

Abstract
Highly sensitive testing of nucleic acids is essential to improve the detection of pathogens, which pose a major threat for public health worldwide. Currently available molecular assays, mainly based on PCR, have a limited utility in point-of-need control or resource-limited settings. Consequently, there is a strong interest in developing cost-effective, robust, and portable platforms for early detection of these harmful microorganisms. Since its description in 2004, isothermal helicase-dependent amplification (HDA) has been successfully applied in the development of novel molecular-based technologies for rapid, sensitive, and selective detection of viruses and bacteria. In this review, we highlight relevant analytical systems using this simple nucleic acid amplification methodology that takes place at a constant temperature and that is readily compatible with microfluidic technologies. Different strategies for monitoring HDA amplification products are described. In addition, we present technological advances for integrating sample preparation, HDA amplification, and detection. Future perspectives and challenges toward point-of-need use not only for clinical diagnosis but also in food safety testing and environmental monitoring are also discussed.
Graphical Abstract.
Expanding the analytical toolbox for the detection of DNA sequences specific of pathogens with isothermal helicase dependent amplification (HDA)
Keywords: Nucleic acid amplification, Helicase, Isothermal, Pathogen detection, Molecular test
Introduction
Pathogens are microorganisms that act as invaders, attack our bodies, and are capable of causing disease. They are responsible for the spread of communicable diseases prone to cause epidemics, and according to the World Health Organization (WHO), an important contributor to human morbidity and mortality [1]. Take, for example, the recent cases of Zika outbreak in America and Ebola epidemics in West Africa. These overlapping global crises were caused by already known pathogens that can now spread around the world much faster than ever as a result of the increased population mobility, the intensified trade in goods and services, the climate change, and other factors linked to globalization [2]. High death rates from infectious diseases commonly appear in the context of poverty, poor diets, and limited infrastructure found in developing countries. Water, food, and environmental quality are key elements to prevent and control infectious diseases in a globalizing world. In this context, rapid and cost-effective detection of pathogens at the “point-of-need” is a critical demand, not only for clinical diagnosis but also for food safety testing and environmental monitoring.
Traditional methods for the detection of pathogens are well established microbiological assays based on culturing and measuring the growth of individual microorganisms, which require viable pathogens for replication in different culture media such as pre-enrichment and selective plating media [3]. Although these methods are usually inexpensive, they are time consuming, space occupying, and laborious, involving 2 to 3 days for preliminary identification and even more than 1 week for confirmation of the specific pathogenic microorganism. As an alternative, there are different molecular methods that entail near-time or real-time pathogen detection, among them immunoassays and tests for nucleic acids specific of the pathogen.
The advent of polymerase chain reaction (PCR) as a tool for in vitro nucleic acid amplification has led to a new paradigm for the direct detection of pathogen-specific nucleic acids, giving rise to assays that are exquisitely sensitive, and relatively rapid. These methods involve multiple thermocycling steps in high-precision instruments difficult to miniaturize, requiring stringent conditions of laboratory compartmentalization and thus almost exclusively performed in centralized laboratories with high-end instrumentation [4]. An ideal test for point-of-need pathogen detection should meet the ASSURED—affordable, sensitive, specific, user-friendly, rapid and robust, equipment-free, and delivered to those who need it—criteria outlined by the WHO [5]. With an incremental knowledge on the complex in vivo nucleic acid amplification processes, which are essential in biological systems [6], novel isothermal nucleic acid amplification methods are gaining their importance for the development of analytical devices aimed to meet those ideal criteria. Isothermal amplification methods can be easily integrated into simple and low-energy consumption microsystems, which may in the future outperform PCR for pathogen detection. These methods include loop-mediated isothermal amplification (LAMP), recombinase polymerase amplification (RPA), nucleic acid sequence-based amplification (NASBA), helicase-dependent amplification (HDA), rolling circle amplification (RCA), signal-mediated amplification of RNA technology (SMART), and strand displacement amplification (SDA). Previously published reviews [6] gave a broad description of these methods and their potential applications. This review focuses in one of these emerging methodologies, HDA, giving a look into the most recent HDA-based analytical methods for the detection of pathogens. We endeavored to highlight new methods for the detection of HDA amplification products as well as relevant systems to integrate sample treatment, amplification, and detection, with the future trends forecasted to conclude.
Principle of helicase-dependent amplification (HDA)
HDA is one of the simplest approaches for isothermal nucleic acid amplification that mimics an in vivo process of DNA replication, using a helicase to isothermally unwind DNA duplexes instead of heat, as is the case in PCR. Vincent et al. developed this method in 2004 [7], which was subsequently patented in 2007 [8]. The initial studies employed UvrD helicase from E. coli, a well-studied helicase II that unwinds DNA in a 3’ to 5’ direction, using the energy from the hydrolysis of adenosine triphosphate to break the hydrogen bonds between complementary bases in duplex DNA [9]. In E. coli cells, this protein is involved in the repair of mismatches in DNA, working in coordination with methyl-directed mismatch repair (MutL) protein that stimulates the helicase activity. Consequently, the original HDA system requires, in addition to the helicase to unwind dsDNA, two accessory proteins: MutL and a single-stranded DNA-binding protein (SSB) to stabilize the ssDNA preventing the re-association of the complementary strands. Once DNA is isothermally unwound and stabilized, two oligonucleotide primers hybridize to the 5′ and 3′ borders of the target sequence and a DNA polymerase extends the annealed primers by adding deoxynucleotide-triphosphates (dNTPs) to generate double-stranded amplification products (Fig. 1), in a scheme very similar to PCR but without thermal cycling. The process is repeated, giving rise to an exponential amplification of the target dsDNA. The dNTPs mixture must be enriched with dATP, which acts as cofactor of helicase, and Mg2+, cofactor for both helicase and polymerase. It is very important that helicase and polymerase work in a coordinated way, preventing polymerase to be displaced by helicase, as the helicase-catalyzed reaction is the rate-limiting step, and unwound DNA strands do not re-anneal before they are copied by polymerase. It is for this reason that a pair helicase/polymerase working together in natural systems must be employed. In the original work, the E. coli UvrD helicase/DNA polymerase I Klenow fragment pair is used [7] to exponentially amplify DNA at 37 °C, mesophilic form of HDA (mHDA). Later, a thermostable helicase Tte-UvrD from Thermoanaerobacter tengcongenesis in combination with Bacillus stearothermophilus DNA polymerase (Bst-DNA polymerase) allowed performing the amplification at higher temperatures (60–65 °C), called the thermophilic form of HDA (tHDA) [10]. Unlike mHDA, the tHDA system does not require the MutL protein, thus simplifying the reaction components. At the same time, the efficiency and selectivity of the amplification reaction is improved.
Fig. 1.
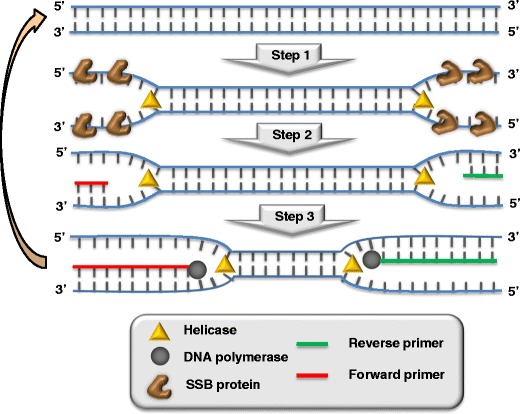
Scheme of the helicase-dependent amplification system, adapted from reference [7]. (1) Unwinding dsDNA by helicase and stabilization of ssDNA by SSB; (2) annealing of primers, and (3) elongation of primers by DNA polymerase
Design of primers
Similar to PCR, HDA amplifies nucleic acids exponentially using two flanking primers. To find the oligonucleotide primers pair that leads to high yield of a target-specific amplicon, several primer combinations are often tailored by using online programs originally devised to design PCR primers (Primer3 [11] and PrimerQuest [12]). For that, specific recommendations for the isothermal technique should be borne in mind, namely: (a) primer size ranging from 24 to 33 nucleotides, (b) melting temperature, Tm, from 60 to 74 °C, and (c) GC% within the interval 35-60% [13]. However, successful HDA amplification has been achieved when relaxing some of these constraints as well. Afterwards, primer candidates are examined for potential dimer and self-dimer formation, first in silico employing DNA folding algorithms [14], and then experimentally screened. Likewise, BLAST analysis [15] must be carried out to exclude homology to nontarget sequences.
Even taking these cautions, the mix of primers along with DNA polymerase and the building blocks (dNTPs) are likely to do mischief, giving rise to high background signal, and low sensitivity and selectivity. These problems, mainly attributed to unwanted primer dimers and off-target interactions, are more prevalent in isothermal amplification techniques due to the lack of an annealing step from high to low temperatures to form Watson-Crick base pairing. A particular feature of HDA is, however, the recommended long size of HDA primers with respect to PCR counterparts, which feeds confusion between specific amplicons and spurious products, and hence false positive results, as a consequence of their similar size [16]. The risk of primer-dimer artifacts would therefore justify the lower primer concentrations used in HDA (75–100 nM) when comparing with PCR (0.1–1 μM).
