

Abstract
Infectious diseases remain a formidable challenge to human health, and understanding pathogen evolution is crucial to designing effective therapeutics and control strategies. Here, we review important evolutionary aspects of HIV infection, highlighting the concept of selection at multiple spatial and temporal scales. At the smallest scale, a single cell may be infected by multiple virions competing for intracellular resources. Recombination and phenotypic mixing introduce novel evolutionary dynamics. As the virus spreads between cells in an infected individual, it continually evolves to circumvent the immune system. We discuss evolutionary mechanisms of HIV pathogenesis and progression to AIDS. Viral spread throughout the human population can lead to changes in virulence and the transmission of immune-evading variation. HIV emerged as a human pathogen due to selection occurring between different species, adapting from related viruses of primates. HIV also evolves resistance to antiretroviral drugs within a single infected host, and we explore the possibility for the spread of these strains between hosts, leading to a drug-resistant epidemic. We investigate the role of latency, drug-protected compartments, and direct cell-to-cell transmission on viral evolution. The introduction of an HIV vaccine may select for viral variants that escape vaccine control, both within an individual and throughout the population. Due to the strong selective pressure exerted by HIV-induced morbidity and mortality in many parts of the world, the human population itself may be co-evolving in response to the HIV pandemic. Throughout the paper, we focus on trade-offs between costs and benefits that constrain viral evolution and accentuate how selection pressures differ at different levels of selection.
Electronic supplementary material
The online version of this article (doi:10.1007/s00109-012-0892-1) contains supplementary material, which is available to authorized users.
Keywords: HIV, Evolutionary dynamics, Drug resistance, Immune escape, Virulence, Coevolution
Introduction
Over the course of the twentieth century, human life spans increased dramatically in many parts of the world. This reduction in mortality is largely attributed to a reduced burden of infectious diseases, due to improved nutrition and sanitation, and the introduction of both antibiotics and vaccines. We were so confident of our domination over the microbial world that in the mid-twentieth century it was common to surmise the end of infectious diseases as a significant health issue [1–3]. This sentiment turned out to be especially ill timed as the last few decades saw the emergence of many novel and extremely dangerous pathogens, including Ebola, SARS, Lyme disease, Legionella, and drug-resistant malaria and tuberculosis. Perhaps no disease shattered this view as much as the outbreak of the human immunodeficiency virus (HIV) and the acquired immune deficiency syndrome (AIDS) that it causes. Reaching 34 million currently infected worldwide [4], a prevalence of 0.3 % in the USA [5], and near 100 % untreated fatality rate, the HIV pandemic exemplifies humanity’s continued vulnerability to pathogens and highlights how much we have yet to learn about infectious diseases.
Evolution has been used to understand human disease processes from allergy to cancer to cystic fibrosis, reviving the field of evolutionary medicine [6, 7]. Nowhere is the connection between evolution and medicine more evident than in the field of infectious diseases. Although we have recently added vaccines and chemotherapy to our arsenal of defenses against microorganisms, augmenting the protection offered by our innate and adaptive immune responses, it is now accepted that the rapid rate of pathogen evolution ensures infectious diseases will inevitably remain important health concerns. Therefore, understanding the evolutionary processes relevant to infectious diseases is absolutely necessary to prevent, control, treat, and ultimately minimize the damage to human health due to pathogens.
HIV infection is a particularly well-suited example to highlight the medical relevance of evolutionary dynamics occurring simultaneously on multiple spatial and temporal scales (Fig. 1), including the evolution of the virus within an individual and at the global level, as well as the coevolution between HIV and the human population. HIV originated from a cross-species transmission of simian immunodeficiency virus (SIV), and it eventually evolved adaptations facilitating productive infection in, and transmission between, human hosts. At the individual level, the virus evolves in response to a dynamic, variably hostile environment presented by the immune system and drug treatment. At the population level, HIV likely evolves to evade immune control, optimizing virulence levels, and possibly transmitting drug resistance. Creation of a vaccine is hindered by rapid and unpredictable evolution of the virus, and if introduced, may further drive viral evolution, potentially causing resistance to the vaccination. Due to the strong selective pressure exerted by HIV-induced morbidity and mortality in many parts of the world, evolution of human populations in response to the virus is likely.
Fig. 1.
