

Abstract
Infection by the Alkhurma virus (ALKV) leading to the Alkhurma hemorrhagic fever is a common thread in Saudi Arabia, with no efficient treatment or prevention available as of yet. Although the rational drug design traditionally uses information on known 3D structures of viral proteins, intrinsically disordered proteins (i.e., functional proteins that do not possess unique 3D structures), with their multitude of disorder-dependent functions, are crucial for the biology of viruses. Here, viruses utilize disordered regions in their invasion of the host organisms and in hijacking and repurposing of different host systems. Furthermore, the ability of viruses to efficiently adjust and accommodate to their hostile habitats is also intrinsic disorder-dependent. However, little is currently known on the level of penetrance and functional utilization of intrinsic disorder in the ALKV proteome. To fill this gap, we used here multiple computational tools to evaluate the abundance of intrinsic disorder in the ALKV genome polyprotein. We also analyzed the peculiarities of intrinsic disorder predisposition of the individual viral proteins, as well as human proteins known to be engaged in interaction with the ALKV proteins. Special attention was paid to finding a correlation between protein functionality and structural disorder. To the best of our knowledge, this work represents the first systematic study of the intrinsic disorder status of ALKV proteome and interactome.
Electronic supplementary material
The online version of this article (10.1007/s00018-018-2968-8) contains supplementary material, which is available to authorized users.
Keywords: Intrinsically disordered protein, Alkhurma virus, Proteome, Protein structure, Protein function, Protein folding, Partially folded conformation, Protein–protein interactions, Interactome
Introduction
Infection with a tick-borne Alkhurma virus (ALKV), which is a representative member of the tick-borne encephalitis virus (TBEV) family of the Flavivirus genus, triggers Alkhurma hemorrhagic fever (AHF) characterized by high mortality rates (up to 25%) [1, 2]. AHF cases appear to peak in spring and summer. It is believed that among the natural hosts of ALKV are camels and sheep possessing Ornithodoros savignyi ticks, which are known to be actively seeking multiple hosts. The first confirmed case of the fatal ALKV infection was reported in 1995, when a Saudi patient, who slaughtered a sheep, died of hemorrhagic fever [3, 4]. This case as well as several subsequent AHF cases reported from 2001 to 2003 were found in a particular region of Saudi Arabia, namely, Alkhumra district, south of Jeddah [5], which gave the name to the virus causing this malady. Subsequently, from 2003 to 2009, about 150 patients in Najran region in the south part of Saudi Arabia were suspected as being infected with ALKV, showing an exponential increase in the number of potentially infected subjects [2] and indicating that AHF should be treated as an emerging infectious disease. In fact, although originally AHF was exclusively found in Saudi Arabia, several tourists in Egypt were reported to have this disease, indicating that the geographic range of the virus distribution is broader than one country and that ALKV might represent a serious global threat. All these clearly indicate that the improvement of public health measures requires in-depth analysis of AHFV.
Although ALKV belongs to the well-known Flavivirus genus of the Flaviviridae family, which, in addition to the tick-borne pathogens, such as ALKV, TBEV, Omsk hemorrhagic fever virus (OHFV), Kyasanur Forest Disease Virus (KFDV), and Powassan virus and others, includes extensively studied human pathogens transmitted by mosquitoes, such as Zika Virus (ZIKV), Dengue Virus (DENV), West Nile Virus (WNV), and Japanese Encephalitis Virus (JEV) [6–9], ALKV (and, as a matter of fact, other tick-borne flaviviral pathogens) are studied to much lesser degree than their mosquito-borne counterparts. As a result, specific characteristics of the tick-borne flaviviruses (including ALKV) continue to be poorly understood, and even for the most studied tick-borne viral pathogen, TBEV, majority of structural information pertaining to its infection is extrapolated from the better-characterized mosquito-borne flaviviral pathogens [10].
Under the electron microscope, ALKV viral particles are characterized by a dark hexagonal core (capsid) and a translucent envelope, and have a mean diameter of ~ 41 nm [11]. Similar to other flaviviruses [12], ALKV has a positive-strand RNA (+RNA) genome that contains an open reading frame (ORF) which is flanked by 5′ and 3′ untranslated regions (UTR), that play important roles in viral transcription and replication. In relation to the protein expression order, ORF of ALKV has the structure similar to ORFs of other flaviviruses: 5′-C-prM-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5-3′. This ALKV ORF encodes a single polyprotein, which is processed during maturation into three structural proteins (capsid protein C, membrane protein prM, and envelope protein E) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). In ALKV, the genetic polyprotein has 3416 residues (UniProt ID: Q91B85), whereas mature proteins and peptides range in length from 23 residues (peptide 2k) to 903 residues (RNA-directed RNA polymerase NS5). Similarly, the length of the polyproteins of four serotypes of the dengue virus [e.g., DENV-1 (UniProt ID: P33478), DENV-2 (UniProt ID: P29990), DENV-3 (UniProt ID: Q99D35), and DENV-4 (UniProt ID: P09866)] ranges between 3387 and 3396 residues [13], whereas ZIKV genomic polyprotein (UniProt ID: Q32ZE1) has a length of 3419 residues [14]. The closest relative of ALKV is KFDV (which is present in the southern Indian state of Karnataka), with these two viruses sharing 89% nucleotide sequence homology [15], and with KFDV polyprotein (UniProt ID: D7RF80) containing 3416 residues.
Despite clear danger of this virus, no efficient treatments or prevention of ALKV infection are available as of yet. Although rational drug design traditionally uses information on known 3D structures of viral proteins, it is recognized now that intrinsically disordered proteins (IDPs) or intrinsically disordered protein regions (IDPRs, i.e., functional proteins or protein regions do not possess unique 3D structures), with their wide spectrum of functions dependent on disorder, are crucial for the biology of viruses. One should keep in mind though that the disorder enrichment of viral proteins (which show the widest variability of the disorder levels [16]) is not an exception, and IDPs/IDPRs are abundantly present in all organisms, for which proteome information is currently available [17–25]. Proteins and protein regions can show different degrees and depth of disorder [20, 22, 26, 27], giving rise to an intricate view of structures of IDPs and IDPRs as highly dynamic conformational ensembles [17, 19, 23, 27–30] with different levels of residual structure that can range from collapsed (molten globule-like), to partially collapsed (pre-molten globule-like), and even highly extended (coil-like) conformations [20, 22, 26, 27]. Some of the major biological functions associated with IDPs/IDPRs are related to the control and regulation of various signaling pathways, and intrinsic disorder is important for recognition and promiscuous binding to multiple partners [19, 22, 24, 27, 31, 32]. On the other hand, IDPs/IDPRs are incapable of catalytic activities, and thereby serve as a crucial complement to the functionality of ordered proteins [29, 33–37]. Pathogenesis of numerous human diseases is associated with the misbehavior of IDPs/IDPRs [38, 39]. Viruses utilize disordered regions of their proteins for invasion of the host organisms and for the hijacking and repurposing of different host systems. Furthermore, the ability of viruses to efficiently adjust and accommodate to their hostile habitats and to evade the immune system of a host is also intrinsic disorder-dependent. It is also likely that the disorder-based lack of structural constraints in viral proteins allows them to resist high rates of spontaneous mutations, which are typically attributed to viruses [40].
Our previous in silico studies showed many viruses (such as HCV [41], HIV-1 [42], various HPVs [43, 44], Zika virus [14, 45], respiratory syncytial virus (RSV) [46], Dengue virus [13], and MERS-CoV [47]) are not only enriched in IDPs and IDPRs, but commonly utilized these structure-less proteins and regions for various purposes [41–44]. Based on these considerations, we hypothesized that IDPRs can be found in key ALKV proteins and that these IDPRs might have important biological functions. Since the currently available information on the commonness and functionality of intrinsic disorder in the ALKV proteome is rather limited, the goal of this study was to fill this gap and to shed new intrinsic disorder-centric light on the biology of this important virus. To check the validity of this hypothesis, we utilized multiple bioinformatics and computational approaches that allowed us to analyze the intrinsic disorder predisposition of the ALKV genome polyprotein. We also looked at the individual ALKV proteins to find and characterize peculiarities of disorder distributions in their amino acid sequences and to see if structural disorder is related to functions of these proteins. We also characterized intrinsic disorder predispositions and disorder-based functionality of human proteins known to be engaged in interaction with the ALKV proteins. To the best of our knowledge, this study represents a first systematic analysis specifically dedicated to the evaluation of the prevalence of intrinsic disorder and assessment of the disorder-based biological functions of the ALKV proteome and interactome. Results of this research will form a foundation for subsequent studies aiming at the development of novel antivirals for the ALKV infection.
Materials and methods
Data set
We collected all complete genomes of the Alkhurma virus from UniProt [48] in July 2018. The query consisted of “Alkhurma virus” keyword as the organism and included both reviewed and unreviewed entries. Although considering reviewed entries assures that the query polyproteins are manually curated and that include functional annotations, there is only one curated entry of ALKV (UniProt ID: Q91B85). Therefore, the search was extended to include unreviewed entries. This extended query returned 66 polyprotein sequences, of which only 19 entries were full-length genome polyproteins (UniProt IDs: Q91B85, V9NZA4, V9NZB5, H8Y6L8, H8Y6M2, H8Y6M0, H8Y6L6, H8Y6L1, H8Y6K8, H8Y6K5, H8Y6L2, U5I905, H8Y6K7, H8Y6K6, H8Y6K4, H8Y6M1, H8Y6K9, U5IAE3, and A0A1S6VSX4), whereas remaining 47 hits were fragments of the viral polyprotein ranging in length from 2455 to 69 residues that were excluded from the subsequent analysis. The selected 19 ALKV polyproteins corresponding to the different ALKV isolates from infected humans have a similar length of 3416 residues. Each polyprotein encodes 12 protein chains, for which cleavage sites were annotated in the UniProt.
We also collected the sequences of genome polyproteins of Kyasanur forest disease virus (KFDV, UniProt ID: D7RF80), Zika virus (strain Mr 766, ZIKV, UniProt ID: Q32ZE1), and Dengue virus type 1 (strain Singapore/S275/1990, DENV, UniProt ID: P33478) from UniProt [48] in July 2018. We found a set of 38 human proteins interacting with ALKV proteins using a protein interaction database and analysis system IntAct (https://www.ebi.ac.uk/intact/) [49] accessed on September 9, 2018. These members of the ALKV interactome include Centrosome-associated protein CEP250 (CEP250, Q9BV73), Myomegalin (PDE4DIP, Q5VU43), Cadherin-11 (CDH11, P55287), Aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL, O00327), TATA-binding protein-associated factor 2N (TAF15, Q92804), Laminin subunit beta-2 (LAMB2, P55268), Mitotic interactor and substrate of PLK1 (MISP, Q8IVT2), Macoilin (MACO1 or Transmembrane protein 57 (TMEM57), Q8N5G2), E3 SUMO-protein ligase PIAS3 (PIAS3, Q9Y6X2), Leucine-rich repeat-containing protein 45 (LRRC45, Q96CN5), Amyloid beta A4 precursor protein-binding family B member 1-interacting protein (APBB1IP, Q7Z5R6), Protein jagged-1 (JAG1, P78504), Protein NipSnap homolog 1 (NIPSNAP1, Q9BPW8), Myosin-9 (MYH9, P35579), Vimentin (VIM, P08670), α-enolase (ENO1, P06733), Protein scribble homolog (SCRIB, Q14160), Zinc finger and BTB domain-containing protein 17 (ZBTB17, Q13105), Vacuolar protein sorting-associated protein 11 homolog (VPS11, Q9H270), Latent-transforming growth factor beta-binding protein 3 (LTBP3, Q9NS15), Protein phosphatase 1 regulatory subunit 3E (PPP1R3E, Q9H7J1), G patch domain-containing protein 2-like (GPATCH2L or C14orf118, Q9NWQ4), Deoxynucleotidyltransferase terminal-interacting protein 2 (DNTTIP2, Q5QJE6), Phosphorylated adapter RNA export protein (PHAX or RNA U small nuclear RNA export adapter protein (RNUXA), Q9H814), AT-rich interactive domain-containing protein 2 (ARID2, Q68CP9), Four and a half LIM domains protein 2 (FHL2, Q14192), Protein melanophilin (MLPH, Q9BV36), Thioredoxin domain-containing protein 9 (TXNDC9, O14530), Zinc finger protein 135 (ZNF135, P52742), Heterogeneous nuclear ribonucleoprotein H3 (HNRNPH3, P31942), Polyhomeotic-like protein 2 (PNC2, Q8IXK0), Protein Spindly (SPDL1 or Coiled-coil domain-containing protein 99 (CCDC99), Q96EA4), MyoD family inhibitor (MDFI, Q99750), Granulins (GRN, P28799), E3 ubiquitin-protein ligase TRIM21 (TRIM21, P19474), Ras association domain-containing protein 7 (RASSF7, Q02833), Calmodulin-binding transcription activator 2 (CAMTA2, O94983), Mitotic interactor and substrate of PLK1 (MISP or C19orf21, Q8IVT2), and Kinesin-like protein KIF3B (KIF3B, O15066).
Amino acid composition analysis of ALKV polyprotein and mature viral proteins
Since contents of amino acids in sequences of IDPs/IDPRs and ordered proteins/domains are characterized by large differences (with disorder-promoting residues A, G, R, D, H, Q, K, S, E, and P being more common in IDPs/IDPRs, which thereby contain less order-promoting residues, such as W, F, Y, I, M, L, V, N, C, and T [50]), some important information on the overall predisposition of a query protein to order or disorder can be retrieved form the comparative analysis of its amino acid composition using a specialized tool, Composition Profiler [17, 51] (http://profiler.cs.ucr.edu). This approach utilizes calculation of a fractional difference between a given protein set (or a query protein), and a set of ordered proteins in terms of their amino acid compositions [17, 51]. This fractional difference is computed for each residue as (C − Corder)/Corder, where C is the fraction of a given amino acid in a given query protein or a query protein set, and Corder is the fraction of this residue in ordered proteins.
Functional and structural annotations of ALKV proteins
We collected 19 types of structural and functional annotations for the ALKV genomic polyprotein. They were collected from a variety of resources including multiple sequence alignment with Clustal Omega [52], UniProt [48], eukaryotic linear motif (ELM) resource server [53], and a set of prediction methods: PONDR® VLXT [54], PONDR® VSL2 [55], PONDR® VL3 [55], PONDR® FIT [56], and IUPred [57, 58] were used to annotate putative intrinsic disorder and ANCHOR was utilized to predict disorder-based protein–protein interaction sites known as molecular recognition features (MoRFs) [59, 60]. These annotations include polymorphisms within and between different virus serotypes; cleavage sites (CLV); transmembrane regions (Trans); intramembrane regions (Intra); topological cytoplasmic, extracellular, and luminal domains (Topo-cy, Topo-ex, Topo-lu); functional sites; eukaryotic linear motifs (ELMs); intrinsically disordered protein regions (IDPRs); molecular recognition features (MoRFs).