To circumvent undesired side products arising from primers, a series of actions have been reported; some of them constitute assay strategies themselves and they will be discussed below, whereas others are more general, involving primer design. Given that DNA polymerase synthesizes new DNA strands in a 5’–3’ direction, interactions between or within the primers are due to overlapping 3’-ends, even little apparent complementarity, and corrective actions have been focused in this direction.
Use of chemically inactivated primers containing a 3’ blocking group as well as a ribonucleotide moiety near the 3’-end has been proposed to prevent elongation by DNA polymerase. Unblockage and activation of the primers only take place once they hybridize with the target DNA and the ribonucleotide linkage is subsequently cleaved by a hot-start RNase H2. At 65 °C, optimal temperature for HDA, RNase H2 becomes fully active and releases a DNA fragment with a 3′-hydroxyl group able to initiate the amplification [17]. With this approach, primer-based side reactions are eliminated, while HDA amplification speed and efficiency are not significantly affected; however, it cannot be implemented in RNA amplification.
Inefficient consumption of primers, not leading to the desired product, can be also attenuated by self-avoiding molecular-recognition system (SAMRS). SAMRS draws on a set of dNTP analogues (A*, T*, G*, and C*) that have been designed to pair with natural nucleobases but not with each other, based on the number of hydrogen bonds [18]. Therefore, primers built from SAMRS hybridize with the target but they do not interact with each other, avoiding primer-dimer artifacts. Thermophilic enzymes (polymerase and helicase) have demonstrated their capability to work with SAMRS-structurally altered nucleotides by just adjusting Mg2+ concentration [19], albeit the best yield has been reported when four SAMRS components are placed at the 3′-end of the primers, followed by one terminal natural nucleotide. Moreover, the addition of a thermostable reverse transcriptase has allowed RNA target amplification to benefit from these chimeric primers.
On the other hand, the influence of the nucleobases placed at the 5′-end of the primers has been also studied, finding an improved efficiency in HDA when these termini are enriched in adenine or cytosine [20].
Many primers designed for PCR amplification do not meet the recommended Tm needed for HDA. In those cases, it is possible to modify the existing primer set by adding a customized 22-nt tail at 5′-end [21]. Though this tail is not specific of the target gene sequence, it increases the Tm after the first amplification and also the length of the amplicon, which is critical in this isothermal technique. These can be considered a general strategy for adapting PCR primers to HDA amplification, especially for comparison with PCR. Under these conditions both sensitivity and selectivity of HDA are improved, and HDA reaches identical limit of quantification (1 f. of plant virus cDNA) to end-point, SYBR Green, and TaqMan PCR.
Improvements in HDA amplification
Despite its simplicity, the utility of HDA is often hampered by template-independent primer interactions that give rise to a nonspecific amplification phenomenon. This causes false positive results and adversely affects the detectability and robustness of the methods based on this amplification reaction. Although a careful primer design can reduce primer-dimer formation, this process and unspecific amplification are more pronounced in HDA than in PCR [16], and fundamentally occur because no hot-start polymerase for HDA is currently available. This is especially a problem for the detection of low copy number targets due to the long time required for amplification, and in the design of multiplex amplifications where various pairs of primers must function together. Besides the HDA modification explained before, termed blocked-primer HDA, in which DNA polymerase cannot extend the primers until they are cleaved by a thermostable RNase [17] giving rise to a quasi-hot-start amplification method, selectivity of the amplification may be modulated by careful optimization of the reaction conditions. The use of additives frequently employed in PCR, such as DMSO, betaine, and sorbitol, which reduce DNA secondary structures facilitating primer annealing, has proven to be effective in HDA amplification [22], improving its yield and specificity. However, these additives can also greatly reduce the activity of polymerase. Therefore, it is important to test different concentrations to find the best conditions for each amplification system. Similarly, macromolecular crowding agents (polyethylene glycol, dextran, Ficoll) that mimic the conditions for amplification in natural systems, increase the speed of polymerase and affect the structure of DNA, improving yield, robustness, and specificity [23]. The optimization of reaction enzymes mixture is another important issue; for example, it has been demonstrated that, surprisingly, lowering the concentration of both ATP and dNTPs with respect to that usually employed improves the efficiency and selectivity of HDA, suppressing primer-dimer formation, and thus reduces the background amplification problems [24]. All these measures may be combined, and it is of paramount importance to experimentally determine the optimal amount of all additives and reagents for each amplification system.
A different strategy that improves the sensitivity and robustness of HDA is the digestion with specific restriction endonucleases during amplification. Mimicking the natural process of mismatch repair in which helicase is involved, restriction enzymes are selected to cleave a sequence near the place where primers anneal, generating blunt ends that help recruiting and loading the helicase [23]. This improves the effective loading of the helicase and thus accelerates the HDA reaction. However, it renders the design more challenging since it is necessary to find an enzyme capable of cutting the dsDNA close to the target sequence, without fragmenting it.
It is also possible to expand the HDA system to RNA amplification by incorporating into the reaction mixture a thermostable reverse transcriptase, RT-HDA [25]. This enzyme converts the RNA target into DNA, which is subsequently exponentially amplified by HDA reaction. Importantly, because the UvrD helicase is able to unwind DNA-RNA hybrids more efficiently than DNA duplex, the RT-HDA system is able to perform one million-fold amplification of RNA in 10 min in a single step at a constant temperature [25].
The efficiency of HDA to amplify long amplicons is low due to the limited speed and processivity of the chosen helicases [7]. This is related to the mechanism of action of UvrD helicase that must be on high molar excess in relation to the dsDNA substrate to unwind it. To increase the speed of DNA synthesis, a new bifunctional protein, termed helimerase, has been engineered by linking the UvrD helicase with Bst-DNA polymerase [26]. This complex demonstrated increased apparent processivity, and allowed the amplification of fragments significantly longer (up to 2.3 kb) than that with the two individual proteins. However, this new enzyme is not commercially available, and further studies should be undertaken to simplify and make its production more cost-effective.
Another possibility to improve the efficiency of HDA is to employ gold nanoparticles (AuNPs) in the reaction media [27]. AuNPs bind to ssDNA with higher affinity than to dsDNA [28], and consequently similar to SSB they may improve the denaturation efficiency of helicases. In this way not only sensitivity but also selectivity of HDA has been improved [27]. This is a very recent discovery that opens new opportunities for improving HDA, and undoubtedly it requires further investigations.
Strategies for monitoring HDA
Similar to end-point PCR, the successful performance of HDA is usually accomplished by agarose gel electrophoresis and staining with fluorescent DNA binders such as ethidium bromide or safer and more environmentally friendly alternatives (SimplySafe, GelRed, or GelGreen). This rapid analysis does not allow either quantitation or discrimination of unspecific amplicons of similar length, a very common pitfall of suboptimal isothermal amplification designs. Nevertheless, HDA is compatible with different detection mechanisms, and significant improvements have been made in monitoring HDA amplicons both at the end-point of the amplification reaction and in real time.
Real-time HDA
Fluorescence detection
Monitoring the amplification in real time is the most straightforward solution. Real-time HDA was first developed using a non-mutagenic non-cytotoxic fluorescent intercalator (EvaGreen) that is only fluorescent when bound to dsDNA, which ensures low background signals [29]. Besides, it shows less amplification inhibition than SYBR Green [30]. Since no temperature cycles are performed, the analytical signal is the time needed to exceed an established threshold in fluorescence intensity. This time decreases with target concentration, as expected, and with the primer concentration used. This means that the amplification speed is boosted by the amount of primers in solution. However, an enhancement in primer-dimer amplification occurs, limiting the improvement in detectability [31]. In fact, detailed comparison of HDA and PCR suggests that the primer concentration to obtain the lowest limit of detection (1 f. for oligonucleotide template) is 75 nM. For PCR, higher primer concentration, above 200 nM, permitted reaching a smaller limit of detection, 100 aM. From real-time experiments, HDA has been claimed to be faster than PCR [16] but no good linearity was obtained. More recently, HDA was proven to be slightly slower than PCR [32] and doubling times (equivalent to one cycle in PCR) are reported to consistently range from 50 s [33] to 78 s [17, 32, 34], sometimes longer than usual PCR cycles. Good linearity achieved for real-time HDA assays allowed the detection of two human papillomavirus types using plasmids in clinical samples [29].
Electrochemical detection
Electrochemical monitoring of the HDA is also possible by replacing the fluorescent probe with an electroactive intercalator [35]. The redox activity decreases after intercalation because of the inability to reach the electrode surface, so this detection strategy is more prone to false positive results. As conventional real-time amplifications, high-throughput measurements can be carried out in disposable electrochemical microplates. Similar detection limits and reproducibility to fluorescent real-time HDA were obtained for a specific sequence of E. coli plasmid but amplification rate was slower, probably due to stronger inhibition by redox probe than by EvaGreen.
