

Abstract
This is a study on the Avian coronavirus IBV and chicken host-relationship from the codon usage point of view based on fifty-nine non-redundant IBV S1 sequences (nt 1–507) from strains detected worldwide and chicken tissue-specific protein genes sequences from IBV-replicating sites. The effective number of codons (ENC) values ranged from 36 to 47.8, indicating a high-to-moderate codon usage bias. The highest IBV codon adaptation index (CAI) value was 0.7, indicating a distant virus versus host synonymous codons usage. The ENC × GC3 % curve indicates that both mutational pressure and natural selection are the driving forces on codon usage pattern in S1. The low CAI values agree with a low S protein expression and considering that S protein is a determinant for attachment and neutralization, this could be a further mechanism besides mRNA transcription attenuation for a low expression of this protein leading to an immune camouflage.
Keywords: Codon usage bias, Avian coronavirus, Gallus gallus, Co-evolution
Introduction
Infectious bronchitis virus (IBV) (Nidovirales: Coronaviridae: Coronavirinae: Gammacoronavirus: Avian coronavirus) is an enveloped single-stranded positive sense RNA virus with circa 27 kb and 120 nm in diameter, with 20 nm spikes formed by trimers of the spike glycoprotein S (Cavanagh 2007; Thiel 2005).
The spike glycoprotein is a class 1 fusion protein with the two major domains S1 and S2, with a function in receptor attachment and membrane fusion, respectively, being the target for neutralizing antibodies and presenting an evolution so sensitively driven by host humoral immune-response that polymorphisms in 10–15 amino acids in S1 might give rise to different serotypes of the virus (Cavanagh 2007; Thiel 2005).
Besides S, IBV genome codes for 15 non-structural proteins in the ORF1 that occupies the 5′ one-third of the genome and are involved in viral replication and pathogenesis while the structural proteins E (envelope) and M (membrane) involved in virion stability and nucleoprotein (N), which associates to the genomic RNA forming the helical nucleocapsid, are coded by the remaining 3′ 7 kb (Thiel 2005).
Different pathotypes of IBV exist that cause disease in the respiratory system, kidneys, reproductive tracts of both males and females and enteritis and the virus occurs worldwide with a massive diversity in terms of serotypes, genotypes, and geographic-specific lineages (Cavanagh 2007; Jones 2010) and thus studies on IBV molecular evolution must necessarily include data sets that represent such diversity.
IBV and chicken host/virus relationship has been comprehensively studied in terms of receptors, immune response, molecular epidemiology, and pathogenesis (De Wit et al. 2011; Jones 2010; Winter et al. 2008; Yang et al. 2009), pointing toward a positive outcome to the virus and a highly negative one to the birds due to productive virus replication, rapid disease spread, severe tissue damage, and immune evasion that relies mainly on antigenic polymorphism.
Nonetheless, in depth studies on virus and host relationship must also take into account that not all synonymous codons for a same amino acid occur in the same frequencies in an mRNA, but are rather used in varying frequencies, what is called codon usage bias. Synonymous codon usage for the Nidovirales has been shown to be virus-specific and conserved in a phylogenetic fashion with no host-specificity though (Gu et al. 2004).
IBV as well as chicken codon usage bias have been studied already (Rao et al. 2011; Woo et al. 2007), but the diversity of IBV types and the use of chicken genes coding for tissue-specific proteins in an integrated way has not been considered thus far.
In the view of the lack of information on the relationship between IBV and Gallus gallus, its natural host, from the codon usage point of view, the aims of this study were thus to understand the relationship of IBV and different chicken tissues based on codon usage bias analyses and to assess the forces that drive such a relationship.
Materials and Methods
DNA Sequences
A survey was carried out in Genbank for IBV S1 sequences and the inclusion criteria were: geographic origin, pathotype, recent detection for field strains and vaccine strains related to archetypical IBVs; redundant sequences, i.e., those with nucleotide identities = 100 %, were excluded from the analysis.
