

Abstract
Bats have increasingly been recognized as the natural reservoir of severe acute respiratory syndrome (SARS), coronavirus, and other coronaviruses found in mammals. However, little research has been conducted on bat coronaviruses in South Korea. In this study, bat samples (332 oral swabs, 245 fecal samples, 38 urine samples, and 57 bat carcasses) were collected at 33 natural bat habitat sites in South Korea. RT-PCR and sequencing were performed for specific coronavirus genes to identify the bat coronaviruses in different bat samples. Coronaviruses were detected in 2.7% (18/672) of the samples: 13 oral swabs from one species of the family Rhinolophidae, and four fecal samples and one carcass (intestine) from three species of the family Vespertiliodae. To determine the genetic relationships of the 18 sequences obtained in this study and previously known coronaviruses, the nucleotide sequences of a 392-nt region of the RNA-dependent RNA polymerase (RdRp) gene were analyzed phylogenetically. Thirteen sequences belonging to SARS-like betacoronaviruses showed the highest nucleotide identity (97.1–99.7%) with Bat-CoV-JTMC15 reported in China. The other five sequences were most similar to MERS-like betacoronaviruses. Four nucleotide sequences displayed the highest identity (94.1–95.1%) with Bat-CoV-HKU5 from Hong Kong. The one sequence from a carcass showed the highest nucleotide identity (99%) with Bat-CoV-SC2013 from China. These results suggest that careful surveillance of coronaviruses from bats should be continued, because animal and human infections may result from the genetic variants present in bat coronavirus reservoirs.
Keywords: Coronavirus, Bats, South Korea, Severe acute respiratory syndrome, Middle East respiratory syndrome, Phylogenetic analysis
Introduction
Coronaviruses are large, pleomorphic, enveloped viruses containing a single linear, positive-sense single-stranded RNA molecule. Coronavirus genomes are approximately 27–32 kb in length, the largest continuous RNA genomes among mammalian viruses [1]. Coronaviruses are classified into four genera: alpha, beta, gamma, and delta [2]. Coronaviruses are the second most prevalent cause of the common cold in humans [3]. In livestock and poultry, coronaviruses are recognized causes of enteric and respiratory infections that are often fatal in young animals [4]. An outbreak of severe acute respiratory syndrome (SARS) in 2002 and 2003 resulted in infection of 8096 people worldwide, with 774 (9.5%) of them dying from this novel human coronavirus [5]. SARS coronavirus is believed to have been acquired from an animal species, and the Chinese horseshoe bat may have been the original source of SARS infection [6–9]. The emergent Middle East respiratory syndrome coronavirus (MERS-CoV) may also have originated in bats, with fragments of MERS-CoV genes being identified in bats from both Saudi Arabia [10] and Africa [11]. More than 200 viruses have been isolated from and detected in bats [12]. The ability of bats to fly and migrate, as well as the large sizes of their social groups, predisposes bats to acquire and maintain viruses [13].
Given the import of MERS into South Korea [14] and the presence of SARS in the relatively close geographic location of China [9] (Fig. 3), together with the fact that bats are a reservoir for coronaviruses, the prevalence of coronavirus infection in Korean bat species should provide valuable information. Although SARS-like and MERS-like bat coronaviruses have already been reported in the feces of Korean bats [14], the prevalence and presence of coronaviruses in other bat samples have not been described. Therefore, we investigated the prevalence of coronaviruses in Korean bat species using various kinds of samples, including oral swabs, fecal and urine samples, and carcasses. The coronaviruses found were characterized for their relationships to each other and to coronaviruses isolated in South Korea and in other countries.
Fig. 3.
