

Abstract
Recombinant vesicular stomatitis virus (rVSV) is currently under evaluation as a human immunodeficiency virus (HIV)-1 vaccine vector. The most compelling reasons to develop rVSV as a vaccine vector include a very low seroprevalence in humans, the ability to infect and robustly express foreign antigens in a broad range of cells, and vigorous growth in continuous cell lines used for vaccine manufacture. Numerous preclinical studies with rVSV vectors expressing antigens from a variety of human pathogens have demonstrated the versatility, flexibility, and potential efficacy of the rVSV vaccine platform. When administered to nonhuman primates (NHPs), rVSV vectors expressing HIV-1 Gag and Env elicited robust HIV-1-specific cellular and humoral immune responses, and animals immunized with rVSV vectors expressing simian immunodeficiency virus (SIV) Gag and HIV Env were protected from AIDS after challenge with a pathogenic SIV/HIV recombinant. However, results from an exploratory neurovirulence study in NHPs indicated that these prototypic rVSV vectors might not be adequately attenuated for widespread use in human populations. To address this safety concern, a variety of different attenuation strategies, designed to produce a range of further attenuated rVSV vectors, are currently under investigation. Additional modifications of further attenuated rVSV vectors to upregulate expression of HIV-1 antigens and coexpress molecular adjuvants are also being developed in an effort to balance immunogenicity and attenuation.
Keywords: VSV, Immunogenicity, Neurovirulence, Safety, AIDS
VSV classification and natural history
Vesicular stomatitis virus (VSV) is a member of the Vesiculovirus genus in the family Rhabdoviridae. Although Rhabdoviridae family members are widely distributed in nature where they infect vertebrates, invertebrates, and plants, those in the Vesiculovirus genus are restricted to biting insects (mosquitoes, sand flies, blackflies, and ticks) and mammals. There are at least 14 phylogenetically and serologically distinct members in the Vesiculovirus genus [108] that separate into two broad groups based on geographic distribution; those found in the Americas, including the Indiana (IND) and New Jersey (NJ) serotypes of VSV, and those found in the eastern hemisphere (India, Eastern Europe, and the Middle East), including Isfahan, Chandipura, and Yug Bogdanovac.
In nature, VSV normally infects insects and livestock, and was named after the associated disease called “vesicular stomatitis” in horses, cattle, and swine. Signs of disease include vesicular lesions around the mouth, nostrils, teats, and coronary bands of the hooves. Although the disease often leads to weight loss due to impaired feeding, symptoms usually resolve in 10–14 days without consequence. VSV infection of livestock is accompanied by a very transient, low-level viremia and the disease is not readily transmitted from animal-to-animal even in a confined setting [45, 104]. Therefore, while livestock may display overt signs of disease, they are likely dead-end hosts without a major role in virus spread. In contrast, there is circumstantial evidence that biting insects are a major virus reservoir and vectors of disease in animals. Many vesiculoviruses have been recovered in nature from blood-sucking insects [17, 102, 107, 109] and bite transmission of VSV IND and Chandipura to laboratory animals by experimentally infected mosquitoes and sandflies has been demonstrated [6, 46, 80, 103, 105, 109]. In addition, natural infection has occurred in sentinel animals exposed in areas of known virus activity [47, 104]. The hypothesis that insects are the major VSV reservoir is further supported by the demonstration of vertical transmission in experimentally infected sand flies that, in nature, would allow virus to propagate and persist in insect populations without the need of an intermediate host [103, 106, 110].
VSV infection in humans is rare, but it can occur when animal handlers and veterinarians come in close contact with infected livestock and through inadvertent exposure of laboratory personnel [24, 42]. The most common portals of infection in humans are skin and mucous membranes of the nose, mouth, and eyes, although there is also serological evidence that suggests that some vesiculoviruses may be directly transmitted to humans through insect bites [7, 12, 47]. Human infection with VSV can either be asymptomatic or may lead to disease symptoms, which include myalgia, headache, and fever that resolve in 3–5 days without complications.
Virus structure and replication
The standard VSV particle is bullet-shaped (65×180 nm), comprising a ribonucleoprotein core surrounded by a lipid envelope that is derived from host cell plasma membrane. The virus genome is an ∼11-kilobase single strand of negative-sense RNA encoding five major viral proteins (Fig. 1). The N protein is the most abundant virus protein expressed in infected cells and associates closely with genomic RNA to form viral nucleocapsid, which is the functional template for both viral transcription and replication [115]. The P and L proteins associate to form the functional viral RNA polymerase that performs both transcriptase and replicase functions [21, 74, 113]. The M protein is the most abundant protein in the virus particle and has multiple functions in the infected cell, including virus budding, inhibition of host cell gene expression [2, 8, 41, 54], and regulation of viral transcription [13, 15]. The G protein is a type I transmembrane glycoprotein that forms trimeric spike-like structures on the virus surface [40, 55, 83, 118], and facilitates virus attachment to host cell receptors and uptake by endocytosis. At pH <6 the G protein undergoes a conformational change resulting in the fusion of viral envelope with the membrane of endosomal vesicles [30], releasing viral nucleocapsid and viral RNA polymerase into the host cell cytoplasm to initiate the replicative cycle.
Fig. 1.
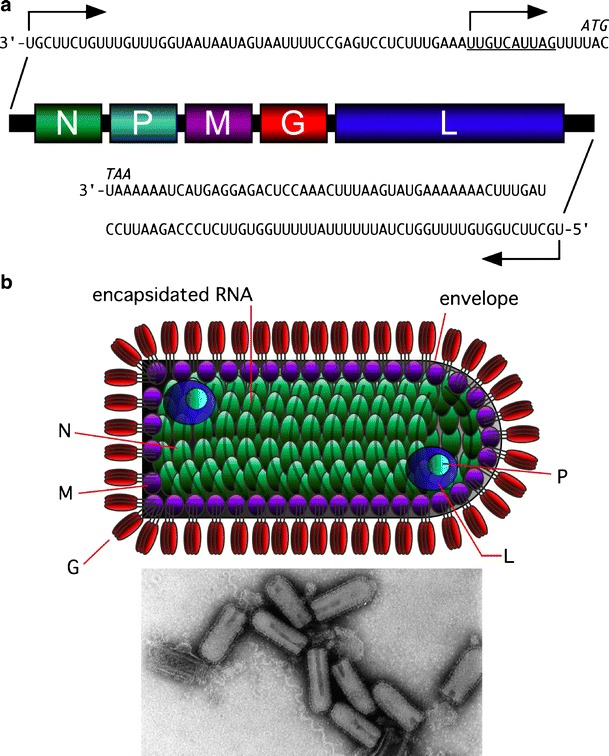
VSV genome organization, particle structure, and morphology. a The VSV genome encodes five major viral proteins, expressed from five discreet TUs. A single 3′ transcription promoter and genome replication promoter reside within the 50-nucleotide-long leader sequence. The antigenome replication promoter resides within the 59-nucleotide-long trailer sequence. Consensus transcription start and stop signals at the beginning and end of each TU, respectively, result in the synthesis of five VmRNAs. Genome replication occurs through a positive-sense antigenome intermediate. b The VSV particle is bullet-shaped, containing a ribonucleoprotein core surrounded by a lipid envelope that is derived from host cell plasma membrane. The N protein is closely associated with genomic RNA forming viral nucleocapsid. Viral RNA polymerase comprises a complex between the P and L proteins and is packaged inside the virus particle. G protein trimers form spike-like projections from the viral envelope that attach the virus particle to host cell receptors. The electron micrograph shows VSV particles stained with phosphotungstic acid (reprinted with permission from Rose and Whitt [87])
After engaging the unique transcription promoter at the 3′-end of the nucleocapsid template, the RNA polymerase initiates mRNA transcription and proceeds toward the genome 5′-end, terminating and reinitiating transcription at each gene junction, resulting in the production of five discreet viral mRNAs (VmRNAs) [1, 4, 39]. During this process the transcriptase complex occasionally detaches from the nucleocapsid template at intergenic junctions and must return to the 3′ promoter to reinitiate transcription, which results in a pronounced 3′ to 5′ gradient of VmRNA expression (Fig. 2). This transcription gradient allows the virus to express individual proteins at optimal levels for viral replication. For example, the L gene encoding the viral RNA polymerase, which is a catalytic protein needed in relatively small amounts, is distal to the 3′ transcription promoter and is transcribed with low efficiency in infected cells. Conversely, the N gene encoding a major structural protein, which is required in abundance for nucleocapsid formation, is immediately adjacent to the 3′ transcription promoter and is efficiently transcribed [112].
Fig. 2.
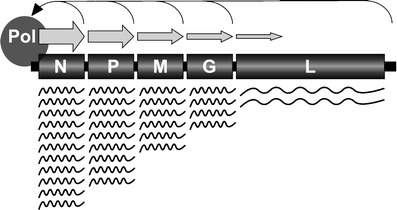
The VSV mRNA transcription gradient. Genes proximal to the 3′ transcription promoter are transcribed more abundantly than 5′ proximal genes. The gradient of mRNA transcription arises because the RNA polymerase complex often detaches from the template at intergenic junctions and can only reinitiate transcription at the 3′ transcription promoter
Some time after initiation of primary mRNA transcription, genome replication also begins. The mechanism responsible for switching RNA polymerase from a transcription to a replication mode is not fully understood, although a number of hypotheses have been proposed [3, 9, 10, 16, 73]. The nucleocapsid is also the functional template for genome replication as it is for transcription. The viral replicase initiates RNA synthesis at the exact genome 3′-end and proceeds to the 5′-end, ignoring transcription stop and start signals to produce a full-length positive sense copy of the viral genome. Nascent positive- and negative-sense genomic RNA is encapsidated with N protein in a cooperative, coreplicational manner [10, 59], and the resulting ribonucleprotein complex subsequently functions as a template for further genome amplification and mRNA transcription. The increased availability of amplified viral genome templates for secondary transcription leads to a rapid increase in the abundance of all viral proteins needed for mature virus particle production.
