

Abstract
Antibodies currently constitute the most rapidly growing class of human therapeutics; however, the high-yield production of recombinant antibodies and antibody fragments is a real challenge. High expression of active single-chain antibody fragment (scFv) in Escherichia coli has not been successful, as the protein contains three intramolecular disulfide bonds that are difficult to form correctly in the bacterial intracellular environment. To solve this problem, we fused the scFv gene against VEGF165 with a small ubiquitin-related modifier gene (SUMO) by synthesizing an artificial SUMO–scFv fusion gene that was highly expressed in the BL21(DE3) strain. The optimal expression level of the soluble fusion protein, SUMO–scFv, was up to 28.5% of the total cellular protein. The fusion protein was purified by Ni nitrilotriacetic acid (NTA) affinity chromatography and cleaved by a SUMO-specific protease to obtain the native scFv, which was further purified by Ni-NTA affinity chromatography. The result of the high-performance liquid chromatography showed that the purity of the recombinant cleaved scFv was greater than 98%. The primary structure of the purified scFv was confirmed by N-terminal amino acid sequencing and matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy analysis. In vitro activity assay demonstrated that the recombinant scFv could dose-dependently inhibit VEGF165-induced human umbilical vein-derived endothelial cell proliferation. The expression strategy presented in this study allows convenient high yield and easy purification of recombinant scFv with native sequences.
Keywords: Vascular endothelial growth factor, scFv, Small ubiquitin-related modifier, Purification
Introduction
Vascular endothelial growth factor (VEGF) is a multifunctional cytokine which plays a major role in angiogenesis. Alternative splicing causes the production of several different isoforms(VEGF121, VEGF145, VEGF165, VEGF183, VEGF189, VEGF206; Byun et al. 2001; Cohen et al. 1999). VEGF is essential for tumor angiogenesis, and several studies have correlated elevated VEGF levels with tumor stage, metastases, and progression (Fernandez et al. 2004). We recently isolated a novel neutralizing human anti-VEGF165 single-chain antibody fragment (scFv) from a phage antibody library (Lin and Lei 2008). Although monoclonal antibody (mAb) possesses a higher specific activity than polyclonal immunoglobulins and is suitable for large-scale production, hybridoma cell lines are not stable, especially after cryopreservation (Rando and Notkins 1994). Some of the limitations of mAb can be overcome by a genetically engineered antibody. scFv is a small antibody-engineered antibody (molecular mass about 30 kDa), in which the variable heavy chain and light chain of the antibody molecule are connected by a short, flexible polypeptide linker (Gly4Ser)3 (Huston et al. 1991). This antibody fragment retains the original antigen-binding site allowing it to maintain its specific affinity for the antigen (Huston and George 2001). The advantages of scFv are its stability as a protein, the fact that it can be produced in large scale in Escherichia coli at low cost. However, as scFv is a small protein with disulfide bonds, it is almost impossible to fold well in the intracellular environment of E. coli. Several protein fusion systems, such as His6-tagged, glutathione-S-transferase and thioredoxin, have been applied to express and purify scFv, but these methods have shortcomings in efficient soluble expression, cleavage, and purification.
Small ubiquitin-related modifier (SUMO) family proteins function as post-translation modifiers by covalently and reversibly attaching to other proteins (Johnson and Blobel 1999). SUMO modifies many proteins which are involved in a wide range of cellular process such as transcriptional regulation, nuclear transportation, chromosome organization, deoxyribonucleic acid (DNA) repair, and signal transduction (Kuliopulos and Walsh 1994). SUMO and its associated enzymes are present in all eukaryotes and are highly conserved from yeast to human but are absent from prokaryotes (Lamsoul et al. 2005). Although SUMO has only 18% sequence identity with ubiquitin, it shares a common three-dimensional structure (Lavallie and McCoy 1995). Similar to ubiquitin, SUMO functions by covalent modification of the target protein via a peptide bond that can be specifically cleaved by SUMO protease. Recently, SUMO, fused at the N terminus with heterologous proteins was found to improve protein folding, to enhance expression level, and to protect the protein from degradation via its chaperoning properties. SUMO fusion protein can be cleaved by the SUMO protease according to SUMO protein three-dimensional structure, and the target protein was obtained with native N terminus (Li and Hochstrasser 1999, 2003). Several proteins including matrix metalloprotease 3, green fluorescent protein, SARS-CoV 3CL, and SARS-CoV Spike C were successfully expressed and purified using this fusion strategy (Liew et al. 2005). In the present study, we report the high-level expression and rapid purification of scFv with a SUMO fusion strategy.