Annotations of polymorphisms, i.e., changes in amino acid type between aligned polyproteins, are derived by running multiple sequence alignment with the Clustal Omega algorithm (https://www.ebi.ac.uk/Tools/msa/clustalo/). Here, we considered three types of annotations: conserved positions, strong polymorphisms, and weak polymorphisms. The conserved positions have identical amino acid type across all aligned sequences. The strong polymorphisms are defined as substitutions that involve amino acid types that are strongly dissimilar based on PAM substitution matrix [61]; these are denoted by space symbol in the Clustal Omega output. The weak polymorphisms involve substitutions of amino acid types that have similar physico-chemical properties; these are denoted by colon and period symbols by Clustal Omega.
Annotations of cleavage sites, transmembrane and intramembrane regions, as well as the three types of topological domains and functional sites were derived directly from UniProt for the reviewed UniProt entry for ALKV polyprotein (Q91B85). The functional sites are a union set of annotations of regions of interest, active sites, binding sites, other functional sites (except for the cleavage sites), and nucleotide binding regions. In other words, a given position is annotated as functional site if any of these annotations is true.
ELMs are short, usually between 3 and 11 residues in length, conserved functional sequence motifs [62] which are often found in the IDPRs [53]. We include annotations of all six types of ELMs as defined by the ELM server [53]. They include motifs that serve as proteolytic cleavage sites (ELM_CLV); post-translational modification sites (ELM_MOD); motifs for recognition and targeting to subcellular compartments (ELM_TRG); generic ligand-binding motifs (ELM_LIG); degron motifs that are involved in polyubiquitylation and targeting the protein to the proteasome for degradation (ELM_DEG); docking motifs that correspond to site of interactions with modifying enzyme that are distinct from active sites (ELM_DOC). The ELM_DEG and ELM_DOC motifs are specific subtypes of the ligand-binding motifs that were introduced recently to improve discrimination of functions of ELMs [53]. These annotations are parsed from the results of ELM motif search after globular domain filtering, structural filtering, and context filtering.
The putative intrinsically disorder residues were predicted using a set of commonly used intrinsic disorder predictors, such as PONDR® VLXT, PONDR® VSL2, PONDR® VL3, PONDR® FIT, and IUPred. PONDR® VLXT is the first computational tool for evaluating the intrinsic disorder predisposition of a query protein. It is a neural network-based disorder predictor with high sensitivity to local peculiarities of the query amino acid sequence [54]. PONDR® VSL2B (which in the following text will be called PONDR® VSL2) is a fastest member of the PONDR® VSL2 predictor family [55], which is among the more accurate intrinsic disorder predictors [56, 63]. Based on the comprehensive assessment of in silico predictors of intrinsic disorder [64, 65], PONDR® VSL2 was shown to perform reasonably well. PONDR® VL3 was designed for accurate prediction of long disordered regions in query proteins [55, 66]. IUPred identifies intrinsically disordered protein regions (IDPRs) from the amino acid sequence alone based on the estimated pairwise energy content [57, 58]. This algorithm has two implementations, IUPred_short and IUPRder_long, designed to predict short and long IDPRs, respectively. A meta-predictor, PONDR® FIT, is based on the combination of the outputs of six individual predictors, such as TopIDP [50], PONDR® VSL2 [67], PONDR® VL3 [55], PONDR® VLXT [54], IUPred [57], and FoldIndex [68]. Although component predictors are characterized by different accuracies, their combination makes PONDR® FIT relatively more accurate than its most accurate components [56].
Furthermore, in addition to the use of this set of six disorder predictors, all predictor-specific per-residue disorder profiles were averaged to generate a mean per-residue intrinsic disorder profile of a given protein, and this mean disorder profile was also added to the corresponding plot. Use of consensus for evaluation of intrinsic disorder is motivated by empirical observations that this approach usually increases the predictive performance compared to the use of a single predictor [65, 69, 70]. The outputs of the evaluation of the per-residue disorder propensity by these tools are represented as the real numbers between 1 (ideal prediction of disorder) and 0 (ideal prediction of order). A threshold of ≥ 0.5 was used to identify disordered residues and regions in query proteins, whereas residues/regions characterized by the disorder scores ranging from 0.2 to 0.5 are classified as flexible.
Analysis of the interactability of the members of ALKV interactome
We analyzed the interactability of human proteins shown to interact with ALKV proteins using the APID (Agile Protein Interactomes DataServer) web server (http://apid.dep.usal.es) [71]. APID has information on 90,379 distinct proteins from more than 400 organisms (including Homo sapiens) and on the 678,441 singular protein–protein interactions. For each protein–protein interaction (PPI), the server provides currently reported information about its experimental validation. For each protein, APID unifies PPIs found in five major primary databases of molecular interactions, such as BioGRID [72], Database of Interacting Proteins (DIP) [73], Human Protein Reference Database (HPRD) [74], IntAct [75], and the Molecular Interaction (MINT) database [76], as well as from the BioPlex (biophysical interactions of ORFeome-based complexes) [77] and from the protein databank (PDB) entries of protein complexes [78]. This server provides a simple way to evaluate the interactability of individual proteins in a given data set and also allows researchers to create a specific protein–protein interaction network in which proteins from the query data set are engaged.
Results and discussion
Intrinsic disorder predisposition and functionality of the ALKV proteins
Similar to all other members of the Flavivirus genus of the Flaviviridae family, translation of the ALKV genome produces a single polypeptide, an ALKV genomic polyprotein containing 3416 residues. This polyprotein is subjected to the proteolytic processing to give rise to three active viral structural (highly basic capsid protein C, membrane precursor protein prM, and envelope protein E) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B/2k, and NS5). All the structural proteins, i.e., proteins playing various roles in the structural organization of the virion, are found within the N-terminal part of the polyprotein. On the other hand, all non-structural proteins, which are crucial for the control, coordination, and regulation of the various intracellular processes associated with the different stages of the virus life cycle are localized within the C-terminal part of the ALKV polyprotein. Since no structural information is currently available about any of the ALKV proteins, we utilized a series of computational tools to gain some knowledge on their structure–function relationship and on the potential roles of IDPRs both in the ALKV genomic polyprotein and mature ALKV proteins.
Amino acid composition peculiarities of the ALKV polyprotein and mature viral proteins
First, we looked at the amino acid compositions of the ALKV polyprotein (UniProt ID: Q91B85) and mature viral proteins. This analysis was conducted based on the important observation that the amino acid compositions of IDPs/IDPRs are noticeably different from the compositions of their ordered counterparts. An illustrative example is given by the so-called natively-unfolded proteins/regions (or extended IDPs/IDPRs), which contain very high disorder levels and lack almost any residual structure [17, 20, 22, 27, 79–81]. Such highly disordered and extended structure originates from the weak hydrophobic attraction and strong electrostatic repulsion caused by the low mean hydropathy and high mean net charge of these proteins and regions [23]. In general, IDPs/IDPRs are significantly depleted in order-promoting residues, such as C, W, Y, F, I, L, V, and N, being instead substantially enriched in disorder-promoting residues, A, R, G, Q, S, P, E, K, and D [17, 34, 54]. This observation provides means for a rather straightforward analysis of protein amino acid compositions, known as composition profiling [17, 51], which is based on evaluation of the fractional difference in composition between a given protein or set of proteins (e.g., ALKV polyprotein, mature ALKV proteins, or a set of disordered proteins from the DisProt database [82, 83]) and a set of reference proteins (e.g., ordered proteins). In this study, the fractional compositional difference was computed using composition profiler [51]. Here, fractional compositional difference is calculated as (C − Corder)/Corder, where C is the content of a given amino acid in a query protein (set), and Corder is the corresponding value for the set of ordered proteins from PDB Select 25 (which is a subset of all proteins in PDB that share below 25% sequence identity) [84], and plotted for each amino acid. In a corresponding plot (see Fig. 1), the amino acids are arranged from the most order-promoting to the most disorder-promoting [34]. The amino acid types were separated into order-promoting residues (C, W, I, F, Y, L, H, V, N, and M) and disorder-promoting residues (R, T, D, G, A, K, Q, S, E, and P) [50], which are shown in Fig. 1a, b, respectively. The bars represent fractional differences between the amino acid composition of the ALKV polyprotein or individual mature ALKV proteins and the ordered proteins from PDB, where positive and negative values indicate enrichment and depletion of a given residue in a query protein when compared to the structured proteins. Since the composition profiler computes these differences via bootstrapping, the corresponding standard deviations are shown in the plot as the error bars. The bars in Fig. 1a, b are color-coded to simplify the identification of various query proteins. The composition profile calculated for known IDPs from the DisProt database [82, 83] is also shown for comparison.
Fig. 1.

Compositional profiling of the ALKV polyprotein and mature viral proteins. a Results for the order-promoting residues, and b represents data for the disorder-promoting residues. This analysis was conducted by the web-based computation tool, fractional compositional difference was computed using Composition Profiler (http://profiler.cs.ucr.edu) [51]. Since this tool generates fractional compositional difference calculated as (C − Corder)/Corder, where C is the content of a given amino acid in a query protein (set), and Corder is the corresponding value for the set of ordered proteins from PDB Select 25 (which is a subset of all proteins in PDB that share below 25% sequence identity) [84], the magnitudes of the bars indicate the fractional difference between the amino acid compositions, where positive (negative) value indicates enrichment (depletion) of the give amino acid type. The error bars indicate the standard deviations of the fractional differences based on the bootstrapping performed by the composition profiler. The proteins are color coded, and the corresponding keys are given in figure legends
This analysis showed that the amino acid compositions of ALKV proteins are characterized by some interesting features. For example, Fig. 1a shows that all these proteins are noticeably enriched in some order-promoting residues, such as W, V, and M (except to envelope protein E, which is depleted in these residues). This enrichment is partially compensated by the almost invariant depletion in the other order-promoting residues (I, Y, F, and N). Other order-promoting residues are rather differently represented in these viral proteins. For example, cysteine is common in ALKV polyprotein, proteins prM and M, envelope protein E, NS1, and NS5, whereas other viral proteins (capsid protein C, and non-structural proteins NS2A, NS2B, NS3, NS4A, and NS4B) are noticeably depleted in this residue. Leucine is commonly present in polyprotein, capsid protein C, proteins M and E, as well as NS2A, NS2B, NS4A, and NS4B, whereas prM, NS1, and NS3 are as depleted in this residue as typical IDPs. Finally, H is not common in proteins C, NS2A, NS4A, and NS5, being rather abundant in proteins M, E, NS3, and NS4B. Similarly, Fig. 1b illustrates that disorder-promoting residues R, T, G, and A are very common in the majority of viral proteins, whereas contents of other disorder-promoting residues (especially D, E, K, and Q) are low. The general enrichment of viral proteins in the positively charged R and depletion in negatively charged D and E defines the overall positive charge of these proteins. In the case of capsid protein C, which is exceptionally enriched in R, resulting high positive charge is needed for the RNA binding. Furthermore, with the exception for capsid protein C, NS1, NS3, and NS4B, mature ALKV proteins are depleted in strong disorder-promoting proline residues, indicating that many ALKV proteins are expected to be possessing rather moderate levels of structural disorder.
Intrinsic disorder in the ALKV genomic polyprotein
Figure 2 represents disorder profile generated for the ALKV genomic polyprotein by a set of commonly used disorder predictors, such as PONDR® VLXT, PONDR® VSL2, PONDR® VL3, PONDR® FIT, and IUPred (Fig. 2a) together with the mean disorder profile calculated by averaging of all predictor-specific per-residue disorder profiles (Fig. 2b). Although polyprotein is predicted to by mostly ordered, it is expected to possess noticeable structural flexibility (its mean disorder score is 0.243 ± 0.153) and have several short IDPs that account for 5.59% of its residues (see Table 1). Figure 2b shows that intrinsic disorder is unevenly distributed within polyprotein, with N-terminal part being more disordered than the C-terminal half. As a result, some of ALKV proteins within polyprotein are expected to be more disordered than others. Relatively high levels of intrinsic disorder are expected to be present in proteins C, M, NS3, and 2k, whereas proteins E, NS2A, and NS4A are predicted to be mostly ordered. Such unevenness of intrinsic disorder predisposition of individual ALKV proteins is further supported by the analysis of their mature forms (see below).
Fig. 2.

Characterizing the intrinsic disorder predisposition of the ALKV genomic polyprotein (UniProt ID: Q91B85). a Disorder profile generated by a set of commonly used disorder predictors, such as PONDR® VLXT (black curve), PONDR® VL3 (red curve), PONDR® VSL2 (green curve), PONDR® FIT (pink curve), IUPred_short (yellow curve), and IUPred_long (blue curve). Light pink shadow around the PONDR® FIT curve represents error distribution for this predictor. b Mean disorder profile calculated by averaging of all predictor-specific per-residue disorder profiles. Light blue shadow around the disorder curve show distribution of standard deviations. Positions of individual viral proteins within the ALKV polyprotein are indicated by differently colored bars, and the corresponding keys are given in figure legend. In both plots, light gray bars show locations of cleavage sites. In these analyses, the predicted intrinsic disorder scores above 0.5 are considered to correspond to the disordered residues/regions, whereas residues and regions with the disorder scores between 0.2 and 0.5 are considered as flexible
Table 1.