In both fluorescent and electrochemical approaches, a melt curve after amplification is compulsory to check the presence of unspecific amplicons. To improve selectivity and allow multiplex assays, specific probes such as molecular beacons, FRET, TaqMan, or Scorpions probes are specifically designed to hybridize with a short fragment of the target strand (Fig. 2a, right panel). TaqMan MGB (minor groove binder) probes were first used in combination with two DNA polymerases with 5′ to 3′ exonuclease activity [36]. These probes are shorter than conventional TaqMan probes because of the special quencher that binds to the minor groove increasing the overall binding energy expected for a probe of the same size. The probes were designed to have a melting temperature close to the HDA optimized one (63–65 °C). The Gst DNA polymerase was very efficient in cleaving the probe, which increased dramatically the signal-to-noise ratio and, as a consequence, reduced the threshold time (transformed into threshold cycle, Ct, by analogy with PCR). Taq polymerase was also efficient under isothermal conditions, even though it is intended to work after each denaturation step at 95 °C. Asymmetric amplification is not needed but it can speed up the reaction when using one of the primers at concentration above the recommended 75 nM provided no unspecific amplification is promoted. Multiplex detection is possible using hydrolysis probes but optimization is critical for target sequences with very different copy number and requires a denaturing step, so it is not entirely isothermal. In any case, the amplification is slower than for individual amplifications.
Fig. 2.
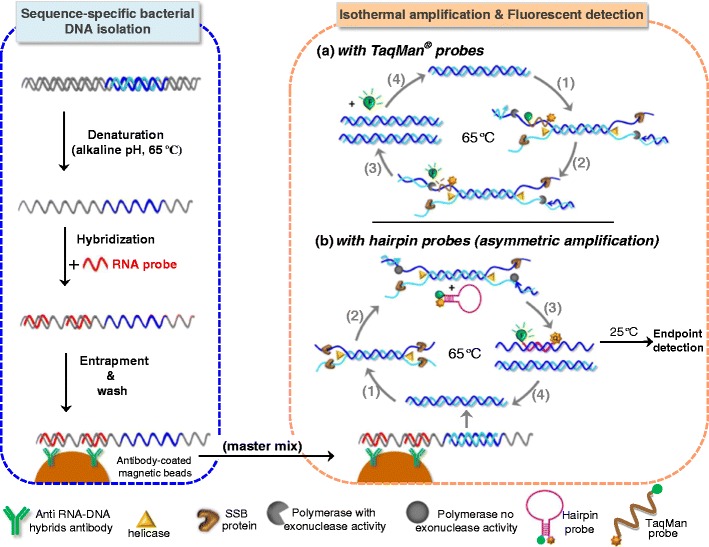
Right panel: schematic representation of HDA assays with homogeneous fluorescent monitoring, by using dual-labeled probes (a), TaqMan (b) hairpin ones. Left panel: a previous sequence-specific sample preparation employing RNA probes along with magnetic particles modified with an antibody specific for RNA-DNA hybrids boosts assay performance in clinical samples
More recently, hairpin probes with a fluorophore and a quencher at each end where also successfully used in a 4-plex real-time HDA assay for two sequences of C. trachomatis and one of N. gonorrhoeae in addition to the internal control [22]. However, end-point detection was finally selected after careful optimization of components in the multiplex assay. The hairpin probe is not fluorescent in solution but it fluoresces after hybridization with the amplified target. Unlike TaqMan probes, polymerases with no exonuclease activity are required in order to maintain the probe hybridized to the amplified strand. Asymmetry ratio of 1:3, with concentration above 25 nM for the limiting one, amplified preferentially the strand complementary to the probe, improving the end-point signal. Initial alkaline denaturation at 65 °C was more effective than the thermal shock at 95 °C, making this method completely isothermal. HDA was inhibited with 50–100 ng of human genomic DNA but this limitation was overcome with a sequence-specific isolation and purification of the bacterial DNA. This method is based on hybridization with RNA oligonucleotides complementary to the analyte far from the target sequence and entrapment and preconcentration on magnetic beads covered with antibodies specific for RNA-DNA hybrids. The purification thus provides ssDNA as an input sequence for HDA using probes with a fluorophore and a quencher for detection (Fig. 2b and left panel). In such a way, as low as 25 cells/mL of two different bacteria were detected simultaneously, 2.5–5 times higher than for individual amplifications, in about 3 h including sample preparation [37].
Dedicated real-time instrumentation for HDA (Solana from Quidel [38]) and other isothermal amplifications are now commercially available although hand-held devices are still under development [39].
Amplicon capturing approaches
Lateral flow devices
In order to achieve low-cost DNA detection assays, real-time monitoring is not adequate. Since the beginning, lateral flow devices (LFD) were developed to reveal the isothermal amplification. In symmetric amplifications a dual-labeled amplicon is obtained by incorporating a biotinylated primer and the other primer bound to a fluorescein tag. The LFD contains streptavidin-coated gold nanoparticles that bind the biotinylated amplicon, and the complex flows until the detection line, where immobilized antibodies capture the fluorescein-labeled amplicons forming a visible red-colored line (Fig. 3).
Fig. 3.
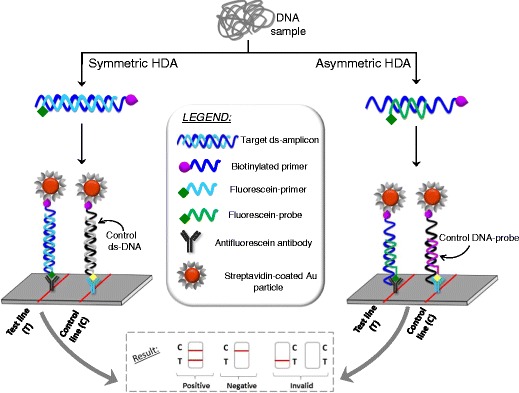
Schematic of lateral flow HDA assay (left), symmetric (right), asymmetric DNA amplification
Asymmetric amplification is more widely used and relies on a preferential amplification of the strand arisen from a biotinylated primer. The limiting primer is not labeled. A specific complementary short probe labeled with a hapten (fluorescein, digoxigenin or 2,4-dinitrophenyl) at the 3’-end to avoid extension is also added to the amplification solution. This probe hybridizes with the biotinylated strand, forming a partial duplex. This particular hybridization decreases the chance of detecting unspecific products. Then, the partial duplex enters the LFD where it binds to streptavidin-conjugated color particles and is finally captured by the appropriate antibody on the detection line (Fig. 3). This way, duplex amplifications (two targets in a single experiment) can be carried out, one of them being the internal control of amplification [40, 41]. Commercial kits for some pathogen are currently marketed [42]. The assay for Herpes simplex virus (HSV)-1 and -2 in genital and oral lesions was cleared by FDA in 2011 and it does not require nucleic acid extraction [43]. A simple dilution allows detection of as low as 5.5 and 34.1 copies of HSV-1 and HSV-2, respectively, per reaction within 1.5 h [44]. Manual extraction amenable to resource-limited places reached 470 copies/reaction of HIV-1 DNA from dried blood spots with LFD [45]. The identification at species level of Plasmodium malaria parasite is possible by careful selection of the hybridization probes and primers [46]. Three probes with different labels were designed for a single HDA assay; one for all Plasmodium species, a second one specific for P. falciparum, and the third as an internal control. A second HDA amplification with a specific primer for P. vivax and the first and third probes allowed the detection of this species in LFD cartridges. No extraction of DNA from the protozoon is needed. The sensitivity is 5 to 10 times lower when applied to patient samples (200 parasites/μL) instead of spiked human blood. Microtiter plate version of these LFD with colorimetric enzymatic detection was also demonstrated for detecting 10 copies of the ureC gen of Helicobacter pylori [47]. Since symmetric amplification was carried out, a step of thermal denaturation was needed before hybridization in a separate tube, which lengthens the assay.
Hybridization-based capture of amplicons
Undoubtedly, an extra level of selectivity is needed when revealing HDA amplicons. Hybridization-based target capture on surfaces is a simpler and cost-effective alternative to immunocapturing. A specific short capture probe partially complementary to asymmetrically amplified biotinylated-strand is usually anchored on the surface. Further enzymatic labeling allows optical measurement (Fig. 4). A variety of surface chemistries have been applied to construct the DNA chip from the simplicity of polypropylene sheets that only require aminated probes and UV-cross-linking [48] to silicon nitride wafers that are not biologically compatible and need two additional layers of polydimethylsiloxane and poly(phenylalanine-lysine), respectively [17, 33]. Those layers introduce a large number of amine groups for further DNA binding, achieving an optimal DNA coverage of 1.5×1012 molecules/cm2 and 100% hybridization efficiency [49]. Hydrazine-terminated DNA capture probes were cross-linked using N-succinimidyl 4-formylbenzoate and these modified surfaces are stable up to 9 months at 37 °C [34]. In both assays, asymmetric HDA is performed (1:4 and 1:2, respectively) and the biotinylated strand is captured on the chip surface. It is worth noting the high concentration of primer used in the second approach (above 100 nM).
Fig. 4.