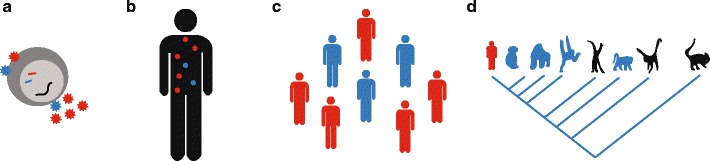
HIV evolution occurs on multiple spatial and temporal scales. a Multiple virions may infect a single cell, competing for cellular resources controlling viral replication. Budding virions may contain RNA, structural proteins, and enzymes from various genetic backgrounds. b During infection of a single individual, HIV diversifies into multiple strains that compete to infect CD4+ T cells and evade immune responses and drug treatment. c As HIV spreads through the human population it may adapt, becoming more or less virulent and accumulating mutations selected by immune responses or even widespread drug use. d HIV arose from a cross-species transmission of simian immunodeficiency virus (SIV), known to infect many primate groups. Primate phylogeny, from left to right: humans, chimpanzees, gorillas, orangutans, gibbons, old world monkeys, new world monkeys, prosimians
In this review, we will summarize the range of evolutionary processes important to HIV and highlight how understanding them can help guide prevention, control, and treatment of AIDS. We first summarize the natural history of the epidemic and the host-driven selective pressures that the virus faces regardless of treatment. We then consider the effect of medical interventions on viral evolution, focusing on recent efforts to understand and quantify the mechanisms that can render treatment ineffective.
Natural history of the HIV pandemic
High retroviral mutation rate facilitates rapid evolution
Evolution requires the generation of variation through mutation. The capacity for HIV to evolve exceeds that of many other pathogens, due in part to the high mutation rate of retroviruses. At ~3 × 10−5 substitutions per base per replication cycle [8] and a genome size of 9,800 base pairs, one in every three replication events is expected to create a mutated genome. This puts the virus very near the error catastrophe threshold: at mutation rates higher than approximately one per genome per replication cycle, it is not possible for a genome to accumulate and maintain beneficial alleles, and a lineage risks extinction [9–12] (Section 3.1.8 of [11]). While the high mutation rate of retroviruses is usually explained by the fact that their virally encoded reverse transcriptase lacks error-correcting domains found in other organisms, it is currently unknown how much of this mutation rate is due to HIV reverse transcriptase and how much is due to host cell RNA polymerase since experiments to date cannot separate out these two steps. High mutation rates are expected to be favored more often in heterogeneous or rapidly changing environments than in stable ones[13], and due to the chronic nature of HIV infection and its ability to reach many tissues, it is possible that its high mutation rate has been favored by natural selection. Recent theoretical modeling work demonstrates that host–pathogen coevolution may also generate selective pressure for high pathogen mutation rates, even in a static, homogeneous environment [13, 14]. Interestingly, mutation in HIV seems to be strongly skewed towards adenosines, much more than is represented by the adenosine bias in its sequence composition [15, 16]. The high mutation rate of HIV leads to tremendous variation in a given viral population; with up to 10 % sequence variation between strains within an individual and up to 50 % between different types circulating over the globe (Fig. 7.10 Section 7.2 of [11, 17]).
Humans and other primates actually have an innate defense targeted towards the high mutation rate of retroviruses. The APOBEC3G enzyme edits the viral cDNA during reverse transcription, likely as part of a host defense mechanism. HIV encodes a protein Vif that facilitates polyubiquitylation and subsequent proteasomal degradation of APOBEC3G, but in the absence of Vif, the enzyme causes sufficient mutation to scramble the viral genome, triggering an “error catastrophe” and preventing productive infection [18–20].
In addition to mutation, the potential of the virus to recombine during replication is high as reverse transcriptase typically jumps templates about ten times per genome [21]. If two distinct viral strains infect the same cell (the likely frequency of such an event is discussed in a later section on within-cell competition), then each one may contribute to one of the two RNA copies in budding virus. When this virion infects a new cell, recombination between the two parental genomes may then occur. Many strains of HIV currently circulating the globe appear to be recombinant forms of divergent virus strains [22].
The emergence of HIV
As HIV spreads between individuals in a population, selection occurs on traits that maximize its transmission. HIV is a zoonotic disease, meaning it had its origins in a closely related infection of animals; in this case, non-human primates. Analysis of molecular phylogenies has traced the origin of HIV-1 (for example, see Section 7.2 in [11]), the viral strain responsible for the largest global epidemic, to the simian immunodeficiency virus infecting chimpanzees [23]. Similar cross-species jumps have happened with other primate species: a smaller epidemic of HIV restricted mainly to western Africa is caused by HIV-2, with origins in the virus of sooty mangabeys [24].