With these criteria, 59 sequences (Fig. 1) for nucleotides 1–507 (regarding strain H120, GU393335) were used for the analyses. Though the inclusion criteria were intended to increase sequence diversity, the resulting sequences were shorter than the whole S gene due to a low availability of complete sequences for this gene, but sequences >150 nt are considered as statistically reliable for codon usage bias analysis (Gu et al. 2004).
Fig. 1.
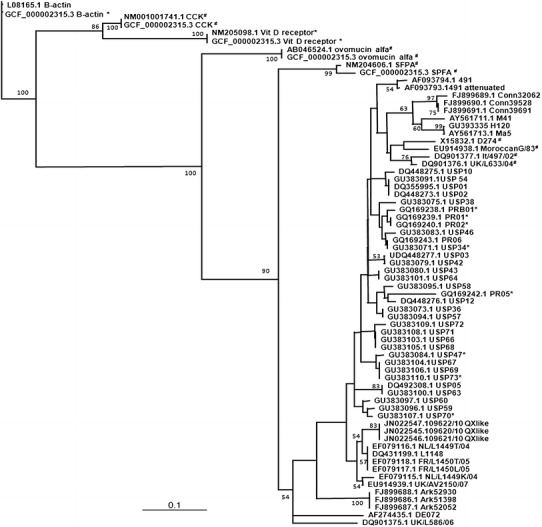
Neighbor-joining tree for binary data for preferred (1) or non-preferred/neutral (0) codons for 59 codons for 59 IBV S1 sequences and 5 G. gallus genes (B-actin β-actin, CCK cholecystokinin, vit D receptor vitamin D receptor, ovomucin alpha ovomucin α-subunit, SPFA surfactant, pulmonary-associated protein A1). Sequences with ENC (effective number of codons) values <40 and >45 are marked with asterisk and hash, respectively; sequences with ENC between 40 and 45 have no marks. Number at each nodes are bootstrap values (only >50 are shown)
Chicken tissue-specific non-redundant genes sequences related to IBV replication sites were retrieved from Genbank for duodenum (cholecystokinin NM001001741.1), lung (surfactant, pulmonary-associated protein A1 NM204606.1), kidney (vitamin D receptor NM205098.1), and oviduct (ovomucin α-subunit AB046524.1) and these same genes were also retrieved from the NCBI chicken genome resources (version GFC_000002315.3). G. gallus β-actin gene (L08165 and GFC_000002315.3) was included in the analyses as a reference, ubiquitary-expressed gene.
Relative Synonymous Codon Usage (RSCU)
RSCU is the ratio between the observed number for a codon and its expected frequency under the random distribution of all its corresponding isoacceptors and was calculated for 59 codons (64 codons minus 3 stop codons and the codons for methionine and tryptophan with a single codon each) using MEGA 5.0 (Tamura et al. 2011) according to the equation RSCUi = x i/(Σi X i/n), where x i is the total count for a given codon, ΣX i is the sum of the count for all isoacceptors related to that amino acid, and n is the number of possible isoacceptors for that amino acid.
A RSCU > 1 means that a given codon is preferential; an RSCU < 1 means that a given codon is not preferential and if RSCU = 1 means that the given codon is neutral.
In order to represent both host and virus codon usage preferences in a unique tree, the following algorithm was developed: first, RSCU continuous variables for both G. gallus and IBV were converted to discrete binary data using 1 for RSCU > 1 (i.e., a given codon is preferred for a specific amino acid) and 0 for RSCU ≤ 1 (i.e., the codon is not preferred or is neutral). Next, a matrix was built using the binary data for the presence or absence of that isoacceptor/allele as a preferred codon and used to build a Neighbor-joining tree (1,000 bootstrap replicates) using PAUP* 4.1b (Swofford 2000).
Effective Number of Codons (ENC)
ENC is similar to the effective number of alleles and measures the departure from the equal use of synonymous codons taking each isoacceptor as an allele and was calculated with ACUA (Vetrivel et al. 2007) with the equation ENCactual = 2 + (9/F2) + (1/F3) + (5/F4) + (3/F6), where Fi is the average homozygosity (assuming equal use of each synonymous codon or allele) estimate for each class of degeneracy ranging from 2 to 6. ENC ranges from 20 to 61, values closer to 61 meaning low bias (Wright 1990).