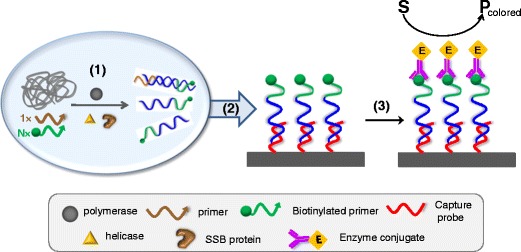
Hybridization-based on-surface capture of HDA amplicons: (1) homogeneous asymmetric HDA, (2) specific capture of biotinylated ss-amplicon, and (3) enzyme labeling and optical detection of the enzymatic product
Polypropylene chips use streptavidin-HRP conjugates for labeling and detect silver enzymatically deposited. In this way, the plant pathogen Phytophthora was detected at the species level using three different sets of primers in individual HDA assays on chips containing probes for each species. All five species were detected but two of them could not be distinguished because they differ in a single nucleotide [50]. Combining hybridization and labeling in a single step, time is reduced to 60 min without loss of detectability [48].
The silicon wafer approach uses anti-biotin antibody-peroxidase conjugate and interference-based detection. This unusual strategy relies on the apparent color change caused by the deposition of the enzymatic product on the thin film. The viability of multiplex HDA was demonstrated for Staphylococcus species [33]. Two genes were amplified in the tube and then detected on a chip containing 13 different probes (1 for genus identification, 11 for each species, and an additional for highly homolog bacteria) after a quick hybridization and labeling steps (5 min each). The limit of detection for pure species is 1 cfu/reaction (about 3000 cfu/mL) but increases 10 to 30 times in the presence of several species. The time turnaround is shortened to 30 min when performing a symmetric HDA in the hybridization chip [34]. Interestingly, unlike HDA in a tube, no artifacts were observed in the chip, suggesting beneficial surface/volume conditions. Blocked primer HDA combined with these chips permitted the detection of one copy of genomic M. tuberculosis in 40 min with single nucleotide polymorphism selectivity [17].
Magnetic particles are very easy to handle and facilitate the washing steps, so they are more and more incorporated into commercial assays. This platform was first used to develop a sandwich assay to capture the denatured and purified amplicons obtained in a symmetric HDA assay [51]. Aminated magnetic beads were modified with thiolated partially complementary probes through a heterobifunctional crosslinker. Then the DNA sandwich assay was completed by hybridization with AuNPs covered by another partially complementary probe. The duplexes on the beads were placed onto disposable carbon electrodes where Au was dissolved and oxidized at 1.25 V. Finally the cathodic current of Au reduction was measured by differential pulse voltammetry (DPV). However, the assay is long (6 h excluded DNA extraction) and semi-quantitative for M. tuberculosis.
More recently this pathogen was detected at attomolar level also using magnetic beads for capturing the unpurified biotinylated strands amplified by asymmetric HDA [31]. Fluorescein-labeled probes hybridize with the anchored amplified strand at the center region and are detected after enzymatic labeling on the disposable carbon electrode by chronoamperometry. Minimization of unspecific products using as low as 5 nM of the limiting primer was essential. Reduction of the salt concentration during the hybridization step from 1 to 0.1 M NaCl was also needed to achieve a limit of detection of 0.5 aM of a synthetic specific sequence (15 copies). This value rivals the detectability of an analogous assay using PCR (11 copies) [32]. Symmetric HDA was faster (30 min versus 90 min) than the asymmetric one but 10 times less sensitive when combined with the hybridization assay even after a denaturation step to separate the duplex. The amplification factor of asymmetric HDA was calculated to be 106 respective to the genomagnetic assay, which explains the ultrasensitivity. Genomic DNA from a variety of clinical samples was also tested with concordant results in comparison with real-time PCR and cultures.
Labels other than enzymes can also be exploited in hybridization assays but until now only fluorophores were applied to HDA assays. Quantum dots covered by DNA short specific capture probes were immobilized on paper after oxidation the cellulosic fibers with periodate to generate aldehyde groups on surface that can react with aminopropylimidazol forming a Schiff base subsequently reduced to yield stable covalent C–N bonds [52]. Imidazol moieties bind to CdSeS/ZnS-probe oligonucleotides to complete the sensing surface. Optimization of quantum dot concentration improved the limit of detection up to 7.5 times. Paper modification is stable for at least 2 wk and reusable up to five times [53]. This platform was combined with a sandwich hybridization assay, which was performed with hybridization probes with an acceptor fluorophore in the nearest end to the quantum dot platform. FRET-based transduction allows the detection of the decreasing emission of the donor QD, the increasing emission of the acceptor fluorophore, or, as in this work, the ratio of both signals, which is more powerful to discriminate low concentrations. SNP selectivity was reached by adjusting the concentration of formamide and the ionic strength in the hybridization step. An amplification factor of 107 was estimated permitting the detection of 37 zmol of a synthetic sequence corresponding to a diagnostic marker of E. coli in 3 h [52]. However, this time could be shortened if asymmetric HDA was performed avoiding the time- and reagent-consuming magnetic separation step employed for generating single stranded sequences from HDA amplicons.
Integration of HDA and other steps in the analytical process
Nucleic acid-based pathogen analysis involves three main steps: sample preparation (cell lysis, nucleic acid extraction and purification), amplification, and detection, all of which are of paramount importance for obtaining reliable results. There are different attempts in the literature toward the design of platforms that integrate HDA amplification and at least one of these steps in order to obtain cost-effective, robust, automated, and user-friendly systems useful for point-of-need pathogen control. The development of such fully integrated, low cost devices to detect DNA from pathogens fuels the development of isothermal amplification schemes as HDA. Beyond the format scheme, determined steps are being taken to make this technology amenable to decentralize analysis in remote locations without electricity supply.
Amplification and detection are easily integrated by performing the amplification on suitable materials. Membrane-based devices are well suited for low cost analysis, and several materials were tested [54]. While glass-fiber and nitrocellulose inhibit both PCR and isothermal amplifications due to strong enzyme adsorption, polycarbonate impeded HDA when carried out within its pores but not when some liquid remains. Polyethersulfone showed the best compatibility with both PCR and isothermal amplification. Cellulose can also be used after BSA treatment to avoid enzyme adsorption [55]. The mixture of enzymes for HDA amplification is stable, after drying on paper, at room temperature for about 1 month, probably due to the presence of high molecular weight carbohydrate in the formulation. A rapid HDA detection of M. tuberculosis (10 min) was performed in this substrate using three times higher concentration of enzymes to compensate adsorption and PicoGreen as fluorescent dye. Amplification occurred even at 50 °C achieved with a hand warmer. This qualitative assay offers a limit of detection of 100 copies. Easy and efficient extraction of DNA from cell lysate on paper inserted on a pipette was also shown [56]. The isothermal amplification of Chlamydia trachomatis was carried out in the sealed pipette and detected in a LFD. In this way, an easily operated and rapid diagnostic platform useful for point-of-care detection of sexually transmitted infectious is presented, with improved limit of detection compared with the immunoassays typically employed for this purpose. Although not truly integrated, sample preparation and amplification are combined by using a syringe or bicycle pump to remove the cellular lysis solution and transfer the HDA solution after amplification. These pumps combined with toe warmers make this approach electricity-free. A pump-based extraction with a hand-warmer heated amplification was also employed to detect Clostridium difficile in stool samples [57]. Positive amplifications were detected by PAGE after on-chip HDA for 45 min, including warming up. Channels were coated with a costly polymer to limit enzyme adsorption. Formation of primer-dimers was not negligible and attributed to the high surface to volume ratio, contrary to the observation of Jenison et al. [34], but might be reduced by adding DMSO. Similarly, a modular device using chip-based DNA hybridization with optical detection was reported without the need of power supply [48].
Only two batteries are the power need of a fully disposable integrated paper-based device [58]. It consists of three modules: the extraction module constructed on paper, the more complex amplification module with very thin ceramic heating plates, and a lateral flow strip based on DNA hybridization for detection. All reagents are dried and stored on the device but long storage requires 4 °C (2 months) or –20 °C (3 months). The extraction is performed in 2 min and the overall turnaround is 77 min.
The simplicity of these extraction methods contrasts with micro solid phase extraction on polymer microchannels. Integration of extraction and HDA on a disposable microfluidic disposable device was first achieved in 2010 [16]. In 2012 a meso-scale fluidic system integrated the entire process: from extraction to optical detection (a digital camera) in an analyzer/cartridge device [59]. It was applied to stool samples for the detection of Clostridium difficile with a limit of detection of 10 cfu using blocked-primer HDA and silicon wafer chips. However, the manual assay is faster because of the motor movements and mechanical calibrations.
Passive mixing is an alternative to pumps for fluid movement. When combined with DNA electrostatic entrapment from cell lysate on chitosan membranes, an 80% extraction efficiency compared with a commercial kit was achieved. After DNA extraction, HDA was performed in a low cost device incorporating simple optical detection [60]. Reducing the cost of optics is also behind the development of the ratiometric FRET assay described above. A UV lamp for excitation and an i-Pad camera permitted obtaining limits of detection in the fM range comparable to the less cost-efficient epifluorescence microscope [52]. Based on the same electrostatic principle, DNA purification was achieved on polycarbonate modified with polyethyleneimine in a valve-free fashion in 30 min [61]. Device rotation sequentially propels the solutions from each of three reservoirs (for extraction, washing, and HDA reagents) to the chamber where DNA is electrostatically entrapped to perform the symmetric HDA amplification of E. coli DNA. However, detection was not integrated.