Materials and methods
Materials
E. coli BL21(DE3), E. coli DH5α, and the expression vector pET28a were purchased from Novagen (Madison, WI, USA). Taq DNA polymerase, T4 DNA ligase, and restriction endonucleases were obtained from Dalian Takara (China). Saccharomyces cerevisiae SUMO complementary DNA (cDNA) was maintained in our laboratory. The oligonucleotides encoding the scFv gene and polymerase chain reaction (PCR) primers were synthesized by Invitrogen (Shanghai, China). The plasmid minikit and gel extraction kit were from Axygen (Hangzhou, USA). SUMO protease 1 containing a histidine tag was purchased from Lifesensors (Malvern, PA, USA). Yeast extract, tryptone, and other chemicals were obtained from Fisher Scientific (Springfield, NJ, USA).
Construction of pET28/SUMO–scFv expression plasmid
In order to produce the fusion gene encoding SUMO–scFv, the SUMO fragment was amplified using primers P1 (5′-CGATATACCATGGAAGTACAACAGGTAGAG-3′) and P2 (5′-CACCAGCTGCACACCTCCAATCTGTTCGCGGTG-3′). In a previous paper (Lin and Lei 2008), we reported the isolation of a novel human anti-VEGF165 scFv, selected from a phage antibody library. The cDNA coding for the anti-VEGF165 scFv was amplified from the vector pHEN2 by PCR using primers P3 (5′-GGAGGTGTGCAGCTGGTG-3′) and P4 (5′-GAAGCTCTCGAGTTATTTACCCGGGGAC-3′). The P1 and P4 had NcoI and XhoI recognition sites (italicized), respectively, for directed cloning into the vector. The P1 and P2 primers were used to amplify the cDNA sequence of SUMO, with an extra NcoI recognition site. The P3 and P4 primers were used to amplify the sequence of scFv. After the first round of PCR using P1/P2 and P3/P4, respectively, gel purification was performed for the two products. A second round of PCR followed using the two resulting PCR products and the P1, P4 primers to obtain the fragment encoding SUMO–scFv. Then, the fragment was digested with NcoI and XhoI and inserted in frame into the pET-28a expression vector (Novagen). DH5α competent cells were transformed firstly and screened in Luria Bertani (LB) medium containing kanamycin (50 μg/ml). The plasmids were then confirmed by endonuclease restriction digestion assay and further confirmed by DNA sequencing. The resulting plasmid was named pET28a/SUMO–scFv (Fig. 2).
Fig. 2.
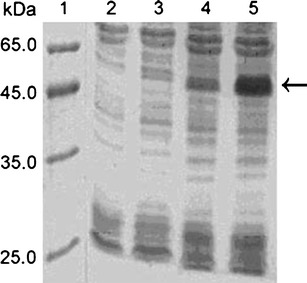
SDS-PAGE analysis of the supernatant of BL21(DE3) containing pET28a/SUMO–scFv induced at different temperatures. Lane 1: LMW marker of protein; lane 2: supernatant of BL21(DE3) containing pET28a/SUMO–scFv induced at 37°C for 4 h; lane 3: supernatant of BL21(DE3) containing pET28a/SUMO–scFv induced at 30°C for 4 h; lane 4: supernatant of BL21(DE3) containing pET28a/SUMO–scFv induced at 18°C for 4 h; lane 5: supernatant of BL21(DE3) containing pET28a/SUMO–scFv induced at 18°C for 24 h
Induction and expression of SUMO-scFv
Plasmid DNA pET28a/SUMO–scFv was transformed into E. coli BL21(DE3); the bacteria were grown at 37°C in 50 ml LB medium (1% Bacto-tryptone, 0.5% yeast extract, 85 mM NaCl) with 50 μg/ml kanamycin; when the optical density at 600 nm reached at 0.6, the culture was divided into four tubes, and isopropyl-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.4 mmol/l. Four divided tubes were continued to grow for 4 h at 37°C, 30°C, and 18°C, respectively, or for 24 h at 18°C to determine the optimal induction conditions. For protein purification, cultures were scaled up to 1.0 l medium.