Natural Selection × Drift Test
To test if the codon usage of IBV S1 is under natural selection or, conversely, mutation pressure is driving codon usage bias, expected and observed ENC and the corresponding GC3 % values were plotted in a same graphic.
Expected ENC, meaning the expected codon usage if it’s influenced only by GC3 %, i.e., the percent of G and C at the third position of all codons, was calculated as ENCexpec = 2 + s + 29[s 2 + (1 − s)2]−1 (Wright 1990), where s is the GC3 %. Then, each ENCexpec was plotted against each respective GC3 % and the actual ENCs were added to the graphic to measure its deviation from the expected values. If an ENCactual plot lies on or just below the ENCexpec curve, this might be interpreted as drift/mutational bias and if a plot is distant from the curve, this means that natural selection is in action and GC3 % does not follow genomic GC3 % (Wright 1990).
Codon Adaptation Index (CAI)
CAI is an estimative of the adaptation of synonymous codons to a given expression system with a set of highly expressed genes (Comeron and Aguadé 1998) and might be used to estimate the adaptation of virus to host codons. A CAI < 1 means low fit and use of codons which are non-preferred by the host while CAI = 1 means high virus × host codon fit. CAI was calculated with ACUA (Vetrivel et al. 2007) with a default set of highly expressed G. gallus genes.
Results
RSCU
IBV S1 codons that differed from all host genes studied in non-variable amino acids positions were UUA (L169), GUA (V49), UCA (S93), and GGU/GGG (G39, 44, 45, and 89) for strains analyzed herein. AUA (I) was also exclusive to the IBV strains and not used by the host, but no 100 % conserved site for this amino acid was found amongst the sequences studied. The codon usage tree (Fig. 1) showed that all IBV strains segregated in a same cluster and close to lung codon usage, in an increasing distance from oviduct to kidney, duodenum, and the β-actin gene. Regarding specifically the IBV strains, the tree topology was similar to that expected for a S1 nucleotide tree, with strains segregating according to established genotypes.
Effective Number of Codons
Observed ENC ranged from 36.04 (strain GQ169238.1IBV/Brazil/PRB01M) to 47.83 (strain EU914938.1MoroccanG/83) for IBVs (mean 42.79, sd 2.25). For G. gallus, mean observed ENCs were 33.59 (vitamin D receptor), 40.03 (β-actin), 46.48 (cholecystokinin), 50.21 (surfactant, pulmonary-associated protein A1), and 53.03 (ovomucin α-subunit). Considering an ENC ≤ 40 as indicative of bias, biased codon usage was found for IBV strains GU383110.1USP73C (39.94), GU383107.1USP70C (38.82), GU383084.1USP47C (38.21), GU383071.1USP34C (38.88), GQ169242.1IBV/Brazil/PR05M (39.14), GQ169239.1IBV/Brazil/PR01M (38.07), GQ169240.1IBV/Brazil/PR02M (37.8), and GQ169238.1IBV/Brazil/PRB01M (36.04), all Brazilian strains.
On the other hand, strains X15832.1 D274, EU914938.1 MoroccanG/83, DQ901377.1 It/497/02, and DQ901376.1 UK/L633/04 from the Netherlands, Morocco, Italy, and the UK showed ENCs > 45 and grouped in a same cluster.
No temporal pattern was found for ENCs, as IBV strains detected decades apart showed similar ENC values.
Natural Selection × Drift Test
The graphic with the expected ENC × expected GC3 % and the observed ENC × observed GC3 % (Fig. 2) showed that some IBV S1 and G. gallus plots are just below the expected curve, while others are more distantly below the curve, indicating that both mutation pressure and natural selection and are driving forces for the observed bias.
Fig. 2.
