

Abstract
The occurrence of microorganisms in water due to contamination is a health risk and control thereof is a necessity. Conventional detection methods may be misleading and do not provide rapid results allowing for immediate action. The quantitative polymerase chain reaction (qPCR) method has proven to be an effective tool to detect and quantify microorganisms in water within a few hours. Quantitative PCR assays have recently been developed for the detection of specific adeno- and polyomaviruses, bacteria and protozoa in different water sources. The technique is highly sensitive and able to detect low numbers of microorganisms. Quantitative PCR can be applied for microbial source tracking in water sources, to determine the efficiency of water and wastewater treatment plants and act as a tool for risk assessment. Different qPCR assays exist depending on whether an internal control is used or whether measurements are taken at the end of the PCR reaction (end-point qPCR) or in the exponential phase (real-time qPCR). Fluorescent probes are used in the PCR reaction to hybridise within the target sequence to generate a signal and, together with specialised systems, quantify the amount of PCR product. Quantitative reverse transcription polymerase chain reaction (q-RT-PCR) is a more sensitive technique that detects low copy number RNA and can be applied to detect, e.g. enteric viruses and viable microorganisms in water, and measure specific gene expression. There is, however, a need to standardise qPCR protocols if this technique is to be used as an analytical diagnostic tool for routine monitoring. This review focuses on the application of qPCR in the detection of microorganisms in water.
Keywords: Water, Waste/sludge, Quality assurance/control, PCR
Introduction
The quality of drinking water derived from water treatment plants or other water sources is constantly measured to prevent waterborne outbreaks caused by bacteria, viruses and protozoa. Risk assessments are therefore implemented to determine point source or non-point source pollution by sewage, farm and/or urban run-off [1].
Conventional methods used to identify microorganisms are laborious and time-consuming and certain microorganisms are not culturable on bacteriological media [2] or do not exist in numbers high enough in water to allow detection [3]. Monitoring of microorganisms in water therefore involves the use of different techniques including the use of indicator and index bacteria such as faecal coliforms [4]. Examples of faecal indicators include Escherichia coli, Enterococci [5], Bacteroidetes spp. [4], Candida spp. [6], adenoviruses and polyomaviruses [7]. However, the reliability of these indicators is questioned. The quantity of indicator microorganisms may differ from the actual number of pathogens [8], infectious doses between the microorganisms may vary [8] and in some cases the microorganisms are viable but nonculturable [9]. Microbial source tracking indicators are for example useful for pinpointing pollution sources of groundwater, used as a drinking water supply.
The introduction of molecular assays has significantly improved and simplified the detection and identification of microorganisms in the environment [7]. Since 1985, the polymerase chain reaction (PCR) is the most widely used method for the amplification of nucleic acid and has played an important role in the characterization of microorganisms [10].
Research has shown that the quantitative polymerase chain reaction (qPCR) method is reliable and adequate for the quantitative measurement of source-specific genetic markers of microorganisms in water [11]. Several attempts have been made to determine the quantity of nucleic acids on the basis of PCR reactions [11] such as qPCR.
The qPCR technique involves the simultaneous amplification, detection and quantification of a specific nucleic acid target in a biological sample [12]. This is accomplished through the monitoring of fluorescently labelled PCR products [13, 14]. The methods of quantitation of the amplicons and type of standard used in the assay determine the strategy for qPCR. Different approaches include the use of internal and external standards for competitive or non-competitive assays, respectively [15]. Two methods are used for qPCR, namely end-point PCR and real-time PCR. During end-point PCR a measurement is taken after completion of the entire PCR reaction to calculate the amount of template present prior to PCR. Real-time PCR takes measurements at the exponential phase of the PCR and is a more sensitive and reproducible method [16]. Different protocols exist for real-time PCR, including relative qPCR and absolute measurements, and small differences in variables may influence the final accumulation and quality of PCR products [17].
Due to the fact that the direct amplification of RNA during a PCR reaction is impossible, the enzyme reverse transcriptase (RT) is required to catalyse the synthesis of a complementary DNA (cDNA) copy from the RNA template. This single-stranded cDNA serves as the target during PCR amplification. This technique is referred to as RT-PCR and is used for the detection of enteric viruses containing RNA genomes [18, 19]. The quantitative reverse transcription polymerase chain reaction (q-RT-PCR) has been used to detect and quantify enteric viruses in environmental water sources. This method can also be used to measure low levels of messenger RNA (mRNA) expression in samples of a small volume, for the comparison of mRNA levels in different samples, characterization of patterns in mRNA expression and discrimination between closely related mRNAs [20]. Messenger RNA is only present in viable organisms and can therefore be used to discriminate between viable and non-viable organisms [21].
Quantitative PCR assays are applied to determine the efficiency of water treatment plants [22] and wastewater treatment plants [23], the quality of drinking water from resources [24] and the safety of water from recreational beaches [25]. The use of qPCR in water analysis allows quick risk assessment, and immediate corrective action to be taken.
In this article, qPCR methods and their use in water analysis will be reviewed.
Waterborne pathogens
Viruses
Enteric viruses are important indicators used for routine monitoring of water quality because these viruses replicate in the human intestine and are secreted in large numbers in human faecal matter. Waterborne enteric viruses include adenoviruses [26], astroviruses [27], enteroviruses [27], hepatitis viruses [27], noroviruses [27], polyomaviruses [28] and rotaviruses [27]. Adenoviruses and polyomaviruses are double-stranded DNA viruses while astroviruses, hepatitis viruses, noroviruses and rotaviruses are single-stranded RNA viruses [26, 28]. Enteric viruses have been detected in different water sources including marine, river, ground, drinking, recreational and wastewater [29–34].
Human adenovirus has been recommended as an indicator virus for human contamination in water due to the high prevalence of this virus in different contaminated water sources in Europe [7, 35–37]. This virus was also observed to be present in wastewater in higher densities than other enteric viruses [38–40] and has been detected more often in environmental water sources than other enteric viruses [41, 42]. Recently it was shown that human adenoviruses can persist, for extensive time periods, in natural environmental conditions [43, 44]. The fact that adenoviruses are double-stranded DNA viruses is related to their prolonged persistence in environmental water sources compared to single-stranded RNA viruses [45, 46]. Polyomaviruses are also double-stranded DNA viruses and have been investigated as a possible viral indicator for monitoring water quality [47, 48].
Astroviruses, adenoviruses, noroviruses and rotaviruses are agents of gastroenteritis and infections typically result in symptoms such as vomiting and diarrhoea. Ineffective treatment of infections caused by astroviruses could lead to life-threatening dehydration [49, 50]. Enterovirus infections are asymptomatic with approximately 1 % presenting clinical symptoms. Depending on the serotype, clinical symptoms can range from flu-like symptoms to diseases that affect the central nervous system [51]. Young children, the elderly and immunocompromised individuals are more susceptible to viral infections. Other factors that play a role in viral disease progression are the route of infection and the infective dose of the viral agent [52].
Bacteria
Traditional indicators for faecal contamination and assessment of the potential presence of human pathogens in different water sources include total coliforms, E. coli and enterococci [53, 54]. A disadvantage of the use of these indicators is the inability to discriminate between different sources of faecal contamination [55]. Host-associated Bacteroidales spp. have been identified as a viable alternative faecal indicator to be used during microbial source tracking (MST). Bacteroidales spp. are present in high numbers in the faeces of both humans and animals [53, 56–58] and have poor survival rates outside the host due to their anaerobic physiology [59, 60].
A range of bacterial pathogens are associated with waterborne diseases. The list includes Salmonella typhi, S. paratyphi, other Salmonella spp., Shigella spp., Vibrio cholerae, E. coli spp., Yersinia enterocolitica, Campylobacter jejuni, Legionella jejuni, L. pneumophila, Leptospira spp., mycobacteria spp. and opportunistic bacteria [61]. Typhoid and paratyphoid fever caused by S. typhi and S. paratyphi respectively are responsible for 17 million cases annually. Humans can be carriers of Salmonella spp. even after recovery from typhoid fever because small numbers of the bacteria can persist [62]. Epidemic cholera is caused by the serogroups V. cholerae O1 and O39 that produce the cholera toxin. Cholera is associated with watery diarrhoea and if left untreated could lead to rapid dehydration and death in 50–70 % of the cases [63]. C. jejuni, C. coli, pathogenic and enterohaemorrhagic E. coli strains, and Y. enterocolitica are all agents for diarrhoea [61]. Legionella spp. are opportunistic human pathogens that proliferate in warm waters which support the growth of certain serogroups. These serogroups, once aerosolised, can be inhaled by humans resulting in subsequent legionnaires’ disease and Pontaic fever [64].
Protozoa
Enteric pathogenic protozoa species have been associated with the outbreak of waterborne infections and may be present in water due to direct or indirect contamination with human or animal faecal matter. Protozoa species have several lifecycles, of which the ingestion of either the cysts (Entamoeba histolytica, Giardia duodenalis and Balantidium coli) or oocysts (Cryptosporidium spp., Sarcocystis spp., Toxoplasma gondii and Cyclospora spp.) are infective to humans [65]. Cysts and oocysts are highly resistant to chlorination [66]. Symptoms of protozoan infections include diarrhoea (Cryptosporidium spp., G. duodenalis, B. coli and Cyclospora spp.), dysentery (E. histolytica, B. coli) and fever (T. gondii).
At least 325 outbreaks of protozoan waterborne diseases have been reported, with the majority recorded in the USA and Europe [65]. G. duodenalis and Cryptosporidium spp. were responsible for the majority of protozoan waterborne outbreaks and therefore are used as indicators for protozoan contamination [65]. E. histolytica has infected 80 % of children over a 4-year period in Dhaka Bangladesh with a 53 % reinfection rate. This protozoon is the causative agent for amoebiasis (amoebic dysentery) [67]. It is speculated that the few cases of protozoan disease outbreaks in the developing world are under-reported [68].
Quantitative PCR assays
Real-time PCR
Real-time qPCR is used for gene expression quantification [69], expression profiling [70], single nucleotide polymorphism analysis [71] and allele discrimination [72], validation of microarray data [73], genetically modified organisms (GMO) testing [74], monitoring of viral load and other pathogen detection applications [75]. Different protocols exist for real-time PCR including relative qPCR and absolute measurements.
Relative quantitative PCR
A comparison of the differences of nucleic acid targets in different samples can be performed by relative qPCR. Temporal and functional variations of mRNA molecules are usually measured [15]. However, in cultured cells, the quantity of total RNA produced is determined by comparing with an internal control for example expression of a reference gene or with a calibrator sample [12]. Reference genes include endogenous [76] or exogenous [77] mRNA targets that are assayed separately or together with the unknown target followed by the evaluation of the final ratio [78]. The two genes should be analysed in the same PCR assay to improve the reliability of the method [12]. The calibration sample is used to evaluate a biological effect in terms of per cent variation in comparison to a basal condition. The main approach is to determine the changes in gene expression by varying the experimental conditions in in vitro studies [12]. In this case the difficult process to develop accurate RNA standards is avoided by using a comparative quantification method. The method is based on the fact that the difference in threshold cycles between the target gene and the housekeeping gene is proportional to the relative gene expression level of the target gene [73].
Relative qPCR is also used for DNA measurements where two genes are co-amplified with two primer pairs to allow measurement of their ratio. The reciprocal variation is then determined by comparing with one or more control genes [12]. Relative qPCR is, however, not a sufficient method to quantify the amount or concentration of nucleic acid targets as only estimated values are determined [12].
Absolute/competitive qPCR
Absolute qPCR, also known as competitive qPCR, is used for the quantitation of a gene in DNA measurements by comparing the levels observed in reference materials containing a known number of target copies of the genes [79]. The reference material has the same prime recognition sites as the target gene and is included in the sample as a competitor (DNA or RNA) [80]. Conventional procedures are used to separate the PCR amplicons and the signal of the specific target is then compared to the known concentration of the internal standard. The most accurate procedure is to add a known amount of the competitor to each assay tube to ensure the amplification of the two species at the same rate. After amplification, the nucleic acid target and competitor are separated analytically to evaluate the ratio between the two species. This final ratio reveals the ratio between the target and competitor in the initial sample [17].