Adaptation of any disease to a new species proceeds through five phases (adapted from Chapters 12 and 16 of [6]): Firstly, there must be contact between the animal reservoir and humans that allows transfer of the pathogen. For SIV, this is hypothesized to have occurred through hunting for and preparing primate bushmeat. The second phase of adaptation—the ability to produce a productive infection in the new human host—is calculated to have occurred at the turn of the twentieth century near Kinshasa in the present-day Democratic Republic of Congo [25]. However, both these first and second phases have occurred more than once: humans in some central African regions may have antibodies to multiple simian viruses [26] (suggesting primary transmissions may be frequent), and the ancestry of HIV-1 subtypes implies distinct human–chimpanzees zoonoses (Section 7.2 in [11]). SIV infection of humans is not able to generate high viral loads needed for transmission. The third phase is for the disease to become transmittable between humans. However, to cause an epidemic, the pathogen must be suitably adapted to the new host so that every infected individual passes the disease on average to at least one other individual [27, 28]. HIV has been in this fourth phase of emergence for the past three decades. This phase may be facilitated by viral adaptations, or by changes in the host population that increase transmission [29], such as higher population density and increase in behaviors that facilitate transmission. The latter is believed to be an important factor for HIV, potentially explaining the delay between the evolution of HIV’s ability to productively infect humans, and the beginning of the global epidemic.
Determining the origins of HIV is not just for scientific and historical interest—hopes are that it could be key to determining how to control the disease. SIV infections are common in many species of primates, and related CD4-tropic lentiviruses are found in many other mammals, with current evidence pointing to a lentiviral origin over 7 million years ago [30]. Most SIV infections in their native hosts do not lead to AIDS-like immunodeficiency illness. Viral loads may be sustained at very high levels, but CD4+ T cell levels remain high. These findings have strongly suggested that HIV pathogenesis is driven by more than direct killing of target cells by the virus [31]. Chronic immune activation is emerging as a potential mechanism for HIV pathogenesis; in natural SIV infection, immune activation is only transient [32]. In SIV infections, there are also strong cellular immune responses that seem to recognize highly conserved regions of the viral genome [33, 34]. The recency of successful immune control and avirulence of SIV in natural host populations is a topic of debate. Rapid host-virus coevolution towards reduced pathogenicity would result in a selective sweep, which would likely be detected by molecular phylogenetic methods as a relative absence of lineages that coalesce prior to the emergence of avirulence [35, 36]. Molecular clock-based techniques suggest that the most recent intraspecies common ancestors of SIV in sooty mangabeys and chimpanzees, respectively, existed just 200 and 500 years ago, consistent with very recent evolution of an avirulent infection in both viral strain [35]. Biogeographical evidence gathered from comparisons of island and mainland viral strains in west Central Africa, however, suggests that SIV evolution proceeds at a rate two orders of magnitude slower than HIV evolution, implying a corresponding increase in the time since a common ancestor [37]. Further supporting this analysis, phylogenetic reconstruction that is unconstrained by geological or historical evidence may systematically underestimate branch lengths when lineages are subject to strong purifying selection, as is generally the case for RNA viruses [38].
While viruses represent only about 15 % of all human pathogens, the majority of emerging diseases are RNA viruses, with high mutation rates (Chapter 16 of [6]). Emerging diseases tend to have reservoirs in animals, and they tend to be adapted to a broad range of hosts prior to arrival in humans. By examining likely patterns of cross-species transmission in HIV/SIV and other widespread RNA viruses [36], we may uncover clues that allow us to identify pathogens at risk for jumping the species barrier into humans.
Frequent viral recombination presents a major obstacle to an accurate understanding of HIV origins. Indeed, the entire scheme of classifying the main group of HIV-1 into nine subtypes has recently been called into question by evidence that subtype G may actually have resulted from recombination between subtypes A and J [39]. The occurrence of recombination, which requires infection of a cell by two strains and so should grow roughly as the square of viral load, may mirror the situation in some bacterial species, in which the frequency of horizontal gene transfer depends on population density [40]. The recent growth and success of “species tree” approaches in molecular systematics provides a powerful method for disentangling the discordant evolutionary histories of multiple loci that are brought together by recombination (reviewed in [41]). Though useful in metazoic taxa, these approaches have not been applied to virus evolution; they require the partitioning of the genome into discrete loci, with recombination occurring between, but not within, loci [41, 42]. The frequency of recombination in HIV may be prohibitively high, and therefore the “effective locus size” prohibitively small, in order to import these methods wholesale to the study of its evolutionary history.