The virus assembly process is not understood completely, but available data indicate that viral nucleocapsid associates with regions of the plasma membrane enriched with M and G proteins. The nucleocapsid then condenses into a form that is transcriptionally inactive, and in a process that appears to be primarily driven by the M protein [36, 48], the plasma membrane containing G protein envelops the underlying viral nucleocapsid and mature virus particles bud from the cell surface. The G protein cytoplasmic tail (CT) does not appear to have an essential role in virus budding [85], but does enhance efficiency of the process [97, 98]. The mature virus particles contain the viral RNA polymerase necessary for further propagation in susceptible cells.
Generation of rVSV vectors
Until recently, directed manipulation of the VSV genome was impossible due to the instability of single-stranded RNA and difficulties in defining conditions that would allow recovery of infectious virus from genomic cDNA. However, as a result of pioneering work carried out in a number of laboratories, a recovery process known as “virus rescue,” was developed approximately 10 years ago [57, 95, 116]. The rescue procedure involves the transfection of susceptible cells with plasmids expressing the virus N, P, and L proteins and a full-length positive sense genomic RNA (Fig. 3). The N, P, and L proteins associate with the nascent genomic RNA to initiate virus replication and transcription, producing infectious virus progeny. The ability to rescue VSV from cloned cDNA has allowed fundamental questions on virus replication to be addressed, and has also enabled the rational design and development of recombinant VSV (rVSV) as a vaccine vector [27, 34, 35, 84, 85, 88, 89, 114].
Fig. 3.
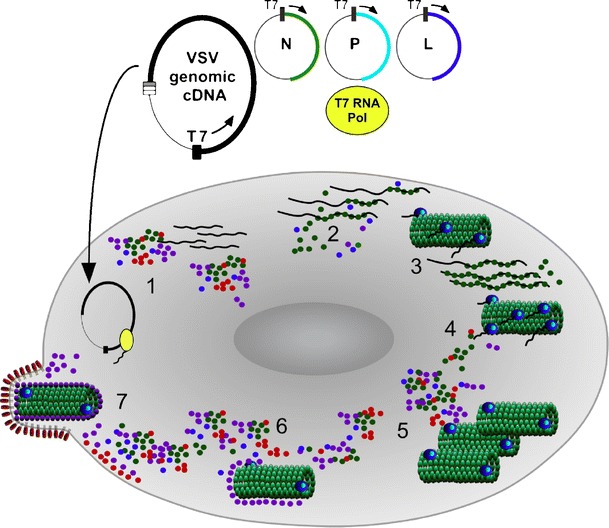
Rescue of rVSV from genomic cDNA. Virus rescue is initiated by cotransfecting plasmids (step 1) encoding the full-length viral genome and transacting viral polypeptides. T7 RNA polymerase, introduced into the cell by a variety of different methods, mediates RNA synthesis in the cell cytoplasm during the earliest stages of rescue, producing copies of the viral genomic RNA and transcripts encoding the transacting polypeptides (N, P, and L) needed to promote de novo assembly of a nucleocapsid (step 2). Functional nucleocapsid subsequently serves as a template for genome replication, transcription of all VmRNAs, and accumulation of viral proteins (steps 3–5) triggering ensuing events in the viral replication cycle including virus assembly (step 6) and budding (step 7)
There are many aspects of VSV biology that support the development of rVSV vaccine vectors for the treatment and prevention of human disease. The virus genome is simply organized into discreet transcriptional units (TUs) for expression of viral proteins, and nucleotide signal sequences involved in the control of viral gene expression have been defined [96]. Accordingly, one or more additional TUs encoding foreign proteins can be added to the VSV genome and robustly expressed under control of the 3′ transcription promoter [82, 84, 85, 89]. Furthermore, the abundance of foreign protein expression can be modulated depending on proximity of the extra TUs relative to the 3′ transcription promoter (unpublished data) [72]. Although the mutation frequency of VSV is high [18, 37] and additional foreign genes are not essential for virus replication, the consensus sequence of foreign protein open reading frames (ORFs) can be maintained over many rounds of amplification [96], ensuring preservation of target antigens during the vaccine manufacturing process. The ability of rVSV vaccine vectors to propagate vigorously in qualified continuous cell lines also facilitates industrial scale manufacture of vaccine preparations.
Because human infection is rare, VSV seroprevalence in the general population is very low; therefore, preexisting immunity will not likely be a factor affecting the efficacy of future rVSV vaccine vectors, as may be the case for some other proposed viral vaccine vectors [14, 61]. Indeed, the existence of many different vesiculovirus serotypes may be exploited to design multiple rVSV vaccine vectors [88] for prevention and treatment of many different diseases. VSV does not cause any obvious signs of disease after parenteral administration to either livestock or nonhuman primates (NHPs) [89], and rVSV vectors are anticipated to be innocuous in humans when administered parenterally. Other desirable safety features include the lack of a DNA intermediate during viral replication and the cytoplasmic site of genome replication, both of which reduce the likelihood of viral nucleic acid integration into host cell DNA. Furthermore, vector genome stability is enhanced because it is encapsidated and cannot undergo recombination.
In the prototypic rVSV vectors, foreign genes were expressed from a TU inserted at position 5 in the viral genome (Fig. 4) between the G and L genes [49, 84, 85, 89]. This insertion site was chosen to minimally perturb the stoichiometry of viral protein expression, which is modulated by distance of each gene from the 3′ transcription promoter. Because the L protein has a catalytic function, displacing the L gene by one additional TU from the 3′ promoter was viewed as a conservative genome alteration that should not adversely affect viral replication, while allowing adequate foreign protein expression to generate an immune response. This rationale was supported by in vitro growth studies showing only minimal reduction in peak virus titers for vectors containing an extra foreign gene between the G and L genes [22, 85]. Furthermore, the level of foreign protein expression was sufficient to elicit robust antigen-specific immune responses after a single inoculation of mice and rabbits [82, 84, 85]. In some cases, primary immune responses could be further boosted with a second dose of homologous vaccine 1 to 2 months later [82, 85]. However, because durable neutralizing antibodies are usually elicited against the VSV surface glycoprotein after a single vaccination with replication-competent rVSV vectors, glycoprotein exchange vectors were designed to allow more effective boosting of immune responses to target antigens. The exchange vectors were generated by switching the IND serotype G gene in priming vectors with either the VSV NJ or Chandipura serotype G gene in boosting vectors [88].
Fig. 4.
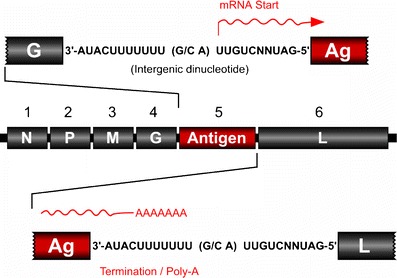
Expression of foreign antigens by prototypic rVSV vectors. Foreign antigens are expressed from a TU inserted between the virus G and L genes. The foreign gene is flanked by VSV-specific consensus transcription start and stop/polyadenylation signals
In view of the flexibility of the rVSV vector platform and the encouraging data from preclinical studies with experimental influenza vaccines [84, 85], rVSV vectors expressing antigens from a variety of other human pathogens were developed and tested in preclinical animal challenge models (Table 1). Other vectors expressing antigens from hepatitis C virus (HCV) [22], hantavirus [58], and mycobacterium tuberculosis [117] were assessed in preclinical immunogenicity studies.
Table 1.
Preclinical efficacy of rVSV vectors
| Immunogen1/Route2 | Challenge1/Route2 | Species | Reference |
|---|---|---|---|
| HIVenv, SIVgag/IN, IM | SHIV/IV, Vaginal | Rhesus macaques | [19, 20, 78, 89] |
| Flu HA/IN | Flu/IN | Mice | [84, 85] |
| RSV F or G/IN | RSV/IN | Mice | [49, 50] |
| SARS S/IN | SARS/IN | Mice | [51] |
| MV HN/IN | MV/IN | Cotton rats | [94] |
| CRPV L1/IN, IM | CRPV/ID | Rabbits | [82] |
| EBOVgp/IM | EBOV/IM | Cynomolgus macaques | [44] |
| MARVgp/IM | MARV/IM | Cynomolgus macaques | [44] |
| LVgp/IM | LV/IM | Cynomolgus macaques | [33] |
| HSV-2gD/IN, IM | HSV-2/Vaginal | Mice, guinea pigs | [69] |
1Flu HA Influenza A hemagglutinin, RSV respiratory syncytial virus, SARS S severe acute respiratory syndrome coronavirus spike protein, MV HN measles virus HA/neuraminidase, CRPV L1 cottontail rabbit papilloma virus L1 protein, EBOVgp Ebola virus glycoprotein, MARV gp Marburg virus glycoprotein, LVgp Lassa fever virus glycoprotein, HSV-2gD herpes simplex type 2 gD glycoprotein
2IN intranasal, IM intramuscular, ID intradermal, and IV intravenous
The first NHP experiments to evaluate rVSV as a vector system for human immunodeficiency virus (HIV) were conducted by Dr. John Rose and colleagues at Yale. In these pioneering studies, rhesus macaques were immunized with a combination of two prototypic rVSV vectors expressing a HIV-189.6 Env gp160/VSV-G fusion polypeptide and simian immunodeficiency virus (SIV) Gag p55 protein. Vaccine vector administration by a combination of intramuscular and intranasal routes clearly demonstrated the ability of rVSV vectors to elicit potent antigen-specific cell-mediated immune responses in NHPs. In addition, this study also clearly demonstrated for the first time the ability of rVSV vectors expressing Env and Gag proteins to provide highly significant protection from AIDS after an intravenous heterologous SIV/HIV (SHIV)89.6P challenge [89]. In this study, all seven macaques receiving rVSV expressing HIVenv and SIVgag remained disease-free after SHIV89.6P challenge for >4 years ([89] and J. Rose, personal communication). By comparison, all eight control macaques [either unimmunized or immunized with lab-adapted rVSV expressing influenza hemagglutinin (HA)] progressed to AIDS with an average time of 8 months ([89] and J. Rose, personal communication). The protection from AIDS in this study correlated with large differences in peak viral loads, low or undetectable viral loads at set point, and with the preservation of CD4+ T cells in the vaccinees relative to controls. This encouraging level of post-challenge vaccine efficacy suggested that rVSV vectors expressing HIV genes might be an effective AIDS vaccine in humans.