DNA and RNA competitors are synthetically constructed for DNA and mRNA measurements in qPCR [81]. In comparison to RNA competitors, DNA competitors are simpler to construct, easier to handle, more stable and provide more accurate quantification results [80]. Different methods exist to synthesise competitors to distinguish them from the target template. Examples are the inclusion of a new restriction site in the native sequence [80] and insertion of a slightly modified nucleotide sequence [82]. However, a simpler method is to modify the size of the competitor in the range of 10–15 % by introducing a short deletion or insertion. The amplification rate of the PCR will thereby not be modified [12]. In general the competitor is then cloned into a plasmid vector, followed by transcription to attain copious amounts of the specific transcript and then finally the determination of the competitor concentration by a precise measurement [12].
Calibration standards
To establish a PCR assay, DNA isolated from the pure culture of the tested microorganisms should be serially diluted so that a calibration curve can be created by the thermocycler instrument. The assay should be repeated up to 10 times to ensure the reproducibility of the curve and thereby to extract highly specific data [83]. A reference DNA sequence is added both to the DNA from the unknown test sample and the calibration sample. The relative quantity of DNA sequences of the two samples can therefore be determined [84]. The cycle threshold (C
T) is determined automatically by the instrument after the threshold fluorescence value is manually set. For every assay the difference in C
T of the reference sequence assay and that of the target sequence assay is determined. The difference in the mean values of the calibration tests and the calibration samples are then calculated (∆∆CT). The amplification efficiency (E) of the target assay can then be determined by the formula 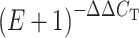 . The estimated number of target organism cells in the test sample is calculated by multiplying these ratios by the known number of target organism cells [25].
. The estimated number of target organism cells in the test sample is calculated by multiplying these ratios by the known number of target organism cells [25].
When a microorganism cannot be cultivated, plasmid DNA standards are preferably used. They are also designed to function as internal amplification controls in multiplex assays to monitor for amplification inhibitors [85]. The advantages of using plasmid preparations are that the standards are of high quality, are pure and can be converted to the number of copies of target DNA. The assumption is made that the amplification efficiency of plasmid and genomic DNA is similar. However, a few strategies have been used to treat the genomic DNA e.g. cocktail of restriction enzymes or by ultrasonification so that the two types of DNA can be equilibrated [86]. The disadvantage of using plasmid DNA is that environmental samples are fully extracted and purified leading to inconsistent DNA recovery due to variance in the replicate extracts and unauthentic results [87].
Multiplex real-time PCR
Multiplex real-time PCR is used to screen large numbers of environmental samples and to detect and discriminate different bacterial strains in a single assay [88, 89]. The advantage over monoplex qPCR is the amplification of several DNA targets in a single reaction that subsequently reduces cost of assays, time and labour [89]. The disadvantages include the selection of primer pairs that must function in the same reaction conditions, the occurrence of primer dimer formation between primer pairs leading to poor sensitivity and the preferential amplification of certain targets that can take place [90]. The assay can only be successful if the concentrations of primers, PCR buffer, magnesium chloride vs. deoxynucleotide, cycling temperatures and amount of DNA template and Taq DNA polymerase are correct [90].
Multiplex PCR assays have been used to detect single strains of pathogens and multiple pathogens in water [91–95]. The following microorganisms were detected in water samples using multiplex real-time PCR: E. coli O157:H7 [88], Aeromonas spp., Salmonella spp., Shigella spp., Vibrio cholerae, V. parahaemolyticus and Y. enterocolitica in a single assay [96]. V. cholerae, Calicivirus and Aureococcus anophagefferens have also been simultaneously detected in a single multiplex PCR assay [97]. These studies found that in combination with a real-time SYBR Green PCR assay, it was 10 times more sensitive and less time consuming than an endpoint PCR protocol.
Detection and measurement of PCR products
Different methods exist to separate and identify PCR products such as the use of agarose or polyacrylamide gels, fluorescence measured by image analysis systems [98], radioactively or fluorescence-labelled dNTPs [99] or oligonucleotides [100] and fluorescently labeled primers [101]. Due to the complexity of these approaches, commercial methods have been developed to improve the convenience of amplification, detection and measurement of PCR products. These methods are based on electrochemiluminescence (QPCR System 5000™, Perkin-Elmer, Foster City, CA, USA) [102] or the combination of paramagnetic microparticles and the avidin–horseradish peroxidase system (COBAS AMPLICOR™, Roche, Basel, Switzerland) [103]. Examples of real-time equipment are the Model 7700 and Model 5700 sequence detectors (PE Applied Biosystems, Foster City, CA, USA), LightCycles (Roche, Basel, Switzerland), Sentinel (Stratagene, La Jolla, CA, USA) and Rotorgene (Corbett Research, Mortlake, Australia) [104–106].
Fluorogenic and fluorescent probes
Fluorogenic probes are based on fluorescent resonance energy transfer (FRET) systems including TaqMan, molecular beacons and SYBR Green I, a dsDNA-binding dye [104–106]. Fluorogenic probes hybridise within the target sequence to generate a signal that accumulates during PCR cycling proportional to the concentration of amplification products. The signal is associated with the amplified target that in turn quantifies the amount of PCR products [107]. Fluorescence can be measured at the end of PCR (end-point fluorescence) or measured during the amplification phase (real-time PCR). In real-time PCR quantification takes place early, in the exponential amplification phase of the reaction. This close-tube system requires no post-treatment of the PCR products that reduces the changes of PCR contamination [14]. The real-time PCR method is therefore a more accurate and less time-consuming method than end-point PCR.
TaqMan principle
The 5′ nuclease activity of Taq polymerase is used to cleave a non-extendable oligonucleotide hybridisation probe during the extension phase of PCR [12, 13]. Dual-labeled fluorogenic hybridisation probes are used, including a reporter dye i.e. FAM (6-carboxyfluorescein) covalently linked to the 5′ end whose emission spectra is quenched by a second dye i.e. TAMRA (6-carboxytetramethylrhodamine), covalently linked to the 3′ end. During a PCR cycle, the probe specifically hybridises to the corresponding template, cleaves via the 5′ to 3′ exonuclease activity of Taq DNA polymerase and subsequently increases the FAM fluorescent emission [108]. The increase in fluorescence is proportional to the amount of specific PCR product as the exonuclease activity of Taq polymerase acts only if the fluorogenic probe is annealed to the target [12] (Fig. 1). The TaqMan assay can be applied for end-point and real-time measurements of PCR products. End-point measurements are only for qualitative applications i.e. presence or absence of a nucleic acid target. In real-time PCR, the fluorescence spectra are continuously measured during PCR amplification by a sequence detector [12, 13].
Fig. 1.
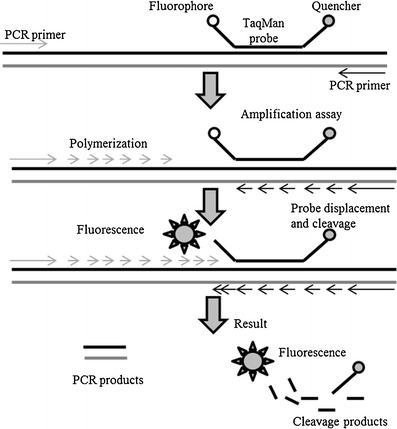
Mechanism of the TaqMan probe. The probes rely on the 5′–3′ nuclease activity of Taq DNA polymerase to cleave a dual-labeled probe during hybridisation to the complementary target sequence (adapted from [213])
SYBR Green I
SYBR Green I Dye is a non-specific DNA-binding fluorophore with the advantage of being used in straightforward and low-cost assays [109] (Fig. 2). Although it is easy to use it binds non-specifically to any dsDNA and makes the quantification of low copy numbers difficult [110]. It prevents the need for target-specific fluorescent probes and the same PCR master mix can be used for many genes of interest [16]. However, the disadvantage of using SYBR Green I is the occurrence of false positives. It is important to carefully optimise PCR conditions and the design of the primers and to prevent contamination from genomic DNA [16].
Fig. 2.
SYBR® Green I detection mechanism. SYBR® Green I is 1,000-fold more fluorescent in the bound state (green star) than in the unbound state (blue circle). The fluorescent signal increases proportionately as the PCR amplification increases (adapted from [214])
Molecular beacons
These structures resemble stem-loop hairpins as they consist of a loop that is complementary to a target nucleic acid and a stem that forms with the annealing of complementary termini [109]. The reporter fluorophore is on one end of the stem and the quencher is on the other. Molecular beacons (MB) fluoresce during the denaturation step when it is in a random-coil configuration and binds to the target amplicons at the annealing temperature [109]. The hairpin is subsequently opened out that separates the fluorophore and quencher resulting in fluorescence. They should only be used in cases where highly conserved regions are known as they have high specificity to recognise nucleotide sequence mismatches [73] (Fig. 3).
Fig. 3.
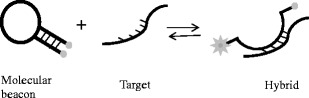
Mechanism of molecular beacon chemistry. The molecular beacon includes a hairpin loop structure, the loop being complementary to a target sequence and the stem formed by the addition of internal complementary sequences. The molecular beacon hybridises to the target and the fluorophore and quencher are far enough apart to allow fluorescence to be detected (adapted from [213])
Scorpions probe chemistry
Scorpions probe chemistry is similar to MB; however, the hairpin structure is incorporated onto one of the primers. Complementary stem sequences are on the 5′ and 3′ ends of the probe with the fluorophore in the immediacy of the quencher [71]. After amplification and incorporation of the hairpin probe, the loop sequence of the resultant amplicon is complementary to the probe. During each cycle of denaturation and annealing, the probe is free to bind to the complementary sequence, leading to the separation of the fluorophore and quencher and resulting in an increase in fluorescence. The PCR stopper in the hairpin prevents the stem-loop structure from being copied during PCR by extension from the other primer [71].
The mechanism of Scorpions is an intramolecular reaction which is more favourable than the bimolecular probing of TaqMan probes [108] and MBs [105] by resulting in a higher fluorescence signal. PCR cycling is rapid because it does not depend on enzymatic cleavage such as in TaqMan probes [111]. The disadvantage of the use of Scorpions over MBs is the challenge to design and optimise the probe structure.
Use of qPCR for the detection and quantification of microorganisms in water
Depending on the source of the water sample and the type of microorganism, certain pretreatment procedures are required for the detection of microorganisms in the sample by qPCR. The number of microorganisms to be monitored in potable and surface waters may be below the detection limit of qPCR. Therefore the microorganisms are normally concentrated by membrane filtration to a detectable number [35]. Tangential-flow, hollow-fibre ultrafiltration has recently showed promising results in concentrating pathogens in one process [112, 113]. Viruses in water samples from wastewater treatments plants have been concentrated through centrifugation steps before the re-dissolved pellets are filtered [23]. Viruses in water samples can be extracted by using glass wool columns [7], pre-acidified glass wool columns [114], ultrafiltration cartridges [115], electropositive cartridges [47] and ultracentrifugation [35]. The first two methods are the most cost-effective with acceptable recoveries of viruses, whereas electropositive cartridges have higher cost and cannot be used for turbid water samples and ultracentrifugation is only applicable for small volumes of heavily polluted water [7]. DNA can be extracted by a variety of DNA kits in combination with bead beating depending on the tested microorganism [4, 24]. Cell lysates can also successfully be used with the same reproducibility of purified DNA and involves less labour [11]. The genomic segment(s) to be amplified is (are) then selected and the optical tubes containing the reaction mixtures are subjected to thermal cycles that vary between 1 h and 15 min to 2 h and 45 min [6, 22, 24, 69, 116]. The amplified genomic segment(s) is (are) then detected and quantified by using specific protocols [36] (Tables 1 and 2). To determine the presence of inhibitors, the tested water sample is autoclaved and a pure culture of the test organism is cultured and inoculated into the water sample [117].
Table 1.