Evaluating intrinsic disorder predisposition of proteins from the ALK proteome and interactome
| Protein | Function | Length | Mean disorder score | Mean PPID (%) | MoRFs | Number of ELMs | Number of ELM instances | NAPID |
|---|---|---|---|---|---|---|---|---|
| Proteome of the Alkhurma virus (ALKV) | ||||||||
| Q91B85, genome polyprotein | Genome polyprotein is a product of the ALKV genome translation that includes all ALKV proteins and is cleaved by viral and host peptidases to generate mature ALKV proteins | 3416 | 0.243 ± 0.153 | 5.59 | 2–27, 606–609, 637–640, 1669–1674, 1685–1693, 1699–1702 | 121 | 1286 | 37 |
| Capsid protein C | One of structural proteins that complexes with viral mRNA to form a nucleocapsid has multiple other functions | 117 | 0.472 ± 0.154 | 39.83 | 2–27 | 28 | 63 | |
| Protein prM | Pre-membrane protein (prM) is an intracellular precursor of membrane protein M that is a component of the mature virion. PrM plays a role in immune evasion | 164 | 0.301 ± 0.137 | 19.51 | Not detected | 39 | 81 | |
| Envelope protein E | Major envelope glycoprotein is responsible for virus entry and serves as a major target of neutralizing antibodies | 496 | 0.235 ± 0.104 | 1.81 | 325–328, 356–359, 369–371 | 51 | 169 | |
| Non-structural protein 1 (NS1) | NS1 plays a role in immune evasion, pathogenesis, and viral replication, being important for the formation of the replication complex | 353 | 0.287 ± 0.132 | 8.78 | Not detected | 58 | 142 | |
| Non-structural protein 2A (NS2A) | Component of the viral RNA replication complex | 230 | 0.149 ± 0.158 | 6.96 | Not detected | 43 | 96 | |
| Serine protease subunit NS2B | NS2B is a crucial cofactor for the serine protease function of NS3 | 131 | 0.222 ± 0.145 | 4.58 | Not detected | 25 | 40 | |
| Serine protease NS3 | NS3 is a mutidomain protein with three enzymatic activities, serine protease (which is required for the autocleavage of the ALKV polyprotein at dibasic sites: C-prM, NS2A-NS2B, NS2B-NS3, NS3-NS4A, NS4A-2K and NS4B-NS5), NTPase (which is required for the 5′-RNA cap formation) and RNA helicase (which binds and unwinds dsRNA) | 621 | 0.333 ± 0.161 | 16.59 | 130–132, 178–183, 194–202, 208–211, 319–321, 474–477 | 71 | 207 | |
| Non-structural protein 4A (NS4A) | NS4A serves as regulator of the ATPase activity of the NS3 helicase | 126 | 0.187 ± 0.161 | 4.76 | Not detected | 26 | 43 | |
| Non-structural protein 4B (NS4B) with peptide 2k | NS4B triggers the formation of ER-derived membrane vesicles where the viral replication takes place | 275 | 0.189 ± 0.117 | 2.18 | Not detected | 59 | 118 | |
| RNA-directed RNA polymerase NS5 | NS5 is a RNA-directed RNA polymerase that replicates the viral RNA genome, and performs the capping of viral genomes | 903 | 0.236 ± 0.122 | 2.66 | Not detected | 72 | 314 | |
| Human proteins interacting with proteins of the Alkhurma virus (ALKV) | ||||||||
| Q92804, TATA-binding protein-associated factor 2N (TAF15) | TAF15 is an RNA and ssDNA-binding protein involved in transcription initiation at specific promoters | 592 | 0.714 ± 0.154 | 89.19 | 1–22, 32–86, 96–222, 233–243, 248–259, 295–303, 313–328, 343–354, 390–401, 585–592 | 22 | 105 | 95 |
| Q9BV73, Centrosome-associated protein CEP250 (CEP250) | CEP250 is needed for the centriole–centriole cohesion during interphase of the cell cycle | 2442 | 0.637 ± 0.134 | 84.64 | 1–13, 317–332, 527–532, 656–683, 705–711, 829–834, 848–852, 873–893, 913–920, 983–1008, 1201–1218, 1309–1330, 1344–1350, 1608–1623, 171–1729, 1839–1864, 1944–1948, 1958–1990, 1996–2008, 2119–2144, 2220–2270, 2294–2298, 2310–2330, 2383–2442 | 56 | 230 | 244 |
| Q5QJE6, Deoxynucleotidyl transferase terminal-interacting protein 2 (DNTTIP2) | DNTTIP2 acts as a chromatin remodeling protein and regulates transcriptional activities of DNA nucleotidylexotransferase (DNTT) and ethylene-responsive transcription factor (ESR1). | 756 | 0.647 ± 0.172 | 78.04 | 1–43, 49–67, 71–76, 92–121, 138–159, 170–234, 243–286, 301–308, 318–361, 371–373, 403–414, 419–427, 438–450, 461–468, 486–492, 498–525, 529–548, 620–630, 644–646, 661–664, 747–756 | 68 | 379 | 34 |
| Q8IXK0, Polyhomeotic-like protein 2 (PHC2) | PCH2 serves as an important component of a Polycomb group (PcG) multiprotein PRC1-like complex that maintains the transcriptionally repressive state of many genes, acting via chromatin remodeling and histone modifications | 858 | 0.677 ± 0.221 | 77.51 | 1–12, 42–50, 58–63, 203–352, 362–553, 562–565, 589–590, 691–735, 739–758, 766–776, 780–785 | 62 | 313 | 107 |
| Q02833, Ras association domain-containing protein 7 (RASSF7) | RASSF7 is a promoter of the MAP2K7 phosphorylation that negatively regulates stress-induced JNK activation and apoptosis | 373 | 0.631 ± 0.229 | 77.48 | 131–157, 162–163, 171–179, 192–248, 259–262, 266–267, 271–284, 291–372 | 31 | 61 | 37 |
| Q9BV36, Protein melanophilin (MLPH) | MLPH links melanosome-bound RAB27A and the motor protein MYO5A, and is involved in melanosome transport | 600 | 0.626 ± 0.247 | 76.00 | 149–154, 167–186, 190–296, 304–312, 318–329, 366–525, 535–544, 551–600 | 63 | 222 | 13 |
| Q9NWQ4, G patch domain-containing protein 2-like (GPATCH2L or C14orf118) | No functional information is currently available for GPATCH2L or C14orf118 | 482 | 0.586 ± 0.158 | 70.75 | 1–13, 22–30, 87–112, 134–135, 247–249, 304–306, 354–356, 415–441, 445–459, 464–475 | 51 | 249 | 42 |
| Q8IVT2, Mitotic interactor and substrate of PLK1 (MISP) | MISP is involved in mitotic spindle orientation and mitotic progression | 679 | 0.611 ± 0.170 | 70.69 | 18–23, 40–112, 121–259, 265–288, 299–328, 333–339, 347–357, 367–480, 490–493, 541–543, 567–579, 591–599, 618–626, 635–641, 649–655, 667–679 | 58 | 298 | 60 |
| Q5VU43, Myomegalin (PDE4DIP) | Myomegalin is a centrosome and cis-Golgi localized protein that is required for the microtubule (MT) growth from the centrosome and Golgi apparatus (GA) | 2346 | 0.567 ± 0.151 | 70.38 | 122–130, 199–223, 235–246, 299–326, 437–442, 663–669, 679–795, 888–892, 936–1018, 1064–1074, 1153–1213, 1299–1303, 1308–1311, 1399–1417, 1560–1568, 1584–1614, 1624–1718, 1742–1769, 1779–1800, 1825–1847, 1886–1890, 1917–1921, 1932–1935, 2083–2170 | 79 | 567 | 171 |
| Q8N5G2, Macoilin (MACO1) | Macoilin has a multitude of neural functions and is related to various processes, such as locomotion and chemotaxis | 664 | 0.466 ± 0.260 | 63.86 | 217–220, 241–253, 266–310, 323–337, 363–399, 414–416, 432–447, 555–561, 644–646 | 57 | 224 | 27 |
| Q99750, MyoD family inhibitor (MDFI) | MDFI serves as an inhibitor of the transactivation activity of the Myod family of myogenic factors and represses myogenesis | 246 | 0.635 ± 0.251 | 62.20 | 1–85, 96–134 | 38 | 86 | 269 |
| Q96EA4, Protein Spindly (SPDL1 or Coiled-coil domain-containing protein 99 (CCDC99)) | Spindly targets dynein/dynactin to kinetochores in mitosis and can activate its motility | 605 | 0.544 ± 0.111 | 61.98 | 166–169, 478–479, 518–523, 543–547, 555–564, 590–595 | 46 | 118 | 37 |
| Q9H814, Phosphorylated adapter RNA export protein (PHAX or RNA U small nuclear RNA export adapter protein (RNUXA)) | PHAX is involved in the XPO1-mediated U snRNA export from the nucleus | 394 | 0.572 ± 0.188 | 60.91 | 1–40, 95–106, 110–119, 131–141, 154–160, 170–185, 197–210, 214–218, 341–343, 354–383 | 45 | 137 | 53 |
| Q96CN5, Leucine-rich repeat-containing protein 45 (LRRC45) | LRRC45 is involved in the formation of the fiber-like linker between two centrioles, required for centrosome cohesion | 670 | 0.502 ± 0.210 | 60.45 | 243–248, 262–264, 277–280, 357–360, 396–411, 433–450, 611–615, 668–670 | 26 | 50 | 12 |
| Q9H7J1, Protein phosphatase 1 regulatory subunit 3E (PPP1R3E) | PPP1R3E is a glycogen-targeting subunit for protein phosphatase 1 (PP1) | 279 | 0.569 ± 0.219 | 58.42 | 1–16, 22–23, 28–35, 43–89, 112–117 | 43 | 98 | 2 |
| Q68CP9, AT-rich interactive domain-containing protein 2 (ARID2) | ARID2 is involved in transcriptional activation and repression of select genes by chromatin remodeling | 1835 | 0.518 ± 0.265 | 57.44 | 1–8, 13–15, 623–670, 673–690, 729–734, 756–781, 807–809, 891–899, 940–951, 954–1024, 1045–1096, 1109–1111, 1132–1134, 1196–1201, 1210–1219, 1239–1245, 1250–1269, 1285–1331, 1341–1475, 1478–1505, 1520–1542, 1550–1604, 1614–1628, 1695–1704, 1727–1733 | 78 | 585 | 40 |
| P35579, Myosin-9 (MYH9) | Myosin-9 is a non-muscle cellular myosin that play a role in cytokinesis, cell shape, and specialized functions such as secretion and capping | 1960 | 0.487 ± 0.251 | 55.71 | 1020–1033, 1059–1064, 1089–1093, 1101–1104, 1116–1120, 1140–1159, 1172–1199, 1233–1237, 1302–1305, 1354–1378, 1477–1523, 1536–1549, 1555–1573, 1603–1629, 1637–1691, 1705–1718, 1746–1751, 1763–1772, 1847–1851, 1864–1875, 1895–1960 | 69 | 472 | 387 |
| Q7Z5R6, Amyloid beta A4 precursor protein-binding family B member 1-interacting protein (APBB1IP) | APBB1IP is required for the signal transduction from Ras activation to actin cytoskeletal remodeling | 666 | 0.553 ± 0.311 | 55.56 | 1–11, 22–47, 55–72, 92–98, 120–158, 445–451, 466–601, 608–666 | 52 | 198 | 22 |
| P08670, Vimentin (VIM) | Vimentin is a class-III intermediate filament found in different non-epithelial cells, e.g. mesenchymal cells. It is attached to the nucleus, ER, and mitochondria | 466 | 0.525 ± 0.162 | 54.29 | 2–19, 40–43, 344–347, 460–466 | 37 | 97 | 333 |
| Q14160, Protein scribble homolog (SCRIB) | SCRIB is an important scaffold protein regulating epithelial and neuronal morphogenesis via involvement in different aspects of the differentiation of polarized cells | 1630 | 0.558 ± 0.265 | 51.53 | 397–402, 413–440, 444–608, 610–711, 715–731, 754–763, 798–800, 822–832, 840–863, 929–932, 946–981, 1131–1138, 1225–1265, 1272–1389, 1397–1630 | 72 | 406 | 133 |
| O00327, Aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL) | ARNTL is a transcriptional activator that forms a core component of the circadian clock | 626 | 0.480 ± 0.282 | 50.63 | 1–34, 45–84, 453–606, 615–625 | 47 | 146 | 70 |
| O15066, Kinesin-like protein KIF3B (KIF3B) | KIF3B is involved in tethering the chromosomes to the spindle pole and in chromosome movement and serves as a microtubule-based anterograde translocator for membranous organelles | 747 | 0.516 ± 0.215 | 50.47 | 334–335, 351–354, 356–358, 425–442, 460–470, 474–481, 631–633, 641–650, 668–669, 673–709, 729–747 | 37 | 96 | 37 |
| P31942, Heterogeneous nuclear ribonucleoprotein H3 (HNRNPH3) | HNRNPH3 is an RNA-binding protein forming complex with heterogeneous nuclear RNA (hnRNA), plays a role in the splicing process, and is involved in early heat shock-induced splicing arrest | 346 | 0.489 ± 0.195 | 50.00 | 100–103, 107–113, 262–266 | 42 | 79 | 128 |
| O94983, Calmodulin-binding transcription activator 2 (CAMTA2) | CAMTA2 is a transcription activator that may act as tumor suppressor | 1202 | 0.503 ± 0.279 | 48.59 | 253–495, 512–521, 660–675, 818–935, 957–966. 999–1000, 1138–1164, 1186–1189, 1197–1200 | 73 | 427 | 12 |
| Q9Y6X2, E3 SUMO-protein ligase PIAS3 (PIAS3) | PIAS3 is an E3-type SUMO (small ubiquitin-like modifier) ligase | 628 | 0.435 ± 0.224 | 40.13 | 76–83, 88–95, 99–101, 107–124, 429–434, 444–464, 481–497, 591–616 | 66 | 232 | 82 |
| O14530, Thioredoxin domain-containing protein 9 (TXNDC9) | TXNDC9 negatively affects protein folding, including folding of actin or tubulin via decreasing the ATPase activity of chaperonin TCP1 complex | 226 | 0.391 ± 0.233 | 34.51 | Not detected | 19 | 26 | 79 |
| Q9NS15, Latent-transforming growth factor beta-binding protein 3 (LTBP3) | LTBP3 is related to the assembly, secretion, and targeting of the transforming growth factor beta-1 (TGFB1) to sites of its storage and/or activation | 1303 | 0.412 ± 0.225 | 30.24 | 158–269, 474–571, 1197–1209 | 56 | 217 | 15 |
| Q13105, Zinc finger and BTB domain-containing protein 17 (ZBTB17) | ZBTB17 is a transcription factor that, depending on its binding partners, may act as an activator or repressor and plays an important role at early stages of lymphocyte development | 803 | 0.433 ± 0.282 | 29.51 | 122–131, 139–297, 322–326, 774–803 | 39 | 122 | 50 |
| P52742, Zinc finger protein 135 (ZNF135) | ZNF135 may regulate transcription and control cell morphology and cytoskeletal organization | 658 | 0.426 ± 0.165 | 29.03 | 1–3, 169–178, 230–233, 258–261, 286–288, 398–400, 426–429, 482–485, 510–512, 538–541, 566–569, 622–628 | 37 | 72 | 7 |
| P55268, Laminin subunit beta-2 (LAMB2) | LAMB2 can bind to cells via a high affinity receptor and interact with other extracellular matrix components, thereby mediating the attachment, migration and organization of cells into tissues during embryonic development | 1798 | 0.351 ± 0.177 | 21.25 | 1–3, 1337–1397, 1452–1466, 1492–1498, 1527–1530, 1640–1642, 1651–1655 | 51 | 187 | 39 |
| P55287, Cadherin-11 (CDH11) | Cadherin-11 is a calcium-dependent cell adhesion protein that contributes to the sorting of heterogeneous cell types | 796 | 0.349 ± 0.143 | 16.58 | 124–150, 157–167, 256–261, 269–279, 284–286, 508–511, 672–679, 702–714, 719–743 | 36 | 78 | 8 |
| P19474, E3 ubiquitin-protein ligase TRIM21 (TRIM21) | TRIM21 is an E3 ubiquitin-protein ligase | 475 | 0.317 ± 0.173 | 17.47 | Not detected | 37 | 71 | 105 |
| P78504, Protein jagged-1 (JAG1) | Jagged-1 is involved in the mediation of Notch signaling, serving as a ligand for multiple Notch receptors | 1218 | 0.273 ± 0.175 | 11.08 | 1120–1184, 1194–1218 | 53 | 170 | 21 |
| P28799, Granulins (GRN) | Granulins have cytokine-like activity and play a role in inflammation, tissue remodeling, and wound repair | 593 | 0.286 ± 0.137 | 10.12 | 346–353 | 31 | 48 | 121 |
| Q9H270, Vacuolar protein sorting-associated protein 11 homolog (VPS11) | VPS11 is related to the vesicle-mediated protein trafficking to lysosomal compartments including the endocytic membrane transport and autophagic pathways | 941 | 0.241 ± 0.150 | 8.11 | 608–611 | 67 | 306 | 59 |
| Q9BPW8, Protein NipSnap homolog 1 (NIPSNAP1) | NIPSNAP1 serves as a regulator of the transient receptor potential vanilloid channel 6 (TRPV6), which is an epithelial Ca2+ channel that mediates Ca2+ uptake in various tissues | 284 | 0.246 ± 0.140 | 7.04 | Not detected | 39 | 78 | 79 |
| P06733, α-Enolase (ENO1) | α-Enolase is a multifunctional enzyme involved in glycolysis and various biological processes, such as growth control, hypoxia tolerance, and allergic responses | 434 | 0.243 ± 0.119 | 3.46 | Not detected | 32 | 66 | 213 |
| Q14192, Four and a half LIM domains protein 2 (FHL2) | FHL2 serves as a molecular transmitter that links various signaling pathways to transcriptional regulation | 279 | 0.162 ± 0.095 | 1.43 | Not detected | 34 | 63 | 194 |
Figure 2 also indicates that the majority of proteolytic cleavage sites utilized by internal and external proteases for the generation of mature ALKV proteins have high intrinsic disorder predispositions, especially in comparison with the neighboring residues. This observation is further illustrated by Fig. 3 that represents zoomed-in regions surrounding all such cleavage sites of the ALKV polyprotein and shows that these sites either located within disordered/flexible regions or in the close proximity to such regions. The only two exceptions from this regularity are sites surrounding the most ordered ALKV protein, envelope protein E. However, even in these cases, cleavage sites are positioned in close proximity to regions with elevated structural mobility. Such localization of cleavage sites within the ALKV polyprotein reflects crucial role of intrinsic disorder and structural flexibility in the polyprotein processing and maturation of individual viral proteins. This correlation is also a reflection of a general phenomenon, where the rates of proteolytic cleavage in unstructured or flexible regions are known to be orders of magnitude higher than those within the structured protein regions [85–90], and thereby showing crucial importance of the localization of cleavage sites within the regions that lack structure or possess high structural flexibility.