Is HIV evolving to be less virulent?
The fifth and final phase of emergence of a new infectious disease is adaptation of the pathogen to optimize transmission between hosts and the switch from an epidemic phase (rapidly increasing prevalence) to an endemic one. Although disease prevalence may remain approximately constant in this phase, the system is by no means static—both pathogen and host populations can evolve continually to circumvent each other, in an evolutionary arms race (also known as “Red Queen” race) [43]. The infectivity of a strain is defined as the rate of infection of susceptible individuals, and the virulence is usually defined as the increased death rate that infected individuals suffer due to the virus. At first glance, it seems that it would be optimal for a disease to evolve to both maximize its infectivity and minimize its virulence in a host, the latter allowing the host to live longer and infect more people. However, there are often trade-offs between these two parameters that complicate the optimization of transmission—for example, high numbers of pathogens within a host may be necessary for infectivity but may contribute to virulence [44]. As well, there are clear conflicts between the role of within-host evolution and population level evolution on virulence-related traits. Competition within a host selects for the fastest replicating strain, which could be more virulent, while at the population level, an intermediate replication rate that maximizes transmission may be favored [45, 46]. Even within a host there is selection to control the death rate of infected cells to maximize transmission between cells. Bottlenecks that occur at transmission may to some extent “reset” viral evolution between hosts [17], with the result being that within-host selection may have little effect on the long-term, population level evolution of HIV. However, recent studies have shown that in the case of HIV, there is heritable variation in virulence (measured by viral load) between 20 and 60 % (reviewed in [47]). There is no clear consensus as to whether HIV is evolving at a population level to become less virulent (summarized in [17, 48]), although a recent meta-analysis has concluded that virulence has actually increased over the past three decades [49]. A recent paper modeling the evolution of HIV virulence based on known viral load–transmission rate and viral load–mortality relations concluded that HIV has already reached the optimal virulence to maximize transmission between hosts [48].
Does competition within a single cell drive evolution?
In HIV infection, there is potential for multiple genetically distinct virions to infect a single cell [50]. This results in a new level of selection for the virus which we call within-cell (as opposed to within-host and population level), which introduces novel viral dynamics. Firstly, competition for resources related to viral replication within the cell can select for an increased production rate of virions and hence greater cytopathicity [51]. Secondly, when multiple virions infect the same cell, the new virions created will have a mix of genetic material from both strains and structural proteins and enzymes (determining “phenotype”) also from both strains. This is termed phenotypic mixing [52]. As a result, deleterious mutations may persist at a higher frequency than expected by mutation–selection balance since they can be shielded by the wild-type phenotype [52, 53]. Virions carry two strands of RNA genome, which may be from different strains. When this virion infects a new cell, these genomes may recombine as reverse transcriptase often jumps between RNA strands [21]. Recombination alters the spread of viral genotypes maintained by mutation and changes the error catastrophe threshold [54]. Together, the effects of co-infection have been shown in models to accelerate the rate of immune escape [55] but may either promote or hinder the development of drug resistance, depending on whether multiple mutations display synergistic or antagonistic epistasis [56]. Importantly, a prerequisite for these dynamics to affect the genetic composition of the population is the existence of a significant number of cells co-infected with genetically distinct virions. Some studies claim that co-infection with genetically distinct virions happens quite frequently in the lymph tissue [57], but much lower rates (1–10 %) have been found in the peripheral blood [58] and have been estimated from effective recombination rates measured from patient sequence data [59, 60].
HIV pathogenesis involves immune-directed evolution
Despite three decades of research on HIV, its mechanism of pathogenesis is still not entirely clear. Evolutionary processes are implicated to a large extent in the hypothesized mechanisms of disease progression [61–64]. It is well understood that over the course of a single infection, HIV continually evolves to circumvent host defenses. HIV infects CD4+ T lymphocytes, also known as helper T cells, using the receptors that they uniquely express to gain access to the cell. After initial HIV infection, patients experience very high viral loads and occasionally the flu-like symptoms of acute viremia, after which viral load declines to a comparatively low level called the “set point,” where it may remain for many years. This decline occurs both due to the limitation of uninfected target CD4+ T cells [65] and by the appearance of cellular immune responses against HIV [66].