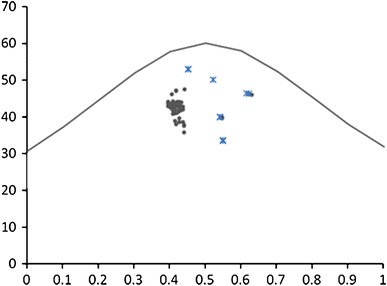
Expected (curve) and observed (points) effective number of codons (Y axis) and GC3 % (X axis) for 59 IBV S1 sequences (dots) and G. gallus β-actin, cholecystokinin, surfactant, pulmonary-associated protein A1, vitamin D and ovomucin genes (asterisks)
Codon Adaptation Index
IBV CAI values ranged from 0.64 (strain GQ169240.1IBV/Brazil/PR02M) to 0.7 (strain AF274435.1DE072) with a mean value of 0.66 (sd 0.01), while G. gallus genes CAIs ranged from 0.71 (surfactant protein A gene NM204606.1) to 0.88 (vitamin D receptors NM205068.1 and GCF_000002315.3), indicating a moderately low adaptation of IBV S1 codons to G. gallus codons.
Discussion
It’s largely known that IBV strains, regardless their specific pathotypes, use the respiratory tract as a first replication site, from where they might spread to kidneys, reproductive system, and the gastroenteric tissues (Cavanagh 2007).
α2,3-Linked sialic acid, a membrane receptor for IBV-spike protein (Winter et al. 2008) is widespread in chicken epithelial cells, rendering a variety of cell types susceptible to IBV infection; it’s noteworthy that IBV of different pathotypes might show no differences in receptor preferences (Abd El Rahman et al. 2009).
Though this fact accounts for a successful replication in the respiratory epithelium and the virus spread to other organs, the events that come after virus attachment have been widely overlooked.
IBV S1 codon usage based on RSCU values has been shown herein to be closer related to respiratory tract codon usage bias than to the values found in the oviduct, duodenum, and kidney. This is evident in Fig. 1 as all IBV strains segregated together with the pulmonary chicken surfactant protein gene with a bootstrap of 90. From the molecular point of view, this means that IBV shares a close relationship with the respiratory tract when it comes to codons and consequently tRNA usage.
The distant relationship between IBV and non-respiratory host tissues in terms of codon usage seen in Fig. 1 does not mean that they are completely opposite, but that one have an ordered, increasing dissimilarity between the virus and the oviduct, kidneys, and duodenum, respectively. It’s interesting to speculate that a successful replication of IBV at the respiratory tract would allow for the emergence of a higher virus diversity and titre with an improved fitness to the other target tissues.
It’s noteworthy that the most distant host genes in the tree are those from β-actin included as an ubiquitary-expressed gene, as the closer proximity of IBV to other host genes is a further evidence of a fine-tuned adaptation of the virus to some specific tissues.
But essential disagreements between virus and host cells emerge from this analysis. RSCU values show that codon usage bias exists amongst the IBV strains studied with a low degree but in a conserved pattern for codons in seven positions. It’s noteworthy that the most frequent of these amino acids was glycine (positions 39, 44, 45, and 89), an amino acid with a short lateral chain that allows for a high sterical plasticity (Berg et al. 2002).
Considering that residues 39, 44, and 45 are within antigenic domain I of S1 (Moore et al. 1997), the use of exclusive codons with no competition with the host would allow for the maintenance of an amino acid at certain strategic positions which sterical plasticity would be translated to a plastic protein structure for S1, increasing the number of possible protein structures and contributing to the huge set of putative epitopes for S1 and the continuous emergence of escape mutants.
Serine, found as a conserved amino acid with an IBV-exclusively preferred codon (UCA) at position 93, is a hydrophilic amino acid in antigenic domain II of S1 (Moore et al. 1997) with a high propensity to turns in protein secondary structure (Berg et al. 2002), what could also be important to keep structural stability in the proteic neighborhood for virus-cell attachment.
Though the tree other 100 % conserved amino acids with IBV-exclusively preferred codons (L169, V49, and G89) are not located exactly inside antigenic domains, the high bias found for these must have some importance for the neighboring structures for aspects of the spike protein not related simply to antigenicity, but to the ignored face of the virus itself, such as protein stability.