In addition to demonstrating postchallenge efficacy in NHPs, rVSV-based vaccines have other advantages that suggest they may be ideally suited for development as a worldwide prophylactic HIV-1 vaccine. Most notably, rVSV-based vaccine vectors, at least in animal models, can be safely and effectively administered via a mucosal route as shown in several animal models. For example, a single intranasal inoculation with an rVSV vector expressing the influenza virus HA protein in mice [84, 85], the herpes simplex virus (HSV) type 2 glycoprotein D in mice [69], or a measles virus HA protein in cotton rats [94] completely protected animals against mucosal challenge with the corresponding wild-type pathogen. Recently, our laboratory systematically compared the immunogenicity of rVSV vaccine vectors when given either as an intramuscular or intranasal inoculation in an effort to determine which route of vaccine administration was optimal for inducing humoral and cellular immune responses in rhesus macaques [19]. Our results demonstrated that Mamu-A*01+ MHC class I-positive macaques, vaccinated with a combination of rVSV vectors expressing HIV-189.6Penv or SIVgag by the intranasal route, developed significantly higher HIVgag and HIVenv peptide-specific cellular immune responses as determined by MHC class I tetramer staining, interferon (IFN)-γ enzyme-linked immunosorbent spot (ELISPOT), and cytotoxic T cell assays. It is interesting to note that systemic and mucosal humoral immune responses did not vary significantly with the route of vaccine administration.
Safety of rVSV vectors
Studies have not yet been conducted that directly evaluate the safety of rVSV vectors when administered to humans. As mentioned previously, infection with field isolates and cell culture-adapted VSV was not associated with significant illness in humans. Furthermore, in the course of numerous immunogenicity and safety studies performed at Wyeth (unpublished data), more than 150 NHPs were inoculated either intranasally or intramuscularly with high doses (107 plaque-forming units) of cell culture-adapted VSV and a variety of rVSV vectors without adverse effects. Animals maintained normal blood chemistry, blood cell count, and body weights after inoculation with virus, and showed no signs of the characteristic vesicular lesions observed in nasal and lingual epithelia.
Although the studies described above indicated that rVSV vaccine vectors could be administered safely to humans, additional safety studies would be required before proceeding with clinical studies. Replication-competent viral vaccines have historically been subjected to stringent safety testing including direct inoculation of virus into the central nervous system of susceptible NHPs to measure neurovirulence (NV) potential [60, 68, 91, 99]. Although the rVSV/HIV-1 vaccine vectors described above are attenuated relative to VSV field isolates, they are replication-competent and subject to the same stringent safety testing used for other live attenuated vaccine viruses. For this reason, the prototypic rVSV vector and a number of closely related rVSV vectors expressing HIV-1 Gag protein were tested in an exploratory NHP NV study. This study was also warranted by historical data showing that VSV field isolates and tissue culture-adapted VSV strains were neurotrophic and neurovirulent in rodents [81, 92, 111], and that VSV serially passaged in mouse brain was found to be neurovirulent in livestock and NHPs inoculated by the intracranial route [29, 70].
The NV potential of rVSV/HIV-1 vaccine vectors was tested by direct intrathalamic inoculation of cynomolgus macaques in accordance with the procedures used to assess production lots of mumps virus vaccine [65, 91]. The results of this exploratory study showed that the prototypic rVSV vector expressing HIV-1 Gag was much more attenuated than cell culture-adapted wild-type VSV, but retained a level of residual NV that would likely be unacceptable to regulatory agencies (J. Erik Johnson et al., submitted for publication).
Attenuation strategies for rVSV vectors
To address the safety concerns raised by the outcome of the exploratory NHP NV study, a variety of distinctive strategies were investigated to further attenuate growth and virulence of rVSV vectors. Multiple approaches were pursued because it was impossible to predict which attenuation strategy would provide an optimal balance between safety and immunogenicity.
The ability to attenuate in vitro growth and virulence of rVSV by moving the N gene from the first position in the genome to downstream locations was clearly demonstrated [27, 28, 112]. The step-wise translocation of the N gene further away from the 3′ transcription promoter leads to incremental reduction in N protein expression, thereby limiting nucleocapsid formation (Figs. 2 and 5). Because nucleocapsid is the functional template for mRNA transcription and genome replication and is part of the virion core, limiting nucleocapsid leads to a decrease in viral protein synthesis, genome amplification, and formation of mature virus particles. In addition to providing a means of attenuating rVSV vectors incrementally, another attractive feature of this attenuation strategy is the known genetic stability of N gene translocations [4, 27], which greatly eliminates the likelihood of reversion.
Fig. 5.
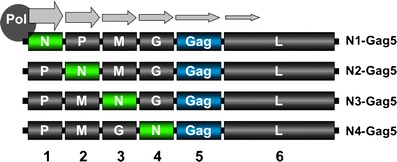
Attenuation of rVSV by gene translocation. Step-wise translocation of the N gene further away from the 3′ transcription promoter leads to incremental attenuation of virus growth in vitro and in vivo. This method of attenuation relies on downregulation of N protein expression due to the 3′ to 5′ transcription gradient
Truncation of the CT region of G protein was also used to attenuate in vitro growth and in vivo virulence of rVSV vectors [85, 98]. The wild-type G protein CT region is 29 amino acids in length. However, when the CT was truncated to either one or nine amino acids (CT1 and CT9, respectively) (Fig. 6), peak virus titers obtained in cell culture were reduced, and the resulting viruses were less pathogenic in mice after intranasal inoculation [85, 98]. The attenuation mechanism likely involves an impaired interaction between the viral ribonucleoprotein core and the truncated G protein CTs during viral morphogenesis [66, 71, 98], leading to a reduction in the efficiency of virus particle assembly. As seen for N gene translocation, G protein CT truncation also gives rise to virus with a very stable attenuation phenotype because replacement of deleted sequence encoding part of a protein is very improbable.
Fig. 6.
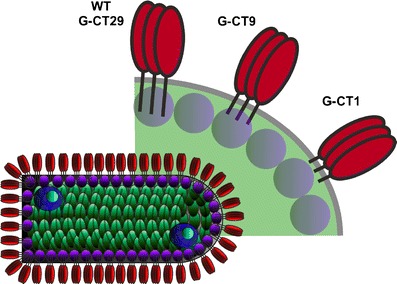
Attenuation of rVSV by G protein truncation. Truncation of the CT region of the rVSV G protein from 29 amino acids to either nine (CT9) or one (CT1) amino acids progressively attenuates virus growth in vitro and in vivo. It is believed that viral morphogenesis is adversely affected due to impaired interaction between shorter CTs and underlying viral core proteins
The VSV M protein plays a key role in the development of virus induced cell cytopathology and cell death [2, 8, 54]. Mutations in the rVSV M gene that delay and diminish cytopathic effect in infected cells were described [41]. These mutations ablate the expression of two overlapping, in-frame polypeptides that are expressed from the M protein mRNA by initiation of protein synthesis at internal AUGs (Fig. 7). Both polypeptides may have a role in the development of cell cytopathology by inhibiting nuclear export of cellular mRNA and disruption of the cell cytoskeleton [23, 41, 91]. These M gene mutants are also much less pathogenic in mice (unpublished data).
Fig. 7.
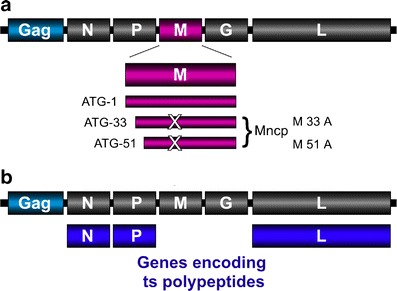
Attenuation of rVSV by incorporation of M gene and ts mutations. a The VSV M gene encodes full-length M protein as well as two smaller in-frame polypeptides. Mutating the internal initiation codons from Met to Ala ablates expression of the smaller polypeptides resulting in a virus that produces minimal cytopathology during infection. These noncytopathic M gene mutations (Mncp) also attenuate virus replication. b One or more viral genes are replaced with those containing ts mutations. Viruses containing ts mutations are growth-attenuated at a defined nonpermissive temperature (typically 37–39°C) but can grow robustly at a lower, permissive temperature (32°C)
Historically, VSV temperature-sensitive (ts) mutants were isolated either as a result of random point mutation or, more commonly, after chemical mutagenesis [26, 76, 77]. These conditional mutants usually propagate robustly at a permissive temperature and poorly or not at all at a defined nonpermissive temperature. Because the temperature of nasal epithelium is typically lower than core body temperature, selective incorporation of ts mutations should produce rVSV vectors that replicate vigorously at 32–35°C but not at 37°C, and serve as vectors for intranasal vaccination (Fig. 7b). The molecular mechanism of ts attenuation is often not clearly defined but probably involves impaired protein folding and instability at the nonpermissive temperature.