Detection of microorganisms in sewage by qPCR assays
| Microorganisms | Country | Type of assay | Gene/genome amplified | Concentration | % Positive samples | Reference |
|---|---|---|---|---|---|---|
| Adenovirus | Spain | AmpliTaq | Hexon gene | 4–7 GC logs/100 mL raw sewage | 100 % | [35] |
| 3 GC logs/100 mL sewage | 100 % | |||||
| JC polyomavirus (JCPyV) | Brazil | Multiplex qPCR | 4–7 GC logs/100 mL in raw sewage and 4–5 GC logs/100 mL in secondary effluent | 96 % for raw sewage and 39 % for secondary effluent | [125] | |
| Spain | AmpliTaq | Regions of early and late viral genes | 5 GC logs/100 mL | 100 % | [35] | |
| Hepatitis A virus | Brazil | TaqMan | 5′ noncoding region of genome | 1–3 GC logs/mL in raw sewage | 58 % in raw sewage | [205] |
| Spain | 4 GC logs/100 mL | [128] | ||||
| Astrovirus | France | TaqMan | 3′ end consensus region of genome | 5–7 GC logs/100 mL | 100 % | [129] |
| Enterovirus | France | RT-qPCR | 5′ noncoding region of genome | 4–5 GC logs/100 mL | [130] | |
| Norovirus | England | SYBR Green | RNA polymerase | 6 GC logs/100 mL | [131] | |
| Brazil | Superscript RT-qPCR | ORF1/ORF2 junction of genome | 2–3 GC logs/L | [206] | ||
| Arcobacter sp. | Spain | SYBR Green | 2–6 cell logs/100 mL | [207] | ||
| Giardia lamblia | France | TaqMan | Elongation factor 1A (efa1a) and triose phosphate isomerase (tpi) | 2–3 log cysts/L | 100 % | [126] |
| Giardia duodenalis | France | TaqMan | Triose phosphate isomerase (tpi) | 670–2,900 cysts/L for assemblage A; 620–1,900 cysts/L for assemblage B (Nancy treatment plant) | 92 % for assemblage A; 42 % for assemblage B (Nancy treatment plant) | [116] |
| Escherichia coli | Canada | TaqMan | Glucuronidase (uidA) | 7 GC logs/100 mL in raw sewage and 5 GC logs/100 mL in effluent | [8] | |
| USA | LightCycler | Glucuronidase (uidA) | 6 CE logs/100 mL in raw sewage and primary effluent and 3 CE logs/100 mL in secondary and 2 CE logs/100 mL tertiary effluent | [127] | ||
| France | TaqMan | Βeta-galactosidase (lacZ) | 7 target gene copies logs/mL in raw sewage and 2–3 target gene copies logs/mL in treated sewage | 100 % for both raw and treated sewage | [23] | |
| Listeria monocytogenes | Canada | TaqMan | Invasion associated protein p60 (iap) | 3 GC logs/100 mL in raw sewage and absent in effluent | [8] | |
| Clostridium perfringens | Canada | TaqMan | Alpha toxin (plc) | 5 GC logs/100 mL in raw sewage and 3 GC logs/100 mL in effluent | [8] | |
| France | TaqMan | Phospholipase C (plc) | 2–3 target gene copies logs/mL in raw sewage and 1–2 target gene copies logs/mL in treated sewage | 100 % for raw sewage and 100 % for treated sewage | [23] | |
| Pseudomonas aeruginosa | Canada | TaqMan | Toxin A synthesis regulating gene (regA) | 4 GC logs/100 mL in raw sewage and 2 GC logs/100 mL in effluent | [8] | |
| Klebsiella pneumoniae | Canada | TaqMan | Outer membrane phosphate porin (phoE) | 6 GC logs/100 mL in raw sewage and 3 GC logs/100 mL in effluent | [8] | |
| Enterococcus faecalis | Canada | TaqMan | Heat-shock protein (groES) | 4 GC logs/100 mL in raw sewage and 3 GC logs/100 mL in effluent | [8] | |
| USA | Lightcycler | 23 rRNA | 6 CE logs/100 mL in raw sewage and primary effluent and 4 CE logs/100 mL in secondary and 3 CE in tertiary effluent | [127] | ||
| Salmonella spp. | France | TaqMan | Invasion protein (invA) | 2–3 target gene copies logs/mL in raw sewage and 2 target gene copies logs/mL in treated sewage | 100 % for raw sewage and 16.67 % for treated sewage | [23] |
| Helicobacter pylori | USA | RT-PCR; SYBR Green | vacA gene | 2–28 cells/mL | 84 % | [22] |
| Campylobacter jejuni | France | TaqMan | VS1 (not described) | 2–3 target gene copies logs/mL in raw sewage and 0 target gene copies logs/mL in treated sewage | 83.33 % for raw sewage and 0 % for treated sewage | [23] |
Results are expressed as genome copy logs (GC logs) or cell logs
Table 2.
Detection of microorganisms in surface water by qPCR assays
| Microorganisms | Country | Type of surface water | Type of assay | Gene/genome amplified | Concentration | % Positive samples | Reference |
|---|---|---|---|---|---|---|---|
| Mycobacterium avium subsp. paratuberculosis | USA | Drinking water | TaqMan | IS900 gene | 82 % < than 10,00 target copies/L and 68 % < 500 target copies/L | 81 % for first-pull water and 88 % for standard water | [208] |
| Salmonella spp. | Australia | Rainwater | SYBR Green | InvA gene | 2 GC logs/100 mL | 3 % | [74] |
| Campylobacter jejuni | Australia | Rainwater | SYBR Green | 16S rRNA | 1 GC logs/100 mL | 25 % | [74] |
| JC polyomavirus (JCPyV) | Spain | River water | Taqman | Regions of early and late viral genes | 0–3 GC logs/L | 90 % | [7] |
| Japan | River water | AmpliTaq | Regions of early and late viral genes | 2–3 logs/L for JCPyV | 11 % | [48] | |
| Adenovirus | Spain | Seawater | Hexon gene | 1–3 GC logs/L | [209] | ||
| Japan | River water | AmpliTaq | Fiber gene | 3–5 GC logs/L | 61 % | [48] | |
| Spain | River water | SYBR Green | Ad hexon gene | 1–4 GC logs/L | 90 % | [7] | |
| Schistosoma japonicum cercariae | China | Surface water | TaqMan | Deoxyribodipyrimidine photolyase (PL) gene | 500 cercariae | 6.48 % | [152] |
| Helicobacter pylori | USA | Groundwater | RT-PCR; SYBR Green | vacA gene | [22] | ||
| Bacteroidetes | Northern Calcareous Alps | Surface water | TaqMan | 16S rRNA | 4.1 × 109 marker equivalents/g | 100 % | [4] |
| Enterococcus spp. | Racine, WI, USA | Lake water | TaqMan | rRNA subunit | 3 GC logs/100 mL | [210] | |
| Lake Michigan | Lake water | TaqMan | rRNA subunit | 27 cells per extract | 98 % and 97 % | [25] | |
| Lake Erie | |||||||
| Candida spp. | Ohio, Lake water, USA | Drinking and surface water | TaqMan | Nuclear large subunit ribosomal gene | [6] | ||
| E. coli spp. | USA | Surface water | Triplex qPCR, minor groove binding probes | Eae, stx1 and stx2 genes | 3–4 cells/L | 100 % | [119] |
| China | Surface water | SYBR Green | 16s rRNA | Low level river pollution 1.8 GC logs/100 mL | [211] | ||
| High level river pollution 3.7 GC logs/100 mL | |||||||
| 2 urban lakes 2.7–3.3 GC logs/100 mL | |||||||
| Vibrio spp. | USA | Seawater | TaqMan | Hemolysin genes (vvhA and vvhB) | 2 and 91 cells/100 mL | [212] | |
| Cryptosporidium spp. | France | River water | TaqMan | Genomic DNA | 8 Oocyts | [135] | |
| Japan | River water | Quenching probe | 18S rRNA | 1 Oocyst | [136] |
Results are expressed as genome copy logs (GC logs) or cell logs
The risk of waterborne disease outbreaks due to contamination of surface water from sewage or run-off from farms can be decreased or prevented by a rapid and sensitive method for detecting pathogens [52]. Studies have confirmed that qPCR is a high precision and sensitive method to detect pathogens in water and that it could be used to evaluate and control water quality and the efficiency of water treatment plants (Table 2).
Adenoviruses and polyomaviruses are transmitted by the faecal-oral route and can therefore act as indicators for water quality. Quantitative PCR was used to analyse the levels of John Cunningham (JC) polyomaviruses and human adenoviruses in three drinking-water treatment plants at several control points over a period of 1 year. The results revealed that qPCR could be used to determine the efficiency of the water treatment plants to remove viruses, to identify Hazard Analysis and Critical Control Points (HACCP) and to determine the quality of the water [7].
The detection and quantification of Candida spp. have been suggested as alternative and potentially improved indicators for faecal contamination. The population ratio of Enterococcus and Candida spp. can serve as an early warning system for faecal contamination over time to indicate the need to change supply of water. Drug resistance in yeasts is increasing and it may be important to monitor the changes in yeast population of faecal origin regularly [6]. Brinkman et al. [6] developed a qPCR method to accurately identify six Candida spp. in different water sources. Untreated effluent and stormwater run-off into the ocean increases the risk of pathogenic Candida spp. on beaches. Quantitative PCR was used to analyse these water samples on the same day to eliminate the possible risk of infection [6].
Knowing the source of microbial contamination is useful for eliminating disease risks. Recently, qPCR has been used for MST. High numbers of Bacteriodetes are found in faeces and serve as a potential faecal indicator. Reischer et al. [4, 24] developed a qPCR assay to detect human-specific and ruminant-specific Bacteriodetes markers (BacH and BacR) in spring water in alpine karstic regions. The BacH assay could detect human faeces as low as 100 pg per volume of water. The detection limit of the BacR assay was 1.7 × 10−9 g per analysed filter. The assay met the requirements for MST in the alpine karstic spring water and was used to pinpoint the source of pollution. The use of qPCR assays can therefore assist in determining critical control points from source to tap to ensure safe drinking water. It is also a tool for quantitative microbial risk assessment and forms part of the World Health Organization (WHO) water safety plan recommendations [24].
Advantages/potential of using qPCR in detection of microorganisms in water
Conventional microbiological analysis requires between 18 and 24 h before the results are analysed. The advantage of molecular techniques is that the results are obtained within a shorter time period. The USA is therefore considering the application of qPCR for the rapid identification of indicator bacteria, such as faecal bacteria, as a national water quality metric [118]. Traditional methods used for the detection of E. coli O157:H7, a bacterial pathogen in water, takes about 3 to 5 days. An alternative method is the use of PCR assays although other pathogenic or non-pathogenic E. coli strains are also detected. A method is therefore required that would ideally be rapid and specific for the viable and stressed pathogenic isolates of E. coli O157:H7. Sen et al. [119] developed a culture-qPCR assay to detect stressed E. coli O157:H7 in source and finished drinking water. The assay takes 24 h from collection to detection and entails an enrichment step to allow growth of stressed cells followed by two triplex qPCR assays.
The US Environmental Protection Agency (EPA) was the first to determine the relationship between illness rates in swimmers and faecal indicator concentrations by using qPCR as one of the methods. Quantitative PCR was compared to EPA Method 1600 membrane filter (MF) analysis to measure the faecal pollution indicator genus Enterococcus at two recreational beaches during the summer of 2003 [25]. The qPCR method showed similar results and could therefore replace the MF method [25]. These results were confirmed in a study that investigated the relationship between qPCR and culturing techniques in the detection of Enterococcus spp. in surface water collected from 37 sites [120]. A linear relationship between the two techniques was observed at high levels of contamination [120]. The reproducibility of qPCR in the detection of the faecal bacteria, Enterococcus and Bacteroidales, was compared between eight different facilities, including the US EPA [118]. Only small variations in results between the laboratories were observed [118]. A qPCR assay for the rapid identification of Bacteroides fragilis has also been developed to indicate faecal pollution in recreational waters [121]. A higher relative error in replicate qPCR assays was observed than during replicate culturing techniques. This emphasizes the need to further optimise qPCR assays for routine analysis of environmental samples, taking into account sample variability, different pollution sources and environmental factors [120]. The relationship between gastrointestinal illnesses among swimmers and the presence of Enterococcus spp. in surface water has been well established with qPCR in fresh [122, 123] and marine water beaches [124] and can assist authorities in preventing potential disease outbreaks in public recreational areas.
Certain microbial pathogens like viruses are difficult to detect with conventional culturing techniques and are either not detected in environmental samples or their detection rates are too low. In this regard qPCR has been used for the detection and quantification of viruses including JC polyomavirus [7, 35, 48, 125], adenoviruses [7, 35, 126, 127], hepatitis A viruses [128, 129], astroviruses [130], enteroviruses [130] and noroviruses [131] from water samples.