Fig. 3.

Disorder propensities of the ALKV polyprotein regions containing cleavage sites between capsid protein C and ER anchor (a), ER anchor and protein prM (b), peptide pr and protein M (c), protein M and envelope protein E (d), proteins E and NS1 (e), NS1 and NS2A (f), NS2A and NS2B (g), NS2A and NS3 (h), NS3 and NS4A (i), protein NS4A and peptide 2k (j), peptide 2k and NS4B (k), and proteins NS4A and NS5 (l). Disorder propensities were evaluated by PONDR® VLXT (black curves), PONDR® VL3 (red curves), PONDR® VSL2 (green curves), PONDR® FIT (pink curves), IUPred_short (yellow curves), and IUPred_long (blue curves). Mean disorder predisposition was calculated by averaging of all predictor-specific per-residue disorder profiles (bold, dashed, dark cyan curves). Light pink shadow around the PONDR® FIT curve represents error distribution for this predictor, whereas light blue shadow around the mean disorder curve show distribution of standard deviations. Cleavage sites are shown as gray bars
Since several ALKV polyproteins were found in different ALKV isolates from infected humans, we also investigated the effect of polymorphism on the intrinsic disorder predisposition of the different variants of the ALKV polyprotein. Results of this analysis are shown in Fig. 4 and further summarized in Table S1 (see Supplementary Materials). Typically, disorder profiles were minimally affected by the majority of mutations, which were predicted to have rather limited local effects, causing moderate increase or decrease in intrinsic disorder predisposition of regions surrounding mutation sites (see Table S1). This is not too surprising, since, although variants of the ALKV polyprotein have several polymorphic sides, the sequence identity of these proteins does not decrease below 99.12% (see Supplementary Materials). However, Fig. 4 also shows that at least three regions with high levels of disorder (residues 175–205, 341–379, and 3202–3222) were rather strongly affected by the polymorphism-related mutations. Although exact functional consequences of these changes in the disorder predisposition of these regions are not known, the first of these regions (residues 175–205) is located in the proximity to a cleavage site leading to the release of the pr peptide from the membrane precursor protein prM, suggesting that polymorphism in this region (that leads to the decrease in local disorder predisposition) can affect the efficiency of the proteolysis, thereby increasing the probability of the partial maturation and existence of the uncleaved prM that can be related to the immune-evasion strategy of flaviviruses [91]. The second polymorphism-affected region (residues 341–379) is located within the extracellular domain of the envelope protein E, and, therefore, can be involved in the regulation of the capability of this viral protein to interact with the host. Finally, the last region strongly affected by polymorphism (residues 3202–3222) is located within the RNA-directed RNA polymerase (RdRp) NS5 in the close proximity to the RdRp catalytic domain of this protein (residues 3042–3191), suggesting that mutations in this region might affect efficiency of the NS5 interaction with its partners.
Fig. 4.

Effect of amino acid polymorphism on the intrinsic disorder predisposition of the different variants of the ALKV polyprotein evaluated by PONDR® VSL2. Different plots show consecutive 500 residue-long fragments of the ALKV polyprotein to better illustrate local changes in the disorder predisposition in regions surrounding polymorphic sites. The proteins are color coded, and the corresponding keys are given in figure legends
Curiously, Fig. 5 shows that the peculiarities of the local intrinsic disorder predisposition are rather well preserved among the genomic polyproteins of the Flavivirus genus of the Flaviviridae family. In fact, polyproteins of closely related ALKV and Kyasanur forest disease virus (KFDV, UniProt ID: D7RF80) that share 97.16% of identical residues possess almost indistinguishable disorder profiles (see Fig. 5). Even disorder profiles of more distant ALKV relatives, Zika virus (strain Mr 766, ZIKV, UniProt ID: Q32ZE1) and Dengue virus (type 1, strain Singapore/S275/1990, DENV, UniProt ID: P33478) with the sequence identities to ALKV polyprotein of 41.63% and 40.88%, respectively, and with the sequence identity to each other of 55.26% (see Supplementary Materials), share many local features with the disorder profiles of ALKV and KFDV. Furthermore, aligned amino acid sequences of these polyproteins shown in Supplementary Materials and their aligned disorder profiles shown in Fig. 5 illustrate remarkable conservation of cleavage sites and their preferential localization within the disordered/flexible regions. These observations are in line with the results of analogous analysis of DENV, where it was shown that IDPRs occurred in cleavage sites 6.2 times more often compared to the overall genome [13]. Therefore, cleavage sites in flaviviruses are substantially enriched in disorder and depleted in polymorphic sites. In general, all these observations clearly show high conservation level of the peculiarities of distribution of intrinsic disorder predisposition within the sequences of studied flaviviral polyproteins, providing strong support for the importance of both ordered and disordered regions for the functionality of viral proteins.
Fig. 5.

Analysis of the evolutionary conservation of the intrinsic disorder predisposition in some flaviviral polyproteins, such as ALKV (UniProt ID: Q91B85, black curves), KFDV (UniProt ID: D7RF80, dashed red curves), ZIKV (strain Mr 766, UniProt ID: Q32ZE1, dashed green curves), and DENV (type 1, strain Singapore/S275/1990, UniProt ID: P33478, dashed yellow curves). Different plots show consecutive 870 residue-long fragments of the aligned polyproteins to better represent some peculiarities in the local distribution of their intrinsic disorder predispositions. Sequences of these polyproteins were aligned using Clustal Omega algorithm [52]. The gaps in sequence alignments are shown as gaps in the corresponding disorder profiles. The original results of multiple sequence alignment including aligned sequences, conservation matrix, and evolutionary tree are shown in Supplementary Materials. Localization of cleavage sites is indicated by gray bars
Intrinsic disorder and functionality of mature ALKV proteins
Viruses are characterized by a rational use of their limited genetic materials and proteins. As a result, many viral proteins have multiple functions and are utilized at different stages of the viral life cycle. Although functions of the ALKV proteins are poorly characterized, and although no structural information is currently available for any ALKV protein, high sequence similarity of ALKV proteins to their homologues from other flaviviruses provides strong grounds for making reliable assumptions on the functionality of these proteins.
As it was already indicated, similar to other flaviviral proteins, all ALKV proteins (three structural proteins, envelope, E; membrane precursor, PrM; capsid C, and seven non-structural (NS) proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B/2k, and NS5) are generated as a result of the co- and post-translational cleavage of the genomic polyprotein encoded by a single ORF. The viral genome is translated into the viral polyprotein on the rough endoplasmic reticulum (ER) [92]. NS1, prM, and E are translated into the ER lumen, the transmembrane domains of NS2A, NS2B, NS4A, and NS4B are translated into the ER membrane, and proteins C, NS3, and NS5 remain in the cytoplasm [12, 93]. Processing of this polyprotein is managed by a combined action of the furin-type or some other Golgi-localized cellular proteases and the viral NS3 serine protease. As it follows from their names, structural proteins form the virus particle. They also play a role in virus entry into the host cell and are engaged in the assembly and release of new virions. For example, capsid C protein is classically depicted as a structural protein that functions by sheltering the viral genome via the assembly of the viral nucleocapsid [94]. The nucleocapsid is surrounded by a host-derived lipid membrane, in which two transmembrane proteins are inserted, the major envelope glycoprotein E (53 kDa) and the membrane protein M (8 kDa). On the envelope of the mature virus particle, there are 180 copies of the E and M proteins, where 90 glycoprotein E dimers form icosahedral scaffold that completely covers the viral surface [95, 96]. Besides their classical structural roles, each of the structural proteins C, prM/M, and E has a multitude of functions that goes beyond the virus assembly process. Complete description of all the functions of the structural proteins is outside the scopes of this study. Therefore, they will be only briefly listed below.
As many of the viral proteins are known to interact with or be embedded into the membrane, we also looked at the presence of transmembrane regions in the ALKV proteins. According to UniProt, the ALKV genomic polyprotein is predicted to contain 18 transmembrane α-helical regions (residues 100–120, 244–261, 263–281, 729–749, 757–777, 1135–1155, 1163–1183, 1190–1210, 1236–1256, 1296–1316, 1362–1379, 1385–1405, 2163–2183, 2213–2233, 2247–2267, 2369–2389, 2433–2453, and 2477–2497) and four intramembrane α-helical regions (residues 1459–1479, 2192–2211, 2302–2322, and 2346–2366). In other words, almost all processed ALKV proteins [except for the non-structural proteins NS1 (residues 778–1130), NS3 (residues 1492–2112), and RNA-directed RNA polymerase NS5 (residues 2514–3416)] are expected to possess membrane-interacting elements. For example, the C-terminal region of capsid protein C contains ER anchor (residues 97–117), which is removed in the mature form of this protein by serine protease NS3. Similarly, small envelope protein M (residues 207–281) and envelope protein E (residues 282–777) each contain two transmembrane α-helices localized in their C-terminal regions. Non-structural protein NS2A (residues 1131–1360) has five transmembrane regions, whereas serine protease subunit NS2B (residues 1361–1491) has two transmembrane and one intramembrane regions. Non-structural protein NS4A (residues 2113–2238) has two transmembrane and one intramembrane regions, and peptide 2k (residues 2239–2261) includes one transmembrane α-helix. Finally, non-structural proteins NS4B (residues 2262–2513) contains three transmembrane and two intramembrane regions. Since these regions of the ALKV proteins are embedded into the membrane, they are characterized by high content of hydrophobic residues and are predicted to be ordered.
Protein C is crucial for virus budding and formation of a nucleocapsid representing the core of a mature virus particle [94]. Here, protein C binds to the membrane of a host cell and gathers the viral RNA into a nucleocapsid. It is also play a role in virus entry by inducing genome penetration into the host cytoplasm after hemifusion. The C protein can migrate to the nucleus of flavivirus-infected cells [97] and modulate functions of host nuclear proteins [94], e.g., acting as a histone mimic, interacting with core histones, and disrupting nucleosome formation [98], or interacting with other host nuclear proteins, such as heterogeneous nuclear ribonucleoprotein K (hnRNP-K) [99], death domain-associated protein 6 (DAXX) [100], and nucleolin (NCL) [101]. Finally, the C protein can be involved in the inhibition of the RNA silencing via interference with the host ribonuclease Dicer [102].
The membrane protein M (also known as small envelope protein M) is produced in the ER of a host cell as a precursor protein, pre-membrane protein (prM). Being inserted, together with the envelope protein E, into the ER membrane, prM plays an important role in acquiring viral envelope during budding. It forms heterodimers with protein E, and newly formed immature viral particle is covered with 60 spikes composed of such heterodimers. The prM protein is also known for acting as a chaperone for envelope protein E during intracellular virion assembly by aiding folding and maturation of E proteins [103, 104], and protecting the E proteins from premature fusion by the low pH conditions in the transport vesicles via masking and inactivating envelope protein E fusion peptide [105–108]. Activation and maturation of the assembled virion are initiated by the cleavage of the prM protein by furin, a protease residing in the trans-Golgi network (TGN) [109]. It was also indicated that many flaviviral virions are only partially matured because of the inefficient prM cleavage, and such partial maturation and existence of the uncleaved prM represents an immune-evasion strategy of flaviviruses [91]. Finally, besides playing a role in the virus budding, mature protein M possesses proapoptotic properties and shows the cytotoxic capability via the activation of a mitochondrial apoptotic pathway [110].
The major functions of the envelope protein E (which is a large membrane-bound glycoprotein) are to mediate virus attachment to cells and entry, as well as to promote fusion between viral and cellular membranes via binding to host cell surface receptors. It also plays a role in virion budding in the ER and serves as a main antigenic target for the development of neutralizing humoral immunity. Although the immature viral particle is characterized by a spiky surface due to the presence of 60 trimeric protrusions containing trimers of PrM-E heterodimers, the low pH environment of the Golgi apparatus causes reorganization of the spiky PrM-E heterodimers into antiparallel E homodimers that lie flat against the surface on the virion [110]. As a result of these structural reorganizations, cleavage site of prM becomes exposed and accessible for digestion by the host furin. Since not all prM can be cleaved [111], uncleaved E-prM heterodimers return back to a spiky immature trimeric structure [112].