Detection of soluble situation of SUMO–scFv
Cells of 50 ml induced at different conditions were collected by centrifugation at 10,000 rpm for 5 min at 4°C. The cell pellets were freeze–thawed once and resuspended in 20 mmol/l phosphate-buffered saline buffer of 7 ml containing 10,000 IU lysozyme for 1 g of the cells. The suspensions were incubated at 30°C for 30 min and then centrifuged at 12,000 rpm for 20 min at 4°C. The pellets were discarded and the supernatant was mixed with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and heated at 95°C for 5 min. The soluble proteins were analyzed by 12% SDS-PAGE, and the expression level of SUMO–scFv was detected by densitometer scanning.
Purification of SUMO–scFv
The cell lysate was applied to a column containing 10 ml Ni nitrilotriacetic acid (NTA) resin equilibrated with 50 mM Tris–HCl buffer. The resin was washed with 100 ml wash buffer I (50 mM Tris–HCl, 150 mM NaCl, and 5 mM imidazole, pH 7.9), until optical density at 280 nm reached baseline values. The contaminated proteins were eluted with 100–150 ml wash buffer II (50 mM Tris–HCl, 150 mM NaCl, and 20 mM imidazole, pH 7.9). Finally, the 6× His-tagged SUMO fusion proteins were eluted with elution buffer (50 mM Tris–HCl, 150 mM NaCl, and 1 M imidazole, pH 7.9). Fractions of purification steps were pooled at the elution peak. The purity of SUMO–scFv was assessed using SDS-PAGE, and the concentration was evaluated by Bradford’s method.
Cleavage of SUMO–scFv and purification of scFv
The purified fusion protein was diluted to the concentration of 1 mg/ml; 10 U SUMO protease was added to the protein, and the mixture was incubated at 30°C for 2 h. The cleaved sample was applied to the Ni-NTA resin to obtain the recombinant scFv, which was further concentrated by using an ultrafilter with 10-kDa cutoff membrane at 4°C. SUMO–scFv, SUMO, and SUMO protease bound to the resin were eluted as described above. The purity of scFv was checked by SDS-PAGE.
N-terminal sequence and mass spectroscopic analysis of scFv
Following SDS-PAGE electrophoresis of the purified scFv, the resolved protein was transferred on a polyvinylidene fluoride (PVDF) membrane. The appropriate PVDF band was subsequently sequenced using Edman degradation with the aid of an Applied Biosystems Model Procise automated protein sequenator 491 (Applied Biosystems, USA). Stepwise liberated phenylthiohydantoin (PTH) derivatives were identified using an online 120A high-performance liquid chromatography (HPLC) system equipped with a PTH C18 (2.1 × 220 mm; 5 μm particle size) column. HPLC analysis of the pure scFv was carried out on a HPLC system (Agilent 1100 LC, USA) using a C18 reverse-phase column; the sample was eluted using a gradient of water and acetonitrile over a period of 70 min. Absorbance was monitored at 280 nm. The peak centered at 26.43 min was collected and lyophilized. The lyophilized material was dissolved in Milli-Q water, and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy was performed on Applied Biosystems Voyager DE Pro (ABI, USA).
Determination of binding parameters by surface plasmon resonance
Binding constants of the interaction between VEGF165 and scFv determined by surface plasmon resonance with a BIAcore X instrument (Biacore, Uppsala, Sweden) according to the methods (Friguet et al. 1985). Purified scFv was coupled to a carboxymethyl dextran (CM5) chip using the N-hydroxysuccinimide/1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride kit. Different concentrations of VEGF165 diluted in the 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer containing 0.001% detergent P-20 was injected onto the sensor surface. The real-time curves were recorded automatically and analyzed using BIA evaluation software 3.0.
Endothelial cell proliferation assay
Human umbilical vein-derived endothelial cells (HUVECs) were plated in 96-well plates at 5 × 103 cells per well and incubated overnight in 100 μl of growth medium containing heparin and supplemented with 10% fetal bovine serum (Gibco-BRL, Gaithersburg, USA). The medium was changed after cell adhesion to endothelial serum-free media and incubated for 24 h. HUVECs were then exposed to VEGF165 (2 μg/ml final concentration), and varying amounts of scFv as indicated were added to wells. The cells were incubated at 37°C in a humidified atmosphere with 5% CO2 for the time indicated in the results of each experiment. Cell proliferation was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method using a Bio-Rad model 550 microplate reader.
Results
pET28/SUMO-scFv plasmid construction and expression
We constructed a plasmid for the expression of scFv fused to SUMO to make a fusion protein (Fig. 1). In addition, there was a sequence of His6 tag for immobilized metal affinity chromatography purification fused upstream of the SUMO–scFv.