Valine and leucine, both non-polar, hydrophobic amino acids, take part more often in α-helixes and β-sheets, respectively (Berg et al. 2002), and their maintenance at those respective positions might also have to do with S1 globular structure stability.
In this study, the highest IBV CAI was 0.7, which is bellow G. gallus lowest CAI (0.71 for surfactant protein A gene NM204606.1), evidencing that for all IBVs and for some G. gallus genes there’s a trend for a low CAI, with the consequent lower efficiency of protein synthesis (Sharp and Li 1987), meaning that IBV-spike gene follows the trend shown by low-expressed genes of its host for a codon deoptimization-based regulation of translation.
Codon bias is stronger in high than in low expression genes in terms of protein synthesis efficiency at the initiation step, meaning that the most 5′ nucleotides of any gene, as the S-region focused in this study, are more critical for protein synthesis efficiency. Thus, in genes with high expression, natural selection acts against codons changes, keeping the correspondence between codons and the tRNAs of higher availability (Bulmer 1991; Ridley 2004).
Coronaviruses mRNA transcription happens in an attenuated form from the smallest to the largest mRNAs from the 3′ to the 5′ end of the genome, with smaller sub-genomic mRNAs being transcribed in higher amounts (Van Marle et al. 1995) and S is the second gene after ORF1, meaning that S is synthesized at a lower amount when compared to the other 3′ coronaviruses proteins.
Spike protein is a major target for neutralizing antibodies and the presentation of this protein to the chicken immune system allows for the production of such antibodies. Thus, a lower amount of S favored by transcription attenuation would allow for a lower exposition to the immune system and a low CAI could make a still unknown but herein mathematically demonstrated mechanism that, associated to mRNA transcription attenuation, allows for a parsimonious spike protein synthesis and immune camouflage for IBV. A similar mechanism has been suggested for Pestiviruses as a consequence of a high number of underrepresented codons leading to decreased protein expression and a less intense host immune-response (Zhou et al. 2012).
Furthermore, if a higher similarity between virus and host codon usage would allow for a higher viral protein expression, it could be that a G. gallus codon-optimized attenuated IBV vaccine would result in an increased immune response due to a higher spike protein expression.
An indication of a geographic pattern for codon usage can be noticed in Fig. 1, as only Brazilian IBV strain showed the lowest ENC values. The significance of this finding in terms of virulence and immunity cannot be understood hitherto as no data on these parameters is available for these strains, but considering the high diversity of IBV in this country (Chacon et al. 2011; Villarreal et al. 2010), low ENCs could be a further mechanism for the emergence of escape mutants.
The highest (>45) ENC values were found for strain from countries as distant as Morocco and The Netherlands, including the UK and Italy, but a very low number of sequences from these areas is available in the Genbank when compared to, e.g., Brazil; thus, instead of a geographic pattern for low codon usage bias in this case, i.e., high ENCs, this could be primarily attributed to a lack of sequence diversity.
The expected versus observed ENC × GC3 % graphic showed that natural selection is not acting alone on the codon usage patterns of G. gallus (as already shown by Rao et al. 2011) and of the IBV strains under analysis, but in association with mutation pressure. As S is expressed in lower amounts when compared to other IBV proteins (as discussed above) and thus its synthesis relies on those tRNAs of lower availability in G. gallus cells, this could be the reason for the presence of drift in a nearly neutral evolution mode, i.e., for some S1 sequences codons there’s no competition with host tRNAs and thus third positions nucleotides are not subjected to selection but might follow the whole genome GC % trend instead.
As a conclusion, IBV types show a concerted codon bias for epitope-important amino acids on the spike protein with a general codon usage pattern of the virus closer to the respiratory tract than other replication sites driven by genetic drift and natural selection.