The volume of sample used is decreasing to the nL range. HDA was performed in 192 nL in a microfluidic device with real-time detection [30]. However, low fluorescence increase and variability in threshold time was higher than when using 5 μL volume. This technology was applied to the amplification of a plasmid containing a fragment of SARS virus.
Finally, surface HDA has also been demonstrated as a step forward to full assay integration on modified glass slides. The efficiency of on-surface amplification has been reported to be poorer than in solution phase because of the steric hindrance of the solid surface that reduces the polymerase extension efficiency, electrostatic repulsion of high-density primers, and attachment of long DNA sequences that prevent access to the anchored primer [62]. Slightly lower detectability for S. aureus, 5×104 versus 2×104 cells, was achieved in the first on-chip HDA automated assay [63]. The aminated primer was linked to an epoxysilanized glass slide. Only one primer was added to a minimum of 150 μL of solution, which consumed 3-fold amount of reagents. Programmed pump mixing during the 120 min of HDA with automatic washing steps at 30 °C leaves the surface ready for laser scanning of the fluorophore-labeled duplex anchored on the surface. The single assay for N. gonorrhoeae was much less sensitive. Multiplex amplification/detection was feasible using the same fluorophore because of the spatial separation of capture probes specific of each bacterium.
In order to promote the on-surface amplification, a limited amount of a shortened version of the anchored primer was added to the solution (1:15 ratio) [64]. Under those conditions, it is believed that amplification initiates in solution until almost complete depletion of the limiting primer. Then the amplification shifts to the surface promoted by the longer anchored primer. This pathway is supported by a delay reported on ITO-modified surfaces, which implied a minimum of 90 min for amplicon detection. No differences with blank experiments were obtained at 75 min. The fluorescein-labeled duplex synthesized on the surface was electrochemically detected by DPV after enzymatic labeling with alkaline phosphatase (ALP). Peroxidase could not be used because the enzymatic product, the oxidized TMB, presents a large overpotential that precludes its detection at usual potentials. Transparent ITO surfaces are also compatible with optical detection but UV-vis detection offered one order of magnitude higher limit of detection even though peroxidase, which has a higher turnover than ALP, was used. Only 10 genomic copies of Salmonella were detected electrochemically, which matches real-time PCR approaches. Interestingly, the ITO modified surface was stable under dry conditions for 9 months provided that a mixture of BSA and glucose, both at 2.5%, was previously added. A rehydration step is also needed before use. This extended storage stability is highly desirable for electrochemical DNA-based sensors mostly based on fairly stable self-assembled monolayers on Au.
Applications
Most applications of HDA are devoted to the clinical field where rapid, cost-efficient devices are more and more in demand to cut the increasing cost of diagnosis, especially in the case of infectious disease. Another market niche is the use at the point of need with special interest in remote locations or in developing countries where electricity is not always taken for granted. However, some applications for animal and plant viruses as well as for food contamination surveillance are starting to gain recognition, including amplification of RNA virus [65]. In Table 1, a summary of the main features of the reported methods is shown.
Table 1.
Summary of published helicase-dependent amplification methods for pathogen detection
| Pathogen | Detection | LD (copies/reaction) | HDA time (min) | HDA T (°C) | Sample | turnaround time (min) | Ref |
|---|---|---|---|---|---|---|---|
| E. coli | Gel electrophoresis | – | 60 | 65 | cultures | 90a | [61] |
| Tomato virus | Gel electrophoresis | 4 pg RNA | 60 | 65 | Leaf tissue | – | [65] |
| L. reticulatis | Gel electrophoresis | 1 ng | 90 | 65 | Insect infected mixed cereals | – | [66] |
| C. difficile | PAGE | 0.0125 pg | 45 | 65 | Stool | – | [57] |
| HPV-16 | Real-time | 1 | 90 | 65 | Cervical swabs | 90 | [29] |
| HPV-18 | Real-time | 10 | 90 | 65 | Cervical swabs | 90 | [29] |
| SARS | Real-time | – | 30 | 62 | Plasmid DNA | 30 | [30] |
| M.T. | Real-time | 300 | 90 | 65 | Plasmid DNA | 90 | [18] |
| E. coli | Real-time | 100 cfu/mL | 60 | 65 | Cultures | 60 | [16] |
| T. vaginalis | Real-time (comercial) | – | 45 | – | Vaginal swab/ urine | 45 | [38] |
| E. coli | Echem real-time | 105 | 80 | 65 | Plasmid DNA | 80 | [35] |
| V. cholerae | TaqMan real-time | 50 | 60 | 65 | Genomic DNA | 60 | [36] |
| B. antracis | TaqMan real-time | 50 (pXO1 gene) | 90 | 63 | Genomic DNA | 90 | [36] |
| B. antracis | TaqMan real-time | 50 (pXO2 gene) | 90 | 63 | Genomic DNA | 90 | [36] |
| B. antracis | TaqMan real-time | 50 (multiplex) | 90 | 63 | Genomic DNA | 90 | [36] |
| M.T. | Fluorescence | 100 | 10 | 65 | Artificial sputum | – | [55] |
| S. aureus | Fluorescence | 5 cfu/mL | 90 | 65 | Spiked milk powder | 120 | [67] |
| 50 cfu/mL | 90 | 65 | Spiked pork meat | 120 | [67] | ||
| N.G. | Fluorescence | 5–10 cells | 60-120 | 65 | Spiked collection media | – | [22] |
| 25 cells (multiplex) | 90-120 | 65 | Spiked collection media | – | [22] | ||
| C.T. | Fluorescence | 5–10 cells | 60-120 | 65 | Spiked collection media | – | [22] |
| 25 cells (multiplex) | 90-120 | 65 | Spiked collection media | – | [22] | ||
| N.G. | LFD | 50 | 20 | 65 | Urine | 35 | [23] |
| C. difficile | LFD | 20 | 60 | 65 | Stool | 75 | [40] |
| S. aureus | LFD | 50 cfu/mL | 60 | 65 | Bacteria blood culture | 80 | [41] |
| Strep Group A | LFD | – | 35 | 64 | pharyngeal swab | 45 | [42] |
| HSV-1 | LFD | 5.5 | 60 | 64 | Genital lesion swab | 75 | [44] |
| HSV-2 | LFD | 34.1 | 60 | 64 | Genital lesion swab | 75 | [44] |
| HIV-1 | LFD | 470 copies/mL | 75 | 64 | Dried blood spots | 95 | [45] |
| Plasmodium | LFD | 50 copies | 90 | 64 | genomic DNA | 105 | [46] |
| Plasmodium | LFD | 200 parasites/μL | 120 | 64 | Blood samples | 135 | [46] |
| C.T. | LFD | 1000 cells | 30 | 65 | Synthetic urine | 50 | [56] |
| S. equi | LFD | 50 | 90 | 65 | Horse nasal swabs | 105 | [68] |
| M. T | LFD | 2 | 30 | 65 | Cultures | 50 | [69] |
| Salmonella | LFD | 35–40 cfu | 60 | 64 | Bacterial culture | 90 | [70] |
| 1.3–1.9 cfu/mL | 60 | 64 | spiked chicken, infant cereal, milk | 90b | [70] | ||
| S. typhymurium | LFD | 100 cfu/mL | 60 | 65 | waswater / eggs | 77a | [58] |
| 1000 cfu/mL | 60 | 65 | milk /juice | ||||
| H. pylori | Hyb Enzym optic | 10 | 60 | 60 | Gastric biopsies | 240 | [47] |
| Staphylococcus | Hyb Enzym optic | 1 (mecA gene) | 45 | 65 | Blood samples | 75 | [33] |
| Staphyloccocus | Hyb Enzym optic | 1–250 cfu (tuf gene) | 45 | 65 | Blood samples | 75 | [33] |
| M. T. | Hyb Enzym optic | 1 | 40 | 65 | Clinical samples | – | [17] |
| S. aureus | Hyb Enzym optic | 10 | 30 | 65 | genomic DNA | 35 | [34] |
| S. aureus | Hyb Enzym optic | – | 40 | 65 | Blood cultures | 60a | [34] |
| Phytophthora | Hyb Enzym optic | – | 90 | 65 | --- | [50] | |
| P. kernoviae | Hyb Enzym optic | 10 pg/μL | 75 | 65 | Artificially infected leaf tissue | 135 | [48] |
| C. difficile | Hyb Enzym optic | 1 cfu (manual) | 45 | 65 | Fecal samples | 60a | [59] |
| 10 cfu (automated) | 45 | 65 | Fecal samples | 90a | [59] | ||
| M. T. | Hyb Enzym chronoA | 15 | 90 | 65 | Clinical samples | – | [31, 32] |
| M.T. | Hibridization DPV | 0.1 ng/μL | 90 | 65 | Synthetic DNA | 360 | [51] |
| E. coli | Hibridization FRET | 37 zmol | 60 | 65 | Synthetic DNA | – | [52] |
| S. aureus | On-surface fluorescence | 50,000 cells | 120 | 65 | Genomic DNA | – | [63] |
| S. aureus | On-surface fluorescence | 5×105 cells (multiplex) | 120 | 65 | Genomic DNA | – | [63] |
| N.G. | On-surface fluorescence | 1.32×105 cells | 120 | 65 | Genomic DNA | – | [63] |
| N.G. | On-surface fluorescence | 1.32×106 cells (multiplex) | 120 | 65 | Genomic DNA | – | [63] |
| Salmonella | On-surface enzym chronoA | 10 | 90 | 65 | Genomic DNA | – | [64] |
| Salmonella | On-surface enzym optic | 100 | 90 | 65 | Genomic DNA | – | [64] |
C.T.: Clamidia trachomatis, N.G.: Neisseria gonorrhoeae, M.T.: Mycobacterium tuberculosis, HSV: herpes simplex virus, HPV: human papiloma virus, HIV: human immunodeficiency virus, Echem: electrochemical, Hyb: hybridization, Enzym: enzymatic, ChronoA: chronoamperometry, DPV: differential pulse voltammetry
aIncluding extraction.
b+2–4 h of enrichment.