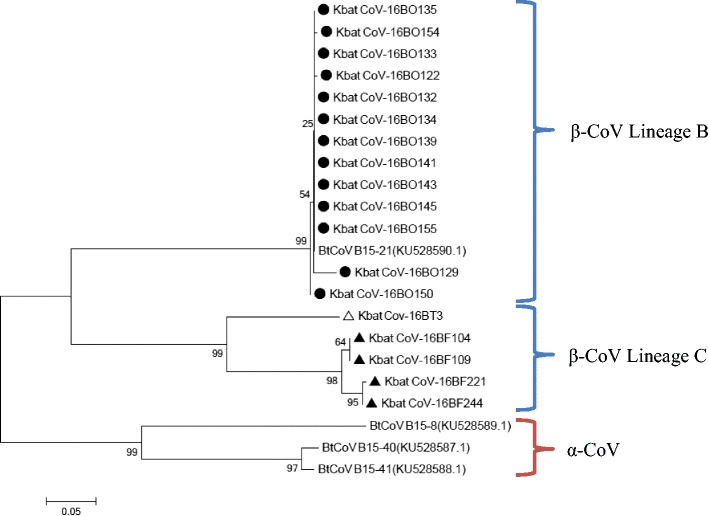
Phylogenetic tree of Korean bat coronaviruses detected by Kim et al. [13] and in this study, based on 355-nt sequences. Phylogenetic trees were constructed by using the neighbor joining method and bootstrap values were determined by 3000 replicates. Scale bar the estimated genetic distance of these viruses. Black circle sequences from oral swabs. Black up-pointing triangle sequences from fecal samples. White up-pointing triangle sequences from carcass
Materials and Methods
Sample Collection
A total of 672 samples (332 oral swabs, 245 fecal samples, 38 urine samples, and 57 bat carcasses) were collected at 33 sites of natural bat habitat from January 2016 to September 2016 (Table 1). Bats were captured by net for collection of oral swabs, fecal samples, and urine samples, and released immediately after sampling. Fecal samples were also collected from guano. The bat carcasses collected were naturally dead bats found at the site where the other samples were taken. Swab samples were kept in viral transport medium at 4 °C, and organs collected from carcasses and other samples were stored at −80 °C before processing.
Table 1.
Bat sample information
| Region 1 | Region 2 | Site name | Site type | Major bat species | No. of samples | ||||
|---|---|---|---|---|---|---|---|---|---|
| Oral swab | Feces | Carcass | Urine | Total | |||||
| Gangwon | Yeongwol | YW 1 | Forest | Eptesicus serotinus | 11 | 18 | 29 | ||
| Gyeongnam | Changwon | CW 1 | Abandoned mine | Rhinolophus ferrumequinum | 7 | 7 | |||
| Gyeongbuk | Gyeongju | GJ 1 | Abandoned mine | Rhinolophus ferrumequinum | 70 | 2 | 6 | 12 | 90 |
| GJ 2 | Forest | Myotis aurascens | 1 | 1 | 2 | ||||
| GJ 3 | In area of private houses | Pipistrelus abramus | 10 | 10 | |||||
| Forest | Pipistrelus abramus | 5 | 5 | ||||||
| Andong | AD 1 | Forest | Eptesicus serotinus | 25 | 18 | 43 | |||
| AD 2 | Forest | Eptesicus serotinus | 6 | 2 | 2 | 10 | |||
| Yeongdeok | YD 1 | Abandoned mine | Myotis macrodactylus | 2 | 3 | 5 | |||
| YD 2 | Forest | Pipistrelus abramus | 5 | 5 | |||||
| Yeongju | YJ 1 | Abandoned mine | Myotis nattereri | 23 | 6 | 1 | 30 | ||
| YJ 2 | Forest, stream | Pipistrelus abramus | 5 | 5 | |||||
| Yeongcheon | YC 1 | Abandoned mine | Miniopterus schreibersii | 12 | 12 | ||||
| Cheongsong | CS 1 | Forest, valley | Myotis petax | 4 | 1 | 5 | |||
| Gwangju | Shinchon | GwJ 1 | Forest | Eptesicus serotinus | 11 | 6 | 1 | 18 | |
| Seokgok | GwJ 2 | Cave | Rhinolophus ferrumequinum | 5 | 5 | ||||
| Daegu | Dalseong | DS 1 | Forest, stream | Pipistrelus abramus | 5 | 5 | |||
| Ulsan | Ulju | US 1 | Abandoned mine | Rhinolophus ferrumequinum | 12 | 2 | 8 | 22 | |
| US 2 | In area of private houses | Eptesicus serotinus | 1 | 1 | 2 | ||||
| Forest | Eptesicus serotinus | 31 | 31 | 62 | |||||
| US 3 | Forest | Myotis aurascens | 9 | 9 | |||||
| Jeonnam | Gurye | GR 1 | Forest | Eptesicus serotinus | 12 | 12 | |||
| Muan | MA 1 | Abandoned mine | Myotis macrodactylus | 7 | 1 | 8 | |||
| MA U | Cave | Myotis formosus chofukusei | 2 | 5 | 7 | ||||
| Jindo | JD 1 | Abandoned mine | Rhinolophus ferrumequinum | 10 | 1 | 1 | 12 | ||