References
- Abd El Rahman S, El-Kenawy AA, Neumann U, Herrler G, Winter C. Comparative analysis of the sialic acid binding activity and the tropism for the respiratory epithelium of four different strains of avian infectious bronchitis virus. Avian Pathol. 2009;38:41–45. doi: 10.1080/03079450802632049. [DOI] [PubMed] [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5. New York: Freeman and Company; 2002. [Google Scholar]
- Bulmer M. The selection-mutation drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet Res. 2007;38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- Chacon JL, Rodrigues JN, Assayag Junior MS, Peloso C, Pedroso AC, Ferreira AJ. Epidemiological survey and molecular characterization of avian infectious bronchitis virus in Brazil between 2003 and 2009. Avian Pathol. 2011;40:153–162. doi: 10.1080/03079457.2010.544641. [DOI] [PubMed] [Google Scholar]
- Comeron JM, Aguadé M. An evaluation of measures of synonymous codon usage bias. J Mol Evol. 1998;47:268–274. doi: 10.1007/PL00006384. [DOI] [PubMed] [Google Scholar]
- De Wit JJ, Cook JK, Van der Heijden HM. Infectious bronchitis virus variants: a review of the history, current situation and control measures. Avian Pathol. 2011;40:223–235. doi: 10.1080/03079457.2011.566260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhou T, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res. 2004;101:155–161. doi: 10.1016/j.virusres.2004.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RC. Viral respiratory diseases (ILT, aMPV infections, IB): are they ever under control? Br Poul Sci. 2010;51:1–11. doi: 10.1080/00071660903541378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KM, Jackwood MW, Hilt DA. Identification of amino acids involved in a serotype and neutralization specific epitope within the S1 subunit of avian infectious bronchitis virus. Arch Virol. 1997;142:2249–2256. doi: 10.1007/s007050050239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Wu G, Wang Z, Chai X, Nie Q, Zhang X. Mutation bias is the driving force of codon usage in the Gallus gallus genome. DNA Res. 2011;18:499–512. doi: 10.1093/dnares/dsr035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley M. Evolution. 3. Oxford: Blackwell Publishing; 2004. [Google Scholar]
- Sharp PM, Li W. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL (2000) PAUP*. Phylogenetic analysis using parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V. Coronaviruses molecular and cellular biology. Norfolk: Caster Academic Press; 2005. [Google Scholar]
- Van Marle G, Luytjes W, Van der Most RG, Van der Straaten T, Spaan WJ. Regulation of coronavirus mRNA transcription. Virology. 1995;69:7851–7856. doi: 10.1128/jvi.69.12.7851-7856.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel U, Arunkumar V, Dorairaj S. ACUA: a software tool for automated codon usage analysis. Bioinformation. 2007;2:62–63. doi: 10.6026/97320630002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal LY, Sandri TL, Souza SP, Richtzenhain LJ, de Wit JJ, Brandao PE. Molecular epidemiology of avian infectious bronchitis in Brazil from 2007 to 2008 in breeders, broilers, and layers. Avian Dis. 2010;54:894–898. doi: 10.1637/9218-121709-Reg.1. [DOI] [PubMed] [Google Scholar]
- Winter C, Herrler G, Neumann U. Infection of the tracheal epithelium by infectious bronchitis virus is sialic acid dependent. Microbes Infect. 2008;10:367–373. doi: 10.1016/j.micinf.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo PC, Wong BH, Huang Y, Lau SK, Yuen KY. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology. 2007;369:431–442. doi: 10.1016/j.virol.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. The “effective number of codons” used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Yang A, Wei L, Zhao W, Xu Y, Rao Z. Expression, crystallization and preliminary X-ray diffraction analysis of the N-terminal domain of nsp2 from avian infectious bronchitis virus. Acta Crystallogr, Sect F. 2009;65:788–790. doi: 10.1107/S1744309109024749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Gao Z, Zhang J, Chen H, Pejsak Z, Ma L, Ding Y, Liu Y. Comparative the codon usage between the three main viruses in pestivirus genus and their natural susceptible livestock. Virus Genes. 2012;44:475–481. doi: 10.1007/s11262-012-0731-z. [DOI] [PubMed] [Google Scholar]