Clinical samples are contaminated more than desired with low levels of ubiquitous bacteria such as Streptoccocus or Staphyloccocus. Elimination of unintended contamination during sample processing is of utmost importance in clinical diagnosis. Addition of a non-target specific sequence at a large copy number or contaminating the sample with a defined amount of bacteria, the amplification of the contaminant species can be completely suppressed because of the limited amount of HDA primers [71]. It is important that the non-target sequences/organisms have some level of homology with the target to consume the primers. The approach is combined with on-chip hybridization. Only the competitive sequence hybridization is visible under conditions where primers are spent in the competitive amplification before detectable levels of the contaminants are reached. A mathematical expression to calculate the appropriate amount of suppressor is derived and tested against the limit of detection of the target bacteria. As expected, it decreases when increasing the suppressor concentration but it remains below the threshold to trigger clinically relevant blood infection. Although this strategy has some limitations, it can be applied to other DNA amplification schemes.
Pros and cons of HDA for pathogen detection
There is no doubt about the urgent need of effective systems for rapid, at the point of need detection of pathogens for a wide variety of applications, including clinical diagnostics, food safety, and environmental monitoring. Nucleic acid amplification by PCR revolutionized the field of microbiology, offering major advantages of speed and sensitivity for pathogen detection. Although it is now possible to use PCR when there are no classic lab benches [72], new isothermal amplification methods have been designed, which, compared with PCR, retain their advantages while reducing the complexity of analysis, thus resulting in a high potential for a simple integration into point-of-care testing and resource-limited places.
The main pros of HDA applied to the detection of pathogens are common to other molecular techniques, both PCR and isothermal amplification methods, i.e., high sensitivity in such a way that organisms that are difficult or impossible to grow in the laboratory can be detected at very low levels, and rapid turnaround time (typically between 30 and 90 min). In addition, HDA is easy to use, especially when lateral flow devices are used for detection. AmpliVue assays, commercialized by Quidel (proprietary for HDA technology), are small hand-held cartridge based on HDA-lateral flow devices that are considered CLIA moderately complex and are available for the rapid detection of pathogens such as Bordella pertusis, Clostridium difficile, Herpes simples virus, and Trichomonas vaginalis [73] without any other equipment than a heated lid. One of the requirements for a test system to be considered a CLIA-waiver is that it must be run using a specimen that does not require processing [74]. This can be considered another advantage of HDA, which is used for a variety of specimen collections such as vaginal, rectal, or nasopharyngeal swabs with a simple heat treatment for lysing. However, the sample treatment depends on the pathogen/sample analyzed. In food analysis, for example, a culture enrichment step is usually required to reach the legal limits, in many cases absence of the pathogen.
Despite the above advantages, HDA still need to overcome some important limitations, again in many cases common to other nucleic acid amplification techniques. Sample contamination is one of the typical concerns when using highly sensitive methods. HDA can be very useful for pathogen detection, but it can also be easily misinterpreted. Closed and disposable devices may contribute to alleviate this problem. The use of a constant temperature of amplification is the origin of another problem, i.e., the non-specific amplification products attributable to mispriming that may lead to false positive. Among isothermal methods, low-temperature amplifications such as RPA (operating at 37 °C) or LAMP that uses six different primers are more prone to this problem. Conversely, false negatives may arise because test inhibition, especially in complex biological matrices. The nature and amount of potential inhibitors is different in different samples and the concentration at which these inhibitors affect HDA must be empirically obtained. The use of an internal control, as is the case in Solana commercially available assays [75], is a way of minimizing and controlling this problem. Finally, lack of validated assays is another important drawback.
Considering the common advantages and disadvantages that isothermal nucleic acid amplification methods possess, it is difficult to recommend one method that may be applied as universal pathogen test. However, the LAMP technique is currently more popular for point of need applications. This is probably related to a better marketing by developers, with different companies commercializing LAMP products. Conversely, a single company is still proprietary of HDA technology, limiting its widespread use. In the future, with a high promotion by developers and advancements in the integration of the entire analytical process in lab-on-a-chip systems and wireless communication, HDA will most likely show a significant increase for point of need pathogen detection. An economic study is also needed to determine whether this approach is going to be truly cost-effective.
Conclusions
Helicase-dependent amplification, a novel nucleic acid amplification strategy inspired by biological processes, is a promising alternative to PCR for pathogen detection in different types of samples. It has been mainly applied in the field of infectious disease diagnosis and treatment monitoring, although it is also earning popularity for foodborne pathogen detection. Important progress has been made in the past few years for developing devices based on HDA amplification and compatible with point-of-need testing useful for resource-limited settings.
One of the major advantages of HDA over other isothermal amplification techniques such as loop mediated amplification (LAMP) is that the reaction configuration is very simple, and its PCR-like reaction scheme allows for adaptation of the current PCR assays to the HDA system. However, it would be necessary to have a systematic study of the modifications in the primers needed to alleviate the non-specific amplification problems, more pronounced in HDA. The availability of specific software to model the primer modification would be particularly useful, accelerating the adoption of the HDA systems at field conditions. Another critical challenge is the improvement in multiplexing capability of paramount importance in infectious disease diagnosis and treatment monitoring, especially to cope with the rapid emergence of antibiotic-resistant bacteria.
Early detection of pathogens is an important issue for controlling the spread of infectious disease and to rapidly respond to outbreaks of contagious and serious diseases. On-surface HDA holds promise in the development of integrated devices where amplification and detection are performed in an automated, miniaturized closed system. However, these platforms have to be integrated with sample preparation systems and further evaluated in both clinical laboratories and different settings at the point of need before they are effectively marketed and deployed in real applications. Different applications have already been described for the detection of common diseases associated with developing countries, such as malaria and AIDS. Nevertheless, the demonstration of cost-effectiveness will become a critical factor for their ultimate success. In this sense, the fact that the methodology is under the protection of a patent and that the necessary enzymes are produced, supplied, and licensed by a single company may be a limiting factor for widespread use of HDA. Once these rights have expired, HDA-based devices have the potential to become a reference for nucleic acid amplification at point of need applications, with a profitable commercialization worldwide.
Acknowledgements
This work was financially supported by the Spanish Ministerio de Economía y Competitividad (Project no. CTQ2015-63567-R), the Principado de Asturias government (Project FC15-GRUPIN14-025), and cofinanced by FEDER funds.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest
Footnotes
Published in the topical collection celebrating ABCs 16th Anniversary.
References
- 1.Disease outbreaks news. www.who.int/csr/don/en. Accessed 1 June 2017.
- 2.Saker L, Lee K, Cannito B, Gilmore A, Campbell-Lendrum D. Globalization and infectious diseases: a review of the linkages. Special Topics No. 3. UNICEF/UNDP/World Bank/WHO 2004; 1–67
- 3.Gracias KS, McKillip JL. A review of conventional detection and enumeration methods for pathogenic bacteria in food. Can J Microbiol. 2004;50:883–890. doi: 10.1139/w04-080. [DOI] [PubMed] [Google Scholar]
- 4.Niemz A, Ferguson TM, Boyle DS. Point-of-care nucleic acid testing for infectious disease. Trends Biotechnol. 2011;29(5):240–250. doi: 10.1016/j.tibtech.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Accessible quality-assured diagnostics. 2009 annual report. Available from: www.who.int/tdr/publications/about-tdr/annual-reports/bl7-annual-report/en/. Accessed 1 June 2017.
- 6.Li J, Macdonald J. Advances in isothermal amplification: novel strategies inspired by biological processes. Biosens Bioelectron. 2015;64:196–211. doi: 10.1016/j.bios.2014.08.069. [DOI] [PubMed] [Google Scholar]
- 7.Vincent M, Xu Y, Kong H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004;5(8):795–800. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kong H, Vincent M, Xu Y. Helicase dependent amplification of nucleic acids. Patent US. 2007;7282328:B2. [Google Scholar]
- 9.Caruthers JM, McKay DB. Helicase structure and mechanism. Curr Opin Struct Biol. 2002;12:123–133. doi: 10.1016/S0959-440X(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 10.An L, Tang W, Ranalli TA, Kim H-J, Wytiaz J, Kong H. Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J Biol Chem. 2005;280(32):28952–28958. doi: 10.1074/jbc.M503096200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Primer3 program. http://primer3.ut.ee/. Accessed 1 June 2017.