| Hampyeong | HP U | Cave | Myotis formosus chofukusei | 3 | 3 | ||||
| Jeonbuk | Sunchang | SC 1 | Abandoned mine | Rhinolophus ferrumequinum | 43 | 3 | 10 | 4 | 60 |
| Chungnam | Seosan | SS U | Abandoned mine | Myotis formosus chofukusei | 3 | 4 | 7 | ||
| Chungbuk | Danyang | DY 1 | Cave | Miniopterus schreibersii | 7 | 65 | 22 | 94 | |
| DY 2 | Cave | Myotis macrodactylus | 2 | 2 | 4 | ||||
| DY 3 | Cave | Miniopterus schreibersii | 18 | 1 | 6 | 25 | |||
| DY 4 | Cave | Miniopterus schreibersii | 25 | 25 | |||||
| DY 5 | Forest, valley | Myotis ikonnikovi | 1 | 1 | |||||
| DY 6 | Forest, valley | Myotis ikonnikovi | 8 | 6 | 2 | 16 | |||
| DY 7 | Forest | Murina leucogaster | 7 | 10 | 17 | ||||
| Total | 332 | 245 | 57 | 38 | 672 | ||||
Genome Detection and Sequencing
All fecal and tissue samples were resuspended in 1% antibiotic-antimycotic solution (Corning, USA) diluted in phosphate-buffered saline (PBS), and clarified by centrifugation at 3500×g for 10 min. RNA from 200 μL sample was extracted with the QIAamp® Viral RNA Mini Kit (Qiagen, Germany) and eluted in 60 μL RNase-free water. cDNA was synthesized by PrimeScript First Strand cDNA Synthesis Kit (TaKaRa, Japan) following the manufacturer’s instructions. Bat-CoV screening was performed by a pan-coronavirus PCR method based on primers used by Poon et al. (Corona 1 forward, 5′-GGTTGGGACTATCCTAAGTGTGA-3′, and Corona 2 reverse, 5′-CCATCATCAGATAGAATCATCATA-3′) [15], followed by sequencing of the amplified product to confirm Bat-CoV identification. The pan-coronavirus primers were used to amplify and sequence a 440-bp segment of the highly conserved RNA-dependent RNA polymerase (RdRp) gene. Cycle sequencing reactions were performed using the BigDye® Terminator Cycle Sequencing kit version 1.1 (Applied Biosystems, Foster City, CA, USA). Reactions were purified using SigmaSpin™ Post-reaction Clean-Up Columns (Sigma, St. Louis, MO, USA) and sequenced on an ABI PRISM 3130 automated capillary DNA sequencer (Applied Biosystems) according to the manufacturer’s instructions. Primer sequences were excluded from the raw sequence data, and the sequences we analyzed were 392-bp long. All sequences obtained in this study were submitted to GenBank (accession numbers KY432454–432471).
Phylogenetic Analysis
The nucleotide sequences were aligned and compared to 52 selected human and animal CoV sequences available from the GenBank database using ClustalW software implemented in BioEdit version 7.0.9.0. The phylogenetic trees were drawn using the neighbor joining method using the maximum composite likelihood model with MEGA 7 software.
Results
Prevalence of Coronavirus Infection in Bats
Coronaviruses were detected in 13 oral swabs, one carcass (intestine), and four fecal samples from a total of 672 samples, giving an overall detection rate of 2.7% (18/672). Four of the 14 bat species tested were found to harbor coronavirus. Thirteen positive oral swabs were detected from one species of the family Rhinolophidae, which accounted for 24.3% (163/672) of the samples, and the other positive samples were from Vespertilionidae bats that comprised 55.5% (373/672) of the samples.