All flaviviral NS proteins are multifunctional and play very diverse roles in the control and orchestration of various stages of viral life, including regulation of viral morphogenesis [113] and formation of the replication complex (RC) [114]. Only NS3 and NS5 possess enzymatic activities (which together account for all enzymatic activities required to amplify the RNA genome and to attach a type 1 cap to its 5′ end), whereas biological roles of other NS proteins are not related to the catalysis. NS1 is a secreted protein with multiple roles in immune evasion, pathogenesis, and viral replication cycle, which exists in multiple oligomeric forms and is found in different cellular locations, such as sites of viral replication within the cell, the cell surface, and secreted into the extracellular space [115]. It can be present as a cell membrane-bound form in association the with virus-induced intracellular vesicular compartments, or on the cell surface, or as a soluble secreted hexameric lipoparticle that plays a role against host immune response [115]. It is needed for the RC formation and recruitment of other NS proteins to the ER-derived membrane structures [115–117]. NS1 (especially its secreted form) interacts with numerous host proteins [115]. NS2A, NS2B, NS4A, and NS4B are transmembrane proteins located within the ER membrane. They serve as important components of the viral RNA RC and possess multiple crucial functions. For example, NS2A plays a role in viral RNA synthesis, viral assembly, inhibition of the interferon α/β response, virus-induced membrane formation, and production of NS1′ through a ribosomal frameshift mechanism [118]. NS2B is known as a regulatory subunit of the flaviviral serine protease NS3, plays a role controlling the localization of NS3 to membranes, possesses membrane-destabilizing activity [114], and may act as a viroporin (a class of small, hydrophobic, viral proteins that have ion-channel activity in vitro, and are involved in enhancing infectivity) [113]. NS3 is a multidomain protein that has three enzymatic activities, such as N-terminal domain with serine protease, and C-terminal domain with RNA helicase and RNA-stimulated nucleoside triphosphate hydrolase and 5′-RNA triphosphatase activities [119]. NS3 serine protease uses NS2B as a cofactor and cleaves the viral polyprotein at several dibasic positions between C and prM, NS2A and NS2B, NS2B and NS3, NS3 and NS4A, NS4A and 2k, and between NS4B and NS5 [120]. After binding to RNA, the helicase activity of NS3 (which is regulated by the NS4A protein) is utilized for the unwinding of the double-stranded (ds) RNA intermediate formed during genome synthesis, whereas the 5′-RNA triphosphatase activity of this polyfunctional protein is required for the 5′-RNA cap formation [121]. NS4B protein contains a 2k peptide that serves as a signal peptide for NS4B, and removal of this peptide is required for the NS4B-triggered formation of ER-derived membrane vesicles where the viral replication takes place [122]. Together with viral NS2A, NS2B, and NS4A proteins NS4B forms a scaffold for the assembly of NS3 and NS5 proteins [119], e.g., NS3 interacts with NS4B through its C-terminal helicase domain [123]. NS4B can also interact with NS1 [124] and counteract innate immune responses such as formation of stress granules, RNA interference, type I interferon signaling, and the unfolded protein response [125]. Finally, a second flaviviral protein with enzymatic activities is NS5, which is the largest protein in flaviviruses that has a multidomain organization and shows an N-terminal methyltransferase (MTase) and a C-terminal RNA-dependent RNA polymerase (RdRp) activities [119]. The MTase domain possesses RNA guanylyltransferase and methyltransferase activities needed for 5′-RNA capping and cap methylations [126, 127], whereas the RdRp domain of NS5 is used for the replication of viral genome and carries out both (−) and (+) strand RNA synthesis [128, 129]. Furthermore, NS5 antagonizes the host type I interferon response by preventing JAK-STAT signaling and prevents the establishment of cellular antiviral state by blocking the interferon-α/β signaling pathway [130].
Since protein multifunctionality is commonly associated with the presence of intrinsic disorder [22, 27, 131–135], and since many viral proteins contain functional IDPRs [13, 14, 41–47], we conducted comprehensive computational analysis of the mature ALKV proteins utilizing a set of commonly used disorder predictors. Results of this analysis are shown in Fig. 6 and further summarized in Table 1. The levels of intrinsic disorder in ALKV proteins range from 39.8% in capsid protein C to 1.8% in envelope protein E (see Table 1). Two more ALKV proteins have PPIDs exceeding 10%, PrM (19.5%) and NS3 (16.6%), whereas PPID values of the remaining proteins are below 10%. Curiously, the mean percent of predicted intrinsic disorder (i.e., percent of residues predicted to have the disorder score above 0.5 in mean disorder profile) does not always correlate with the mean disorder score of a query protein (see Table 1 and Fig. 7A). In fact, although E protein is characterized by the lowest PPID of 1.8%, it has the mean disorder score of 0.235 ± 0.104, whereas the lowest mean disorder score of 0.149 ± 0.158 is found for NS2A, which is characterized by the PPID of 7%.
Fig. 6.

Intrinsic disorder predisposition of mature ALKV proteins: a Capsid protein C; b Protein prM; c Envelope protein E; d non-structural protein 1, NS1; e non-structural protein 2A, NS2A; f serine protease subunit NS2B; g serine protease NS3; h non-structural protein 4A, NS4A; i non-structural protein 4B, NS4B; j RNA-directed RNA polymerase NS5. Disorder propensities were evaluated by PONDR® VLXT (black curves), PONDR® VL3 (red curves), PONDR® VSL2 (green curves), PONDR® FIT (pink curves), IUPred_short (yellow curves), and IUPred_long (blue curves). Mean disorder predisposition was calculated by averaging of all predictor-specific per-residue disorder profiles (bold, dashed, dark cyan curves). Light pink shadow around the PONDR® FIT curve represents error distribution for this predictor, whereas light blue shadow around the mean disorder curve shows distribution of standard deviations
Fig. 7.

Intrinsic disorder predisposition of the members of ALKV interactome: A Q92804, TATA-binding protein-associated factor 2N (TAF15); B Q9BV73, Centrosome-associated protein CEP250 (CEP250); C Q5QJE6, Deoxynucleotidyl transferase terminal-interacting protein 2 (DNTTIP2); D Q8IXK0, Polyhomeotic-like protein 2 (PHC2); E Q02833, Ras association domain-containing protein 7 (RASSF7); F Q9BV36, Protein melanophilin (MLPH); G Q9NWQ4, G patch domain-containing protein 2-like (GPATCH2L or C14orf118); H Q8IVT2, Mitotic interactor and substrate of PLK1 (MISP); I Q5VU43, Myomegalin (PDE4DIP); J Q8N5G2, Macoilin (MACO1); K Q99750, MyoD family inhibitor (MDFI); L Q96EA4, Protein Spindly (SPDL1 or Coiled-coil domain-containing protein 99 (CCDC99)); M Q9H814, Phosphorylated adapter RNA export protein [PHAX or RNA U small nuclear RNA export adapter protein (RNUXA)]; N Q96CN5, Leucine-rich repeat-containing protein 45 (LRRC45); O Q9H7J1, Protein phosphatase 1 regulatory subunit 3E (PPP1R3E); P Q68CP9, AT-rich interactive domain-containing protein 2 (ARID2); Q P35579, Myosin-9 (MYH9); R Q7Z5R6, Amyloid beta A4 precursor protein-binding family B member 1-interacting protein (APBB1IP); S P08670, Vimentin (VIM); T Q14160, Protein scribble homolog (SCRIB); U O00327, Aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL); V O15066, Kinesin-like protein KIF3B (KIF3B); W P31942, Heterogeneous nuclear ribonucleoprotein H3 (HNRNPH3); X O94983, Calmodulin-binding transcription activator 2 (CAMTA2); Y Q9Y6X2, E3 SUMO-protein ligase PIAS3 (PIAS3); Z O14530, Thioredoxin domain-containing protein 9 (TXNDC9); a Q9NS15, Latent-transforming growth factor beta-binding protein 3 (LTBP3); b Q13105, Zinc finger and BTB domain-containing protein 17 (ZBTB17); c P52742, Zinc finger protein 135 (ZNF135); d P55268, Laminin subunit beta-2 (LAMB2); e P55287, Cadherin-11 (CDH11); f P19474, E3 ubiquitin-protein ligase TRIM21 (TRIM21); g P78504, Protein jagged-1 (JAG1); h P28799, Granulins (GRN); i Q9H270, Vacuolar protein sorting-associated protein 11 homolog (VPS11); j Q9BPW8, Protein NipSnap homolog 1 (NIPSNAP1); k P06733, α-Enolase (ENO1); l Q14192, Four and a half LIM domains protein 2 (FHL2). Each plot contains an area graph representing the distribution of mean disorder predisposition calculated for a given protein by averaging of the per-residue disorder profiles generated by PONDR® VLXT, PONDR® VL3, PONDR® VSL2, PONDR® FIT, IUPred_short, and IUPred_long. The corresponding disorder profiles generated for query proteins by these six per-residue disorder predictors are assembled in Supplementary Materials
Next, we look at the presence in ALKV proteins molecular recognition features (MoRFs), which are short disordered regions that undergo disorder-to-order transition at binding to their protein partners, and which are commonly related to signaling and regulatory functions of their carriers [136–139]. Table 1 shows that only three ALKV proteins (C, E, and NS3) contain MoRFs. Besides MoRFs, IDPRs are known to contain short (3–11 residue-long) conserved functional sequence motifs [62]. Table 1 shows that the ALKV polyprotein contains 1286 ELMs belonging to 121 types, and all ALKV proteins have ELMs. Mature proteins contain from 40 (as in NS2B) to 314 (as in NS5) ELM instances per protein, with the number of different ELM types per ALKV protein ranging from 25 to 72. Data assembled in Supplementary materials show that ELMs found in ALKV proteins correspond to the motifs that serve as proteolytic cleavage sites, docking motifs corresponding to the interaction sites that are distinct from active sites of the modifying enzyme, post-translational modification sites, ligand-binding motifs, motifs for recognition and targeting to subcellular compartments, as well as degron motifs that are involved in polyubiquitylation and targeting the protein to the proteasome for degradation.
Intrinsic disorder in human proteins interacting with ALKV proteins
Although numerous human proteins targeted by various flaviviruses are known, information pertaining to the ALKV interactome is rather limited. According to the IntAct protein interaction database and analysis system (https://www.ebi.ac.uk/intact/) [49], 38 human proteins were shown to interact with ALKV proteins. These proteins were identified in a high-throughput yeast two-hybrid (Y2H) screen of host proteins interacting with the NS3 and NS5 proteins of various flaviviruses, which established a set of 108 human proteins was found to interact with NS3 or NS5 proteins or both [140]. The authors of that study also emphasized that only one-third of the cellular targeted proteins was able to interact with two or more flaviviruses, with only three human proteins (APBB1IP, ARID2, and ENO1) being able to interact with at least four viruses, and with only five cellular proteins (CAMTA2, CEP250, SSB, ENO1, and FAM184A) being able to interact with both NS3 and NS5 proteins [140]. Among the 38 human proteins interacting with NS3 and NS5 of ALKV, only four proteins (APBB1IP, CAMTA2, FHL2, and KIF3B) were engaged in binding to the NS3 protein (mostly to its helicase domain), whereas the vast majority of human proteins were interacting with the methyltransferase domain of NS5 from ALKV [140].
We looked at the intrinsic disorder predisposition of these 38 proteins using a set of the disorder predictors utilized in the analysis of ALKV proteins. Results of these analyses are shown in Figs. 7 and 8 and further summarized in Table 1. Note that area plots shown in Fig. 7 are used here for the demonstration purpose only (to illustrate the distribution of mean intrinsic disorder propensity in the members of ALKV interactome calculated for each protein by averaging of predictor-specific per-residue disorder profiles generated PONDR® VLXT, PONDR® VL3, PONDR® VSL2, PONDR® FIT, IUPred_short, and IUPred_long), whereas detailed graphs showing the results of the multiparametric intrinsic disorder analysis in these proteins are shown in Supplementary Materials. This analysis revealed the presence of very high levels of intrinsic disorder in the majority of the ALKV interactome members. In fact, according to the accepted classification of proteins based on their PPID values as highly ordered, moderately disordered, or highly disordered if their PPID < 10%, 10% ≤ PPID < 30%, or PPID ≥ 30%, respectively [141], 27 of 38 human proteins shown to interact with ALKV proteins are predicted to by highly disordered, 7 members of the ALKV interactome are moderately disordered, and only 4 these proteins are predicted to be highly ordered. Table 1 also shows that 23 ALKV-binding human proteins are expected to contain at least 50% disordered residues, with the overall disorder level can be as high 89.19%. The conclusion on the high disorder predisposition of these proteins is further illustrated by Fig. 8, where the dependence of the mean disorder score on the corresponding mean PPID for the 38 members of the ALKV interactome are shown. Analogous data for the mature ALKV proteins are also presented for the comparison (Fig. 8a). Figure 8b shows that the vast majority of human proteins interacting with ALKV are exceedingly disordered.
Fig. 8.

Global disorder propensity of the members of ALKV proteome (a) and interactome (b) presented as dependence of the percentages of residues in these proteins predicted to be disordered by averaging the outputs of PONDR® VLXT, PONDR® VL3, PONDR® VSL2, PONDR® FIT, IUPred_short, and IUPred_long on their mean disorder scores
Furthermore, all members of the ALKV interactome analyzed in this study are expected to have multiple ELMs and almost all these proteins are expected to contain multiple MoRFs, indicating that high levels of intrinsic disorder in these proteins are of functional importance, with their IDPRs being extensively utilized in protein–protein interactions. In agreement with this hypothesis, Table 1 represents the results of the analysis of the interactivity of these proteins by the APID web server (http://apid.dep.usal.es) [71]. This analysis revealed that, on average, the ALKV interactome members are engaged in interaction with > 90 partners each, with the number of known partners for these proteins ranging from 2 (for PPP1R3E) to 387 (for myosin-9). Furthermore, more than half of these proteins (55%) are able to interact with more than 50 partners each, and the vast majority of the ALKV interactome members (92%) are interacting with more than ten partners each. These observations suggest that many of the human proteins interacting with ALKV can be considered as hubs. Both high binding promiscuity and ability to serve as hubs are considered as important consequences of the high disorder levels in such proteins. In fact, recent bioinformatics studies clearly showed that the common structural feature of many hub proteins is their intrinsically disordered nature or their ability to interact with intrinsically disordered partners [142–146].
All these ALKV-interacting human proteins were originally discovered in a large high-throughput yeast two-hybrid screen aimed at building of the flavivirus NS3 and NS5 proteins interaction network for dengue virus serotype 1 (strain D1/H/IMTSSA/98/606, DENV1), Alkhurma virus (strain 1176, ALKV), West Nile virus (Strain paAn001, WNV), Japanese Encephalitis virus (strain Beijing1, JEV), Kunjin Australian variant virus (MRM61C, KUNV), and Tick-borne encephalitis virus (strain 263, TBEV) [140]. These pathogens were selected based on their belonging to the major flavivirus evolutionary lineages, such as (a) aedes-borne pathogen: DENV; (b) culex-borne pathogens: WNV (including the Kunjin Australian variant (KUNV)) and JEV; (c) tick-borne pathogens: TBEV and ALKV [140]. Based on the enrichment analysis conducted in [140] where Gene Ontology (GO) database was used, the most over-represented molecular functions of human proteins interacting with ALKV NS3 and NS5 proteins were RNA binding (GO:0003723; proteins RNUXA, TAF15, TRIM21, and HNRNPH3), structural constituent of cytoskeleton (GO:0005200; proteins ZNF135, KIF3B, MYH9, and VIM), transcription factor binding (GO:0008134; proteins ARNTL and CAMTA2), and transcription corepressor activity (GO:0003714; protein ENO1). The authors of that study also indicated that flaviviral NS3 and NS5 proteins can interact with the structural components of the cytoskeleton and with human cellular proteins involved in the Golgi vesicle transport and in the nuclear transport [140]. Furthermore, it was also shown that the human proteins targeted by the flavivirus NS3 and NS5 were highly over-represented in the human interactome, suggesting that the most of the human proteins targeted by the flaviviral NS3 and NS5 proteins are connected with other human proteins. These observations indicated that flavivirus can affect multiple cellular functions. As it was pointed out in this study, many of the ALKV-interacting human proteins are involved in interactions with each other [140]. To understand how common this phenomenon is, we used the ability of the APID web server (http://apid.dep.usal.es) to build a specific PPI network between proteins included in a query list [71]. Figure 9 represents the results of application of this tool to the ALKV interactome and shows that many of these proteins interact with each other. Therefore, both internal (interactions with other proteins from the ALKV interactome) and external connectivities (interaction with other human proteins) are high for many proteins engaged in interaction with ALKV proteins.