Immune control of HIV is largely due to CD8+ T cells [67–69] (also called “killer” T cells or cytotoxic T lymphocytes (CTLs)), though antibodies [70] and innate immunity [71] also play a role. Small virus-derived peptides called epitopes are presented on the surface of infected cells by the human leukocyte antigen (HLA) class 1 molecules, which then allows CTLs to recognize and kill these cells. Throughout the course of chronic infection, which may last from a few years to a decade, viral diversity and fitness gradually increase [17, 72, 73]. Antigenic diversity also increases, as HIV continuously generates escape mutations in CTL epitopes, preventing them from being presented or recognized [63, 72, 74–76]. About 2/3 of mutations acquired over the course of HIV infection have been attributed to CTL selection pressure [77]. These mutants are subsequently selected and may grow to a high frequency. The immune response is further weakened by the fact that HIV is infecting and destroying cells involved in immune control.
While evolution is typically thought of as an open-ended process that may follow many paths, certain aspects of HIV evolution are very predictable. For a given HLA allele present in an individual, certain escape mutations in epitope regions are highly consistent between patients [78, 79]. Another predictable evolutionary event is a switch in the virus’s machinery for entering its target cells (reviewed in [80, 81]). HIV enters cells by binding to the CD4+ cell surface receptor, but also requires a co-receptor. Early in infection, the viral population seems to be composed of strains that preferentially use the CCR5 co-receptor, termed “R5” virus. Later on in infection, about 50 % of patients experience a switch in the tropism of the dominant virus population to an “X4” virus that instead uses another co-receptor, CXCR4, for entry. The factors determining this switch are not completely understood. It seems that R5 viruses have a selective advantage early in infection and dominate the viral population after sexual, mother-to-child, or direct blood-to-blood transmission, though individuals homozygous for the Δccr5 mutation can be infected with X4 virus [82]. Later in infection, selection pressure seems to change to favor X4 virus. This is hypothesized to be a result of the change in the population composition of T cells over the course of chronic infection. X4 virus seems to be more susceptible to antiviral immunity but able to infect a broader range of target cells, including resting T cells, and so as immune function declines, it may outcompete R5 viruses. The reason only 50 % of patients experience this switch is unclear, though could be a result of the chance accumulation of multiple mutations required to make the tropism switch. Patients with X4 virus tend to experience more rapid decline in CD4+ T cells, and so this phenotype switch may be facilitated by immune deficiency but then lead to accelerating deficiency. The co-receptor switch is less common in treated patients [83].
Mathematical models have demonstrated that the accumulation of escape mutations can explain the clinical course of HIV infection [61, 84, 85] (Fig. 2). Viral diversity increases as HIV progressively escapes CTL responses, resulting in higher viral load, and due to HIV-induced damage of CD4+ T cell populations, a progressively weaker immune response. There is an asymmetry in the HIV–T cell interaction since any strain of HIV can infect and damage any CD4+ T cell, but each CTL can only target the HIV strain it is specific for. Eventually, infection reaches a point where the immune response is unable to control the variable strains of HIV, and viral loads rapidly increase as CD4+ T cell levels fall. This is called the “diversity threshold,” and could explain the onset of the clinical symptoms of immune deficiency, called AIDS. This theory highlights that the two specific features of HIV, which make it especially harmful to the host, are the fact that its target cells are involved in orchestrating the immune response and its high rate of evolution in a single infected host.
Fig. 2.