The 13 coronavirus-positive oral swabs were collected from bats at an abandoned mine in Jeonbuk province (Fig. 1). Oral swabs and other samples (n = 60) were obtained from three species of bats, Rhinolophus ferrumequinum, Miniopterus schreibersii, and Myotis macrodactylus, but coronaviruses were only detected in samples from R. ferrumequinum (Table 2). One carcass was collected from a cave in Chungbuk province (Fig. 1). In total, 94 samples were obtained from six bat species (M. schreibersii, Myotis petax, Vespertilio sinensis, Murina leucogaster, R. ferrumequinum, and M. macrodactylus), and coronavirus was detected from a V. sinensis sample. Four coronavirus-positive fecal samples were collected in four different regions, in Gyeongbuk province (Andong, Yeongdeok, and Gyeongju) and in the metropolitan city Gwangju (Fig. 1). In the forest of Andong, 43 samples in total were collected from three species of bats (Eptesicus serotinus, M. petax, and Pipistrelus abramus), and coronavirus was detected from one P. abramus bat. In Yeongdeok, a total of five samples was collected from P. abramus living in the forest, and one sample was positive for coronavirus. In the forest of Gyeongju, five samples in total were collected only from P. abramus bats, and one of the samples was positive for coronavirus. In the forest of Gwangju, 18 samples in total were collected from two species of bats (E. serotinus and Myotis aurascens) and coronavirus was detected from one E. serotinus bat (Table 2).
Fig. 1.
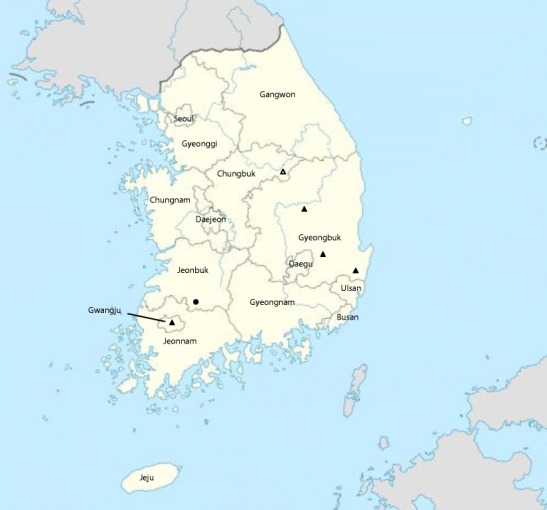
Location of South Korea and geographical location of coronavirus-positive bat samples. Black circle Bat-CoV from oral swabsm. Black up-pointing triangle Bat-CoV from fecal samples. White up-pointing triangle Bat-CoV from a bat carcass
Table 2.
Coronavirus distribution in different bat species and provinces/metropolitan cities
| Family | Species | Gangwon | Gyeongnam | Gyeongbuk | Gwangju | Daegu | Ulsan | Jeonnam | Jeonbuk | Chungnam | Chungbuk | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Miniopteridae | Miniopterus schreibersii | 0/6 | 0/19 | 0/102 | 0/127 | |||||||
| Rhinolophidae | Rhinolophus ferrumequinum | 0/7 | 0/79 | 0/3 | 0/20 | 13/24 (54.2%)b | 0/20 | 13/163 (8.0%) | ||||
| Vespertilionidae | Eptesicus serotinus | 0/24 | 0/48 | 1/17 (5.9%)a | 0/62 | 0/12 | 1/163 (0.6%) | |||||
| Murina leucogaster | 0/18 | 0/15 | 0/33 | |||||||||
| Murina ussuriensis | 0/3 | 0/3 | ||||||||||
| Myotis aurascens | 0/2 | 0/1 | 0/7 | 0/1 | 0/11 | |||||||
| Myotis formosus chofukusei | 0/5 | 0/3 | 0/8 | |||||||||
| Myotis ikonnikovi | 0/14 | 0/14 | ||||||||||
| Myotis macrodactylus | 0/4 | 0/9 | 0/17 | 0/4 | 0/34 | |||||||
| Myotis nattereri | 0/29 | 0/29 | ||||||||||
| Myotis petax | 0/8 | 0/4 | 0/15 | 0/27 | ||||||||
| Pipistrellus alaschanicus | 0/1 | 0/2 | 0/3 | |||||||||
| Pipistrelus abramus | 0/5 | 3/27 (11.1%)a | 0/5 | 0/2 | 3/39 (7.7%) | |||||||
| Vespertilio sinensis | 1/9 (11.1%)c | 1/9 (11.1%) | ||||||||||
| Unknown | Unknown | 0/5 | 0/4 | 0/9 | ||||||||
| Total | 0/29 | 0/7 | 3/222 (1.4%) | 1/23 (4.3%) | 0/5 | 0/95 | 0/42 | 13/60 (21.7%) | 0/7 | 1/182 (0.5%) | 18/672 (2.7%) |
aFecal samples
bOral swabs
cCarcass (intestine)
Phylogenetic Characterization of Bat Coronaviruses Isolated in Korea
To determine the genetic relationships between the 18 bat coronaviruses obtained in this study and previously known coronaviruses, the 392-nt RdRp sequences were analyzed phylogenetically. All the sequences obtained in this study belonged to the betacoronavirus genus (Fig. 2). The sequences from 13 oral swabs (16BO122, 129, 132, 133, 134, 135, 139, 141, 143, 145, 150, 154, and 155) were grouped with the SARS-related bat betacoronaviruses, lineage B. Sequences from a carcass (16BT3) and four fecal samples (16BF104, 109, 211, and 244) were grouped with MERS-like bat betacoronaviruses, lineage C (Fig. 2).