Fig. 9.

Evaluation of the interset interactivity of human proteins involved in interaction with the ALKV proteins using the APID web server (http://apid.dep.usal.es). This tool builds a PPI network between proteins included in a query list. Here, each node corresponds to a protein, and where PPIs are shown as corresponding edges, thickness of which reflects the reliability of a given interaction
Since, contrarily to the ALKV proteins, many human proteins are rather well studied, we had an opportunity to use the outputs of the D2P2 database (http://d2p2.pro/) [147]. D2P2 is a database of predicted disorder for a large set of proteins from the completely sequenced genomes, which provides disorder evaluations together with important disorder-related functional information for query proteins [147]. This database uses outputs of IUPred [57], PONDR® VLXT [54], PrDOS [148], PONDR® VSL2B [55, 149], PV2 [147], and ESpritz [150] and is supplemented by data concerning location of various curated post-translational modifications (PTMs) and predicted disorder-based protein-binding sites. Figure 10 represents a set of the D2P2-generated profiles for six most disordered human proteins (i.e., proteins with PPID > 75%, which are TAF15, CEP250, DNTTIP2, PHC2, RASSF7, and MLPH) interacting with ALKV proteins, whereas analogous profiles for the remaining members of the ALKV interactome are shown in Supplementary Materials. These data illustrate the abundance of functional disorder in human proteins interacting with ALKV and also show the abundant presence in these proteins of numerous sites of various PTMs and the existence of numerous MoRFs. These observations provide strong support to the crucial roles of intrinsic disorder in functionality of these proteins, which undergo extensive post-translational modifications (phosphorylation, acetylation, glycosylation, ubiquitination, nitrosylation, methylation, and SUMOylation) needed for regulation of their multiple functions, including binding to various partners. High prevalence of PTMs protein–protein-binding sites in the intrinsically disordered members of the ALKV interactome is in line with the previous studies, where it was indicated that the structural ‘floppiness’ defines the ability of IDP/IDPR to be controlled and regulated at multiple levels [27, 81, 151–153], with PTMs being one of the most important means of regulation [154, 155]. Furthermore, among several consequences of the presence of intrinsic disorder in proteins is their multifunctionality, which is mostly determined by the mosaic architecture of IDPs/IDPRs, where multiple relatively short functional elements are spread within the amino acid sequences [27]. The aforementioned multifunctionality is a specific feature of ‘moonlighting’ proteins, many of which were shown to be either completely disordered or possess long IDPRs [156].
Fig. 10.

Evaluating functional intrinsic disorder in six most disordered members of the ALKV interactome, a Q92804, TATA-binding protein-associated factor 2N (TAF15); b Q9BV73, Centrosome-associated protein CEP250 (CEP250); c Q5QJE6, Deoxynucleotidyl transferase terminal-interacting protein 2 (DNTTIP2); d Q8IXK0, Polyhomeotic-like protein 2 (PHC2); e Q02833, Ras association domain-containing protein 7 (RASSF7); f Q9BV36, Protein melanophilin (MLPH), using the D2P2 database (http://d2p2.pro/) [147]. Here, the green-and-white bar in the middle of each plot shows the predicted disorder agreement between nine predictors, with green parts corresponding to disordered regions by consensus. Yellow bar shows the location of the predicted disorder-based binding sites (molecular recognition features, MoRFs), whereas colored circles at the bottom of the plot show location of various PTMs: phosphorylation (red), acetylation (yellow), glycosylation (orange), ubiquitination (violet), nitrosylation (light blue), methylation (dark blue), and SUMOylation (green)
Our study shows that although the ALKV proteins contain low-to-moderate levels of intrinsic disorder, their IDPRs have multiple important functions. In ALKV genome polyprotein, IDPRs or regions with increased flexibility serve as sites of the proteolytic attack leading to the generation of the mature viral proteins. IDPRs and flexible regions in the mature ALKV proteins are utilized for protein–protein interactions and as also serve as PTM sites. One should remember that the biological significance of intrinsic disorder does not necessarily correlate with the overall disorder level of a protein or the length of its IDPRs. In fact, multiple instances can be found where functionality of a protein is critically dependent on relatively short intrinsically disordered or flexible regions. For example, the V3 loop (residues 424–461) of the envelope glycoprotein gp120 of human immunodeficiency virus type 1 (HIV-1) is crucial for the virus entry to the host immune cell and plays a primary role in determining HIV-1 cell tropism and co-receptor specificity [157]. In fact, gp120 triggers HIV-1 cell entry by binding to two host immune cell membrane proteins, the CD4 receptor and the CCR5 or/and CXCR4 co-receptors [158]. HIV-1 cell tropism is defined by the different usage of co-repressors, with R5 and X4 tropic viruses exclusively using the chemokine receptors CCR5 (R5) and CXCR4 (X4) as co-receptors, respectively, and with dual tropic viruses (R5X4) using both co-receptors for cell entry. It was also pointed out that disease progression and pathogenesis are correlated with the switch from CCR5 to CXCR4 [159, 160]. A recent study showed that the levels of intrinsic disorder in the V3 loop of gp120 are correlated with the HIV-1 cell tropism, where the highest intrinsic disorder tendency is present in the V3 loop of X4 virus, whereas R5X4 virus is characterized by the lowest disorder propensity in this loop [161]. Therefore, these data clearly indicated that structural disorder in the V3 loop that constitutes just 3.6% of the gp120 plays an important role in HIV-1 cell tropism and CXCR4 binding [161].
One can argue that HIV-1 is not a flavivirus, and therefore, these observations are not related to potential functionality of IDPRs in ALKV proteins. However, it was pointed out that the presence of the noticeable conformational dynamics of the virion leading to the existence of an ensemble of conformations at equilibrium defines the antigenic structure of flaviviruses [162]. For example, the fact that ZIKV is poorly neutralized by the antibodies against the DII fusion loop (DII-FL) was attributed to the increased length of its intrinsically disordered glycan loop, which, in comparison with the similar region of DENV, extends further towards the DII-FL of the neighboring E proteins within E dimers on mature viruses [162]. Furthermore, the ability of a member of the Flaviviridae family, Dengue virus type 2 (DENV-2), to interact with and enter the host cells, was ascribed to the flexible/disordered loops I (amino acids 297–312) and II (amino acids 379–385) of the viral envelope protein E [163]. Similarly, a flexible loop 3 (residues 362–370) of the Japanese encephalitis virus (JEV) envelope protein E was shown to be important to entry of this virus into the host cells [164].
Our analysis revealed that the vast majority of human proteins involved in the interaction with ALKV proteins are expected to be highly disordered. In fact, the mean content of predicted disordered residues evaluated for these proteins via averaging the outputs of PONDR® VLXT, PONDR® VL3, PONDR® VSL2, PONDR® FIT, IUPred_short, and IUPred_long is really very high, being equal to 47.3 ± 4.1%. Therefore, it is not surprising that the structural coverage of these proteins, which is the percent of residues with known structure, is relatively low (16.2 ± 4.5%), with no structural information being currently available for 23 members of the ALK interactome [CEP250 (PPID = 84.64%), DNTTIP2 (PPID = 78.04%), PHC2 (PPID = 77.51%), RASSF7 (PPID = 77.48%), MLPH (PPID = 76.00%), GPATCH2L (PPID = 70.75%), MISP (PPID = 70.69%), PDE4DIP (PPID = 70.38%), MACO1 (PPID = 63.86%), MDFI (PPID = 62.20%), SPDL1 (PPID = 61.98%), LRRC45 (PPID = 60.45%), PPP1R3E (PPID = 58.42%), ARID2 (PPID = 57.44%), HNRNPH3 (PPID = 50.00%), CAMTA2 (PPID = 48.59%), TXNDC9 (PPID = 34.51%), LTBP3 (PPID = 30.24%), ZNF135 (PPID = 29.03%), LAMB2 (PPID = 21.25%), CDH11 (PPID = 16.58%), VPS11 (PPID = 8.11%), and NIPSNAP1 (PPID = 7.04%)]. According to their structural coverage, the remaining proteins from this set can be arranged in the following order: MYH9 (2.3%) < APBB1IP (4.7%) < ARNTL (10.1%) < TAF15 (15.7%) < PHAX (25.6%) < SCRIB (25.8%) < JAG1 (25.9%) < GRN (28.3%) < KIF3B (47.4%) < ZBTB17 (52.2%) < PIAS3 (56.7%) < VIM (60.7%) < TRIM21 (65.1%) < FHL2 (97.5%) < ENO1 (99.5%). In other words, only two of 15 proteins have almost complete structural coverage (about 100%), whereas, for the remaining proteins, structural information is available only for some of their parts. Therefore, even if one would not consider 23 proteins with unknown structure, the structural coverage of the remaining 15 proteins is of 41.2 ± 7.9%. Among two obvious reasons for the lack of structural coverage for a given protein are the lack of current interest in this protein (no attempts were made to solve its structure) and the presence of some serious obstacles in protein crystallization experiments. However, the “lack of interest” has to be excluded for proteins with partial structural coverage, since structure was determined for a part (often, several parts) of protein sequence. It is known that among bottlenecks in protein structural characterization is the presence of intrinsic disorder in a query protein [165]. In fact, in all stages, the structure determination pipeline, including protein expression, stability [166, 167], solubility [168, 169], and crystallization [89, 165, 168, 170–173], can be dramatically affected by intrinsic disorder. Therefore, highly disordered proteins are commonly excluded from the target list of many structural biologists, and long IDPRs (which can prohibit crystallization) are typically removed from the proteins submitted to the structure determination pipeline. In line with these considerations, 78% of human proteins interacting with ALKV for which structural information is currently unavailable are highly disordered (they have PPIDs exceeding 30%). The mean PPID of this set of proteins is 52.8 ± 4.9%, which is almost 1.5 times higher than the mean PPID of ALKV-interacting proteins with some structural coverage (38.8 ± 6.6%). Furthermore, Fig. 11 illustrates the obvious negative correlation between the structural coverage and mean PPID, with more disordered proteins possessing less structural coverage, thereby visualizing the idea that proteins with more disorder are more difficult to crystallize.
Fig. 11.
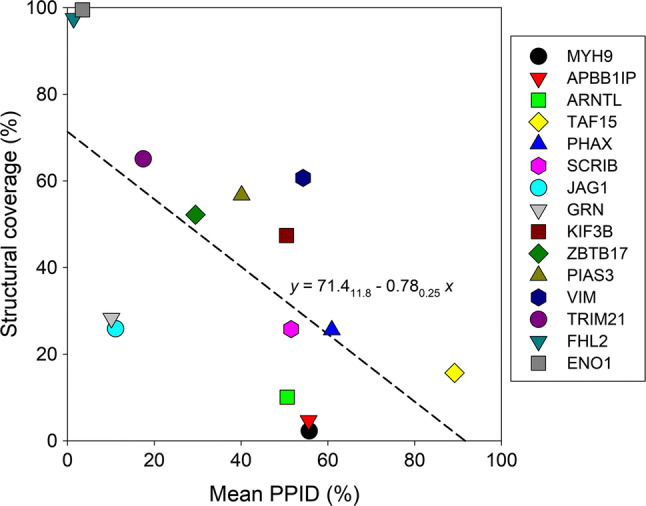
Comparison of structural coverage and intrinsic disorder propensity of human proteins involved in interaction with ALKV proteins. The structural coverage of a protein is the percent of its residues with known structure, whereas disorder propensity is the mean content of disordered residues evaluated for each of these proteins by averaging the outputs of the per-residue disorder predictors, such as PONDR® VLXT, PONDR® VL3, PONDR® VSL2, PONDR® FIT, IUPred_short, and IUPred_long. Dashed line represents the linear fit of data for proteins with known structure
Results of our study suggest that intrinsic disorder in ALKV proteins and human proteins interacting with this tick-transmitted virus can be related to the ALKV pathogenesis. Our work represents the first systematic analysis of the functional intrinsic disorder in members of the ALKV proteome and interactome. We show here that intrinsic disorder is crucial for various functional aspects of these proteins, with IDPRs being used for protein–protein interactions, defining binding promiscuity of their carriers, and serving as sites of various PTMs. Therefore, intrinsic disorder should be taken into account while conducting structural and functional analyses of these viral proteins and their human interactors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (DOCX 18295 kb)
Acknowledgements
This work was supported by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under Grant no. D1439-128-130. The authors, therefore, gratefully acknowledge the DSR technical and financial support.