Giardia and Cryptosporidium are protozoan parasites that are important waterborne disease-causing organisms in surface water in the USA [132] and Canada [133]. Action should be taken if more than 5 Giardia cysts and 10–30 oocysts of Cryptosporidium in 100 L of water are detected. However, it is difficult to detect such low numbers and enrichment techniques cannot be used [134]. Several researchers have confirmed that qPCR is currently a reliable application to detect Giardia and Cryptosporidium in water and sewage and understand their distribution and abundance [3, 116, 126, 135, 136].
Schistosoma japonicum cercariae is the cause of serious human illnesses in China [137] leading to poor child growth and development [138, 139] and socioeconomic impacts [140, 141]. The mouse bioassay is a common method used for the detection of S. japonicum cercariae in surface water. Worrell et al. [142] showed that qPCR was a more sensitive method than the mouse bioassay for detection of S. japonicum. The time for the analysis was reduced from 6 weeks to 14 h for the qPCR assay and the cost was reduced from US$100 to US$15 per sample.
Sludge produced by wastewater treatment plants is recycled and used as an organic fertilizer. However, sludge should be decomposed as pathogens are present in high numbers and may survive several months in the environment. Quantitative PCR makes it possible to determine the fate of these pathogens during the wastewater treatment process and sludge composting and therefore to determine the efficiency of these processes [8, 23].
Disadvantages/limitations of using qPCR in detection of microorganisms in water
Numerous studies have found differences in the levels of microbial cells in water samples when comparing qPCR with conventional cultivation methods [23, 127, 143]. Srinivasan et al. [127] observed that after chlorination of wastewater, the levels of E. coli and enterococci detected by qPCR and cultivation methods differed significantly. Whether the difference could be attributed to dead cells or cells that have entered a viable but not cultivable (VNBC) state remains unclear. Low concentrations of chlorine could have led to cell membrane damage without necessarily leading to cell inactivation and damage to nucleic acids [144]. Wéry et al. [23] monitored Salmonella spp., C. jejuni, E. coli and C. perfringens during wastewater treatment and sludge composting. Differences between qPCR values and cultivation techniques were dependent on the bacteria and the matrix studied. Discrepancies between qPCR and cultivation techniques were larger for E. coli and Salmonella spp. than for C. perfringens. Discrepancies were also smaller for E. coli and Salmonella spp. in dewatered sludge and in the supernatant after centrifugation than in the wastewater, pretreated wastewater and treated water [24].
Reischer et al. [4, 24] developed human-specific and ruminant-specific Bacteroidetes markers (BacH and BacR) to be used in qPCR assays for MST studies. Although the results of these assays could successfully be used to pinpoint sources of pollution, the persistence of these faecal DNA markers in different environments should be tested in future studies. The detectable marker may decay in aquatic systems with a higher temperature and trophic status. The presence of the marker in soil and sediment that may influence the water body of interest should also be determined [24].
Substances in environmental water samples could lead to the inhibition of DNA or RNA amplification during conventional and qPCR. PCR inhibitors in environmental water samples, including heavy metals, humic acids and phenolic compounds, could lead to the underestimation of microorganisms present. This is especially a problem when microorganisms are present in low concentrations [145]. Diluting DNA or RNA concentrations before qPCR has been used to remove PCR inhibitors in water collected from various sources including surface water, wastewater and drinking water [146]. In the qPCR assay for the detection of Candida spp. in water, Brinkman et al. [6] found that samples with high turbidity should be diluted up to a 100 times to overcome PCR inhibition. In some cases additional DNA purification was required that caused a tenfold loss of DNA. Water samples from different sources may therefore affect the sensitivity of qPCR. However, this could lead to increased variations in gene copy numbers or false negatives if the DNA or RNA concentrations are diluted below their detection limit [147, 148]. Researchers have developed protocols that remove PCR inhibitors during extraction including methods utilizing phenol–chloroform–isoamyl alcohol [149], AlNH4(SO4)2 [150] and the polymeric absorbent Superlite™ DAX-8 [151]. Commercial DNA extraction kits have also been shown to remove PCR inhibitors in environmental water samples for successful qPCR [83]. Magnetic separation beads coated with antibodies reduced the effect of inhibitors during the isolation and qPCR of RNA from rotaviruses [152].
Live/dead discrimination
The viability of a pathogenic microorganism has a direct impact on its pathogenicity and therefore plays an important role in the potential risks associated with the presence of the organism in a water source. Quantitative PCR, as with other molecular techniques, cannot discriminate between viable and non-viable microorganisms. The inability of qPCR to differentiate between live and dead microbial cells therefore remains a major limitation. This is for instance the case in the detection of S. japonicum cercariae in water where the disadvantage of qPCR over the traditional mouse bioassay was that it could not discriminate between live or dead cercariae. Inactive cercariae, not detected by qPCR, may still be a risk for infection [142].
Ethidium monoazide (EMA) and propidium monoazide (PMA), dyes that bind to DNA of membrane-damaged cells, have been used in conjunction with qPCR to quantify viable organisms in water. The drawback of this method is that viability is based only on membrane integrity. The efficiency of viable qPCR methods utilizing EMA and PMA dyes in the detection of bacteria in wastewater samples before and after chlorination treatment has been investigated. After disinfection, qPCR results were not comparable with results obtained with cultivation methods. The researchers suggested that EMA could not penetrate some of the cells in the chlorinated samples [127, 153]. EMA-qPCR assays to detect viable Legionella species in spa water [154], tap water [155] and cooling water [156] have also been developed. EMA-qPCR was more suited for quantifying viable microorganisms because PMA-qPCR led to an overestimation of viable Legionella species in cooling water samples. Important factors to consider during quantifying viable organisms are the determination of the optimal concentration of EMA and exposure time [156].
Furthermore, no direct correlation can be made between viral genome copies detected and quantified by qPCR and viral infectivity. This problem can be overcome by combining cell culturing with qPCR, known as integrated cell culture PCR (ICC-PCR) or integrated cell culture quantitative PCR (ICC-qPCR). Quantitative PCR is performed after the initial biological amplification of viral nucleic acids during ICC-qPCR. Combining cell culturing techniques with molecular techniques not only results in time savings but also increases sensitivity. ICC-qPCR has been used to detect polioviruses, astroviruses, enteroviruses and adenovirus [146, 157–159] in environmental water samples.
Certain microorganisms can enter a VBNC state in different water sources [2]. Conventional culturing techniques are unable to detect VBNC microorganisms giving water authorities a false sense of security and thereby increasing the risk of infection. Helicobacter pylori, a gram-negative microaerophilic bacterium, has been associated with peptic ulcers and chronic gastritis [160]. H. pylori enters a VBNC state after exposure to tap water for about 120 h [22]. These bacteria were detected using scanning electron microscopy, most probable number PCR (MPN-PCR) and RT-qPCR. MPN-PCR was a 100-fold more sensitive than culture techniques and RT-qPCR was a 100-fold more sensitive than MPN-PCR [22]. Legionellae and legionellae-like amoeba organisms are difficult to quantify by culturing on agar plates [161]. These organisms are commonly associated with cooling towers and have been linked with severe outbreaks of legionellosis [162, 163]. Quantitative PCR assays have been developed that can detect and quantify viable cells of legionellae and legionellae-like amoeba in water by utilizing EMA dyes [154–156].
Standardisation of protocols
There is a need for the standardisation of protocols for the development and implementation of qPCR as a tool for determining the microbial quality of water. Currently there is a lack of information on the reproducibility of qPCR and the factors associated with variation in qPCR protocols are mostly unknown. Two key issues that need to be resolved are the influence of DNA isolation protocols on qPCR and the use of simplex or multiplex amplification protocols.
Bacterial DNA can be isolated through two strategies during water quality testing. This can be done either by bead milling, dilution and amplification of the crude lysate or by purifying and concentrating the DNA in the lysate with commercial kits. Shanks et al. [118] compared the interlaboratory variability of the two DNA isolation strategies for the qPCR measurement of enterococci and Bacteroidales concentrations from standardised, spiked and environmental water sources. The study was performed between eight facilities in the USA, including federal, state, city and academic laboratories. No significant difference between the two DNA isolation approaches was observed [118].
Viruses present in environmental water sources need to be concentrated before qPCR can be performed. The use of different viral concentration methods influences the outcome of virus recovery and qPCR results [7, 125, 152]. Viral concentration methods therefore need to be optimised and standardised. However, Rodriguez et al. [164] observed that for the detection of adenovirus and norovirus in recreational waters the method should be optimised for each sampling site and for each virus.
The interlaboratory variability between simplex and multiplex amplification protocols has also been investigated. No significant variability was detected when the two approaches were investigated in eight different laboratories. The advantages of using the multiplex amplification protocol are cost savings due to multiple assays being performed simultaneously and allowance for the inclusion of an internal control [118].
Finally the specific PCR conditions, primers and probes used need to be standardised to reduce variability between laboratories [35]. Standardised qPCR kits are commercially available and include kits for the detection of Salmonella spp. (based on the invasion protein gene), Shigella spp. (based on the virulence plasmid pCP301) and E. coli strains (based on the glucuronidase gene). These kits have been optimised and are provided with internal controls. Quantitative PCR kits have also been developed for the monitoring of Legionella pneumophila isolates in water samples [165].
Standard procedures used by different organizations
Once a qPCR protocol for the detection of a specific microorganism has been standardised and the reproducibility and sensitivity of the assay validated, it can be used for routine monitoring of water samples by an organisation. The EPA approved the TaqMan® qPCR method for the detection of Enterococci and Bacteroidales in water samples. The water sample is filtered through a membrane filter and the filter is then agitated in a microcentrifuge tube with glass beads and buffer to extract the DNA into the solution. The TaqMan® Universal Master Mix PCR reagent and probe system is then used for PCR amplification and detection of target sequences in the supernatant [166]. The Environmental Agency for England and Wales (EA) has also developed and accredited a qPCR analysis method for the detection of the faecal indicator Bacteroidales [167].
Although the USA are using molecular techniques such as qPCR for routine monitoring of recreational water samples, these techniques have not been widely implemented in the policy frameworks for water regulation of the European Union and other countries. One major limitation that hinders the implementation of qPCR for the use in environmental water analysis is that the qPCR signal does not decrease after common disinfection methods. In other words, as previously discussed, it cannot discriminate between viable and non-viable microorganisms. The EA and academic institutions compared the efficiency of conventional faecal indicators with the accredited EA qPCR method to determine whether the data obtained from this qPCR method could be solely used for making regulatory decisions. Crude sewage, secondary treated sewage and UV-disinfected sewage were collected from Scarborough Wastewater Treatment Works and analysed using both methods. After UV treatment a significant decrease in the colony forming units (CFU) of faecal coliforms and presumptive intestinal enterococci was observed. However, no significant decrease was observed in the genome copies of Bacteroidales after UV treatment (Fig. 4). Researchers therefore advise that qPCR cannot be used as the sole detection method for routine analysis of environmental water samples [168].
Fig. 4.
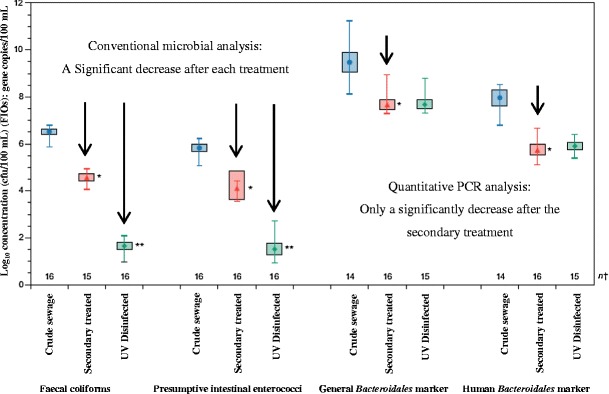
Mean, range and 95 % confidence intervals of the mean for faecal indicator organisms (FIOs) (expressed as log10 CFU/100 mL) and Bacteroidales marker concentrations (gene copies/100 mL) in crude sewage (blue circle), secondary treated sewage (red triangle) and UV-disinfected sewage (green dot). The secondary treatment sewage that was significantly different from the mean of the crude extract (*) and UV-disinfected sewage that was significantly different from the secondary treated sewage (**) are also indicated in the figure (adapted from [168])
Alternative quantification and detection methods for waterborne pathogens
Isothermal amplification assays
Isothermal amplification assays that exclude PCR methods and the use of thermocycler machines have been developed. Such methods are based on DNA/RNA synthesis and the mimicking action of accessory proteins in vitro for nucleic acid amplification. The best known assays include transcription-mediated amplification (TMA), self-sustained sequence replication, nucleic acid sequence-based amplification (NASBA), signal-mediated amplification of RNA technology, strand displacement amplification, rolling circle amplification, loop-mediated isothermal amplification of DNA (LAMP), isothermal multiple displacement amplification (IMDA), helicase-dependent amplification (HDA), single primer isothermal amplification (SPIA) and circular helicase-dependent amplification (cHDA). LAMP and NASBA are mostly used to detect waterborne pathogens and will be described in more detail [169].