In case replication-competent rVSV vectors prove to be inadequately attenuated for use in humans, propagation-incompetent rVSV vectors are also being developed. These vectors are restricted to a single round of replication and are unable to spread beyond primary infected cells. One such vector has the entire G gene deleted (ΔG), and therefore requires G protein transcomplementation for propagation of infectious virus particles in vitro (Fig. 8) [85]. Another related vector has most of the G protein ectodomain deleted (G stem), retaining the CT region, transmembrane domain, and 42 amino acids of the membrane proximal ectodomain (Fig. 8). This vector is also propagation-defective, requiring G protein in trans for production of infectious particles in vitro. Expression of the truncated form of G protein (G stem) enhances the overall yield of progeny virus particles in vitro compared to vectors with the entire G gene deleted, perhaps due to increased concentration of G protein CTs at the cell plasma membrane, facilitating virus assembly and budding [86]. Although propagation-defective virus offers obvious safety advantages, difficulties in providing adequate quantities of complementing G protein to allow efficient vector amplification during industrial scale manufacture are a possible limitation.
Fig. 8.
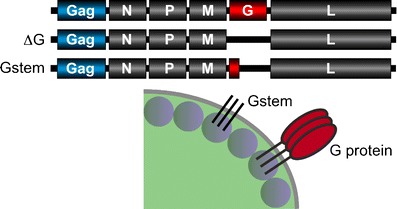
Propagation defective rVSV vectors. Deletion of sequence encoding either all of the G protein (ΔG) or most of the G protein ectodomain (Gstem) results in virus that can propagate only in the presence of transcomplementing G protein. Both vectors are unable to spread beyond primary infected cells in vivo
Further attenuation of rVSV vectors
The above strategies gave rise to rVSV vectors showing a range of growth attenuation in vitro and a marked reduction in pathogenicity after intranasal inoculation of mice [77, 85, 86, 114]. However, some vectors still displayed levels of NV that were almost equivalent to unaltered prototypic rVSV vector after intracranial inoculation of mice [28, 77, 114]. In addition, one of the attenuated vectors containing a G protein CT truncation that was highly attenuated in vitro and after intranasal inoculation of mice still caused moderate levels of neuropathology in the NHP NV model (J. Erik Johnson et al., submitted for publication). These observations indicated that the previously described attenuation strategies might not be sufficient to reduce rVSV vector NV in animal models to a level that would be acceptable to regulatory agencies. Therefore, additional strategies to further attenuate rVSV vectors are being explored, including combinations of CT truncations with either N gene rearrangements or Mncp mutations (Fig. 9). It was thought that these mutation combinations would increase vector attenuation beyond levels obtained using single attenuation strategies. Other types of gene rearrangement, including M and G gene shuffling, singly and in combination, are also under investigation [5, 25]. Specific combinations of ts mutations are also being developed in an effort to ensure tight shut-off of viral replication at the nonpermissive temperature while retaining high levels of transcriptional activity (Fig. 9).
Fig. 9.
Future approaches to rVSV attenuation. Combination of attenuation strategies may further extend the range of attenuated rVSV vectors to be tested to achieve an optimum balance of safety and immunogenicity. N gene shuffles may be combined with either G protein CT truncations or Mncp mutations. Other individual gene translocations (M and G) may, on their own and in combination with other attenuation strategies, provide an optimal attenuation phenotype. Novel combinations of ts mutations may also produce rVSV vectors that can replicate well at 34–35°C, but not at core body temperature, and may be suited to intranasal vaccine delivery
As with other attenuated rVSV vectors, plaque size and one-step growth kinetics will serve as an initial measure of replication efficiency of further attenuated combination mutants. Those vectors showing significant growth attenuation in vitro will then be tested for NV in mice after intracranial inoculation. This route of inoculation was chosen because significant differences in virulence between highly attenuated vectors are often not apparent after intranasal inoculation, but can be measured after intracranial inoculation due to the exquisite sensitivity to VSV infection by this route [38, 67, 75, 81, 114]. The resulting mortality and morbidity can be used to calculate a 50% lethal dose (LD50) for each vector, and these data will then be used to define a rank order of vector attenuation.
Although the murine NV model is tractable and provides a very sensitive means of ranking rVSV vector NV, additional NV studies also will be carried out in ferrets and NHPs to further evaluate vector safety. Ferrets were used as a disease model to study influenza virus because they are naturally susceptible to infection and display many of the disease symptoms seen in humans [63, 64, 90, 93, 100]. A ferret NV model was recently developed because components of the influenza virus particle can induce significant neuropathology in NHPs after intracranial inoculation in the absence of viral replication (unpublished data). Because the ferret NV model might be a better indicator of NV and neurotropism potential in NHPs and humans, rVSV vector safety also will be assessed after intranasal and intracranial inoculation of ferrets. In the absence of a clearly defined marker of ferret NV (LD50), changes in brain histopathology profiles will be used to assess the extent of injury associated with each rVSV vector.
Data from murine NV and immunogenicity studies and ferret NV studies will then be used to identify the most promising candidate vectors for further NV testing in NHPs. These studies will be performed as before for the prototypic rVSV vectors. For the NHP NV studies, UV-inactivated rVSV will be used as a control to measure baseline pathology resulting from the injection procedure and potential injurious effects of viral components in the inoculum. These controls are important in stringent NV testing of novel replication-competent vaccine candidates because injection of even the most innocuous material directly into the brain can lead to cell necrosis around the needle track and low-level inflammatory responses throughout the CNS.
Typically, the two major types of injury resulting from intrathalamic inoculation of replication-competent virus are inflammatory responses stimulated by the presence of foreign material in the brain, mediated by cytokines and infiltrating leukocytes, and tissue necrosis resulting from cell death caused by viral replication. More specifically, the distinction between neuronal necrosis and inflammatory responses is important because neuronal cell necrosis is thought to be an irreversible process resulting in permanent neurological deficit, while inflammatory responses typically resolve over time, usually without permanent sequelae. Therefore, rVSV vectors that produce only minimal to mild inflammatory responses in the brain would be strongly favored over those causing necrotic lesions. If, as expected, some of the further attenuated rVSV vectors currently under development cause only very minimal inflammatory responses in the NHP CNS after intrathalamic inoculation and retain HIV-1-specific immunogenicity, then one or more of these vectors will be considered for future clinical evaluation.
Enhancement of the immunogenicity of rVSV vectors
As rVSV vectors are further attenuated to enhance their safety, additional measures to maintain or increase immunogenicity may be required. The relative flexibility of the rVSV vector system and its relative ease of genetic manipulation allow for potential improvements in the immunogenicity of these vectors. There are several approaches that can be exploited to increase immunogenicity, including enhancement of foreign antigen expression, reduction of antivector immunity, coexpression of cytokines or immunomodulators, in vivo targeting of dendritic cells, and heterologous prime/boost vaccination regimens.
Enhancement of foreign antigen expression
Ideally, viral vectors used as vaccines can be rationally designed such that immunogenicity can be directed away from vector-specific antigens and toward the expressed foreign antigen, thereby maximizing immunogenicity of the target antigen and minimizing immunogenicity of the vector antigens. Antivector immunity can limit the ability to boost responses, and may limit the use of the same vector backbone for several vaccine targets. This might be especially relevant to immunotherapeutic approaches for persistent infections such as HIV, HSV, and HCV where repetitive boosting may be required to control infection.
One potential approach to shift the focus of the immune response toward the expressed foreign antigen and away from VSV antigens is to take advantage of the transcriptional gradient of VSV described earlier. The prototype rVSV vectors developed by Rose et al. [89] positioned the gene expressing the foreign antigen toward the 5′-end of the viral genome. This was done to promote efficient rescue from cDNA and maintain viral replicative fitness. However, due to the 3′ to 5′ transcription gradient, positioning the foreign gene toward the 5′-end of the genome reduces the expression of the foreign antigen, relative to the four VSV 3′ proximal gene products. Therefore, future rVSV vectors will be designed with the foreign antigen gene at the 3′-end of the VSV genome (position 1), thereby maximizing the expression of the foreign antigen. Positioning the foreign gene in the first position in the genome should also have an attenuating effect on virus growth as all viral genes will be translocated further away from the 3′ transcription promoter, reducing their expression.
Heterologous prime/boosting of rVSV vectors
One potential limitation of viral vectors is preexisting immunity to the vector. In addition, the primary immune response to the vector often limits the effectiveness of subsequent booster immunizations. In both small animal models and NHPs, a strong serotype-specific neutralizing antibody response is generated against the G protein of VSV after a single inoculation. The presence of these neutralizing antibodies precludes using the identical rVSV vector as a boosting vector in animals and will presumably have the same effect after vaccination in humans. To circumvent neutralizing antibodies and create vectors for boosting, Rose et al. [88] developed rVSV glycoprotein exchange vectors that encode the N, P, M, and L genes of the VSV IND serotype, but have the gene expressing the VSV IND serotype G protein substituted with either the gene expressing the VSV NJ serotype G protein or the gene for the G protein of the related Vesiculovirus Chandipura virus [88]. While these glycoprotein exchange vectors circumvent the neutralizing antibody response to the G protein, it is possible that T cell responses to the N, P, M, and L proteins might restrict virus replication and limit the ability of these vectors to boost responses. Generation of a complete rVSV NJ serotype vector was not attempted due to the potential for cross-reactive cytotoxic T-lymphocyte responses between IND and NJ VSV serotypes. However, there are additional viruses in the Vesiculovirus genus that could be used to supply G genes for future rVSV G protein exchange vectors if multiple boosting vectors are desired. Attachment glycoprotein exchange of this manner does introduce a new gene into the vector, may alter the vector phenotype, and therefore careful safety testing must be conducted before advancing these vectors to clinical trials.