Contributor Information
Elrashdy M. Redwan, Email: lradwan@kau.edu.sa
Vladimir N. Uversky, Email: vuversky@health.usf.edu
References
- 1.Alzahrani AG, Al Shaiban HM, Al Mazroa MA, Al-Hayani O, Macneil A, Rollin PE, Memish ZA. Alkhurma hemorrhagic fever in humans, Najran, Saudi Arabia. Emerg Infect Dis. 2010;16:1882–1888. doi: 10.3201/eid1612.100417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Madani TA, Azhar EI, Abuelzein el TM, Kao M, Al-Bar HM, Abu-Araki H, Niedrig M, Ksiazek TG. Alkhumra (Alkhurma) virus outbreak in Najran, Saudi Arabia: epidemiological, clinical, and laboratory characteristics. J Infect. 2011;62:67–76. doi: 10.1016/j.jinf.2010.09.032. [DOI] [PubMed] [Google Scholar]
- 3.Qattan I, Akbar N, Afif H, Azmah SA, Khateeb T, Zaki A. A novel flavivirus: Makkah region 1994–1996. Saudi Epidemiol Bull. 1996;1:2–3. [Google Scholar]
- 4.Zaki AM. Isolation of a flavivirus related to the tick-borne encephalitis complex from human cases in Saudi Arabia. Trans R Soc Trop Med Hyg. 1997;91:179–181. doi: 10.1016/S0035-9203(97)90215-7. [DOI] [PubMed] [Google Scholar]
- 5.Madani TA. Alkhumra virus infection, a new viral hemorrhagic fever in Saudi Arabia. J Infect. 2005;51:91–97. doi: 10.1016/j.jinf.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Gaunt MW, Sall AA, de Lamballerie X, Falconar AK, Dzhivanian TI, Gould EA. Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. J Gen Virol. 2001;82:1867–1876. doi: 10.1099/0022-1317-82-8-1867. [DOI] [PubMed] [Google Scholar]
- 7.Grard G, Moureau G, Charrel RN, Lemasson JJ, Gonzalez JP, Gallian P, Gritsun TS, Holmes EC, Gould EA, de Lamballerie X. Genetic characterization of tick-borne flaviviruses: new insights into evolution, pathogenetic determinants and taxonomy. Virology. 2007;361:80–92. doi: 10.1016/j.virol.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 8.Heinze DM, Gould EA, Forrester NL. Revisiting the clinal concept of evolution and dispersal for the tick-borne flaviviruses by using phylogenetic and biogeographic analyses. J Virol. 2012;86:8663–8671. doi: 10.1128/JVI.01013-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moureau G, Cook S, Lemey P, Nougairede A, Forrester NL, Khasnatinov M, Charrel RN, Firth AE, Gould EA, de Lamballerie X. New insights into flavivirus evolution, taxonomy and biogeographic history, extended by analysis of canonical and alternative coding sequences. PLoS One. 2015;10:e0117849. doi: 10.1371/journal.pone.0117849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pulkkinen LIA, Butcher SJ, Anastasina M. Tick-borne encephalitis virus: a structural view. Viruses. 2018;10:E350. doi: 10.3390/v10070350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madani TA, Abuelzein EM, Jalalah SM, Abu-Araki H, Azhar EI, Hassan AM, Al-Bar HM. Electron microscopy of Alkhumra hemorrhagic fever virus. Vector Borne Zoonotic Dis. 2017;17:195–199. doi: 10.1089/vbz.2016.2064. [DOI] [PubMed] [Google Scholar]
- 12.Lindenbach BD, Thiel H-J, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe D, Howley P, editors. Fields virology. Philadelphia: Lippincott-Raven Publishers; 2007. pp. 1101–1152. [Google Scholar]
- 13.Meng F, Badierah RA, Almehdar HA, Redwan EM, Kurgan L, Uversky VN. Unstructural biology of the Dengue virus proteins. FEBS J. 2015;282:3368–3394. doi: 10.1111/febs.13349. [DOI] [PubMed] [Google Scholar]
- 14.Giri R, Kumar D, Sharma N, Uversky VN. Intrinsically disordered side of the Zika virus proteome. Front Cell Infect Microbiol. 2016;6:144. doi: 10.3389/fcimb.2016.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deresinski S. Alkhurma hemorrhagic fever virus: an emerging pathogen. Clin Infect Dis. 2010;51:vi. [Google Scholar]
- 16.Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30:137–149. doi: 10.1080/07391102.2012.675145. [DOI] [PubMed] [Google Scholar]
- 17.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 18.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 19.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/S0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 20.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uversky VN. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol. 2010;2010:568068. doi: 10.1155/2010/568068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 1804;2010:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 24.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 25.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Dunker AK, Obradovic Z. The protein trinity—linking function and disorder. Nat Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 27.Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta. 1834;2013:932–951. doi: 10.1016/j.bbapap.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 28.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998;1998:473–484. [PubMed] [Google Scholar]
- 29.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure–function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 30.Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. Natively disordered proteins. In: Buchner J, Kiefhaber T, editors. Handbook of protein folding. New York: Wiley-VCH; 2005. pp. 271–353. [Google Scholar]
- 31.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/S0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 32.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 33.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 34.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–1916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 39.Vacic V, Markwick PR, Oldfield CJ, Zhao X, Haynes C, Uversky VN, Iakoucheva LM. Disease-associated mutations disrupt functionally important regions of intrinsic protein disorder. PLoS Comput Biol. 2012;8:e1002709. doi: 10.1371/journal.pcbi.1002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, Tawfik DS. Do viral proteins possess unique biophysical features? Trends Biochem Sci. 2009;34:53–59. doi: 10.1016/j.tibs.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 41.Fan X, Xue B, Dolan PT, LaCount DJ, Kurgan L, Uversky VN. The intrinsic disorder status of the human hepatitis C virus proteome. Mol BioSyst. 2014;10:1345–1363. doi: 10.1039/C4MB00027G. [DOI] [PubMed] [Google Scholar]
- 42.Xue B, Mizianty MJ, Kurgan L, Uversky VN. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell Mol Life Sci. 2012;69:1211–1259. doi: 10.1007/s00018-011-0859-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uversky VN, Roman A, Oldfield CJ, Dunker AK. Protein intrinsic disorder and human papillomaviruses: increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J Proteome Res. 2006;5:1829–1842. doi: 10.1021/pr0602388. [DOI] [PubMed] [Google Scholar]
- 44.Xue B, Ganti K, Rabionet A, Banks L, Uversky VN. Disordered interactome of human papillomavirus. Curr Pharm Des. 2014;20:1274–1292. doi: 10.2174/13816128113199990072. [DOI] [PubMed] [Google Scholar]
- 45.Mishra PM, Uversky VN, Giri R. Molecular recognition features in Zika virus proteome. J Mol Biol. 2018;430:2372–2388. doi: 10.1016/j.jmb.2017.10.018. [DOI] [PubMed] [Google Scholar]
- 46.Whelan JN, Reddy KD, Uversky VN, Teng MN. Functional correlations of respiratory syncytial virus proteins to intrinsic disorder. Mol BioSyst. 2016;12:1507–1526. doi: 10.1039/C6MB00122J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goh GK, Dunker AK, Uversky V. Prediction of intrinsic disorder in MERS-CoV/HCoV-EMC supports a high oral-fecal transmission. PLoS Curr. 2013;5:1–22. doi: 10.1371/currents.outbreaks.22254b58675cdebc256dbe3c5aa6498b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.T.U. Consortium Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, Duesbury M, Dumousseau M, Galeota E, Hinz U, Iannuccelli M, Jagannathan S, Jimenez R, Khadake J, Lagreid A, Licata L, Lovering RC, Meldal B, Melidoni AN, Milagros M, Peluso D, Perfetto L, Porras P, Raghunath A, Ricard-Blum S, Roechert B, Stutz A, Tognolli M, van Roey K, Cesareni G, Hermjakob H. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett. 2008;15:956–963. doi: 10.2174/092986608785849164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vacic V, Uversky VN, Dunker AK, Lonardi S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007;8:211. doi: 10.1186/1471-2105-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dinkel H, Van Roey K, Michael S, Davey NE, Weatheritt RJ, Born D, Speck T, Krüger D, Grebnev G, Kubań M, Strumillo M, Uyar B, Budd A, Altenberg B, Seiler M, Chemes LB, Glavina J, Sánchez IE, Diella F, Gibson TJ. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 2014;42:D259–D266. doi: 10.1093/nar/gkt1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 55.Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 1804;2010:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 58.Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347:827–839. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 59.Meszaros B, Simon I, Dosztanyi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol. 2009;5:e1000376. doi: 10.1371/journal.pcbi.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dosztanyi Z, Meszaros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–2746. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dayhoff MO, Schwartz RM, Orcutt BC. A model of evolutionary change in proteins. Atlas Protein Seq Struct. 1978;5:345–351. [Google Scholar]
- 62.Gould CM, Diella F, Vian A, Puntervoll P, Gemünd C, Chabanis-Davidson S, Michael S, Sayadi A, Bryne JC, Chica C, Seiler M, Davey NE, Haslam N, Weatheritt RJ, Budd A, Hughes T, Paś J, Rychlewski L, Travé G, Aasland R, Helmer-Citterich M, Linding R, Gibson TJ. ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010;38:D167–D180. doi: 10.1093/nar/gkp1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mizianty MJ, Zhang T, Xue B, Zhou Y, Dunker AK, Uversky VN, Kurgan L. In-silico prediction of disorder content using hybrid sequence representation. BMC Bioinform. 2011;12:245. doi: 10.1186/1471-2105-12-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peng ZL, Kurgan L. Comprehensive comparative assessment of in silico predictors of disordered regions. Curr Protein Pept Sci. 2012;13:6–18. doi: 10.2174/138920312799277938. [DOI] [PubMed] [Google Scholar]
- 65.Fan X, Kurgan L. Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J Biomol Struct Dyn. 2014;32:448–464. doi: 10.1080/07391102.2013.775969. [DOI] [PubMed] [Google Scholar]
- 66.Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003;53(Suppl 6):566–572. doi: 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 67.Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Obradovic Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol. 2005;3:35–60. doi: 10.1142/S0219720005000886. [DOI] [PubMed] [Google Scholar]
- 68.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 69.Walsh I, Giollo M, Di Domenico T, Ferrari C, Zimmermann O, Tosatto SC. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics. 2015;31:201–208. doi: 10.1093/bioinformatics/btu625. [DOI] [PubMed] [Google Scholar]
- 70.Peng Z, Kurgan L. On the complementarity of the consensus-based disorder prediction. Pac Symp Biocomput. 2012;2012:176–187. [PubMed] [Google Scholar]
- 71.Alonso-Lopez D, Gutierrez MA, Lopes KP, Prieto C, Santamaria R, De Las Rivas J. APID interactomes: providing proteome-based interactomes with controlled quality for multiple species and derived networks. Nucleic Acids Res. 2016;44:W529–W535. doi: 10.1093/nar/gkw363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A, Kolas N, O’Donnell L, Reguly T, Nixon J, Ramage L, Winter A, Sellam A, Chang C, Hirschman J, Theesfeld C, Rust J, Livstone MS, Dolinski K, Tyers M. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015;43(2015):D470–D478. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32(2004):D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, Balakrishnan L, Marimuthu A, Banerjee S, Somanathan DS, Sebastian A, Rani S, Ray S, Harrys Kishore CJ, Kanth S, Ahmed M, Kashyap MK, Mohmood R, Ramachandra YL, Krishna V, Rahiman BA, Mohan S, Ranganathan P, Ramabadran S, Chaerkady R, Pandey A. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, Duesbury M, Dumousseau M, Feuermann M, Hinz U, Jandrasits C, Jimenez RC, Khadake J, Mahadevan U, Masson P, Pedruzzi I, Pfeiffenberger E, Porras P, Raghunath A, Roechert B, Orchard S, Hermjakob H. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40:D841–D846. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, Galeota E, Sacco F, Palma A, Nardozza AP, Santonico E, Castagnoli L, Cesareni G. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40(2012):D857–D861. doi: 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP. The BioPlex Network: a systematic exploration of the human interactome. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rose PW, Prlic A, Bi C, Bluhm WF, Christie CH, Dutta S, Green RK, Goodsell DS, Westbrook JD, Woo J, Young J, Zardecki C, Berman HM, Bourne PE, Burley SK. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015;43:D345–D356. doi: 10.1093/nar/gku1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 80.Uversky VN. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci. 2003;60:1852–1871. doi: 10.1007/s00018-003-3096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Uversky VN. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 2013;22:693–724. doi: 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vucetic S, Obradovic Z, Vacic V, Radivojac P, Peng K, Iakoucheva LM, Cortese MS, Lawson JD, Brown CJ, Sikes JG, Newton CD, Dunker AK. DisProt: a database of protein disorder. Bioinformatics. 2005;21:137–140. doi: 10.1093/bioinformatics/bth476. [DOI] [PubMed] [Google Scholar]
- 83.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35:D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de Laureto PP, Tosatto L, Frare E, Marin O, Uversky VN, Fontana A. Conformational properties of the SDS-bound state of alpha-synuclein probed by limited proteolysis: unexpected rigidity of the acidic C-terminal tail. Biochemistry. 2006;45:11523–11531. doi: 10.1021/bi052614s. [DOI] [PubMed] [Google Scholar]
- 86.Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Polon. 2004;51:299–321. [PubMed] [Google Scholar]
- 87.Fontana A, Fassina G, Vita C, Dalzoppo D, Zamai M, Zambonin M. Correlation between sites of limited proteolysis and segmental mobility in thermolysin. Biochemistry. 1986;25:1847–1851. doi: 10.1021/bi00356a001. [DOI] [PubMed] [Google Scholar]
- 88.Fontana A, Polverino de Laureto P, De Filippis V, Scaramella E, Zambonin M. Probing the partly folded states of proteins by limited proteolysis. Fold Des. 1997;2:R17–R26. doi: 10.1016/S1359-0278(97)00010-2. [DOI] [PubMed] [Google Scholar]
- 89.Iakoucheva LM, Kimzey AL, Masselon CD, Bruce JE, Garner EC, Brown CJ, Dunker AK, Smith RD, Ackerman EJ. Identification of intrinsic order and disorder in the DNA repair protein XPA. Protein Sci. 2001;10:560–571. doi: 10.1110/ps.29401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Polverino de Laureto P, De Filippis V, Di Bello M, Zambonin M, Fontana A. Probing the molten globule state of alpha-lactalbumin by limited proteolysis. Biochemistry. 1995;34:12596–12604. doi: 10.1021/bi00039a015. [DOI] [PubMed] [Google Scholar]
- 91.Rodenhuis-Zybert IA, Wilschut J, Smit JM. Partial maturation: an immune-evasion strategy of dengue virus? Trends Microbiol. 2011;19:248–254. doi: 10.1016/j.tim.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 92.Stohlman SA, Wisseman CL, Jr, Eylar OR, Silverman DJ. Dengue virus-induced modifications of host cell membranes. J Virol. 1975;16:1017–1026. doi: 10.1128/jvi.16.4.1017-1026.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Perera R, Kuhn RJ. Structural proteomics of dengue virus. Curr Opin Microbiol. 2008;11:369–377. doi: 10.1016/j.mib.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Oliveira ERA, Mohana-Borges R, de Alencastro RB, Horta BAC. The flavivirus capsid protein: structure, function and perspectives towards drug design. Virus Res. 2017;227:115–123. doi: 10.1016/j.virusres.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 95.Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, Schein S, Zhou ZH. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat Struct Mol Biol. 2013;20:105–110. doi: 10.1038/nsmb.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108:717–725. doi: 10.1016/S0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bhuvanakantham R, Chong MK, Ng ML. Specific interaction of capsid protein and importin-alpha/beta influences West Nile virus production. Biochem Biophys Res Commun. 2009;389:63–69. doi: 10.1016/j.bbrc.2009.08.108. [DOI] [PubMed] [Google Scholar]