Loop-mediated isothermal amplification assays
Loop-mediated isothermal amplification (LAMP) was developed to detect a specific DNA sequence with specific characteristics [170]. Autocycling strand displacement DNA synthesis is performed by the Bst DNA polymerase large fragment [170]. The products synthesised after amplification include stem-loop DNA structures with many inverted repeats of the target and cauliflower-like structure with multiple loops [171]. The advantages of LAMP include a single reaction temperature ranging between 60 and 65 °C, high specificity by using six primers that can recognise eight distinct regions and lack of DNA purification due to tolerance to inhibitory materials [172–174]. Detection of the products is visually assessed as a large amount of DNA is synthesised in a short time resulting in the production of pyrophosphate that forms white precipitates of magnesium pyrophosphate leading to turbidity [175]. An increase in turbidity indicates an increase in precipitate production and correlates with the amount of DNA [172, 174]. The method is therefore cost-effective as expensive equipment is not necessary [172, 176, 177]. LAMP products have been detected in research studies by using gel electrophoresis, real-time turbidimetry and fluorescence probes [178, 179]. Pathogenic bacteria such as E. coli [172], C. coli, C. jejuni [174], S. aureus [176], Salmonella [177], H. pylori [180], Shigella [181] and V. cholerae [174] have been detected in water samples by LAMP assays.
Nucleic acid sequence-based amplification
Nucleic acid sequence-based amplification (NASBA) is similar to the transcription-mediated amplification (TMA) that makes RNA from a promoter engineered in the primer region, using the RNA polymerase function and DNA from RNA templates by reverse transcriptase [182]. This technology was improved by using RNase H that removes RNA from cDNA without heat-denaturation and therefore eliminating the thermocycler step. This method is also named self-sustained sequence replication (3SR) [183]. Gel electrophoresis, fluorescence probes (real-time NASBA) and a colorimetric assay (NASBA ELISA 6) are used to detect NASBA products [184–186]. The Food and Drug Administration office of United States of America (FDA) approved the technique for the molecular detection of some microorganisms such as HCV and HIV-1 [187, 188].
Nested and semi-nested PCR
Nested and semi-nested PCR is a modified PCR method that utilises additional primers to perform a second PCR reaction. Research studies have showed that the detection efficiency is significantly increased by using the original primers (semi-nested) or a different set of selective primers (nested PCR) in the second reaction [189–192]. Semi-nested PCR was successfully used to detect Cryptosporidium oocysts in wastewater after treatment [192], Y. enterocolitica in water [193] and Legionella species in hospital cooling tower water [194].
Phylochips and pyrosequencing
High-density phylogenetic microarrays can analyse many taxa simultaneously in complex environmental samples. The Phylochip is a phylogenetic microarray that can determine a microbial community structure [195]. The latest version can detect up to 50,000 bacterial, archael and microalgal taxa within the 16S rRNA gene [196]. This technology has been used in biological wastewater treatment systems to analyse the microbial community [197] and in the MST of pathogens in coastal urban watershed [198]. Another novel tool that has emerged in the detection of waterborne pathogens is pyrosequencing. Pyrosequencing is based on the sequencing-by-synthesis principle that utilizes enzyme-coupled reactions and bioluminescence to monitor the release of pyrophosphate and nucleotide incorporation in real time [199]. A large number of parallel sequencing reactions can be carried out e.g. the GX FLX+ produces a million reads within 23 h [200]. Pyrosequencing has been applied to detect Clostridium, Mycobacterium, parechoviruses, coronaviruses, adenoviruses, aichi viruses and herpes viruses in wastewater biosolids [201, 202], Aeromonas and Clostridium in wastewaters [203] and bacteriophages in potable waters [204].
Conclusion
Quantitative PCR assays are applied to determine the quality of drinking water straight from resources [24] and after treatment [22], the efficiency of wastewater treatment plants [23] and the safety of water from recreational beaches [25]. Research showed that qPCR is a specific, sensitive and rapid tool to determine the presence and numbers of microorganisms in water. It has also proven to be useful for reducing the health risks associated with microorganisms in water and to assist in ensuring a safe supply of water [6, 22, 142]. The US EPA is currently considering qPCR as a rapid analytical tool to detect and quantify faecal indicators in recreational waters [118]. Limitations of using qPCR as an analytical tool in routine monitoring include the inability of qPCR to differentiate between live and dead microbial cells. The presence of PCR inhibitors in environmental water samples and the need for standardized qPCR protocols remain a challenge. There is potential for the application of high-throughput analytical systems in detection of waterborne pathogens; however, the technology is still in its infancy. Many challenges still remain to validate the methods in order to use one common protocol. Other challenges in the application of these technologies are the complexity of the assays and the time spent to train laboratory personnel. The high cost involved to do water analysis on a routine basis versus the high sensitivity and reduced time of the assays should also be considered. It is clear from the limitations described that the qPCR method still needs improvement before it can be applied for routine analysis of water.
Acknowledgments
The authors would like to thank the National Research Foundation (NRF) of South Africa and ESKOM South Africa for funding.
References
- 1.Szewzyk U, Szewzyk R, Manz W, Schleifer KH. Annu Rev Microbiol. 2000;54(1):81–127. doi: 10.1146/annurev.micro.54.1.81. [DOI] [PubMed] [Google Scholar]
- 2.Oliver JD. J Microbiol. 2002;43(5):93–100. [PubMed] [Google Scholar]
- 3.Guy RA, Payment P, Krull UJ, Horgen PA. Appl Environ Microbiol. 2003;69(9):5178–5185. doi: 10.1128/AEM.69.9.5178-5185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reischer GH, Kasper DC, Steinborn R, Mach RL, Farnleitner AH. Appl Environ Microbiol. 2006;72(8):5610–5614. doi: 10.1128/AEM.00364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farnleitner AH, Mach RL, Burtscher MM, Reischer GH, Ryzinska G, Keiblinger K, Rudnicki S, Knetsch S (2005) In: Proceedings of the 13th international symposium on health related water microbiology, Swansea, 5–9 Sept 2005
- 6.Brinkman NE, Haugland RA, Wymer LJ, Byappanahalli M, Whitman RL, Vesper SJ. Appl Environ Microbiol. 2003;69(3):1775–1782. doi: 10.1128/AEM.69.3.1775-1782.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albinana-Gimenez N, Clemente-Casares P, Calgua B, Huguet JM, Courtois S, Girones R. J Virol Methods. 2009;158(1–2):104–109. doi: 10.1016/j.jviromet.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Shannon KE, Lee DY, Trevors JT, Beaudette LA. Sci Total Environ. 2007;382(1):121–129. doi: 10.1016/j.scitotenv.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 9.Alexandrino M, Grohmann E, Szewzyk U. Water Res. 2004;38(5):1340–1346. doi: 10.1016/j.watres.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 10.Saiki RK, Scharf S, Faloona F, Mullins KB, Horn GT, Erlich HA, Arnhrim N. Science. 1985;230(4732):1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 11.Zimmermann K, Mannhalter JW. BioTechniques. 1996;21(2):268–279. doi: 10.2144/96212rv01. [DOI] [PubMed] [Google Scholar]
- 12.Orlando C, Pinzani P, Pazzagli M. Clin Chem Lab Med. 1998;36(5):255–269. doi: 10.1515/CCLM.1998.045. [DOI] [PubMed] [Google Scholar]
- 13.Heid CA, Stevens J, Livak KJ, Williams PM. Genome Res. 1996;6(10):986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 14.Mackay IM. Clin Microbiol Infect. 2004;10(3):190–212. doi: 10.1111/j.1198-743X.2004.00722.x. [DOI] [PubMed] [Google Scholar]
- 15.Haberhausen G, Pinsl J, Kuhn CC, Markert-Hahn C. J Clin Microbiol. 1998;36(3):628–633. doi: 10.1128/jcm.36.3.628-633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leong DT, Gupta A, Bai HF, Wan G, Yoong LF, Too HP, Chew FT, Hutmacher DW. Biomaterials. 2007;28(2):203–210. doi: 10.1016/j.biomaterials.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 17.Diviacco S, Norio P, Zentilin L, Menzo S, Clementi M, Biamonti G, Riva S, Falaschi A, Giacca M. Gene. 1992;122(2):313–320. doi: 10.1016/0378-1119(92)90220-J. [DOI] [PubMed] [Google Scholar]
- 18.Schwab KJ, Neill FH, Estes MK, Atmar RL. Water Sci Technol. 1998;38(12):83–86. doi: 10.1016/S0273-1223(98)00805-1. [DOI] [Google Scholar]
- 19.Huang PW, Laborde D, Land VR, Matson DO, Smith AW, Jiang X. Appl Environ Microbiol. 2000;66(10):4383–4388. doi: 10.1128/AEM.66.10.4383-4388.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dallman MJ, Montgomery RA, Larsen CP, Wanders A, Wells AF. Immunol Rev. 1991;119(1):163–179. doi: 10.1111/j.1600-065X.1991.tb00583.x. [DOI] [PubMed] [Google Scholar]
- 21.Baeumner AJ, Humiston MC, Montagna RA, Durst RA. Anal Chem. 2001;73(6):1176–1180. doi: 10.1021/ac001293h. [DOI] [PubMed] [Google Scholar]
- 22.Nayak AK, Rose JB. J Appl Microbiol. 2007;103(5):1931–1941. doi: 10.1111/j.1365-2672.2007.03435.x. [DOI] [PubMed] [Google Scholar]
- 23.Wéry N, Lhoutellier C, Ducray F, Delgenès JP, Godon JJ. Water Res. 2008;42(1–2):53–62. doi: 10.1016/j.watres.2007.06.048. [DOI] [PubMed] [Google Scholar]
- 24.Reischer GH, Kasper DC, Steinborn R, et al. Lett Appl Microbiol. 2007;44:351. doi: 10.1111/j.1472-765X.2006.02094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haugland RA, Siefring SC, Wymer LJ, Brenner KP, Dufour AP. Water Res. 2005;39(4):559–568. doi: 10.1016/j.watres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Thomas JJ, Bothner B, Traina J, Benner WH, Siuzdak G. Spectroscopy Int J. 2004;18(1):31–36. doi: 10.1155/2004/376572. [DOI] [Google Scholar]
- 27.Bosch A. Internatl Microbiol. 1998;1(3):191–196. [PubMed] [Google Scholar]