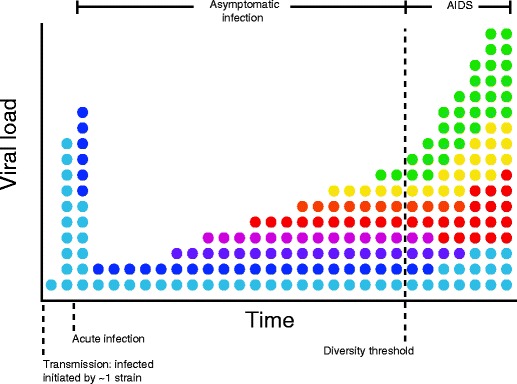
Hypothetical evolutionary mechanism for HIV pathogenesis. Colors schematically represent diversity of viral strains. Bottlenecks during transmission lead to acute infection growing from approximately a single viral strain. Viral loads peak and decline rapidly during acute infection, with diversity remaining low. During asymptomatic infection, which may last from months to decades, viral loads remain near a lower set point. Diversity gradually increases, potentially as mutants that escape cytotoxic T cells are generated. At a certain point, helper (CD4+ T cells) reach a critically low level, due to direct and indirect effects of HIV, leading to a rapid rise in viral load and progression to the clinical symptoms of AIDS. Mathematical models suggest that crossing a diversity threshold could cause progression to disease. At the final stages of disease, diversity begins to decrease, possibly due to reduced selection pressure from a failing immune system. Figure adapted from [17, 169–172]
Despite its elegance, the evolutionary model for HIV pathogenesis may not be the whole story. Various lines of evidence suggest that CTL responses are important for viral control, including the observation that the selective advantage of escape mutations correlates with rate of disease progression (specifically, the portion of this rate that is statistically associated with particular HLA alleles) [86]. However, it has been observed that only a small fraction of CD4+ T cells are infected with HIV, making it difficult to explain how either direct viral killing or CTL killing, for which death rates have been measured, could be responsible for such a large decline in their numbers. During infection, there is a large increase in the death rate of uninfected (termed “bystander”) CD4+ T cells. Multiple, though not mutually exclusive, mechanisms have been proposed to explain this observation, including (1) direct cytotoxicity of free-floating HIV proteins on uninfected cells, (2) an increase in apoptotic signals, or (3) an increase in the fraction of activated, short-lived T cells due to HIV-induced immune activation [81, 87, 88]. While the importance of each of these mechanisms remains to be determined, it is interesting to note that models for many potential pathogenic processes have found that the extremely slow decline of CD4+ T cells is difficult to explain by any process that does not, at some level, involve gradual evolution and an accompanying change in the balance between the virus and host defenses [89]. However, evolution could be either viral or related to somatic evolution of T cells: an increased turnover rate of cells could lead to the accumulation of deleterious mutations and eventual senescence of immune cells [90]. The ability of HIV to induce immune activation may itself be subject to selection. The direction and strength of selection depend on the trade-off between creating more target cells and increased virulence. However, the ability to cause immune activation is only under selection if it preferentially changes the fitness of a particular viral genotype, that is, if it is local. The ability to cause systemic immune activation affecting all viral genotypes is a selectively neutral trait [91].
Clues to the virus’s mechanism of harm can be deduced from a rare group of patients who become infected with HIV, but maintain lifelong low viral loads and do not seem to progress to AIDS [92]. Existence of these “elite controllers” suggests that immune control of the virus is possible in humans, though how this control works is still under investigation. This subpopulation of individuals is enriched for certain HLA alleles, and their CTL responses tend to have higher HLA avidity, proliferate more, kill better, and preferentially recognize epitopes from the HIV capsid protein Gag (reviewed in [93]). Immune activation levels remain relatively low in elite controllers. Despite very low viral loads, they do seem to experience ongoing viral replication, and even continual evolution, including escape mutations [94]. How then, do they avoid increasing viral loads and decreasing immune function? When HIV mutates to avoid immune recognition, these mutations often harm native functions of the virus, leading to a fitness cost (defined as a reduction in replication rate as compared to a wild-type virus in a laboratory environment, free of immune response). The selective advantage of an escape mutant depends on a trade-off between the benefit attained from escaping CTL killing and the fitness cost of the mutation, and it in elite controllers, the cost of escape may be so great and the advantage so low that viral loads remain approximately constant [95–97]. The “diversity threshold” model of HIV pathogenesis mentioned earlier does include specific parameter regimes where indefinite control of the virus occurs [98], but it has yet to be determined if these parameter regimes are quantitatively consistent with elite controllers.
The ability of HIV to rapidly escape from immune control has important implications for vaccine development. There are currently effective vaccines for many diseases, including measles, smallpox, and polio. For each of these diseases, a single natural infection provides lasting immunity. Vaccine-induced immunity is similarly effective. In contrast, individuals infected with HIV can be superinfected with other strains and effective immune control is rarely achieved. Designing an HIV vaccine requires first determining the unknown correlates of immune control. Such a vaccine will have to elicit immune responses to an extremely diverse viral population. All currently used protective vaccines elicit antibodies, which until recently were believed to be generally ineffective in controlling HIV, leading to much focus on CTL-based vaccines [99]. However, recent years have seen advances in the characterization of “elite neutralizers,” individuals capable of naturally controlling HIV infection with potent broadly neutralizing antibody responses, which has renewed confidence in the future of antibody-based vaccines (reviewed in [100]). This strategy is complicated by the finding that elite neutralizers can produce these antibodies only after extensive somatic mutation of B cells, suggesting that a vaccine may have to elicit the “ancestral” antibodies and then direct evolution through particular affinity maturation pathways. Studying the mechanisms of immune control in elite controllers and elite neutralizers will likely continue to be an important step in research towards a vaccine [101].