Fig. 1.
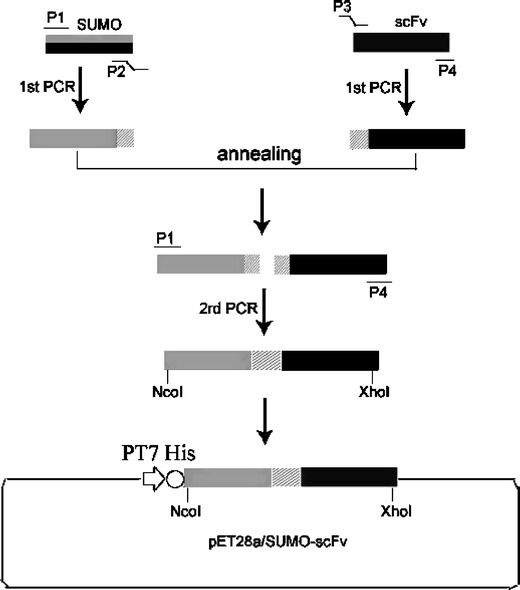
Schematic illustration of chimeric construct of SUMO and scFv against VEGF165 in a pET28a prokaryotic expression vector with the kanamycin-resistant gene
The E. coli BL21 (DE3) cells harboring pET28a/SUMO–scFv were induced by IPTG, and the expression of the SUMO–scFv protein was analyzed by SDS-PAGE with Coomassie brilliant blue R-250 staining. A predominant protein at 50 kDa, corresponding to the expected molecular size of SUMO–scFv, was observed (Fig. 3, lane 3). The soluble expression levels of SUMO–scFv were 1.3% at 37°C, 9.3% at 30°C, 14.5% at 18°C for 4 h, and 33.2% at 18°C for 24 h, respectively (Fig. 2). The optimal soluble expression condition was conducted at 18°C for 24 h.
Fig. 3.
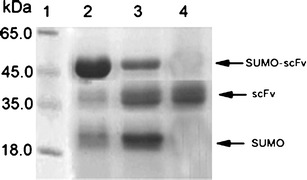
SDS-PAGE analysis of SUMO–scFv digested by SUMO protease and purified scFv. Lane 1: purified fusion SUMO–scFv; lane 2: digestion of purified fusion SUMO–scFv using SUMO protease at 37°C for 2 h; lane 3: the purified scFv obtained after reloading the digested sample thought the Ni-NTA column
Purification of SUMO–scFv
The cells lysate was applied to an affinity column. Proteins without 6× His tags were removed from the Ni-NTA resin using Tris–HCl containing 10 and 20 mmol/l imidazole, and the SUMO–scFv was eluted using Tris–HCl containing 300 mM imidazole. The result of SDS-PAGE electrophoresis showed that the purity of SUMO–scFv was 88.1% (Fig. 3).
Cleavage of SUMO–scFv
About 4 g cell pellets were collected from 1 l of culture with 0.5 mM IPTG inducement. The pellets were treated according to “Materials and methods.” About 50.3 mg of the fusion protein was obtained from 1 l culture. The fusion protein was diluted and cleaved by SUMO protease. The result of SDS-PAGE showed that about 90% of SUMO–scFv protein was cleaved within 2 h at 30°C (data not shown). The cleaved SUMO fusion sample was then reloaded to the Ni-NTA resin to obtain the recombinant scFv. The purity of scFv detected by SDS-PAGE was higher than 95%. After concentration and desalting, the recombinant scFv was further purified by HPLC. The HPLC chromatograms of the recombinant were the same as the expected, and the purity of the recombinant scFv was higher than 98%. The final yield of purified recombinant scFv from 1 l of culture was about 16.53 mg.
Mass spectrometric and N-terminal amino acid sequence analysis
The molecular weight of cleaved recombinant scFv analyzed by MALDI-TOF mass spectroscopy was 32,766 Da, which was similar to the theoretical molecular weight of scFv (Fig. 4).In order to confirm its authenticity, recombinant scFv was subjected to N-terminal amino acid sequencing, according to the sequencing result; the first six amino acid of N-terminal of recombinant scFv was E-V-Q-L-V-E, which was the same as the native scFv sequence.
Fig. 4.