- 12.PrimerQuest program. https://eu.idtdna.com/Primerquest/Home/Index. Accessed 1 June 2017.
- 13.Biohelix Corporation. http://biohelix.com. Accessed 1 June 2017.
- 14.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Primer-BLAST. https://www.ncbi.nlm.nih.gov/tools/primer-blast/. Accessed 1 June 2017.
- 16.Mahalanabis M, Do J, Almuayad H, Zhang JY, Klapperich CM. Erratum to: An integrated disposable device for DNA extraction and helicase dependent amplification. Biomed Microdevices. 2011;13:599–602. doi: 10.1007/s10544-011-9518-6. [DOI] [PubMed] [Google Scholar]
- 17.Ao W, Aldous S, Woodruff E, Hicke B, Rea L, Kreiswirth B, et al. Rapid detection of rpoB gene mutations conferring rifampin resistance in Mycobacterium tuberculosis. J Clin Microbiol. 2012;50(7):2433–2440. doi: 10.1128/JCM.00208-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoshika S, Chen F, Leal NA, Benner SA. Artificial genetic systems: self-avoiding DNA in PCR and multiplexed PCR. Angew Chem Int Ed. 2010;49:5554–5557. doi: 10.1002/anie.201001977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z, McLenddon C, Hutter D, Bradlwy DM, Hoshika S, Frye CB, et al. Helicase-dependent isothermal amplification of DNA and RNA by using self-avoiding molecular recognition systems. ChemBioChem. 2015;16:1365–1370. doi: 10.1002/cbic.201500135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korfhage C, van Ooyen S. Primers for helicase dependent amplification and their methods of use. Patent US. 2012;9074248:B1. [Google Scholar]
- 21.Arif M, Aguilar-Moreno GS, Wayadande A, Fletcher J, Ochoa-Corona FM. Primer modification improves rapid and sensitive in vitro and field-deployable assays for detection of high plains virus variants. Appl Environ Microbiol. 2014;80(1):320–327. doi: 10.1128/AEM.02340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doseeva V, Forbes T, Wolff J, Khripin Y, O’Neil D, Rothmann T, et al. Multiplex isothermal helicase-dependent amplification assay for detection of Chlamydia trachomatis and Neisseria gonorrhoeae. Diagn Microbiol Infect Dis. 2011;71:354–365. doi: 10.1016/j.diagmicrobio.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 23.Tong Y, Lemieux B, Kong H. Multiple strategies to improve sensitivity, speed, and robustness of isothermal nucleic acid amplification for rapid pathogen detection. BMC Biotechnol. 2011;11:50. doi: 10.1186/1472-6750-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen F, Zhang D, Zhang Q, Zuo X, Fan C, Zhao Y. Zero-background helicase-dependent amplification and its application to reliable assay of telomerase activity in cancer cell by eliminating primer-dimer artifacts. ChemBioChem. 2016;17:1171–1176. doi: 10.1002/cbic.201500605. [DOI] [PubMed] [Google Scholar]
- 25.Goldmeyer J, Kong H, Tang W. Development of a novel one-tube isothermal reverse transcription thermophilic helicase-dependent amplification platform for rapid RNA detection. J Mol Diagn. 2007;9(5):639–644. doi: 10.2353/jmoldx.2007.070012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Motré A, Li Y, Kong H. Enhancing helicase-dependent amplification by fusing the helicase with the DNA polymerase. Gene. 2008;420:17–22. doi: 10.1016/j.gene.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 27.Sedighi A, Oberc C, Whitehall V, Li PCH. NanoHDA: a nanoparticle-assisted isothermal amplification technique for genotyping assays. Nano Res. 2017;10(1):12–21. doi: 10.1007/s12274-016-1262-z. [DOI] [Google Scholar]
- 28.Li H, Huang J, Lv J, An H, Zhang X, Zhang Z, et al. Nanoparticle PCR: nanogold-assisted PCR with enhanced specificity. Angew Chem Int Ed. 2005;44:5100–5103. doi: 10.1002/anie.200500403. [DOI] [PubMed] [Google Scholar]
- 29.Barbieri D, Venturoli S, Rosl F, Rincon-Orozco B. Detection of high-risk human papillomavirus type 16 and 18 using isothermal helicase-dependent amplification. Diagn Microbiol Infect Dis. 2014;79(2):178–182. doi: 10.1016/j.diagmicrobio.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 30.Ramalingam N, Liu HB, Dai CC, Jiang Y, Wang H, Wang Q, K MH, Gong HQ (2009) Real-time PCR array chip with capillary-driven sample loading and reactor sealing for point-of-care applications. Biomed Microdevices 11(5):1007–1020. [DOI] [PubMed]
- 31.Barreda-García S, González-Álvarez MJ, de-los-Santos-Álvarez N, Palacios-Gutiérrez JJ, Miranda-Ordieres AJ, Lobo-Castañón MJ. Attomolar quantitation of Mycobacterium tuberculosis by asymmetric helicase-dependent isothermal DNA-amplification and electrochemical detection. Biosens Bioelectron. 2015;68:122–8. [DOI] [PubMed]
- 32.Barreda-García S, Miranda-Castro R, de-los-Santos-Álvarez N, Miranda-Ordieres AJ, Lobo-Castañón MJ. Comparison of isothermal helicase-dependent amplification and PCR for the detection of Mycobacterium tuberculosis by an electrochemical genomagnetic assay. Anal Bioanal Chem. 2016;408(30):8603–8610. doi: 10.1007/s00216-016-9514-z. [DOI] [PubMed] [Google Scholar]
- 33.Pasko C, Hicke B, Dunn J, Jaeckel H, Nieuwlandt D, Weed D, et al. Staph ID/R: a rapid method for determining Staphylococcus species identity and detecting the mecA gene directly from positive blood culture. J Clin Microbiol. 2012;50(3):810–817. doi: 10.1128/JCM.05534-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jenison R, Jaeckel H, Klonoski J, Latorra D, Wiens J. Rapid amplification/detection of nucleic acid targets utilizing a HDA/thin film biosensor. Analyst. 2014;139(15):3763–3769. doi: 10.1039/C4AN00418C. [DOI] [PubMed] [Google Scholar]
- 35.Kivlehan F, Mavre F, Talini L, Limoges B, Marchal D. Real-time electrochemical monitoring of isothermal helicase-dependent amplification of nucleic acids. Analyst. 2011;136(18):3635–3642. doi: 10.1039/c1an15289k. [DOI] [PubMed] [Google Scholar]
- 36.Tong Y, Tang W, Kim HJ, Pan X, Ranalli T, Kong H. Development of isothermal TaqMan assays for detection of biothreat organisms. BioTechniques. 2008;45(5):543–557. doi: 10.2144/000112959. [DOI] [PubMed] [Google Scholar]
- 37.O'Neil D, Doseeva V, Rothmann T, Wolff J, Nazarenko I. Evaluation of Chlamydia trachomatis and Neisseria gonorrhoeae detection in urine, endocervical, and vaginal specimens by a multiplexed isothermal thermophilic helicase-dependent amplification (tHDA) assay. J Clin Microbiol. 2011;49(12):4121–4125. doi: 10.1128/JCM.00952-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaydos CA, Schwebke J, Dombrowski J, Marrazzo J, Coleman J, Silver B, et al. Clinical performance of the Solana® point-of-care trichomonas assay from clinician-collected vaginal swabs and urine specimens from symptomatic and asymptomatic women. Expert Rev Mol Diagn. 2017;17(3):303–6. [DOI] [PMC free article] [PubMed]
- 39.Craw P, Mackay RE, Naveenathayalan A, Hudson C, Branavan M, Sadiq ST, et al. A simple, low-cost platform for real-time isothermal nucleic acid amplification. Sensors. 2015;15(9):23418–23430. doi: 10.3390/s150923418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chow WH, McCloskey C, Tong Y, Hu L, You Q, Kelly CP, et al. Application of isothermal helicase-dependent amplification with a disposable detection device in a simple sensitive stool test for toxigenic Clostridium difficile. J Mol Diagn. 2008;10(5):452–458. doi: 10.2353/jmoldx.2008.080008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldmeyer J, Li H, McCormac M, Cook S, Stratton C, Lemieux B, et al. Identification of Staphylococcus aureus and determination of methicillin resistance directly from positive blood cultures by isothermal amplification and a disposable detection device. J Clin Microbiol. 2008;46(4):1534–1536. doi: 10.1128/JCM.02234-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faron ML, Ledeboer NA, Granato P, Daly JA, Pierce K, Pancholi P, et al. Detection of Group A Streptococcus in pharyngeal swab specimens by use of the AmpliVue GAS isothermal helicase-dependent amplification assay. J Clin Microbiol. 2015;53(7):2365–2367. doi: 10.1128/JCM.01085-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemieux B, Li Y, Kong H, Tang YW. Near instrument-free, simple molecular device for rapid detection of herpes simplex viruses. Expert Rev Mol Diagn. 2012;12(5):437–443. doi: 10.1586/erm.12.34. [DOI] [PubMed] [Google Scholar]