Fig. 2.
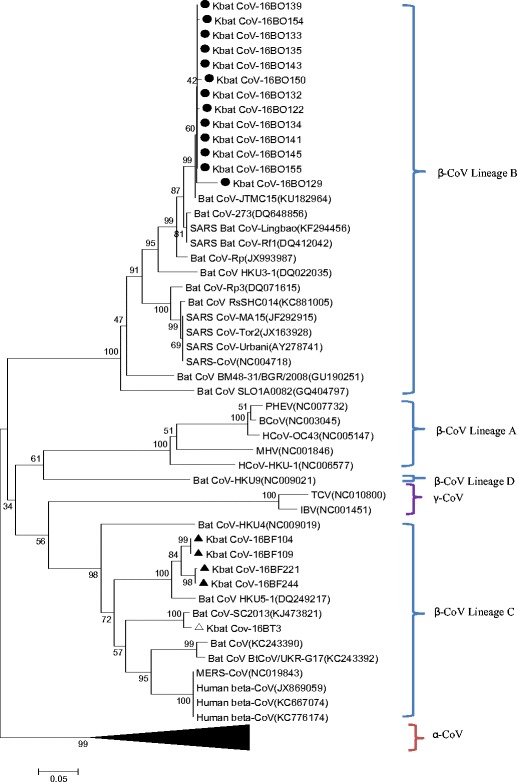
Phylogenetic tree constructed from 392-nt RdRp sequences of Korean bat coronaviruses detected in this study and from other coronaviruses. Phylogenetic trees were constructed by using the neighbor joining method and bootstrap values were determined by 3000 replicates. Scale bar the estimated genetic distance of these viruses. Black circle sequences from oral swabs Black up-pointing triangle sequences from fecal samples. White up-pointing triangle sequences from carcass
The 13 sequences from oral swabs were 97.1–100.0% identical with each other, and the closest known CoV strain, with 97.1–99.7% nucleotide identity, was Bat-CoV-JTMC15 that was recently isolated from R. ferrumequinum in China. The nucleotide identity with SARS-CoV was only 86.7–89.0%. One sequence from a carcass showed the highest identity (99%) with Bat-CoV-SC2013 that was isolated from Vespertilio superans (synonym of V. sinensis), also from China [16]. The nucleotide identity with MERS-CoV was 83.9%. Four sequences from fecal samples shared 96.9–100.0% identity with each other and showed 94.1–95.1% identity with Bat-CoV HKU5 [17]. The nucleotide identity with MERS-CoV was 84.1–84.4%.
Due to shorter reported sequences from Kim et al. [14], another phylogenetic tree was constructed from the 355-bp RdRp sequences of Korean bat coronaviruses obtained by Kim et al. [14] and in this study to analyze the genetic relation between Bat-CoVs in South Korea (Fig. 3). Thirteen sequences from oral swabs were clustered with Bat-CoV B15–21, which was detected in fecal bat samples collected from an abandoned mine in Gangwon province. The major bat species present there was R. ferrumequinum, but the coronavirus-positive bat species were not specified [14]. Nucleotide identity between Bat-CoV B15–21 and the 13 clustered sequences was 97.7–100.0%. The other three Korean bat coronaviruses (Bat-CoV B15–8, B15–40, and B15–41) belonged to the alphacoronavirus genus, but we did not detect coronaviruses belonging to this group in our study.