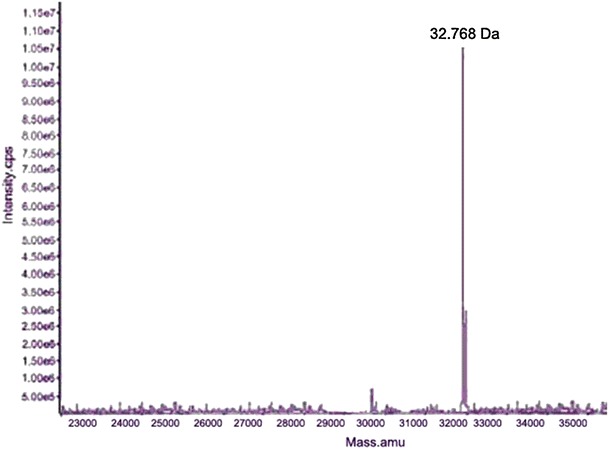
Mass spectroscopic analysis of scFv in agreement with the theoretically calculated mass of 32.766 kDa
Antibody affinity measurement
Purified scFv immobilized on the chip surface and was challenged with VEGF165 at five different concentrations. The K D was 1.65 × 10−8 M, suggesting a sufficient affinity for VEGF165.
Bioactivity assay
In order to investigate the functional potential of the purified scFv, a HUVEC proliferation assay was used. As shown in Fig. 5, HUVECs proliferate in response to the costimulation by VEGF165 signaling. In contrast, a dose-dependent inhibition effect was obtained with the purified scFv.
Fig. 5.
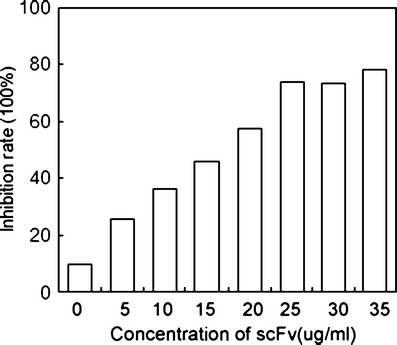
Inhibition of purified scFv to the HUVEC proliferation assay induced by VEGF165. HUVECs (105) were cultured in triplicate in 96-well flat-bottom plates and stimulated with 2 μg/ml VEGF165 and various concentrations of scFv. Cell growth was then evaluated by MTT assay. The experiment was done in triplicate, and the average value is shown in the figure
Discussion
The production of active antibody fragments using recombinant DNA technology and bacteria expression system is very attractive compared to antibody production by hybridoma technology. Recombinant antibody fragments such as Fab and scFv have smaller sizes than the whole antibody and have identical antibody-binding activities with similar affinity. These smaller antibody fragments can be effectively used for diagnostic and therapeutic applications because of having less immunogenicity and better tissue penetration (Yokota et al. 1992).
In the constructed expression system, the recombinant antibody fragments without leader sequence have been expressed at a high level in the form of inclusion bodies under the T7 promoter. Reducing temperature of cultivation or the concentration of IPTG had no effect on the formation of these inclusion bodies. The reason the expressed recombinant protein scFv accumulated in inclusion bodies may be that the overexpression of recombinant protein in prokaryotes and the direct expression without a leader sequence.
Ubiquitin has been reported to exert chaperoning effects on fused proteins, thus increasing expression of proteins in E. coli and yeast (Loll 2003; Malakhov et al. 2004, Marblestone et al. 2006). SUMO is a ubiquitin-like protein containing approximately 100 amino acids, which is highly conserved in eukaryotes and absent from prokaryotes. We hypothesize that the attachment of a highly stable and compact SUMO structure to the N terminus of the scFv proteins will facilitate correct protein folding and enhance solubility and expression. A rationale for the role of SUMO in promoting solubility of insoluble proteins is that the inner SUMO core is a dense hydrophobic globular structure and the protein surface is hydrophilic and highly water soluble (similar to amphipathic detergents).
In this study, we expressed scFv as SUMO fusions in E. coli to evaluate the roles of SUMO and SUMO protease 1 on the production of the proteins. Eukaryotic cells do not produce sufficient quantities of heterogonous proteins; therefore, we have chosen to work in E. coli. In addition, E. coli is easier to grow in vitro, less expensive, and produces recombinant proteins more rapidly as compared to alternative systems (Chaudhuri 1994). When SUMO fusions are expressed as His6–SUMO, rapid purification is possible by using nickel affinity chromatography.
In summary, the data provided here demonstrate that fusion with SUMO is able to enhance expression of membrane proteins in E. coli. After the SUMO tag is removed by SUMO protease 1, The VEGF165-specific functional scFv with high purity and quantity may be useful in diagnostic application for the prediction of VEGF165-relevant vascular diseases in humans.