While changing the rVSV serotype may provide the ability to boost with related vectors, more robust boosting may be possible with completely heterologous viral vectors or nonviral delivery systems such as plasmid DNA, protein subunit, or bacterial vectors. Studies in mice conducted with vectors expressing HIV Env or Gag showed that boosting rVSV-primed animals with vaccinia-based vectors produced a fivefold increase in peak HIV-specific CD8+ T cell responses relative to mice boosted with rVSV glycoprotein exchange vectors [35]. As a follow up study, Ramsburg et al. [78] compared the effectiveness of a single prime-boost immunization regimen in macaques consisting of lab-adapted G protein exchange VSV vectors expressing SHIV env, gag, and pol proteins to that of a protocol consisting of a lab-adapted rVSV vector prime followed by a single boost with modified vaccinia virus Ankara (MVA) expressing the same SHIV proteins. The macaques primed with a rVSV vectors expressing SHIV proteins and boosted once with a MVA vector expressing SHIV proteins mounted increased immune responses relative to animals boosted with VSV exchange vectors; however, due to the small numbers of macaques in each group, this difference did not achieve statistical significance. Nevertheless, after intravenous SHIV89.6P challenge, MVA-boosted macaques demonstrated a further 1 log10 reduction in peak viral loads, cleared challenge virus faster, and preserved CD4+-T cell counts to a greater extent than macaques receiving VSV G protein exchange vectors. Collectively, these data suggest that while G protein exchange rVSV vectors can circumvent VSV-specific neutralizing antibody and boost vaccine-specific immune responses, vaccine-specific immunogenicity and efficacy are substantially improved by boosting with a heterologous viral vector.
Recently, we demonstrated that priming with a series of intramuscular injections of plasmid DNA encoding interleukin-12 (IL-12) and SIVgag effectively enhanced the immunogenicity and postchallenge efficacy of two intranasal doses of rVSV expressing HIVenv and SIVgag in rhesus macaques [20]. The results demonstrated that the plasmid DNA priming vaccination regimen significantly increased SIVgag-specific cell-mediated and humoral immune responses, and significantly lowered viral loads post-SHIV89.6P challenge relative to macaques receiving only the rVSV vectored immunizations. The addition of the plasmid DNA priming regimen also tended to increase the preservation of peripheral blood CD4+ cells and reduce the incidence of AIDS-like disease symptoms and death associated with SHIV89.6P infection, although this did not achieve statistical significance due to the relatively small number of macaques used per group.
An analysis of immune correlates of protection after SHIV89.6P challenge revealed that the SHIV-specific IFN-γ ELISPOT responses elicited by vaccination correlated with postchallenge clinical outcome. Thus, the higher SHIV-specific cell-mediated immune responses elicited by the DNA prime/rVSV boost vaccination regimen was associated with an increased maintenance of CD4+ cells and a reduction in plasma SHIV viral loads. When the rVSV HIV-1envG/SIVgag vaccines were given alone, substantially lower SHIV-specific cell-mediated immune responses were elicited and the postchallenge protection afforded by immunization was reduced.
Once vectors based on rVSV and other platforms have demonstrated safety and immunogenicity in clinical trials, it will be important to assess their combined use in heterologous prime/boost vaccination regimens to maximize their potential.
Redirecting the tropism of rVSV vectors
There may be additional benefit gained by creating rVSV vectors expressing heterologous viral attachment glycoproteins. In the effort to use rVSV as a vaccine vector for human diseases, many groups have either replaced the VSV G protein or coexpressed it with glycoproteins from targeted human pathogens including hantavirus [58], HIV [43], influenza virus [56, 84], HCV [11], respiratory syncytial virus [49, 50], severe acute respiratory syndrome (SARS) coronavirus [31, 51], Marburg virus [32, 44], Ebola virus [32, 44], and Lassa virus [32, 33]. When tested in animal models, these vectors elicited immune responses directed against the expressed foreign glycoproteins. More recently, NHPs immunized with rVSV vectors expressing glycoproteins from either Marburg or Ebola viruses were protected from disease after challenge with these pathogens [44]. Moreover, it is likely that replacing the VSV G protein with filoviruses glycoproteins altered rVSV vector tissue tropism such that they could readily target dendritic cells, as filoviruses are known to do. Such retargeted rVSV vectors could be designed to express additional foreign antigens from unrelated pathogens and serve as either priming or boosting vectors along with rVSV IND and NJ serotype vectors. Because retargeting by glycoprotein exchange can markedly alter virus phenotype and cell tropism, any new rVSV vector of this type will require extensive safety studies before clinical assessment.
Use of adjuvants and immunomodulators with rVSV vectors
One advantage of using viral vectors as vaccines is that they are believed to act as their own adjuvant by stimulating the innate immune response through the binding of viral components to pathogen recognition receptors of the host cells. Specifically, VSV single-stranded RNA is believed to bind Toll-like receptor-7 in infected plasmacytoid dendritic cells, and is essential to the initiation of a type I IFN response and other innate responses [62]. Furthermore, the ribonucleoprotein complex of VSV was shown to activate cellular kinases TBK1 and IKKɛ that are involved in the production of type I IFNs [101]. In spite of this adjuvant activity of VSV, and because rVSV vectors will need to be further attenuated, it may be helpful to enhance their immunogenicity through the expression of proinflammatory cytokines, or the coadministration of small molecule adjuvants and immunomodulators.
Several rVSV vectors have already been generated that express various cytokines. While most of these vectors have had a weak adjuvant effect, positive results from rVSV vectors expressing IL-12 [52, 53] or granulocyte-macrophage colony-stimulating factor (GM-CSF) [79] have been reported. IL-12 is a proinflammatory cytokine expressed by antigen-presenting cells as a heterodimer, and drives the development of Th1-like responses. The ORF for an IL-12 fusion protein was inserted into a replication-restricted rVSV vector lacking the gene for the G protein (VSVΔG-IL12F) [52]. When VSVΔG-IL12F was coadministered with a weakly antigenic listerial antigen, enhanced Th1-polarized T cell responses (determined by listerial-specific IL-2 and IFN-γ secretion) and B cell responses (determined by listeria-specific IgG titers) were generated in mice. Moreover, coadministration of VSVΔG-IL12F and listerial antigen conferred protection from a lethal intraperitoneal listerial challenge [53].
GM-CSF recruits and activates antigen-presenting cells, including macrophages and dendritic cells. An rVSV vector expressing murine GM-CSF from the first position in the viral genome (VSV-GMCSF1) was shown to attenuate viral pathogenesis in mice, while retaining an immune response to the N protein of VSV [79]. Memory responses to VSV N were also slightly enhanced in mice immunized with VSV-GMCSF1 compared to VSV N responses in mice immunized with rVSV expressing green fluorescent protein. It is still to be determined how coadministration of VSV-GMCSF1 along with a rVSV vector expressing a foreign antigen will modulate the response to that antigen.
Conclusions
The development of procedures for the recovery of viruses in the order Mononegavirales from genomic cDNA has created the opportunity to utilize this group of viruses as vaccine vectors for the prevention and treatment of a wide range of human diseases. VSV is one of the most-studied of the nonsegmented negative strand RNA viruses and is foremost among potential vaccine vectors in this group of viruses. VSV is naturally found in insects and livestock, is not considered a human pathogen, and has a very low seroprevalence in human populations. The small, simply organized viral genome can be altered to stably express one or more foreign proteins, and the resulting viral vectors can be amplified to high a titer in approved cell lines. The virus has a very broad host range and rVSV vector serotype can be changed simply by switching the surface glycoprotein with that from other vesiculovirus serotypes. VSV replicates exclusively in the cytoplasm of infected cells, does not persist in vivo, and viral nucleic acid does not integrate into host genomic DNA. These desirable properties have led to the development of rVSV as a potential HIV-1 vaccine vector.
Signs of VSV infection in livestock are usually restricted to the occurrence of vesicular lesions around the mouth, nose, teats, and coronary bands of the hooves that typically resolve in 7–10 days without serious consequence. However, historical data showed that VSV could be lethal in mice, and was neurovirulent when injected directly into the brain of mice, livestock, and NHPs. Therefore, to investigate the potential neurotropism and NV of prototypic rVSV/HIV-1 vaccine vectors, exploratory NV testing in NHPs was carried out. The results of these tests indicated that the prototypic rVSV/HIV-1 vaccine vectors required further attenuation before clinical evaluation. A variety of different attenuation strategies and combinations thereof are being evaluated to define a more acceptable attenuation phenotype. A very sensitive mouse LD50 NV model can be used to rank order rVSV vector attenuation level. Vectors that cause minimal morbidity and mortality in the mouse model will then be further tested in ferret and NHP NV models to assess residual vector virulence. Vectors that cause only a minimal degree of neuropathology will be targeted for clinical evaluation.
Balancing satisfactory levels of safety with adequate immunogenicity of further attenuated rVSV vectors will be challenging. However, upregulation of HIV-1 antigen expression, through gene positioning effects, may enhance immunogenicity and compensate for increased attenuation of virus replicative ability. Coexpression of molecular adjuvants by rVSV vectors may further improve antigenicity of HIV-1 proteins. The combination of rVSV/HIV-1 vaccine vectors with other HIV-1 vaccine vectors, including plasmid DNA, in heterologous prime-boost regimens also provides another promising approach to enhance HIV-1-specific immune responses.
Acknowledgements
This work was sponsored by a HIV-1 Vaccine Design and Development Team contract from the National Institutes of Health and National Institute of Allergy and Infectious Diseases (HVDDT NO1-A1-25458).