- 28.De Ligny BH, Etienne I, Francois A, Toupance O, Buchler M, Touchard G, Lepogamp P, Comoz F, Lobbedez T, Godin M, Ryckelynck JP, Lebranchu Y. Transplant Proc. 2000;32(8):2760–2761. doi: 10.1016/S0041-1345(00)01869-8. [DOI] [PubMed] [Google Scholar]
- 29.Borchardt MA, Bertz PD, Spencer SK, Battigelli DA. Appl Environ Microbiol. 2003;69(2):1172–1180. doi: 10.1128/AEM.69.2.1172-1180.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fong TT, Mansfield LS, Wilson DL, Schwab DJ, Molloy SL, Rose JB. Environ Health Perspect. 2007;115(6):856–864. doi: 10.1289/ehp.9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haramoto E, Katayama H, Oguma K, Ohgaki S. Appl Environ Microbiol. 2005;71(5):2403–2411. doi: 10.1128/AEM.71.5.2403-2411.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katayama H, Shimasaki A, Ohgaki S. Appl Environ Microbiol. 2002;68(3):1033–1039. doi: 10.1128/AEM.68.3.1033-1039.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuo DHW, Simmons FJ, Blair S, Hart E, Rose JB, Xagoraraki I. Water Res. 2010;44(5):1520–1530. doi: 10.1016/j.watres.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 34.Xagoraraki I, Kuo DHW, Wong K, Wong M, Rose JB. Appl Environ Microbiol. 2007;73(24):7874–7881. doi: 10.1128/AEM.01239-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bofill-Mas S, Albinana-Gimenez N, Clemente-Casares P, Hundesa A, Rodriguez-Manzano J, Allard A, Calvo M, Girones R. Appl Environ Microbiol. 2006;72(12):7894–7896. doi: 10.1128/AEM.00965-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pina S, Puig M, Lucena F, Jofre J, Girones R. Appl Environ Microbiol. 1998;64(9):3376–3382. doi: 10.1128/aem.64.9.3376-3382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyn-Jones AP, Carducci A, Cook N, D’Agostino M, Divizia M, Fleischer J, Gantzer C, Gawler A, Girones R, Holler C, Husman AMD, Kay D, Kozyra I, Lopez-Pila J, Muscillo M, Nascimento MS, Papageorgiou G, Rutjes S, Sellwood J, Szewzyk R, Wyer M. Water Res. 2011;45(3):1025–1038. doi: 10.1016/j.watres.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katayama H, Haramoto E, Oguma K, Yamashita H, Tajima A, Nakajima H, Ohgaki S. Water Res. 2008;42(6–7):1441–1448. doi: 10.1016/j.watres.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 39.Simmons FJ, Kuo DHW, Xagoraraki I. Water Res. 2011;45(9):2739–2750. doi: 10.1016/j.watres.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 40.Wong K, Onan BM, Xagoraraki I. Appl Environ Microbiol. 2010;76(19):6441–6448. doi: 10.1128/AEM.02685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Futch JC, Griffin DW, Lipp EK. Environ Microbiol. 2010;12(4):964–974. doi: 10.1111/j.1462-2920.2010.02141.x. [DOI] [PubMed] [Google Scholar]
- 42.Lipp EK, Futch IC, Griffin DW. Mar Pollut Bull. 2007;54(12):1897–1902. doi: 10.1016/j.marpolbul.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Ogorzaly L, Bertrand I, Paris M, Maul A, Gantzer C. Appl Environ Microbiol. 2010;76(24):8019–8025. doi: 10.1128/AEM.00917-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rigotto C, Hanley K, Rochelle PA, De Leon R, Barardi CRM, Yates MV. Environ Sci Technol. 2011;45(9):4145–4150. doi: 10.1021/es103922r. [DOI] [PubMed] [Google Scholar]
- 45.Love DC, Silverman A, Nelson KL. Environ Sci Technol. 2010;44(18):6965–6970. doi: 10.1021/es1001924. [DOI] [PubMed] [Google Scholar]
- 46.Mena KD, Gerba CP. Rev Environ Contam Toxicol. 2009;198:133–167. doi: 10.1007/978-0-387-09647-6_4. [DOI] [PubMed] [Google Scholar]
- 47.Albinana-Gimenez N, Clemente-Casares P, Bofill-Mas S, Hundesa A, Ribas F, Girones R. Environ Sci Technol. 2006;40(23):7416–7422. doi: 10.1021/es060343i. [DOI] [PubMed] [Google Scholar]
- 48.Haramoto E, Kitajima M, Katayama H, Ohgaki S. Water Res. 2010;44(6):1747–1752. doi: 10.1016/j.watres.2009.11.043. [DOI] [PubMed] [Google Scholar]
- 49.Kurtz JB, Lee TW, Pickering D. J Clin Pathol. 1977;30(10):948–952. doi: 10.1136/jcp.30.10.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones EL, Gaither M, Kramer A, Gerba CP. Wildernes Environ Med. 2009;20(1):6–13. doi: 10.1580/06-WEME-OR-43.1. [DOI] [PubMed] [Google Scholar]
- 51.Grist NR, Bell EJ, Assaad F. Prog Med Virol. 1978;24:114–157. [PubMed] [Google Scholar]
- 52.Girones R. J Virol Methods. 2008;153(2):79–83. doi: 10.1016/j.jviromet.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Savichtcheva O, Okabe S. Water Res. 2006;40(13):2463–2476. doi: 10.1016/j.watres.2006.04.040. [DOI] [PubMed] [Google Scholar]
- 54.Roslev P, Bukh AS. Appl Microbiol Biotechnol. 2011;89(5):1341–1355. doi: 10.1007/s00253-010-3080-7. [DOI] [PubMed] [Google Scholar]
- 55.Field KG, Bernhard AE, Brodeur TJ. Environ Monit Assess. 2003;81(1–3):313–326. doi: 10.1023/A:1021349629950. [DOI] [PubMed] [Google Scholar]
- 56.Daly K, Stewart CS, Flint HJ, Shirazi-Beechey SP. FEMS Microbiol Ecol. 2001;38(2–3):141–151. doi: 10.1111/j.1574-6941.2001.tb00892.x. [DOI] [Google Scholar]
- 57.Hold GL, Pryde SE, Russell VJ, Furrie E, Flint HJ. FEMS Microbiol Ecol. 2002;39(1):33–39. doi: 10.1111/j.1574-6941.2002.tb00904.x. [DOI] [PubMed] [Google Scholar]
- 58.Leser TD, Amenuvor JZ, Jensen TK, Lindecrona RH, Boye M, Moller K. Appl Environ Microbiol. 2002;68(2):673–690. doi: 10.1128/AEM.68.2.673-690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scott TM, Rose JB, Jenkins TM, Farrah SR, Lukasik J. Appl Environ Microbiol. 2002;68(12):5796–5803. doi: 10.1128/AEM.68.12.5796-5803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tambalo DD, Fremaux B, Boa T, Yost CK. Water Res. 2012;46(9):2891–2904. doi: 10.1016/j.watres.2012.02.048. [DOI] [PubMed] [Google Scholar]
- 61.Ashbolt NJ. Toxicology. 2004;198(1–3):229–238. doi: 10.1016/j.tox.2004.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kindhauser MK (ed) (2003) Communicable diseases 2002: global defence against the infectious disease threat. World Health Organization, Geneva
- 63.Faruque SM, Albert MJ, Mekalanos JJ. Microbiol Mol Biol Rev. 1998;62(4):1301–1314. doi: 10.1128/mmbr.62.4.1301-1314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Atlas RM. Environ Microbiol. 1999;1(4):283–293. doi: 10.1046/j.1462-2920.1999.00046.x. [DOI] [PubMed] [Google Scholar]
- 65.Karanis P, Kourenti C, Smith H. J Water Health. 2007;5(1):1–38. doi: 10.2166/wh.2006.002. [DOI] [PubMed] [Google Scholar]
- 66.Marshall MA, Naumovitz D, Ortega Y, Sterling C. Clin Microbiol Rev. 1997;10(1):67–85. doi: 10.1128/cmr.10.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haque R, Mondal D, Duggal P, Kabir M, Roy S, Farr BM, Sack B, Petri WA. Infect Immun. 2006;74(2):904–909. doi: 10.1128/IAI.74.2.904-909.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woodall CJ. Desalination. 2009;248(1–3):616–621. doi: 10.1016/j.desal.2008.05.110. [DOI] [Google Scholar]
- 69.Bustin SA. J Mol Endocrinol. 2000;25(2):169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 70.Hurley J, Roberts D, Bond A, Keys D, Chen C. Methods Mol Biol. 2012;822(33–52):33–52. doi: 10.1007/978-1-61779-427-8_3. [DOI] [PubMed] [Google Scholar]
- 71.Solinas A, Brown LJ, McKeen C, Mellor JM, Nicol J, Thelwell N, Brown T. Nucleic Acids Res. 2001;29(20):e96. doi: 10.1093/nar/29.20.e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang C-J, Smith BF. Am J Vet Res. 2007;68(3):231–235. doi: 10.2460/ajvr.68.3.231. [DOI] [PubMed] [Google Scholar]
- 73.Klein D. Trends Mol Med. 2002;8(6):257–260. doi: 10.1016/S1471-4914(02)02355-9. [DOI] [PubMed] [Google Scholar]
- 74.Ahmed W, Sawant S, Huygens F, Goonetilleke A, Gardner T. Water Res. 2009;43(19):4918–4928. doi: 10.1016/j.watres.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 75.Mackay IM, Arden KE, Nitsche A. Real-time PCR in virology. Nucleic Acids Res. 2002;30(6):1292–1305. doi: 10.1093/nar/30.6.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park O, Mayo KE. Mol Endocrinol. 1991;5(7):967–978. doi: 10.1210/mend-5-7-967. [DOI] [PubMed] [Google Scholar]
- 77.Chelly J, Montarras D, Pinset C, Berwald-Netter Y, Kaplan JC, Kahn A. Eur J Biochem. 1990;187(3):691–698. doi: 10.1111/j.1432-1033.1990.tb15355.x. [DOI] [PubMed] [Google Scholar]
- 78.Ferré F, Marchese A, Pezzoli P, Griffon S, Buxton E, Bover V. Quantitative PCR: an overview. The polymerase chain reaction. Boston: Birkhauser; 1994. p. 67. [Google Scholar]
- 79.Billadeau D, Quam L, Thomas W, Kay N, Greipp P, Kyle R, Oken MM, Van Ness B. Blood. 1992;80(7):1818–1824. [PubMed] [Google Scholar]
- 80.Gilliland G, Perrin S, Blanchard K, Bunn HF. Proc Natl Acad Sci. 1990;87(7):2725–2729. doi: 10.1073/pnas.87.7.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sestini R, Orlando C, Zentilin L, Lami D, Gelmini S, Pinzani P, Giacca M, Pazzagli M. Clin Chem. 1995;41(6):826–832. [PubMed] [Google Scholar]
- 82.Ruster B, Zeuzem S, Roth WK. Anal Biochem. 1995;224(2):597–600. doi: 10.1006/abio.1995.1092. [DOI] [PubMed] [Google Scholar]
- 83.Behets J, Declecrk P, Delaedt Y, Creemers B, Ollevier F. J Microbiol Methods. 2007;68(1):137–144. doi: 10.1016/j.mimet.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 84.Haugland RA, Vesper SJ, Wymer LJ. Mol Cell Probes. 1999;13(5):329–340. doi: 10.1006/mcpr.1999.0258. [DOI] [PubMed] [Google Scholar]
- 85.Shanks OC, Atikovic E, Blackwood AD, Lu J, Noble RT, Domingo JS, Seifring S, Sivaganesan M, Haugland RA. Appl Environ Microbiol. 2008;74(3):745–752. doi: 10.1128/AEM.01843-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sivaganesan M, Seifring S, Varma M, Haugland RA, Shanks OC. BMC Bioinformatics. 2008;9:120–132. doi: 10.1186/1471-2105-9-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Countway PD, Caron DA. Appl Environ Microbiol. 2006;72(4):2496–2506. doi: 10.1128/AEM.72.4.2496-2506.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ibekwe AM, Watt PM, Grieve CM, Sharma VK, Lyons SR. Appl Environ Microbiol. 2002;68(10):4853. doi: 10.1128/AEM.68.10.4853-4862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Toplak N, Kovač M, Piskernik S, Možina SS, Jeršek B. J Appl Microbiol. 2012;112(4):752–764. doi: 10.1111/j.1365-2672.2012.05235.x. [DOI] [PubMed] [Google Scholar]
- 90.Markoulatos P, Siafakas N, Moncany M. J Clin Lab Anal. 2002;16(1):47–51. doi: 10.1002/jcla.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shangkuan YH, Show YS, Wang TM. J Appl Bacteriol. 1995;79(3):264–273. doi: 10.1111/j.1365-2672.1995.tb03136.x. [DOI] [PubMed] [Google Scholar]