- 98.Colpitts TM, Barthel S, Wang P, Fikrig E. Dengue virus capsid protein binds core histones and inhibits nucleosome formation in human liver cells. PLoS One. 2011;6:e24365. doi: 10.1371/journal.pone.0024365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Poenisch M, Metz P, Blankenburg H, Ruggieri A, Lee JY, Rupp D, Rebhan I, Diederich K, Kaderali L, Domingues FS, Albrecht M, Lohmann V, Erfle H, Bartenschlager R. Identification of HNRNPK as regulator of hepatitis C virus particle production. PLoS Pathog. 2015;11:e1004573. doi: 10.1371/journal.ppat.1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Netsawang J, Noisakran S, Puttikhunt C, Kasinrerk W, Wongwiwat W, Malasit P, Yenchitsomanus PT, Limjindaporn T. Nuclear localization of dengue virus capsid protein is required for DAXX interaction and apoptosis. Virus Res. 2010;147:275–283. doi: 10.1016/j.virusres.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 101.Balinsky CA, Schmeisser H, Ganesan S, Singh K, Pierson TC, Zoon KC. Nucleolin interacts with the dengue virus capsid protein and plays a role in formation of infectious virus particles. J Virol. 2013;87:13094–13106. doi: 10.1128/JVI.00704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Samuel GH, Wiley MR, Badawi A, Adelman ZN, Myles KM. Yellow fever virus capsid protein is a potent suppressor of RNA silencing that binds double-stranded RNA. Proc Natl Acad Sci USA. 2016;113:13863–13868. doi: 10.1073/pnas.1600544113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Konishi E, Mason PW. Proper maturation of the Japanese encephalitis virus envelope glycoprotein requires cosynthesis with the premembrane protein. J Virol. 1993;67:1672–1675. doi: 10.1128/jvi.67.3.1672-1675.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lorenz IC, Allison SL, Heinz FX, Helenius A. Folding and dimerization of tick-borne encephalitis virus envelope proteins prM and E in the endoplasmic reticulum. J Virol. 2002;76:5480–5491. doi: 10.1128/JVI.76.11.5480-5491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guirakhoo F, Heinz FX, Mandl CW, Holzmann H, Kunz C. Fusion activity of flaviviruses: comparison of mature and immature (prM-containing) tick-borne encephalitis virions. J Gen Virol. 1991;72(Pt 6):1323–1329. doi: 10.1099/0022-1317-72-6-1323. [DOI] [PubMed] [Google Scholar]
- 106.Zhang Y, Corver J, Chipman PR, Zhang W, Pletnev SV, Sedlak D, Baker TS, Strauss JH, Kuhn RJ, Rossmann MG. Structures of immature flavivirus particles. EMBO J. 2003;22:2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heinz FX, Stiasny K, Puschner-Auer G, Holzmann H, Allison SL, Mandl CW, Kunz C. Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with protein prM. Virology. 1994;198:109–117. doi: 10.1006/viro.1994.1013. [DOI] [PubMed] [Google Scholar]
- 108.Guirakhoo F, Bolin RA, Roehrig JT. The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology. 1992;191:921–931. doi: 10.1016/0042-6822(92)90267-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roby JA, Setoh YX, Hall RA, Khromykh AA. Post-translational regulation and modifications of flavivirus structural proteins. J Gen Virol. 2015;96:1551–1569. doi: 10.1099/vir.0.000097. [DOI] [PubMed] [Google Scholar]
- 110.Catteau A, Roue G, Yuste VJ, Susin SA, Despres P. Expression of dengue ApoptoM sequence results in disruption of mitochondrial potential and caspase activation. Biochimie. 2003;85:789–793. doi: 10.1016/S0300-9084(03)00139-1. [DOI] [PubMed] [Google Scholar]
- 111.Pierson TC, Diamond MS. Degrees of maturity: the complex structure and biology of flaviviruses. Curr Opin Virol. 2012;2:168–175. doi: 10.1016/j.coviro.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science. 2008;319:1834–1837. doi: 10.1126/science.1153264. [DOI] [PubMed] [Google Scholar]
- 113.Murray CL, Jones CT, Rice CM. Architects of assembly: roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat Rev Microbiol. 2008;6:699–708. doi: 10.1038/nrmicro1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brand C, Bisaillon M, Geiss BJ. Organization of the Flavivirus RNA replicase complex. Wiley Interdiscip Rev RNA. 2017;8:e1437. doi: 10.1002/wrna.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Muller DA, Young PR. The flavivirus NS1 protein: molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res. 2013;98:192–208. doi: 10.1016/j.antiviral.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 116.Mackenzie JM, Jones MK, Young PR. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology. 1996;220:232–240. doi: 10.1006/viro.1996.0307. [DOI] [PubMed] [Google Scholar]
- 117.Muylaert IR, Chambers TJ, Galler R, Rice CM. Mutagenesis of the N-linked glycosylation sites of the yellow fever virus NS1 protein: effects on virus replication and mouse neurovirulence. Virology. 1996;222:159–168. doi: 10.1006/viro.1996.0406. [DOI] [PubMed] [Google Scholar]
- 118.Xie X, Gayen S, Kang C, Yuan Z, Shi PY. Membrane topology and function of dengue virus NS2A protein. J Virol. 2013;87:4609–4622. doi: 10.1128/JVI.02424-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Klema VJ, Padmanabhan R, Choi KH. Flaviviral replication complex: coordination between RNA Synthesis and 5′-RNA Capping. Viruses. 2015;7:4640–4656. doi: 10.3390/v7082837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Falgout B, Pethel M, Zhang YM, Lai CJ. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol. 1991;65:2467–2475. doi: 10.1128/jvi.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang CC, Huang ZS, Chiang PL, Chen CT, Wu HN. Analysis of the nucleoside triphosphatase, RNA triphosphatase, and unwinding activities of the helicase domain of dengue virus NS3 protein. FEBS Lett. 2009;583:691–696. doi: 10.1016/j.febslet.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 122.Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J Biol Chem. 2007;282:8873–8882. doi: 10.1074/jbc.M609919200. [DOI] [PubMed] [Google Scholar]
- 123.Umareddy I, Chao A, Sampath A, Gu F, Vasudevan SG. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J Gen Virol. 2006;87:2605–2614. doi: 10.1099/vir.0.81844-0. [DOI] [PubMed] [Google Scholar]
- 124.Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol. 2012;86:7360–7371. doi: 10.1128/JVI.00157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zmurko J, Neyts J, Dallmeier K. Flaviviral NS4b, chameleon and jack-in-the-box roles in viral replication and pathogenesis, and a molecular target for antiviral intervention. Rev Med Virol. 2015;25:205–223. doi: 10.1002/rmv.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Issur M, Geiss BJ, Bougie I, Picard-Jean F, Despins S, Mayette J, Hobdey SE, Bisaillon M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA. 2009;15:2340–2350. doi: 10.1261/rna.1609709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chu PW, Westaway EG. Replication strategy of Kunjin virus: evidence for recycling role of replicative form RNA as template in semiconservative and asymmetric replication. Virology. 1985;140:68–79. doi: 10.1016/0042-6822(85)90446-5. [DOI] [PubMed] [Google Scholar]
- 129.Bollati M, Alvarez K, Assenberg R, Baronti C, Canard B, Cook S, Coutard B, Decroly E, de Lamballerie X, Gould EA, Grard G, Grimes JM, Hilgenfeld R, Jansson AM, Malet H, Mancini EJ, Mastrangelo E, Mattevi A, Milani M, Moureau G, Neyts J, Owens RJ, Ren J, Selisko B, Speroni S, Steuber H, Stuart DI, Unge T, Bolognesi M. Structure and functionality in flavivirus NS-proteins: perspectives for drug design. Antiviral Res. 2010;87:125–148. doi: 10.1016/j.antiviral.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Best SM. The many faces of the flavivirus NS5 protein in antagonism of type I interferon signaling. J Virol. 2017;91:e1970-16. doi: 10.1128/JVI.01970-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Uversky VN. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des. 2013;19:4191–4213. doi: 10.2174/1381612811319230005. [DOI] [PubMed] [Google Scholar]
- 132.Uversky VN. Looking at the recent advances in understanding alpha-synuclein and its aggregation through the proteoform prism. F1000Res. 2017;6:525. doi: 10.12688/f1000research.10536.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Uversky VN. p53 proteoforms and intrinsic disorder: an illustration of the protein structure–function continuum concept. Int J Mol Sci. 2016;17:E1874. doi: 10.3390/ijms17111874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.DeForte S, Uversky VN. Order, disorder, and everything in between, molecules. Molecules. 2016;21:E1090. doi: 10.3390/molecules21081090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Uversky VN. Dancing protein clouds: the strange biology and chaotic physics of intrinsically disordered proteins. J Biol Chem. 2016;291:6681–6688. doi: 10.1074/jbc.R115.685859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 137.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 138.Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–13477. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Le Breton M, Meyniel-Schicklin L, Deloire A, Coutard B, Canard B, de Lamballerie X, Andre P, Rabourdin-Combe C, Lotteau V, Davoust N. Flavivirus NS3 and NS5 proteins interaction network: a high-throughput yeast two-hybrid screen. BMC Microbiol. 2011;11:234. doi: 10.1186/1471-2180-11-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rajagopalan K, Mooney SM, Parekh N, Getzenberg RH, Kulkarni P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem. 2011;112:3256–3267. doi: 10.1002/jcb.23252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006;580:2041–2045. doi: 10.1016/j.febslet.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 143.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ekman D, Light S, Bjorklund AK, Elofsson A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 2006;7:R45. doi: 10.1186/gb-2006-7-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dosztanyi Z, Chen J, Dunker AK, Simon I, Tompa P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res. 2006;5:2985–2995. doi: 10.1021/pr060171o. [DOI] [PubMed] [Google Scholar]
- 146.Singh GP, Ganapathi M, Sandhu KS, Dash D. Intrinsic unstructuredness and abundance of PEST motifs in eukaryotic proteomes. Proteins. 2006;62:309–315. doi: 10.1002/prot.20746. [DOI] [PubMed] [Google Scholar]
- 147.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J. D(2)P(2): database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–D516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ishida T, Kinoshita K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007;35:W460–W464. doi: 10.1093/nar/gkm363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Obradovic Z, Peng K, Vucetic S, Radivojac P, Dunker AK. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61(Suppl 7):176–182. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- 150.Walsh I, Martin AJ, Di Domenico T, Tosatto SC. ESpritz: accurate and fast prediction of protein disorder. Bioinformatics. 2012;28:503–509. doi: 10.1093/bioinformatics/btr682. [DOI] [PubMed] [Google Scholar]
- 151.Habchi J, Tompa P, Longhi S, Uversky VN. Introducing protein intrinsic disorder. Chem Rev. 2014;114:6561–6588. doi: 10.1021/cr400514h. [DOI] [PubMed] [Google Scholar]
- 152.van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev. 2011;40:1623–1634. doi: 10.1039/C0CS00057D. [DOI] [PubMed] [Google Scholar]
- 154.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pejaver V, Hsu WL, Xin F, Dunker AK, Uversky VN, Radivojac P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014;23:1077–1093. doi: 10.1002/pro.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tompa P, Szasz C, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005;30:484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 157.Hwang SS, Boyle TJ, Lyerly HK, Cullen BR. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253:71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- 158.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 159.Gorry PR, Ancuta P. Coreceptors and HIV-1 pathogenesis. Curr HIV/AIDS Rep. 2011;8:45–53. doi: 10.1007/s11904-010-0069-x. [DOI] [PubMed] [Google Scholar]
- 160.Regoes RR, Bonhoeffer S. The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol. 2005;13:269–277. doi: 10.1016/j.tim.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 161.Jiang X, Feyertag F, Robertson DL. Protein structural disorder of the envelope V3 loop contributes to the switch in human immunodeficiency virus type 1 cell tropism. PLoS One. 2017;12:e0185790. doi: 10.1371/journal.pone.0185790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Goo L, DeMaso CR, Pelc RS, Ledgerwood JE, Graham BS, Kuhn RJ, Pierson TC. The Zika virus envelope protein glycan loop regulates virion antigenicity. Virology. 2018;515:191–202. doi: 10.1016/j.virol.2017.12.032. [DOI] [PubMed] [Google Scholar]
- 163.Abd-Jamil J, Cheah CY, AbuBakar S. Dengue virus type 2 envelope protein displayed as recombinant phage attachment protein reveals potential cell binding sites. Protein Eng Des Sel. 2008;21:605–611. doi: 10.1093/protein/gzn041. [DOI] [PubMed] [Google Scholar]
- 164.Li C, Zhang LY, Sun MX, Li PP, Huang L, Wei JC, Yao YL, Isahg H, Chen PY, Mao X. Inhibition of Japanese encephalitis virus entry into the cells by the envelope glycoprotein domain III (EDIII) and the loop3 peptide derived from EDIII. Antiviral Res. 2012;94:179–183. doi: 10.1016/j.antiviral.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 165.Oldfield CJ, Xue B, Van YY, Ulrich EL, Markley JL, Dunker AK, Uversky VN. Utilization of protein intrinsic disorder knowledge in structural proteomics. Biochim Biophys Acta. 1834;2012:487–498. doi: 10.1016/j.bbapap.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Reeves R, Nissen MS. Purification and assays for high mobility group HMG-I(Y) protein function. Methods Enzymol. 1999;304:155–188. doi: 10.1016/S0076-6879(99)04011-2. [DOI] [PubMed] [Google Scholar]
- 167.Stewart AA, Ingebritsen TS, Cohen P. The protein phosphatases involved in cellular regulation. 5. Purification and properties of a Ca2+/calmodulin-dependent protein phosphatase (2B) from rabbit skeletal muscle. Eur J Biochem. 1983;132:289–295. doi: 10.1111/j.1432-1033.1983.tb07361.x. [DOI] [PubMed] [Google Scholar]
- 168.Bandaru V, Cooper W, Wallace SS, Doublie S. Overproduction, crystallization and preliminary crystallographic analysis of a novel human DNA-repair enzyme that recognizes oxidative DNA damage. Acta Crystallogr D Biol Crystallogr. 2004;60:1142–1144. doi: 10.1107/S0907444904007929. [DOI] [PubMed] [Google Scholar]
- 169.Petros AM, Medek A, Nettesheim DG, Kim DH, Yoon HS, Swift K, Matayoshi ED, Oltersdorf T, Fesik SW. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci USA. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Harauz G, Ishiyama N, Hill CM, Bates IR, Libich DS, Fares C. Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 2004;35:503–542. doi: 10.1016/j.micron.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 171.Bailey RW, Dunker AK, Brown CJ, Garner EC, Griswold MD. Clusterin, a binding protein with a molten globule-like region. Biochemistry. 2001;40:11828–11840. doi: 10.1021/bi010135x. [DOI] [PubMed] [Google Scholar]
- 172.Daughdrill GW, Chadsey MS, Karlinsey JE, Hughes KT, Dahlquist FW. The C-terminal half of the anti-sigma factor, FlgM, becomes structured when bound to its target, sigma 28. Nat Struct Biol. 1997;4:285–291. doi: 10.1038/nsb0497-285. [DOI] [PubMed] [Google Scholar]
- 173.Cary PD, King DS, Crane-Robinson C, Bradbury EM, Rabbani A, Goodwin GH, Johns EW. Structural studies on two high-mobility-group proteins from calf thymus, HMG-14 and HMG-20 (ubiquitin), and their interaction with DNA. Eur J Biochem. 1980;112:577–580. doi: 10.1111/j.1432-1033.1980.tb06123.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1 (DOCX 18295 kb)