References
- 1.Abraham G, Banerjee AK. Sequential transcription of the genes of vesicular stomatitis virus. Proc Natl Acad Sci USA. 1976;73:1504. doi: 10.1073/pnas.73.5.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed M, McKenzie MO, Puckett S, et al. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol. 2003;77:4646. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnheiter H, Davis NL, Wertz G, et al. Role of the nucleocapsid protein in regulating vesicular stomatitis virus RNA synthesis. Cell. 1985;41:259. doi: 10.1016/0092-8674(85)90079-0. [DOI] [PubMed] [Google Scholar]
- 4.Ball LA, White CN. Order of transcription of genes of vesicular stomatitis virus. Proc Natl Acad Sci USA. 1976;73:442. doi: 10.1073/pnas.73.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ball LA, Pringle CR, Flanagan B, et al. Phenotypic consequences of rearranging the P, M, and G genes of vesicular stomatitis virus. J Virol. 1999;73:4705. doi: 10.1128/jvi.73.6.4705-4712.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergold GH, Suarez OM, Munz K. Multiplication in and transmission by Aedes aegypti of vesicular stomatitis virus. J Invertebr Pathol. 1968;11:406. doi: 10.1016/0022-2011(68)90190-0. [DOI] [PubMed] [Google Scholar]
- 7.Bhatt PN, Rodrigues FM. Chandipura: a new Arbovirus isolated in India from patients with febrile illness. Indian J Med Res. 1967;55:1295. [PubMed] [Google Scholar]
- 8.Black BL, Lyles DS. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J Virol. 1992;66:4058. doi: 10.1128/jvi.66.7.4058-4064.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blumberg BM, Leppert M, Kolakofsky D. Interaction of VSV leader RNA and nucleocapsid protein may control VSV genome replication. Cell. 1981;23:837. doi: 10.1016/0092-8674(81)90448-7. [DOI] [PubMed] [Google Scholar]
- 10.Blumberg BM, Giorgi C, Kolakofsky D. N protein of vesicular stomatitis virus selectively encapsidates leader RNA in vitro. Cell. 1983;32:559. doi: 10.1016/0092-8674(83)90475-0. [DOI] [PubMed] [Google Scholar]
- 11.Buonocore L, Blight KJ, Rice CM, et al. Characterization of vesicular stomatitis virus recombinants that express and incorporate high levels of hepatitis C virus glycoproteins. J Virol. 2002;76:6865. doi: 10.1128/JVI.76.14.6865-6872.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calisher CH, Monath TP, Sabattini MS, et al. A newly recognized vesiculovirus, Calchaqui virus, and subtypes of Melao and Maguari viruses from Argentina, with serologic evidence for infections of humans and horses. Am J Trop Med Hyg. 1987;36:114. doi: 10.4269/ajtmh.1987.36.114. [DOI] [PubMed] [Google Scholar]
- 13.Carroll AR, Wagner RR. Role of the membrane (M) protein in endogenous inhibition of in vitro transcription by vesicular stomatitis virus. J Virol. 1979;29:134. doi: 10.1128/jvi.29.1.134-142.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casimiro DR, Chen L, Fu TM, et al. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J Virol. 2003;77:6305. doi: 10.1128/JVI.77.11.6305-6313.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clinton GM, Little SP, Hagen FS, et al. The matrix (M) protein of vesicular stomatitis virus regulates transcription. Cell. 1978;15:1455. doi: 10.1016/0092-8674(78)90069-7. [DOI] [PubMed] [Google Scholar]
- 16.Das T, Pattnaik AK, Takacs AM, et al. Basic amino acid residues at the carboxy-terminal eleven amino acid region of the phosphoprotein (P) are required for transcription but not for replication of vesicular stomatitis virus genome RNA. Virology. 1997;238:103. doi: 10.1006/viro.1997.8823. [DOI] [PubMed] [Google Scholar]
- 17.Dhanda V, Rodrigues FM, Ghosh SN. Isolation of Chandipura virus from sandflies in Aurangabad. Indian J Med Res. 1970;58:179. [PubMed] [Google Scholar]
- 18.Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu Rev Microbiol. 1997;51:151. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- 19.Egan MA, Chong SY, Rose NF, et al. Immunogenicity of attenuated vesicular stomatitis virus vectors expressing HIV type 1 Env and SIV Gag proteins: comparison of intranasal and intramuscular vaccination routes. AIDS Res Hum Retroviruses. 2004;20:989. doi: 10.1089/aid.2004.20.989. [DOI] [PubMed] [Google Scholar]
- 20.Egan MA, Chong SY, Megati S, et al. Priming with plasmid DNAs expressing interleukin-12 and simian immunodeficiency virus gag enhances the immunogenicity and efficacy of an experimental AIDS vaccine based on recombinant vesicular stomatitis virus. AIDS Res Hum Retroviruses. 2005;21:629. doi: 10.1089/aid.2005.21.629. [DOI] [PubMed] [Google Scholar]
- 21.Emerson SU, Yu Y. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J Virol. 1975;15:1348. doi: 10.1128/jvi.15.6.1348-1356.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ezelle HJ, Markovic D, Barber GN. Generation of hepatitis C virus-like particles by use of a recombinant vesicular stomatitis virus vector. J Virol. 2002;76:12325. doi: 10.1128/JVI.76.23.12325-12334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faria PA, Chakraborty P, Levay A, et al. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol Cell. 2005;17:93. doi: 10.1016/j.molcel.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 24.Fields BN, Hawkins K. Human infection with the virus of vesicular stomatitis during an epizootic. New Engl J Med. 1967;277:989. doi: 10.1056/NEJM196711092771901. [DOI] [PubMed] [Google Scholar]
- 25.Finke S, Conzelmann KK. Recombinant rhabdoviruses: vectors for vaccine development and gene therapy. Curr Top Microbiol Immunol. 2005;292:165. doi: 10.1007/3-540-27485-5_8. [DOI] [PubMed] [Google Scholar]
- 26.Flamand A. Genetic study of vesicular stomatitis virus: classification of spontaneous thermosensitive mutants into complementation groups (in French) J Gen Virol. 1970;8:187. doi: 10.1099/0022-1317-8-3-187. [DOI] [PubMed] [Google Scholar]
- 27.Flanagan EB, Zamparo JM, Ball LA, et al. Rearrangement of the genes of vesicular stomatitis virus eliminates clinical disease in the natural host: new strategy for vaccine development. J Virol. 2001;75:6107. doi: 10.1128/JVI.75.13.6107-6114.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flanagan EB, Schoeb TR, Wertz GW. Vesicular stomatitis viruses with rearranged genomes have altered invasiveness and neuropathogenesis in mice. J Virol. 2003;77:5740. doi: 10.1128/JVI.77.10.5740-5748.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frank AH, Appleby A, Seibold HR (1945) Experimental intracerebral infection of horses, cattle, and sheep with the virus of vesicular stomatitis. Am J Vet Res 28
- 30.Fredericksen BL, Whitt MA. Attenuation of recombinant vesicular stomatitis viruses encoding mutant glycoproteins demonstrate a critical role for maintaining a high pH threshold for membrane fusion in viral fitness. Virology. 1998;240:349. doi: 10.1006/viro.1997.8921. [DOI] [PubMed] [Google Scholar]
- 31.Fukushi S, Mizutani T, Saijo M, et al. Vesicular stomatitis virus pseudotyped with severe acute respiratory syndrome coronavirus spike protein. J Gen Virol. 2005;86:2269. doi: 10.1099/vir.0.80955-0. [DOI] [PubMed] [Google Scholar]
- 32.Garbutt M, Liebscher R, Wahl-Jensen V, et al. Properties of replication-competent vesicular stomatitis virus vectors expressing glycoproteins of filoviruses and arenaviruses. J Virol. 2004;78:5458. doi: 10.1128/JVI.78.10.5458-5465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geisbert TW, Jones S, Fritz EA, et al. Development of a new vaccine for the prevention of Lassa fever. PLoS Med. 2005;2:e183. doi: 10.1371/journal.pmed.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haglund K, Forman J, Krausslich HG, et al. Expression of human immunodeficiency virus type 1 Gag protein precursor and envelope proteins from a vesicular stomatitis virus recombinant: high-level production of virus-like particles containing HIV envelope. Virology. 2000;268:112. doi: 10.1006/viro.1999.0120. [DOI] [PubMed] [Google Scholar]
- 35.Haglund K, Leiner I, Kerksiek K, et al. Robust recall and long-term memory T-cell responses induced by prime-boost regimens with heterologous live viral vectors expressing human immunodeficiency virus type 1 Gag and Env proteins. J Virol. 2002;76:7506. doi: 10.1128/JVI.76.15.7506-7517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harty RN, Paragas J, Sudol M, et al. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J Virol. 1999;73:2921. doi: 10.1128/jvi.73.4.2921-2929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holland J, Spindler K, Horodyski F, et al. Rapid evolution of RNA genomes. Science. 1982;215:1577. doi: 10.1126/science.7041255. [DOI] [PubMed] [Google Scholar]
- 38.Huneycutt BS, Plakhov IV, Shusterman Z, et al. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res. 1994;635:81. doi: 10.1016/0006-8993(94)91426-5. [DOI] [PubMed] [Google Scholar]
- 39.Iverson LE, Rose JK. Localized attenuation and discontinuous synthesis during vesicular stomatitis virus transcription. Cell. 1981;23:477. doi: 10.1016/0092-8674(81)90143-4. [DOI] [PubMed] [Google Scholar]