- 92.Kong RYC, Dung WF, Vrijmoed LLP, Wu RSS. Mar Pollut Bull. 1995;31(4–12):317–324. doi: 10.1016/0025-326X(95)00139-E. [DOI] [Google Scholar]
- 93.Kong RYC, So CL, Law WF, Wu RSS. Mar Pollut Bull. 1999;38(12):1207–1215. doi: 10.1016/S0025-326X(99)00164-2. [DOI] [Google Scholar]
- 94.Way JS, Josephson KL, Pillai SD, Abaszadegan M, Gerba CP, Pepper IL. Appl Environ Microbiol. 1993;59(5):1473–1479. doi: 10.1128/aem.59.5.1473-1479.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.LaGier MJ, Joseph LA, Passaretti TV, Musser KA, Cirino NM. Mol Cell Probes. 2004;18:275–282. doi: 10.1016/j.mcp.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 96.Kong RYC, Lee SKY, Law TWF, Law SHW, Wu RSS. Water Res. 2002;36(11):2802–2812. doi: 10.1016/S0043-1354(01)00503-6. [DOI] [PubMed] [Google Scholar]
- 97.Aridgides LJ, Doblin MA, Berke T, Dobbs FC, Matson DO, Drake LA. Mar Pollut Bull. 2004;48(11–12):1096–1101. doi: 10.1016/j.marpolbul.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 98.Chehadeh HE, Zerlauth G, Mannhalter JW. BioTechniques. 1995;18(1):26–28. [PubMed] [Google Scholar]
- 99.Jin CF, Mata M, Fink DJ. PCR Methods Appl. 1994;3:252–255. doi: 10.1101/gr.3.4.252. [DOI] [PubMed] [Google Scholar]
- 100.Arnold BL, Itakura K, Rossi JJ. Genet Anal Biomol Eng. 1992;9(4):113–116. doi: 10.1016/1050-3862(92)90050-F. [DOI] [PubMed] [Google Scholar]
- 101.Cottrez F, Auriault C, Capron A, Groux H. Nucleic Acids Res. 1994;22(13):2712–2713. doi: 10.1093/nar/22.13.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wilkinson ET, Cheifetz S, De Grandis S. Genome Res. 1995;4(6):363–367. doi: 10.1101/gr.4.6.363. [DOI] [PubMed] [Google Scholar]
- 103.DiDomenico N, Link H, Knobel R, Caratsch T, Weschler W, Loewy ZG, Rosenstraus M. Clin Chem. 1996;42(12):1915–1923. [PubMed] [Google Scholar]
- 104.Gibson U, Heid CA, Williams PM. Genome Res. 1996;69(10):995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- 105.Morrison TB, Weis JJ, Wittwer CT. BioTechniques. 1998;24(6):954–962. [PubMed] [Google Scholar]
- 106.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14(3):303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 107.Lee LG, Connell CR, Bloch W. Nucleic Acids Res. 1993;21(16):3761–3766. doi: 10.1093/nar/21.16.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Holland PM, Abramson RD, Watson R, Gelfand DH. Proc Natl Acad Sci U S A. 1991;88(16):7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bustin SA (2005) In: Fuchs J, Podda M (eds) Encyclopedia of diagnostic genomics and proteomics. Marcel Dekker, New York pp 1117–1125
- 110.Rasmussen R, Morrison T, Herrmann M, Wittwer C. Biochemica. 1998;2:8–11. [Google Scholar]
- 111.Thelwell N, Millington S, Solinas A, Booth J, Brown T. Nucleic Acids Res. 2000;28(19):3752–3761. doi: 10.1093/nar/28.19.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Smith CM, Hill VR. Appl Environ Microbiol. 2009;75(16):5284–5289. doi: 10.1128/AEM.00456-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gibson KE, Schwab KJ. Appl Environ Microbiol. 2011;77(1):385–391. doi: 10.1128/AEM.01164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vilaginès P, Sarrette B, Husson G, Vilaginès R. Water Sci Technol. 1993;27:199–306. [Google Scholar]
- 115.Rajal VB, McSwain BS, Thompson DE, Leutenegger CM, Kildare BJ, Wuertz S. Water Res. 2007;41(7):1411–1422. doi: 10.1016/j.watres.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 116.Bertrand I, Schwartzbrod J. Water Res. 2007;41(16):3675–3682. doi: 10.1016/j.watres.2007.02.043. [DOI] [PubMed] [Google Scholar]
- 117.Khan IUH, Gannon V, Kent R, Koning W, Lapen DR, Miller J, Neumann N, Phillips R, Robertson W, Topp E, van Bochove E, Edge TA. J Microbiol Methods. 2007;69(3):480–488. doi: 10.1016/j.mimet.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 118.Shanks OC, Sivaganesan M, Peed L, Kelty CA, Blackwood AD, Greene MR, Noble RT, Bushon RN, Stelzer EA, Kinzelman J, Anan’eva T, Sinigalliano C, Wanless D, Griffith J, Cao Y, Weisberg S, Harwood VJ, Staley C, Oshima KH, Varma M, Haugland RA. Environ Sci Technol. 2012;46(2):945–953. doi: 10.1021/es2031455. [DOI] [PubMed] [Google Scholar]
- 119.Sen K, Sinclair JL, Boczek L, Rice EW. Environ Sci Technol. 2011;45(6):2250–2256. doi: 10.1021/es103365b. [DOI] [PubMed] [Google Scholar]
- 120.Whitman RL, Ge Z, Nevers MB, Boehm AB, Chern EC, Haugland RA, Lukasik AM, Molina M, Przybyla-Kelly K, Shively DA, White EM, Zepp RG, Byappanahalli MN. Environ Sci Technol. 2010;44(13):5049–5054. doi: 10.1021/es9028974. [DOI] [PubMed] [Google Scholar]
- 121.Lee CS, Marion JW, Lee J. J Water Health. 2011;9(2):253–264. doi: 10.2166/wh.2011.120. [DOI] [PubMed] [Google Scholar]
- 122.Wade TJ, Calderon RL, Sams E, Beach M, Brenner KP, Williams AH. Dufour AP. 2006;114(1):24–28. doi: 10.1289/ehp.8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wade TJ, Calderon RL, Brenner KP, Sams E, Beach M, Haugland R, Wymer L. Dufour AP. 2008;19(3):375–383. doi: 10.1097/EDE.0b013e318169cc87. [DOI] [PubMed] [Google Scholar]
- 124.Wade TJ, Sams E, Brenner KP, Haugland R, Chern E, Beach M, Wymer L, Rankin CC, Love D, Li Q, Noble R, Dufour AP. Environ Health. 2010;9(66):1–14. doi: 10.1186/1476-069X-9-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fumian TM, Guimarães FR, Vaz BJP, Silva MTT, Muylaert FF, Bofill-Mas S, Gironés R, Leite JPG, Miagostovich MP. J Water Health. 2010;8(3):438–455. doi: 10.2166/wh.2010.090. [DOI] [PubMed] [Google Scholar]
- 126.Bertrand I, Gantzer C, Chesnot T, Schwartzbrod J. J Microbiol Methods. 2004;57(1):41–53. doi: 10.1016/j.mimet.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 127.Srinivasan S, Aslan A, Xagoraraki I, Alocilja E, Rose JB. Water Res. 2011;45(8):2561–2572. doi: 10.1016/j.watres.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 128.Rodriguez-Manzano J, Miagostovich M, Hundesa A, Clemente-Casares P, Carratala A, Buti A, Jardí R, Girones R. J Water Health. 2010;8(2):346–354. doi: 10.2166/wh.2009.042. [DOI] [PubMed] [Google Scholar]
- 129.Le Cann P, Ranarijaona S, Monpoeho S, Le Guyader F, Ferré V. Res Microbiol. 2004;155(1):11–15. doi: 10.1016/j.resmic.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 130.Schvoever E, Ventura M, Dubos O, Cazaux G, Serceau R, Gournier N, Dubois V, Caminade P, Fluery HJ, Lafon ME. Res Microbiol. 2001;152(2):179–186. doi: 10.1016/S0923-2508(01)01190-1. [DOI] [PubMed] [Google Scholar]
- 131.Laverick MA, Wyn-Jones AP, Carter MJ. Lett Appl Microbiol. 2004;39(2):127–136. doi: 10.1111/j.1472-765X.2004.01534.x. [DOI] [PubMed] [Google Scholar]
- 132.LeChevallier MW, Norton WD, Ross A. Appl Environ Microbiol. 1991;57(9):2610–2616. doi: 10.1128/aem.57.9.2610-2616.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wallis PM, Erlandsen SL, Isaac-Renton ME, Olson WJ, Robertson WJ, van Heulen H. Appl Environ Microbiol. 1996;62(8):2789–2797. doi: 10.1128/aem.62.8.2789-2797.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rose JB, Slifko TR. J Food Prot. 1999;62:1059–1070. doi: 10.4315/0362-028x-62.9.1059. [DOI] [PubMed] [Google Scholar]
- 135.Fontaine M, Guillot E. J Microbiol Methods. 2003;54(1):29–36. doi: 10.1016/S0167-7012(03)00005-8. [DOI] [PubMed] [Google Scholar]
- 136.Masago Y, Oguma K, Katayama H, Ohgaki S. Water Sci Technol. 2006;54(3):119–126. doi: 10.2166/wst.2006.457. [DOI] [PubMed] [Google Scholar]
- 137.Leenstra T, Coutinho HM, Acosta LP, Langdon GC, Su L, Olveda RM, McGarvey ST, Kurtis JD, Friedman JF. Infect Immun. 2006;74(11):6398–6407. doi: 10.1128/IAI.00757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jukes MCH, Nokes CA, Alcock KJ, Lambo JK, Kihamia C, Ngorosho N, Mbise A, Lorri W, Yona E, Mwanri L, Baddeley AD, Hall A, Bundy DAP. Trop Med Int Health. 2002;7(2):104–117. doi: 10.1046/j.1365-3156.2002.00843.x. [DOI] [PubMed] [Google Scholar]
- 139.McGarvey ST, Wu G, Zhang S, Wang Y, Peters P, Olds GR, Wiest PM. J Trop Med Hyg. 1993;48(4):547–553. doi: 10.4269/ajtmh.1993.48.547. [DOI] [PubMed] [Google Scholar]
- 140.Li Y, Zhao Z, Ellis M, McManus DP. Acta Trop. 2005;96(2–3):266. doi: 10.1016/j.actatropica.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 141.Wu X, Wang T, Lu D. Acta Trop. 2002;82(2):247–252. doi: 10.1016/S0001-706X(02)00016-5. [DOI] [PubMed] [Google Scholar]
- 142.Worrell C, Xiao N, Vidal JE, Chen L, Zhong B, Remais J. Appl Environ Microbiol. 2011;77(6):2192–2195. doi: 10.1128/AEM.01561-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Byappanahalli MN, Whitman RL, Shively DA, Nevers MB. Sci Total Environ. 2010;44(13):5049–5054. doi: 10.1021/es9028974. [DOI] [PubMed] [Google Scholar]
- 144.Virto R, Manas P, Alvarez I, Condon S, Raso J. Appl Environ Microbiol. 2005;71(9):5022–5028. doi: 10.1128/AEM.71.9.5022-5028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wilson IG. Appl Environ Microbiol. 1997;63(10):3741–3751. doi: 10.1128/aem.63.10.3741-3751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rigotto C, Victoria M, Moresco V, Kolesnikovas CK, Corrêa AA, Souza DSM, Miagostovich MP, Simões CMO, Barardi CRM. J Appl Microbiol. 2010;109(6):1979–1987. doi: 10.1111/j.1365-2672.2010.04827.x. [DOI] [PubMed] [Google Scholar]
- 147.Kreader CA. Appl Environ Microbiol. 1996;62(2):1102–1106. doi: 10.1128/aem.62.3.1102-1106.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tsai YL, Olsen BH. Appl Environ Microbiol. 1992;58(2):754–757. doi: 10.1128/aem.58.2.754-757.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tsai YL, Olsen BH. Appl Environ Microbiol. 1991;57(4):1070–1074. doi: 10.1128/aem.57.4.1070-1074.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Braid MD, Daniels LM, Kitts CL. J Microbiol Methods. 2003;52(3):389–393. doi: 10.1016/S0167-7012(02)00210-5. [DOI] [PubMed] [Google Scholar]
- 151.Schriewer A, Wehlmann A, Wuertz S. J Microbiol Methods. 2011;85(1):16–21. doi: 10.1016/j.mimet.2010.12.027. [DOI] [PubMed] [Google Scholar]