Discussion
There are approximately 24 species of bats in South Korea, and most are insectivorous and relatively small [18]. Bats in South Korea usually do not migrate over distances greater than 1000 km [19], but apart from two species found only in South Korea, they are generally distributed in neighboring countries such as China, East Siberia, Japan, and Taiwan. Although it is not clear if bats go between China and other countries, some species that migrate over 500 km can carry pathogens to South Korea. The fact that the 18 bat coronaviruses (Bat-CoVs) detected in this study were all closely related to the Bat-CoVs identified from bats in China suggests the possibility that bat coronaviruses have been introduced from China and other countries into South Korea, or vice versa. This also can indicate the migration of bats over borders between countries.
In this study, the prevalence of coronavirus in Korean bat samples was 2.7% overall. The prevalence by sample type was 5.4% (13/332) for oral swabs, 1.8% (1/57) for carcasses, 1.6% (4/245) for fecal samples, and 0.0% (0/38) for urine samples. Reported rates from other countries for coronavirus prevalence in bats were 6.5% (64/985) and 5.3% (50/951) for China, 8.2% (16/195) for Italy, and 5.3% (32/606) for Mexico [20–23], not greatly different from our figure of 2.7% for prevalence in South Korea.
Fourteen out of 24 Korean bat species were screened for CoVs in this study. The species positive for Bat-CoVs were R. ferrumequinum, V. sinensis, P. abramus, and E. serotinus. The Bat-CoVs that showed the highest nucleotide identity with our isolates were obtained from the same species that we identified as positive, except for E. serotinus. To our knowledge, this is the first time a Bat-CoV has been detected from E. serotinus in Asia. In Europe (Italy), a lineage C betacoronavirus was detected from E. serotinus in 2014 [24].
Bat-CoVs were usually detected at higher rates in alimentary samples (fecal samples, rectal swabs, and intestine) than in respiratory samples (oral and throat swabs) [7, 15, 17, 22, 25, 26]. We collected alimentary and respiratory samples and urine samples. However, Bat-CoVs were not detected in urine samples. Animal coronaviruses usually produce either enteric or respiratory infections. Animal models show similar clinical features to SARS and MERS patients; the majority of whom presented with respiratory symptoms but in some cases also suffered from enteric complications [27]. Unlike other human coronavirus infections, a number of MERS cases were associated with acute renal failure. MERS-CoV replication in the human kidney suggests the potential of shedding viruses in urine [28]. Bat-CoV isolate Bat SL-CoV-WIV1 (KF367457) showed infectivity to Rhinolophus sinicus kidney cell lines, which means it may be possible to detect Bat-CoV in urine [6].
The current study revealed that Korean bats have SARS- and MERS-related Bat-CoVs, consistent with earlier work [14]. The previous study detected alpha- and betacoronaviruses only from fecal samples in four regions, two in Gangwon, one in Chungbuk, and one in Gyeongbuk [14]. The only betacoronavirus RdRp sequence Bat-CoV B15–21 (KU528590.1) was clustered with 13 Bat-CoVs (16BO122, 129, 132, 133, 134, 135, 139, 141, 143, 145, 150, 154, and 155) detected from Danyang, Chungbuk province. The identity between the 14 sequences was very high at 97.7–100%, even though the sample types and collection sites were different. The species of bat infected with Bat-CoV B15-21 was not specified in the paper, but the major bat species of the sample collection site was R. ferrumequinum; the same species in which we detected 13 Bat-CoVs. The same Bat-CoVs were detected from fecal samples in the prior study [14] and from oral swabs in our study. Spike proteins, which are translated from spike genes, define viral tropism by receptor specificity and by membrane fusion activity during entry into cells [29]. Although the spike genes were not identified in this study, full spike gene analysis of Bat-CoV B15-21 suggested a low potential for human emergence. One MERS-like betacoronavirus, Bat-CoV B15-1-6 (KU528586.1, KU528585.1), was identified by Kim et al. [14], but the similarity of this isolate to our isolates could not be analyzed because the RdRp sequence was not obtained. High nucleotide identity between Bat-CoV B15-1-6 and Bat-CoV-SC2013 (KJ473821) suggests that Bat-CoV B15-1-6 and 16BT3 might be very similar.