- 40.Jacobs BL, Penhoet EE. Assembly of vesicular stomatitis virus: distribution of the glycoprotein on the surface of infected cells. J Virol. 1982;44:1047. doi: 10.1128/jvi.44.3.1047-1055.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jayakar HR, Whitt MA. Identification of two additional translation products from the matrix (M) gene that contribute to vesicular stomatitis virus cytopathology. J Virol. 2002;76:8011. doi: 10.1128/JVI.76.16.8011-8018.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson KM, Vogel JE, Peralta PH. Clinical and serological response to laboratory-acquired human infection by Indiana type vesicular stomatitis virus (VSV) Am J Trop Med Hyg. 1966;15:244. doi: 10.4269/ajtmh.1966.15.244. [DOI] [PubMed] [Google Scholar]
- 43.Johnson JE, Schnell MJ, Buonocore L, et al. Specific targeting to CD4+ cells of recombinant vesicular stomatitis viruses encoding human immunodeficiency virus envelope proteins. J Virol. 1997;71:5060. doi: 10.1128/jvi.71.7.5060-5068.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones SM, Feldmann H, Stroher U, et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med. 2005;11:786. doi: 10.1038/nm1258. [DOI] [PubMed] [Google Scholar]
- 45.Jonkers AH. The epizootiology of the vesicular stomatitis viruses: a reappraisal. Am J Epidemiol. 1967;86:286. doi: 10.1093/oxfordjournals.aje.a120738. [DOI] [PubMed] [Google Scholar]
- 46.Jonkers AH, Shope RE, Aitken TH, et al. Cocal virus, a new agent in Trinidad related to vesicular stomatitis virus, type Indiana. Am J Vet Res. 1964;25:236. [PubMed] [Google Scholar]
- 47.Jonkers AH, Spence L, Aitken TH, et al. Cocal virus epizootiology in Bush Bush Forest and the Nariva Swamp, Trinidad, W.I.: further studies. Am J Vet Res. 1965;26:758. [PubMed] [Google Scholar]
- 48.Justice PA, Sun W, Li Y, et al. Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J Virol. 1995;69:3156. doi: 10.1128/jvi.69.5.3156-3160.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kahn JS, Schnell MJ, Buonocore L, et al. Recombinant vesicular stomatitis virus expressing respiratory syncytial virus (RSV) glycoproteins: RSV fusion protein can mediate infection and cell fusion. Virology. 1999;254:81. doi: 10.1006/viro.1998.9535. [DOI] [PubMed] [Google Scholar]
- 50.Kahn JS, Roberts A, Weibel C, et al. Replication-competent or attenuated, nonpropagating vesicular stomatitis viruses expressing respiratory syncytial virus (RSV) antigens protect mice against RSV challenge. J Virol. 2001;75:11079. doi: 10.1128/JVI.75.22.11079-11087.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kapadia SU, Rose JK, Lamirande E, et al. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology. 2005;340:174. doi: 10.1016/j.virol.2005.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klas SD, Robison CS, Whitt MA, et al. Adjuvanticity of an IL-12 fusion protein expressed by recombinant deltaG-vesicular stomatitis virus. Cell Immunol. 2002;218:59. doi: 10.1016/s0008-8749(02)00575-0. [DOI] [PubMed] [Google Scholar]
- 53.Klas SD, Lavine CL, Whitt MA, et al. IL-12-assisted immunization against listeria monocytogenes using replication-restricted VSV-based vectors. Vaccine. 2006;24:1451. doi: 10.1016/j.vaccine.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 54.Kopecky SA, Lyles DS. The cell-rounding activity of the vesicular stomatitis virus matrix protein is due to the induction of cell death. J Virol. 2003;77:5524. doi: 10.1128/JVI.77.9.5524-5528.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kreis TE, Lodish HF. Oligomerization is essential for transport of vesicular stomatitis viral glycoprotein to the cell surface. Cell. 1986;46:929. doi: 10.1016/0092-8674(86)90075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kretzschmar E, Buonocore L, Schnell MJ, et al. High-efficiency incorporation of functional influenza virus glycoproteins into recombinant vesicular stomatitis viruses. J Virol. 1997;71:5982. doi: 10.1128/jvi.71.8.5982-5989.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lawson ND, Stillman EA, Whitt MA, et al. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA. 1995;92:4477. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee BH, Yoshimatsu K, Araki K, et al. A pseudotype vesicular stomatitis virus containing Hantaan virus envelope glycoproteins G1 and G2 as an alternative to hantavirus vaccine in mice. Vaccine. 2006;24:2928. doi: 10.1016/j.vaccine.2005.12.040. [DOI] [PubMed] [Google Scholar]
- 59.Leppert M, Rittenhouse L, Perrault J, et al. Plus and minus strand leader RNAs in negative strand virus-infected cells. Cell. 1979;18:735. doi: 10.1016/0092-8674(79)90127-2. [DOI] [PubMed] [Google Scholar]
- 60.Levenbook IS, Pelleu LJ, Elisberg BL, et al. The monkey safety test for neurovirulence of yellow fever vaccines: the utility of quantitative clinical evaluation and histological examination. J Biol Stand. 1987;15:305. doi: 10.1016/s0092-1157(87)80003-3. [DOI] [PubMed] [Google Scholar]
- 61.Lorin C, Mollet L, Delebecque F, et al. A single injection of recombinant measles virus vaccines expressing human immunodeficiency virus (HIV) type 1 clade B envelope glycoproteins induces neutralizing antibodies and cellular immune responses to HIV. J Virol. 2004;78:146. doi: 10.1128/JVI.78.1.146-157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7 (see comment) Proc Natl Acad Sci USA. 2004;101:5598. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maher JA, DeStefano J. The ferret: an animal model to study influenza virus. Lab Anim (NY) 2004;33:50. doi: 10.1038/laban1004-50. [DOI] [PubMed] [Google Scholar]
- 64.Maines TR, Lu XH, Erb SM, et al. Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J Virol. 2005;79:11788. doi: 10.1128/JVI.79.18.11788-11800.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maximova O, Dragunsky E, Taffs R, et al. Monkey neurovirulence test for live mumps vaccine. Biologicals. 1996;24:223. doi: 10.1006/biol.1996.0030. [DOI] [PubMed] [Google Scholar]
- 66.Metsikko K, Simons K. The budding mechanism of spikeless vesicular stomatitis virus particles. EMBO J. 1986;5:1913. doi: 10.1002/j.1460-2075.1986.tb04444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyoshi K, Harter DH, Hsu KC. Neuropathological and immunofluorescence studies of experimental vesicular stomatitis virus encephalitis in mice. J Neuropathol Exp Neurol. 1971;30:266. doi: 10.1097/00005072-197104000-00008. [DOI] [PubMed] [Google Scholar]
- 68.Nathanson N, Horn SD. Neurovirulence tests of type 3 oral poliovirus vaccine manufactured by Lederle Laboratories, 1964–1988. Vaccine. 1992;10:469. doi: 10.1016/0264-410x(92)90396-2. [DOI] [PubMed] [Google Scholar]
- 69.Natuk RJ, Cooper D, Guo M, et al. Recombinant vesicular stomatitis virus vectors expressing herpes simplex virus type 2 gD elicit robust CD4+ Th1 immune responses and are protective in mouse and guinea pig models of vaginal challenge. J Virol. 2006;80:4447. doi: 10.1128/JVI.80.9.4447-4457.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Olitsky PK, Cox HR, Syverton JT. Comparative studies on the viruses of vesicular stomatitis and equine encephalomyelitis. J Exp Med. 1934;59:159. doi: 10.1084/jem.59.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Owens RJ, Rose JK. Cytoplasmic domain requirement for incorporation of a foreign envelope protein into vesicular stomatitis virus. J Virol. 1993;67:360. doi: 10.1128/jvi.67.1.360-365.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palin A, Chattopadhyay A, Park S et al (2006) An optimized vaccine vector based on recombinant vesicular stomatitis virus gives high-level, long-term protection against Yersinia pestis challenge. Vaccine (in press). DOI 10.1016/j.vaccine.2006.08.010 [DOI] [PubMed]
- 73.Pattnaik AK, Hwang L, Li T, et al. Phosphorylation within the amino-terminal acidic domain I of the phosphoprotein of vesicular stomatitis virus is required for transcription but not for replication. J Virol. 1997;71:8167. doi: 10.1128/jvi.71.11.8167-8175.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patton JT, Davis NL, Wertz GW. Cell-free synthesis and assembly of vesicular stomatitis virus nucleocapsids. J Virol. 1983;45:155. doi: 10.1128/jvi.45.1.155-164.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Plakhov IV, Arlund EE, Aoki C, et al. The earliest events in vesicular stomatitis virus infection of the murine olfactory neuroepithelium and entry of the central nervous system. Virology. 1995;209:257. doi: 10.1006/viro.1995.1252. [DOI] [PubMed] [Google Scholar]
- 76.Pringle CR. Genetic characteristics of conditional lethal mutants of vesicular stomatitis virus induced by 5-fluorouracil, 5-azacytidine, and ethyl methane sulfonate. J Virol. 1970;5:559. doi: 10.1128/jvi.5.5.559-567.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pringle CR. The genetics of vesiculoviruses. Arch Virol. 1982;72:1. doi: 10.1007/BF01314447. [DOI] [PubMed] [Google Scholar]
- 78.Ramsburg E, Rose NF, Marx PA, et al. Highly effective control of an AIDS virus challenge in macaques by using vesicular stomatitis virus and modified vaccinia virus Ankara vaccine vectors in a single-boost protocol. J Virol. 2004;78:3930. doi: 10.1128/JVI.78.8.3930-3940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramsburg E, Publicover J, Buonocore L, et al. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J Virol. 2005;79:15043. doi: 10.1128/JVI.79.24.15043-15053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rao TR, Singh KR, Dhanda V, et al. Experimental transmission of Chandipura virus by mosquitoes. Indian J Med Res. 1967;55:1306. [PubMed] [Google Scholar]