- 44.Kim HJ, Tong Y, Tang W, Quimson L, Cope VA, Pan X, et al. A rapid and simple isothermal nucleic acid amplification test for detection of herpes simplex virus types 1 and 2. J Clin Virol. 2011;50(1):26–30. doi: 10.1016/j.jcv.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jordan JA, Ibe CO, Moore MS, Host C, Simon GL. Evaluation of a manual DNA extraction protocol and an isothermal amplification assay for detecting HIV-1 DNA from dried blood spots for use in resource-limited settings. J Clin Virol. 2012;54(1):11–14. doi: 10.1016/j.jcv.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Kumar N, Gopalakrishnan A, Ginocchio C, Manji R, Bythrow M, et al. Detection and species identification of malaria parasites by isothermal tHDA amplification directly from human blood without sample preparation. J Mol Diagn. 2013;15(5):634–641. doi: 10.1016/j.jmoldx.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gill P, Amini M, Ghaemi A, Shokouhizadeh L, Abdul-Tehrani H, Karami A, et al. Detection of Helicobacter pylori by enzyme-linked immunosorbent assay of thermophilic helicase-dependent isothermal DNA amplification. Diagn Microbiol Infect Dis. 2007;59(3):243–249. doi: 10.1016/j.diagmicrobio.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Schwenkbier L, Pollok S, Rudloff A, Sailer S, Cialla-May D, Weber K, et al. Non-instrumented DNA isolation, amplification and microarray-based hybridization for a rapid on-site detection of devastating Phytophthora kernoviae. Analyst. 2015;140(19):6610–6618. doi: 10.1039/C5AN00855G. [DOI] [PubMed] [Google Scholar]
- 49.Jenison R, Yang S, Haeberli A, Polisky B. Interference-based detection of nucleic acid targets on optically coated silicon. Nat Biotechnol. 2001;19(1):62–65. doi: 10.1038/83530. [DOI] [PubMed] [Google Scholar]
- 50.Schwenkbier L, Pollok S, Koenig S, Urban M, Werres S, Cialla-May D, et al. Towards on-site testing of Phytophthora species. Anal Methods. 2015;7(1):211–217. doi: 10.1039/C4AY02287D. [DOI] [Google Scholar]
- 51.Torres-Chavolla E, Alocilja EC. Nanoparticle based DNA biosensor for tuberculosis detection using thermophilic helicase-dependent isothermal amplification. Biosens Bioelectron. 2011;26(11):4614–4618. doi: 10.1016/j.bios.2011.04.055. [DOI] [PubMed] [Google Scholar]
- 52.Noor MO, Hrovat D, Moazami-Goudarzi M, Espie GS, Krull UJ. Ratiometric fluorescence transduction by hybridization after isothermal amplification for determination of zeptomole quantities of oligonucleotide biomarkers with a paper-based platform and camera-based detection. Anal Chim Acta. 2015;885:156–165. doi: 10.1016/j.aca.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 53.Noor MO, Shahmuradyan A, Krull UJ. Paper-based solid-phase nucleic acid hybridization assay using immobilized quantum dots as donors in fluorescence resonance energy transfer. Anal Chem. 2013;85(3):1860–1867. doi: 10.1021/ac3032383. [DOI] [PubMed] [Google Scholar]
- 54.Linnes JC, Rodriguez NM, Liu L, Klapperich CM. Polyethersulfone improves isothermal nucleic acid amplification compared to current paper-based diagnostics. Biomed Microdevices. 2016;18(2):30. doi: 10.1007/s10544-016-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shetty P, Ghosh D, Singh M, Tripathi A, Paul D. Rapid amplification of Mycobacterium tuberculosis DNA on a paper substrate. RSC Adv. 2016;6(61):56205–56212. doi: 10.1039/C6RA07529K. [DOI] [Google Scholar]
- 56.Linnes JC, Fan A, Rodriguez NM, Lemieux B, Kong H, Klapperich CM. Paper-based molecular diagnostic for Chlamydia trachomatis. RSC Adv. 2014;4(80):42245–42251. doi: 10.1039/C4RA07911F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang S, Do J, Mahalanabis M, Fan A, Zhao L, Jepeal L, et al. Low cost extraction and isothermal amplification of DNA for infectious diarrhea diagnosis. Plos One. 2013;8(3):e60059. doi: 10.1371/journal.pone.0060059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang R, Yang H, Gong Y, You M, Liu Z, Choi JR, et al. A fully disposable and integrated paper-based device for nucleic acid extraction, amplification and detection. Lab Chip. 2017;17(7):1270–1279. doi: 10.1039/C6LC01586G. [DOI] [PubMed] [Google Scholar]
- 59.Hicke B, Pasko C, Groves B, Ager E, Corpuz M, Frech G, et al. Automated detection of toxigenic Clostridium difficile in clinical samples: isothermal tcdB amplification coupled to array-based detection. J Clin Microbiol. 2012;50(8):2681–2687. doi: 10.1128/JCM.00621-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Branavan M, Mackay RE, Craw P, Naveenathayalan A, Ahern JC, Sivanesan T, et al. Modular development of a prototype point of care molecular diagnostic platform for sexually transmitted infections. Med Eng Phys. 2016;38(8):741–748. doi: 10.1016/j.medengphy.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ha ML, Zhang Y, Lee NY. A functionally integrated thermoplastic microdevice for one-step solid-phase-based nucleic acid purification and isothermal amplification for facile detection of foodborne pathogen. Biotechnol Bioeng. 2016;113(12):2614–2623. doi: 10.1002/bit.26027. [DOI] [PubMed] [Google Scholar]
- 62.Andresen D, von Nickisch-Rosenegk M, Bier FF. Helicase-dependent OnChip- amplification and its use in multiplex pathogenic detection. Clin Chim Acta. 2009;403:244–8. [DOI] [PubMed]
- 63.Andresen D, von Nickisch-Rosenegk M, Bier FF. Helicase-dependent amplification: use in on-chip amplification and potential for point-of-care diagnostics. Expert Rev Mol Diagn. 2009;9(7):645–50. [DOI] [PubMed]
- 64.Barreda-García S, Miranda-Castro R, de-los-Santos-Álvarez N, Miranda-Ordieres AJ, Lobo-Castañón MJ. Solid-phase helicase-dependent amplification and electrochemical detection of Salmonella on highly stable oligonucleotide-modified ITO electrodes. Chem Commun. 2017;53:9721–4. [DOI] [PubMed]
- 65.Wu X, Chen C, Xiao X, Deng MJ. Development of reverse transcription thermostable helicase-dependent DNA amplification for the detection of tomato spotted wilt virus. J AOAC Int. 2016;99(6):1596–1599. doi: 10.5740/jaoacint.16-0132. [DOI] [PubMed] [Google Scholar]
- 66.Arif M, Ochoa-Corona FM, Opit GP, Li Z-H, Kucerova Z, Stejskal V, et al. PCR and isothermal-based molecular identification of the stored-product psocid pest Lepinotus reticulatus (Psocoptera: Trogiidae). J Stor Prod Res. 2012;49:184–8.
- 67.Chen X, Wu X, Gan M, Xu F, He L, Yang D, et al. Rapid detection of Staphylococcus aureus in dairy and meat foods by combination of capture with silica-coated magnetic nanoparticles and thermophilic helicase-dependent isothermal amplification. J Dairy Sci. 2015;98(3):1563–70. [DOI] [PubMed]
- 68.Artiushin S, Tong Y, Timoney J, Lemieux B, Schlegel A, Kong H. Thermophilic helicase-dependent DNA amplification using the IsoAmp™ SE experimental kit for rapid detection of Streptococcus equi subspecies equi in clinical samples. J Vet Diagn Invest. 2011;23(5):909–14. [DOI] [PubMed]
- 69.Motre A, Kong R, Li Y. Improving isothermal DNA amplification speed for the rapid detection of Mycobacterium tuberculosis. J Microbiol Methods. 2011;84(2):343–345. doi: 10.1016/j.mimet.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 70.Du X-J, Zhou T-J, Li P, Wang S. A rapid Salmonella detection method involving thermophilic helicase-dependent amplification and a lateral flow assay. Mol Cell Probes 2017;34:37–44. [DOI] [PubMed]
- 71.Ao W, Clifford A, Corpuz M, Jenison R. A novel approach to eliminate detection of contaminating Staphylococcal species introduced during clinical testing. Plos One. 2017;12(2):e0171915. doi: 10.1371/journal.pone.0171915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marx V. PCR heads into the field. Nature Methods. 2015;12(5):393–397. doi: 10.1038/nmeth.3369. [DOI] [PubMed] [Google Scholar]
- 73.Quidel AmpliVue products. http://www.quidel.com/molecular-diagnostics/amplivue-products. Accessed 16 August 2017.
- 74.Weber NC, Klepser ME, Akers JM, Klepser DG, Adams AJ. Use of CLIA-waived point-of-care tests for infectious diseases in community pharmacies in the United States. Expert Rev Mol Diagn. 2016;16(2):253–264. doi: 10.1586/14737159.2015.1116388. [DOI] [PubMed] [Google Scholar]
- 75.Quidel Solana products. http://www.quidel.com/molecular-diagnostics/solana-products. Accessed 16 August 2017.