Bat-CoVs have been identified in many countries including the Australasian region [10, 11, 14–16, 20–26, 30, 31]. But Bat-CoVs identified in Korea were genetically close to Bat-CoVs from China, a close neighbor of Korea. According to the results of Bat-CoV research in China, the degree of host restriction for coronaviruses in bat populations was high. For example, similar viruses were detected in the same species of bat in different regions, approximately 1600 km apart, while two different bat species in the same habitat had different coronaviruses [22]. In our results, three Bat-CoVs (16BF109, 211, and 244) from P. abramus were clustered with each other, even though samples were collected from different habitats in different regions (Andong, Yeongdeok, and Gyeongju). The Bat-CoV detected from E. serotinus bat in the metropolitan city Gwangju (16BF104) was also clustered with these three Bat-CoVs mentioned above. However, there was no matching virus for 16BF104 found in the same species of bat in other locations, nor in other species in the region where samples were collected. Our data could support the possibility of relationships between genetic variation, geographic locations where Bat-CoVs were isolated, and bat host species, but more data on Bat-CoVs in Korea would be needed to address this.
In summary, the prevalence of coronavirus in Korean bats was determined and found to be comparable to or lower than the rates in other countries. We detected SARS-related and MERS-like Bat-CoVs that were genetically very similar to Bat-CoVs identified in China. For the study of Bat-CoV seasonal occurrence and transmission, continuous monitoring during all seasons is required, and ideally alimentary, respiratory, and other samples need to be collected from each bat. Future work to determine the complete genome sequences of Bat-CoVs from South Korea would give a more complete picture of their pathogenicity and the possibility of infectious spread to other animals and humans.
Acknowledgements
We thank Dr. C.W. Jeong and his colleagues for their efforts in the collection of wild bat carcasses and samples. This research was supported by grant no. 2016-01-01-033 from the NIER of the Republic of Korea. The funders had no roles in the study design, data collection and analysis, decision to publish, or the preparation of the manuscript.
References
- 1.Brian DA, Baric RS. Coronavirus genome structure and replication. Curr Top Microbiol Immunol. 2005;287:1–30. doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez J, Gomez-Puertas P, Cavanagh D, Gorbalenya A, Enjuanes L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch Virol. 2003;148:2207–2235. doi: 10.1007/s00705-003-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray PR, Rosenthal KS, Pfaller MA. Medical microbiology. Philadelphia: MOSBY Elsevier; 2009. [Google Scholar]
- 4.Saif LJ. Animal coronaviruses: what can they teach us about the severe acute respiratory syndrome? Rev Sci Tech. 2004;23:643–660. doi: 10.20506/rst.23.2.1513. [DOI] [PubMed] [Google Scholar]
- 5.Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir Res. 2014;101:45. doi: 10.1016/j.antiviral.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ge X, Li J, Yang X, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 8.Quinn PJ, Markey BK, Leonard FC, FitzPatrick ES, Fanning S, Hartigan P. Veterinary microbiology and microbial disease. Hoboken: John Wiley & Sons; 2011. [Google Scholar]
- 9.Shi Z, Hu Z. A review of studies on animal reservoirs of the SARS coronavirus. Virus Res. 2008;133:74–87. doi: 10.1016/j.virusres.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, AlHakeem R, Al Asmari M, Islam A, Kapoor A. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg Infect Dis. 2013;19:1819–1823. doi: 10.3201/eid1911.131172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, Drosten C, Drexler JF, Preiser W. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg Infect Dis. 2013;19:1697–1699. doi: 10.3201/eid1910.130946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moratelli R, Calisher CH. Bats and zoonotic viruses: can we confidently link bats with emerging deadly viruses? Mem Inst Oswaldo Cruz. 2015;110:1–22. doi: 10.1590/0074-02760150048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H, Yoon S, Kim D, Koo B, Noh J, Kim J, Choi Y, Na W, Chang K, Song D. Detection of severe acute respiratory syndrome-like, Middle East respiratory syndrome-like bat coronaviruses and group H rotavirus in Faeces of Korean bats. Transbound Emerg Dis. 2016;63:365–372. doi: 10.1111/tbed.