- 152.Yang W, Gu AZ, Zeng S, Li D, He M, Shi H. J Microbiol Methods. 2011;84(3):447–453. doi: 10.1016/j.mimet.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 153.Varma M, Field R, Stinson M, Rukovets B, Wymer L, Haugland R. Water Res. 2009;43(19):4790–4801. doi: 10.1016/j.watres.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 154.Chang B, Sugiyama K, Taguri T, Amemura-Maekawa J, Kura F, Watanabe H. Appl Environ Microbiol. 2009;75(1):147–153. doi: 10.1128/AEM.00604-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Delgado-Viscogliosi P, Solignac L, Delattre JM. Appl Environ Microbiol. 2009;75(1):3502–3512. doi: 10.1128/AEM.02878-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen NT, Chang CW. J Appl Microbiol. 2010;109(2):623–634. doi: 10.1111/j.1365-2672.2010.04678.x. [DOI] [PubMed] [Google Scholar]
- 157.Chapron CD, Ballester NA, Fontaine JH, Frades CN, Margolin AB. Appl Environ Microbiol. 2000;66(6):2520–2525. doi: 10.1128/AEM.66.6.2520-2525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Balkin HB, Margolin AB. Virol J. 2010;25(7):282. doi: 10.1186/1743-422X-7-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Reynolds KA. Methods Mol Biol. 2004;268:69–78. doi: 10.1385/1-59259-766-1:069. [DOI] [PubMed] [Google Scholar]
- 160.Majumdar D, Bedd J, Atherton J. Medicine. 2011;39(3):154–161. doi: 10.1016/j.mpmed.2010.12.003. [DOI] [Google Scholar]
- 161.Adeleke A, Pruckler J, Benson R, Rowbotham T, Halablab M, Fields B. Emerg Infect Dis. 1996;2(3):225–230. doi: 10.3201/eid0203.960311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Garcia-Fulgueiras A, Navarro C, Fenoll D, Garcia J, Gonzalez-Diego P, Jimenez-Bunuales T, Rodriquez M, Lopez R, Pacheco F, Ruiz J, Segovia M, Baladron B, Pelaz C. Emerg Infect Dis. 2003;9(8):915–921. doi: 10.3201/eid0908.030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gilmour MW, Bernard K, Tracz DM, Olson AB, Corbett CR, Burdz T, Ng B, Wiebe D, Broukhanski G, Boleszczuk P, Tang P, Jamieson F, van Domselaar G, Plummer FA, Berry JD. J. Med Microbiol. 2007;56(3):336–341. doi: 10.1099/jmm.0.46738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rodriguez RA, Thie L, Gibbons CD, Sobsey MD. J Virol Methods. 2012;181(1):43–50. doi: 10.1016/j.jviromet.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 165.Association Française de Normalisation (2005) XP T 90–471. Association Française de Normalisation, Paris, France
- 166.US EPA (2010) Method A: enterococci in water byTaqMan® quantitative polymerase chainreaction (qPCR) assay. http://water.epa.gov/scitech/methods/cwa/bioindicators/upload/rapid1.pdf. Accessed April 2010
- 167.Layton A, McKay L, Williams D, Garrett V, Gentry R, Sayler G. Appl Environ Microbiol. 2006;72(6):4214–4224. doi: 10.1128/AEM.01036-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Stapleton CM, Kay D, Wyer MD, Davies C, Watkins J, Kay C, McDonald AT, Porter J, Gawler A. Water Res. 2009;43(19):4888–4899. doi: 10.1016/j.watres.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 169.Sen K (2011) In: Sen K, Ashbolt NJ (eds) Environmental microbiology. Current technology and water application. Caister, Norfolk, pp 1–39
- 170.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Nucleic Acids Res. 2000;28(12):e63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gill P, Ghaemi A. Nucleos Nucleot Nucleic Acids. 2008;27(3):224–243. doi: 10.1080/15257770701845204. [DOI] [PubMed] [Google Scholar]
- 172.Hara-Kudo Y, Nemoto J, Ohtsuka K, Segawa Y, Takatori K, Kojima T, Ikedo M. J Med Microbiol. 2007;56(3):398–406. doi: 10.1099/jmm.0.46819-0. [DOI] [PubMed] [Google Scholar]
- 173.Kaneko H, Kawana T, Fukushima E, Suzutani T. J Biochem Biophys Methods. 2007;70(3):499–501. doi: 10.1016/j.jbbm.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 174.Yamazaki W, Taguchi M, Ishibashi M, Kitazato M, Nukina M, Misawa N, Inoue KJ. J Med Microbiol. 2008;57(4):444–451. doi: 10.1099/jmm.0.47688-0. [DOI] [PubMed] [Google Scholar]
- 175.Mori Y, Nagamine K, Tomita N, Notomi T. Biochem Biophys Res Commun. 2001;289(1):150–154. doi: 10.1006/bbrc.2001.5921. [DOI] [PubMed] [Google Scholar]
- 176.Goto M, Hayashidani H, Takatori K, Hara-Kudo Y. Lett Appl Microbiol. 2007;45(1):100–107. doi: 10.1111/j.1472-765X.2007.02142.x. [DOI] [PubMed] [Google Scholar]
- 177.Hara-Kudo Y, Yoshino M, Kojima T, Ikedo M. FEMS Microbiol Lett. 2005;253(1):155–161. doi: 10.1016/j.femsle.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 178.Mori Y, Kitao M, Tomita N, Notomi T. J Biochem Biophys Methods. 2004;59(2):145–157. doi: 10.1016/j.jbbm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 179.Mori Y, Hirano T, Notomi T. BMC Biotechnol. 2007;6:1–10. doi: 10.1186/1472-6750-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Minami M, Ohta M, Ohkura T, Ando T, Torii K, Hasegawa T, Goto H. J Clin Microbiol. 2006;44(11):4032–4037. doi: 10.1128/JCM.00898-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Song T, Toma C, Nakasone N, Iwanaga M, Mori Y, Nagamine K, Tomita N, Notomi T. Biochem Biophys Res Commun. 2001;289(1):150–154. doi: 10.1006/bbrc.2001.5921. [DOI] [PubMed] [Google Scholar]
- 182.Compton J. Nature. 1991;350(6313):91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- 183.Guatelli JC, Whitfield KM, Kwoh DY, Barringer KJ, Richman DD, Gingeras TR. Natl Acad Sci U S A. 1990;87(5):1874–1878. doi: 10.1073/pnas.87.5.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Abdel-Galil KH, El-Sokkary MA, Kheira SM, Salazar AM, Yates MV, Chen W, Mulchandani A. Appl Environ Microbiol. 2005;71(11):7113–7116. doi: 10.1128/AEM.71.11.7113-7116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gill P, Ramezani R, Amiri MV-P, Ghaemi A, Hashempour T, Eshraghi N, Ghalami M, Tehrani HA. Biochem Biophys Res Commun. 2006;347(4):1151–1157. doi: 10.1016/j.bbrc.2006.07.039. [DOI] [PubMed] [Google Scholar]
- 186.Polstra AM, Goudsmit J, Cornelissen M. BMC Infect Dis. 2002;2:18–27. doi: 10.1186/1471-2334-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Guichon A, Chiparelli H, Martinez A, Rodriguez C, Trento A, Russi JC, Carballal G. J Clin Virol. 2004;29(2):84–91. doi: 10.1016/S1386-6532(03)00091-X. [DOI] [PubMed] [Google Scholar]
- 188.Barker CS, Griffin C, Dolganov GM, Hanspers K, Yang JY, Erle DJ. J Clin Microbiol. 2000;38(7):2665–2669. [Google Scholar]
- 189.Gajardo R, Pinto RM, Bosch A. Water Sci Technol. 1995;31(5–6):371–374. [Google Scholar]
- 190.Le Guyader F, Menard D, Pommepuy M, Kopecka H. Water Sci Technol. 1995;319(5–6):391–394. [Google Scholar]
- 191.Straub TM, Pepper IL, Gerba CP. Appl Environ Microbiol. 1995;61(6):2066–2068. doi: 10.1128/aem.61.5.2066-2068.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mayer CL, Palmer CJ. Appl Environ Microbiol. 1996;62(6):2081–2085. doi: 10.1128/aem.62.6.2081-2085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kapperud G, Vardund T, Skjeve E, Hornes E, Michaelsen TE. Appl. Environ. Microbiol. 1993;59(9):2938–2944. doi: 10.1128/aem.59.9.2938-2944.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Myamoto H, Yamamoto H, Arima K, Fujii J, Maruta K, Isu K, Shiomori T, Yoshida A. Appl. Environ. Microbiol. 1997;63(7):2489–2494. doi: 10.1128/aem.63.7.2489-2494.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Desantis TZ, Brodie EL, Moberg JP, Zubieta IZ, Piceno YM, Andersen GL. Microb Ecol. 2007;53(3):371–383. doi: 10.1007/s00248-006-9134-9. [DOI] [PubMed] [Google Scholar]
- 196.Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N, Jansson JK, Probst A, Borglin SE, Fortney JL. Science. 2010;330(6001):204–208. doi: 10.1126/science.1195979. [DOI] [PubMed] [Google Scholar]
- 197.Xia S, Duan L, Song Y, Li J, Piceno YM, Andersen GL, Alvarez-Chohen L, Moreno-Andrade I, Huang C-L, Hermanaowicz SW. Environ Sci Technol. 2010;44(19):7391–7396. doi: 10.1021/es101554m. [DOI] [PubMed] [Google Scholar]
- 198.Wu CH, Sercu B, Van de Werfhorst LC, Wong J, DeSantis TZ, Brodie EL, Hazen TC, Holden PA, Andersen GL. PLoS ONE. 2010;5:e11285. doi: 10.1371/journal.pone.0011285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ahmadian A, Ehn M, Hober S. Clin Chim Acta. 2006;363(1–2):83–94. doi: 10.1016/j.cccn.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 200.Rothberg JM, Leamon JH. Nat Biotechnol. 2008;26(10):1117–1124. doi: 10.1038/nbt1485. [DOI] [PubMed] [Google Scholar]
- 201.Bibby K, Viau E, Peccia J. Water Res. 2010;44(14):4252–4260. doi: 10.1016/j.watres.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 202.Bibby K, Viau E, Peccia J. Lett Appl Microbiol. 2011;52(4):386–392. doi: 10.1111/j.1472-765X.2011.03014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ye L, Zhuang T. Environ Sci Technol. 2011;45(17):7173–7179. doi: 10.1021/es201045e. [DOI] [PubMed] [Google Scholar]
- 204.Rosario K, Nilsson C, Lim YW, Ruan YJ, Breibart M. Environ Microbiol. 2009;11(11):2806–2820. doi: 10.1111/j.1462-2920.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 205.Prado T, Fumain TM, Marize P, Miagostovich MP, Gaspar AMC. Trans R Soc Trop Med. 2012;106(2):104–109. doi: 10.1016/j.trstmh.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 206.Victoria M, Rigotto C, Moresco V, de Abreu Corrêa A, Kolesnikovas C, Leite JPG, Miagostovich MP, Barardi CRM. J Appl Microbiol. 2010;109(1):231–238. doi: 10.1111/j.1365-2672.2009.04646.x. [DOI] [PubMed] [Google Scholar]
- 207.González A, Botella S, Montes RM, Moreno Y, Ferrús MA. J Food Prot. 2007;70:341–347. doi: 10.4315/0362-028x-70.2.341. [DOI] [PubMed] [Google Scholar]
- 208.Beumer A, King D, Donohue M, Mistry J, Covert T, Pfaller S. Appl Environ Microbiol. 2010;76(1):7367–7370. doi: 10.1128/AEM.00730-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Calgua B, Mengewein A, Grünert A, Bofill-Mas S, Clemente-Casares P, Hundesa A, Wyn-Jones AP, López-Pila JM. J Virol Methods. 2008;153(2):79–83. doi: 10.1016/j.jviromet.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 210.Lavender JS, Kinzelman JL. Water Res. 2009;43(19):4967–4979. doi: 10.1016/j.watres.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 211.Liu YJ, Zhang CM, Wang XC. Environ Monit Assess. 2009;158(1–4):535–544. doi: 10.1007/s10661-008-0602-1. [DOI] [PubMed] [Google Scholar]
- 212.Wetz J, Blackwood AD, Fries JS, Williams ZF, Noble RT. Aquat Microb Ecol. 2008;53(1):141–149. doi: 10.3354/ame01223. [DOI] [Google Scholar]
- 213.Stratagene (2012) http://www.stratagene.com. Accessed 23 May 2012
- 214.Fraga D, Meulia T, Fenster S (2008) Current protocols essential laboratory techniques 10.3.1–10.3.34. Wiley, Weinheim