Acknowledgments
This work was supported by grants from Scientific and Technological Program of Zhejiang Province (2006C33075; to Prof. Zhihua Lin).
References
- Byun J, Heard JM, Huh JE, Park SJ, Jung EA, Jeong JO, Gwon HC, Kim DK. Efficient expression of the vascular endothelial growth factor gene in vitro and in vivo, using an adeno-associated virus vector. J Mol Cell Cardiol. 2001;33(2):295–305. doi: 10.1006/jmcc.2000.1301. [DOI] [PubMed] [Google Scholar]
- Chaudhuri JB. Refolding recombinant proteins: process strategies and novel approaches. Ann NY Acad Sci. 1994;721:374–385. doi: 10.1111/j.1749-6632.1994.tb47409.x. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Carbajal JM, Schaeffer RC., Jr VEGF stimulates tyrosine phosphorylation of beta-catenin and small-pore endothelial barrier dysfunction. Am J Physiol. 1999;277:2038–2049. doi: 10.1152/ajpheart.1999.277.5.H2038. [DOI] [PubMed] [Google Scholar]
- Fernandez M, Vizzutti F, Garcia-Pagan JC, Rodes J, Bosch J. Anti-VEGF receptor-2 monoclonal antibody prevents portal-systemic collateral vessel formation in portal hypertensive mice. Gastroenterology. 2004;126:886–894. doi: 10.1053/j.gastro.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods. 1985;77(2):305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- Huston JS, George AJ. Engineered antibodies take center stage. Hum Antib. 2001;10:127–142. doi: 10.3233/HAB-2001-103-405. [DOI] [PubMed] [Google Scholar]
- Huston JS, Mudgett-Hunter M, Tai MS, McCartney J, Warren F, Haber E, Oppermann H. Protein engineering of single-chain Fv analogs and fusion proteins. Methods Enzymol. 1991;203:46–88. doi: 10.1016/0076-6879(91)03005-2. [DOI] [PubMed] [Google Scholar]
- Johnson ES, Blobel G. Cell cycle-regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. J Cell Biol. 1999;147:981–994. doi: 10.1083/jcb.147.5.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuliopulos A, Walsh J. Production, purification and cleavage of tandem repeats of tandem proteins. J Am Chem Soc. 1994;116:4599–4607. doi: 10.1021/ja00090a008. [DOI] [Google Scholar]
- Lamsoul I, Lodewick J, Lebrun S, Brasseur R, Burny A, Gaynor RB, Bex F. Exclusive ubiquitination and sumoylation on overlapping lysine residues mediate NF-kappaB activation by the human T-cell leukemia virus tax oncoprotein. Mol Cell Biol. 2005;25:10391–10406. doi: 10.1128/MCB.25.23.10391-10406.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavallie ER, McCoy JM. Gene fusion expression systems in Escherichia coli. Curr Opin Biotechnol. 1995;6:501–506. doi: 10.1016/0958-1669(95)80083-2. [DOI] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M. The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J Cell Biol. 2003;160:1069–1081. doi: 10.1083/jcb.200212052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Lei H. Identification of a neutralizing scFv binding to human vascular endothelial growth factor 165 (VEGF165) using a phage display antibody library. Appl Biochem Biotechnol. 2008;144:15–26. doi: 10.1007/s12010-007-8011-4. [DOI] [PubMed] [Google Scholar]
- Liew OW, Ching Chong JP, Yandle TG, Brennan SO. Preparation of recombinant thioredoxin fused N-terminal proCNP: analysis of enterokinase cleavage products reveals new enterokinase cleavage sites. Protein Expr Purif. 2005;41:332–340. doi: 10.1016/j.pep.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Loll PJ. Membrane protein structural biology: the high throughput challenge. J Struct Biol. 2003;142:144–153. doi: 10.1016/S1047-8477(03)00045-5. [DOI] [PubMed] [Google Scholar]
- Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- Marblestone JG, Edavettal SC, Lim Y, Lim P, Zuo X, Butt TR. Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 2006;15:182–189. doi: 10.1110/ps.051812706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando RF, Notkins AL. Production of human monoclonal antibodies against rabies virus. Lyssaviruses. Berlin: Springer; 1994. pp. 195–205. [DOI] [PubMed] [Google Scholar]
- Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992;52:3402–3408. [PubMed] [Google Scholar]