budding: assay, mechanisms, and cholesterol requirement. J Virol. 2000;74:7708–7719. doi: 10.1128/JVI.74.17.7708-7719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frolova EI, Gorchakov R, Pereboeva L, Atasheva S, Frolov I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J Virol. 2010;84:11679–11695. doi: 10.1128/JVI.01441-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spuul P, Balistreri G, Kaariainen L, Ahola T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest Virus replication complexes from the plasma membrane to modified lysosomes. J Virol. 2010;84:7543–7557. doi: 10.1128/JVI.00477-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorchakov R, Garmashova N, Frolova E, Frolov I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J Virol. 2008;82:10088–10101. doi: 10.1128/JVI.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kujala P, Ikaheimonen A, Ehsani N, Vihinen H, Auvinen P, Kaariainen L. Biogenesis of the Semliki Forest virus RNA replication complex. J Virol. 2001;75:3873–3884. doi: 10.1128/JVI.75.8.3873-3884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fox SM, Birnie GD, Harvey DR, Martin EM, Sonnabend JA. The use of batch-type zonal ultracentrifuge rotors for the isolation and purification of viruses. J Gen Virol. 1968;2:455–459. doi: 10.1099/0022-1317-2-3-455. [DOI] [PubMed] [Google Scholar]
- 42.Singh IP, Coppenhaver DH, Sarzotti M, Sriyuktasuth P, Poast J, Levy HB, Baron S. Postinfection therapy of arbovirus infections in mice. Antimicrob Agents Chemother. 1989;33:2126–2131. doi: 10.1128/AAC.33.12.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garoff H, Sjoberg M, Cheng RH. Budding of alphaviruses. Virus Res. 2004;106:103–116. doi: 10.1016/j.virusres.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 44.Liljestrom P, Garoff H. Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J Virol. 1991;65:147–154. doi: 10.1128/jvi.65.1.147-154.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White J, Matlin K, Helenius A. Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. J Cell Biol. 1981;89:674–679. doi: 10.1083/jcb.89.3.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez MG, Snapp EL, Perumal GS, Macaluso FP, Kielian M. Imaging the alphavirus exit pathway. J Virol. 2014;88:6922–6933. doi: 10.1128/JVI.00592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davey MW, Dennett DP, Dalgarno L. The growth of two togaviruses in cultured mosquito and vertebrate cells. J Gen Virol. 1973;20:225–232. doi: 10.1099/0022-1317-20-2-225. [DOI] [PubMed] [Google Scholar]
- 48.Rolls MM, Webster P, Balba NH, Rose JK. Novel infectious particles generated by expression of the vesicular stomatitis virus glycoprotein from a self-replicating RNA. Cell. 1994;79:497–506. doi: 10.1016/0092-8674(94)90258-5. [DOI] [PubMed] [Google Scholar]
- 49.Rose NF, Buonocore L, Schell JB, Chattopadhyay A, Bahl K, Liu X, Rose JK. In vitro evolution of high-titer, virus-like vesicles containing a single structural protein. Proc Natl Acad Sci USA. 2014;111:16866–16871. doi: 10.1073/pnas.1414991111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meckes DG, Jr, Raab-Traub N. Microvesicles and viral infection. J Virol. 2011;85:12844–12854. doi: 10.1128/JVI.05853-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JA, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus HW, Haagmans BL, Baumert TF, van der Laan LJ. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci USA. 2013;110:13109–13113. doi: 10.1073/pnas.1221899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe. 2012;12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conde-Vancells J, Rodriguez-Suarez E, Embade N, Gil D, Matthiesen R, Valle M, Elortza F, Lu SC, Mato JM, Falcon-Perez JM. Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J Proteome Res. 2008;7:5157–5166. doi: 10.1021/pr8004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, Laszlo V, Pallinger E, Pap E, Kittel A, Nagy G, Falus A, Buzas EI. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68:2667–2688. doi: 10.1007/s00018-011-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 56.Zhao H, Garoff H. Role of cell surface spikes in alphavirus budding. J Virol. 1992;66:7089–7095. doi: 10.1128/jvi.66.12.7089-7095.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forsell K, Xing L, Kozlovska T, Cheng RH, Garoff H. Membrane proteins organize a symmetrical virus. EMBO J. 2000;19:5081–5091. doi: 10.1093/emboj/19.19.5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Rey FA. The Fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 2001;105:137–148. doi: 10.1016/S0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- 59.Welsch S, Muller B, Krausslich HG. More than one door—budding of enveloped viruses through cellular membranes. FEBS Lett. 2007;581:2089–2097. doi: 10.1016/j.febslet.2007.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pietila MK, Laurinavicius S, Sund J, Roine E, Bamford DH. The single-stranded DNA genome of novel archaeal virus halorubrum pleomorphic virus 1 is enclosed in the envelope decorated with glycoprotein spikes. J Virol. 2010;84:788–798. doi: 10.1128/JVI.01347-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hallengard D, Lum FM, Kummerer BM, Lulla A, Lulla V, Garcia-Arriaza J, Fazakerley JK, Roques P, Le Grand R, Merits A, Ng LF, Esteban M, Liljestrom P. Prime-boost immunization strategies against Chikungunya virus. J Virol. 2014;88:13333–13343. doi: 10.1128/JVI.01926-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Knudsen ML, Ljungberg K, Kakoulidou M, Kostic L, Hallengard D, Garcia-Arriaza J, Merits A, Esteban M, Liljestrom P. Kinetic and phenotypic analysis of CD8+ T cell responses after priming with alphavirus replicons and homologous or heterologous booster immunizations. J Virol. 2014;88:12438–12451. doi: 10.1128/JVI.02223-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.