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, Lau SK, Woo PC, Suen KY, Yuen KY, Guan Y, Peiris JS. Identification of a novel coronavirus in bats. J Virol. 2005;79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L, Wu Z, Ren X, Yang F, Zhang J, He G, Dong J, Sun L, Zhu Y, Zhang S, Jin Q. MERS-related betacoronavirus in Vespertilio superans bats, China. Emerg Infect Dis. 2014;20:1260–1262. doi: 10.3201/eid2007.140318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau SK, Li KS, Tsang AK, Lam CS, Ahmed S, Chen H, Chan KH, Woo PC, Yuen KY. Genetic characterization of betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J Virol. 2013;87:8638–8650. doi: 10.1128/JVI.01055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Son SW, Choi BJ. Bats. Seoul: Moonji publishing; 2001. [Google Scholar]
- 19.Fleming TH, Eby P, Kunz T, Fenton M. Ecology of bat migration. In: Kunz T, Fenton M, editors. Bat ecology. Chicago: The University of Chicago Press; 2003. pp. 156–208. [Google Scholar]
- 20.Anthony S, Ojeda-Flores R, Rico-Chavez O, Navarrete-Macias I, Zambrana-Torrelio C, Rostal M, Epstein J, Tipps T, Liang E, Sanchez-Leon M. Coronaviruses in bats from Mexico. J Gen Virol. 2013;94:1028–1038. doi: 10.1099/vir.0.049759-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lelli D, Papetti A, Sabelli C, Rosti E, Moreno A, Boniotti MB. Detection of coronaviruses in bats of various species in Italy. Viruses. 2013;5:2679–2689. doi: 10.3390/v5112679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, Li G, Dong BQ, Liu W, Cheung CL, Xu KM, Song WJ, Vijaykrishna D, Poon LL, Peiris JS, Smith GJ, Chen H, Guan Y. Prevalence and genetic diversity of coronaviruses in bats from China. J Virol. 2006;80:7481–7490. doi: 10.1128/JVI.00697-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu L, Zhang F, Yang W, Jiang T, Lu G, He B, Li X, Hu T, Chen G, Feng Y. Detection and characterization of diverse alpha-and betacoronaviruses from bats in China. Virol Sin. 2016;31:69–77. doi: 10.1007/s12250-016-3727-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Benedictis P, Marciano S, Scaravelli D, Priori P, Zecchin B, Capua I, Monne I, Cattoli G. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes. 2014;48:366–371. doi: 10.1007/s11262-013-1008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu D, Poon L, Chan K, Chen H, Guan Y, Yuen K, Peiris J. Coronaviruses in bent-winged bats (Miniopterus spp.) J Gen Virol. 2006;87:2461–2466. doi: 10.1099/vir.0.82203-0. [DOI] [PubMed] [Google Scholar]
- 26.Falcón A, Vázquez-Morón S, Casas I, Aznar C, Ruiz G, Pozo F, Perez-Brena P, Juste J, Ibáñez C, Garin I. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch Virol. 2011;156:1883–1890. doi: 10.1007/s00705-011-1057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knobler AMS, Lemon S, Mack A, Sivitz L, Oberholtzer K. Microbiology, ecology, and natural history of coronaviruses. In: Knobler AMS, Lemon S, Mack A, Sivitz L, Oberholtzer K, editors. Learning from SARS: preparing for the next disease outbreak-workshop summary. Washington DC: National Academies Press; 2004. pp. 137–172. [PubMed] [Google Scholar]
- 28.Eckerle I, Müller MA, Kallies S, Gotthardt DN, Drosten C. In-vitro renal epithelial cell infection reveals a viral kidney tropism as a potential mechanism for acute renal failure during Middle East respiratory syndrome (MERS) coronavirus infection. Virol J. 2013;10:359. doi: 10.1186/1743-422X-10-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallagher TM, Buchmeier MJ. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279:371. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith C, de Jong C, Meers J, Henning J, Wang L, Field H. Coronavirus infection and diversity in bats in the Australasian region. EcoHealth. 2016;13:72. doi: 10.1007/s10393-016-1116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dominguez SR, O'Shea TJ, Oko LM, Holmes KV. Detection of group 1 coronaviruses in bats in North America. Emerg. Infect Dis. 2007;13:1295. doi: 10.3201/eid1309.070491. [DOI] [PMC free article] [PubMed] [Google Scholar]