- 81.Reiss CS, Plakhov IV, Komatsu T. Viral replication in olfactory receptor neurons and entry into the olfactory bulb and brain. Ann N Y Acad Sci. 1998;30:752. doi: 10.1111/j.1749-6632.1998.tb10655.x. [DOI] [PubMed] [Google Scholar]
- 82.Reuter JD, Vivas-Gonzalez BE, Gomez D, et al. Intranasal vaccination with a recombinant vesicular stomatitis virus expressing cottontail rabbit papillomavirus L1 protein provides complete protection against papillomavirus-induced disease. J Virol. 2002;76:8900. doi: 10.1128/JVI.76.17.8900-8909.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Riedel H, Kondor-Koch C, Garoff H. Cell surface expression of fusogenic vesicular stomatitis virus G protein from cloned cDNA. EMBO J. 1984;3:1477. doi: 10.1002/j.1460-2075.1984.tb01999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roberts A, Kretzschmar E, Perkins AS, et al. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J Virol. 1998;72:4704. doi: 10.1128/jvi.72.6.4704-4711.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roberts A, Buonocore L, Price R, et al. Attenuated vesicular stomatitis viruses as vaccine vectors. J Virol. 1999;73:3723. doi: 10.1128/jvi.73.5.3723-3732.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Robison CS, Whitt MA. The membrane-proximal stem region of vesicular stomatitis virus G protein confers efficient virus assembly. J Virol. 2000;74:2239. doi: 10.1128/jvi.74.5.2239-2246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rose JK, Whitt MA. Rhabdoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields virology. 4. Philadelphia, PA: Lippincott Williams and Wilkins; 2001. p. 1223. [Google Scholar]
- 88.Rose NF, Roberts A, Buonocore L, et al. Glycoprotein exchange vectors based on vesicular stomatitis virus allow effective boosting and generation of neutralizing antibodies to a primary isolate of human immunodeficiency virus type 1. J Virol. 2000;74:10903. doi: 10.1128/jvi.74.23.10903-10910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rose NF, Marx PA, Luckay A, et al. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell. 2001;106:539. doi: 10.1016/s0092-8674(01)00482-2. [DOI] [PubMed] [Google Scholar]
- 90.Rowe T, Cho DS, Bright RA, et al. Neurological manifestations of avian influenza viruses in mammals. Avian Dis. 2003;47:1122. doi: 10.1637/0005-2086-47.s3.1122. [DOI] [PubMed] [Google Scholar]
- 91.Rubin SA, Snoy PJ, Wright KE, et al. The mumps virus neurovirulence safety test in rhesus monkeys: a comparison of mumps virus strains. J Infect Dis. 1999;180:521. doi: 10.1086/314905. [DOI] [PubMed] [Google Scholar]
- 92.Sabin AB, Olitsky PK. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus: I. Effect of age on the invasion of the brain by virus instilled in the nose. J Exp Med. 1937;66:15. doi: 10.1084/jem.66.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Scheiblauer H, Kendal AP, Rott R. Pathogenicity of influenza A/Seal/Mass/1/80 virus mutants for mammalian species. Arch Virol. 1995;140:341. doi: 10.1007/BF01309867. [DOI] [PubMed] [Google Scholar]
- 94.Schlereth B, Rose JK, Buonocore L, et al. Successful vaccine-induced seroconversion by single-dose immunization in the presence of measles virus-specific maternal antibodies. J Virol. 2000;74:4652. doi: 10.1128/jvi.74.10.4652-4657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schnell MJ, Mebatsion T, Conzelmann KK. Infectious rabies viruses from cloned cDNA. EMBO J. 1994;13:4195. doi: 10.1002/j.1460-2075.1994.tb06739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schnell MJ, Buonocore L, Whitt MA, et al. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J Virol. 1996;70:2318. doi: 10.1128/jvi.70.4.2318-2323.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schnell MJ, Johnson JE, Buonocore L, et al. Construction of a novel virus that targets HIV-1-infected cells and controls HIV-1 infection. Cell. 1997;90:849. doi: 10.1016/s0092-8674(00)80350-5. [DOI] [PubMed] [Google Scholar]
- 98.Schnell MJ, Buonocore L, Boritz E, et al. Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J. 1998;17:1289. doi: 10.1093/emboj/17.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sharova OK, Rozina EE, Shteinberg LS, et al. Morphological characterisitcs of the pathological process in the central nervous system of monkeys infected with variants of measles virus strain L-16. Acta Virol. 1979;23:393. [PubMed] [Google Scholar]
- 100.Shinya K, Hatta M, Yamada S, et al. Characterization of a human H5N1 influenza A virus isolated in 2003. J Virol. 2005;79:9926. doi: 10.1128/JVI.79.15.9926-9932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.tenOever BR, Sharma S, Zou W, et al. Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol. 2004;78:10636. doi: 10.1128/JVI.78.19.10636-10649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tesh RB, Johnson KM. Vesicular stomatitis. In: Hubbert WT, McCulloch WF, Schnurrenberger PR, editors. Diseases transmitted from animals to man. 6. Springfield, IL: C. C. Thomas; 1975. p. 897. [Google Scholar]
- 103.Tesh RB, Modi GB. Growth and transovarial transmission of Chandipura virus (Rhabdoviridae: Vesiculovirus) in Phlebotomus papatasi. Am J Trop Med Hyg. 1983;32:621. doi: 10.4269/ajtmh.1983.32.621. [DOI] [PubMed] [Google Scholar]
- 104.Tesh RB, Peralta PH, Johnson KM. Ecologic studies of vesicular stomatitis virus. II. Results of experimental infection in Panamanian wild animals. Am J Epidemiol. 1970;91:216. doi: 10.1093/oxfordjournals.aje.a121130. [DOI] [PubMed] [Google Scholar]
- 105.Tesh RB, Chaniotis BN, Johnson KM. Vesicular stomatitis virus, Indiana serotype: multiplication in and transmission by experimentally infected phlebotomine sandflies (Lutzomyia trapidoi) Am J Epidemiol. 1971;93:491. doi: 10.1093/oxfordjournals.aje.a121284. [DOI] [PubMed] [Google Scholar]
- 106.Tesh RB, Chaniotis BN, Johnson KM. Vesicular stomatitis virus (Indiana serotype): transovarial transmission by phlebotomine sandflies. Science. 1972;175:1477. doi: 10.1126/science.175.4029.1477. [DOI] [PubMed] [Google Scholar]
- 107.Tesh R, Saidi S, Javadian E, et al. Isfahan virus, a new vesiculovirus infecting humans, gerbils, and sandflies in Iran. Am J Trop Med Hyg. 1977;26:299. doi: 10.4269/ajtmh.1977.26.299. [DOI] [PubMed] [Google Scholar]
- 108.Tesh RB, Travassos Da Rosa AP, Travassos Da Rosa JS. Antigenic relationship among rhabdoviruses infecting terrestrial vertebrates. J Gen Virol. 1983;64(Pt 1):169. doi: 10.1099/0022-1317-64-1-169. [DOI] [PubMed] [Google Scholar]
- 109.Tesh RB, Boshell J, Modi GB, et al. Natural infection of humans, animals, and phlebotomine sand flies with the Alagoas serotype of vesicular stomatitis virus in Colombia. Am J Trop Med Hyg. 1987;36:653. doi: 10.4269/ajtmh.1987.36.653. [DOI] [PubMed] [Google Scholar]
- 110.Travassos da Rosa AP, Tesh RB, Travassos da Rosa JF, et al. Carajas and Maraba viruses, two new vesiculoviruses isolated from phlebotomine sand flies in Brazil. Am J Trop Med Hyg. 1984;33:999. doi: 10.4269/ajtmh.1984.33.999. [DOI] [PubMed] [Google Scholar]
- 111.van den Pol AN, Dalton KP, Rose JK. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J Virol. 2002;76:1309. doi: 10.1128/JVI.76.3.1309-1327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Villarreal LP, Breindl M, Holland JJ. Determination of molar ratios of vesicular stomatitis virus induced RNA species in BHK21 cells. Biochemistry. 1976;15:1663. doi: 10.1021/bi00653a012. [DOI] [PubMed] [Google Scholar]
- 113.Wertz GW. Replication of vesicular stomatitis virus defective interfering particle RNA in vitro: transition from synthesis of defective interfering leader RNA to synthesis of full-length defective interfering RNA. J Virol. 1983;46:513. doi: 10.1128/jvi.46.2.513-522.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wertz GW, Perepelitsa VP, Ball LA. Gene rearrangement attenuates expression and lethality of a nonsegmented negative strand RNA virus. Proc Natl Acad Sci USA. 1998;95:3501. doi: 10.1073/pnas.95.7.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Whelan SP, Wertz GW. Regulation of RNA synthesis by the genomic termini of vesicular stomatitis virus: identification of distinct sequences essential for transcription but not replication. J Virol. 1999;73:297. doi: 10.1128/jvi.73.1.297-306.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Whelan SP, Ball LA, Barr JN, et al. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci USA. 1995;92:8388. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xing Z, Lichty BD. Use of recombinant virus-vectored tuberculosis vaccines for respiratory mucosal immunization. Tuberculosis (Edinb) 2006;86:211. doi: 10.1016/j.tube.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 118.Zagouras P, Rose JK. Dynamic equilibrium between vesicular stomatitis virus glycoprotein monomers and trimers in the Golgi and at the cell surface. J Virol. 1993;67:7533. doi: 10.1128/jvi.67.12.7533-7538.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]