HIV’s ability to evade immune responses also emphasizes the importance of trade-offs in evolution [102]. Overall, HIV and other lentiviruses have evolved an infection strategy that evades many defenses of the immune response, allowing the virus to persist and establish a chronic infection lasting many years. One of the particular adaptations enabling this strategy is the arrangement of the cell-binding proteins in the viral envelope. These proteins are placed so that conserved regions are inaccessible to antibody recognition, explaining the scarcity of broadly neutralizing antibodies against HIV. However, as a result, HIV’s ability to infect cells is compromised. In immune-free cell culture, strains quickly evolve which have higher replication rates but are more susceptible to antibodies. Other viruses infecting humans take a different strategy; replicating very quickly and relying on most transmission to occur before the host mounts an immune response, which will then likely clear the infection. This trade-off between the “acute infector” and “persistor” viral lifecycle strategies is reminiscent of life history theory [103, 104], which posits that trade-offs constrain fertility and that organisms respond to dynamic environments with a variety of reproductive strategies, e.g., producing few offspring and investing heavily in each, or producing many offspring each with a low chance of survival.
Is HIV evolving at the population level response to human immune control?
There is evidence that HIV may be evolving at the population level, evading immune responses characteristic of specific human subpopulations. Escape mutations prevent the virus from being recognized by a certain individual host’s immune response, and when the virus is transmitted to another host, they may revert back to wild type [105] or be maintained [74]. Studies have shown that in various populations, the prevalence of escape mutations in particular epitopes was highly correlated with the prevalence of the HLA allele presenting that epitope, even when only individuals without the allele were considered. Particular escape mutants also appeared to be increasing in prevalence over time [106, 107]. It has been suggested that in populations where there are HLA alleles common to many individuals, CTL escape mutations may be retained upon transmission and contribute to increasing virulence of the epidemic, while in populations with high HLA diversity (many African populations), it is more likely for virulence to decrease [17].
Host–pathogen coevolution: are humans evolving in response to HIV?
We have so far discussed evolution of HIV in response to selection pressure imposed by host immune defenses and by potential vaccines. It is however also possible that the human population may be evolving in responding to the virus. Given the high prevalence of HIV in certain regions, and the severe decrease in reproductive success incurred by untreated HIV-positive individuals, a strong selective pressure for protective alleles is expected and changes in allele frequency should be apparent in several generations. Since severe disease burdens have only existed for one to two generations so far, these changes may not yet be detectable.
Studies have identified various sources of natural genetic variation to HIV infection in humans [108–110]. Heterozygosity at the HLA loci, and certain HLA alleles, especially at the HLA-B locus, are strongly related to slower disease progression. Two cellular proteins involved in innate antiviral immunity show strong signatures of positive selection in human populations, and polymorphisms have been implicated in differential susceptibility to HIV infection. TRIM5α interferes with the uncoating of the viral capsid upon entry into a host cell, and APOBEC3G, as discussed earlier, can edit viral cDNA during reverse transcription, leading to hypermutation of viral genomes. Variation in cytokine loci is also implicated in HIV control. About 1 % of the European population is homozygous for a 32-base pair deletion in the CCR5 gene (Δccr5), rendering them resistant to infection by HIV, which requires the CCR5 protein as a co-receptor during initial infection. To the extent that these polymorphisms are prevalent in regions with a high burden of HIV-induced mortality, their frequency may change in response to the epidemic.
HIV evolution in response to medical interventions
How might an HIV vaccine drive population level virulence?
Attempts to make a vaccine against HIV have so far been unsuccessful, largely due to the enormous genetic variation of HIV, both within an individual, starting early in infection, and worldwide. Due to the fact that HIV is only rarely controlled by antibodies, which are induced by most vaccines to date, and that the virus quickly establishes long-lived reservoirs in latently infected cells, it has been suggested that a potential vaccine may not be able to prevent infection, but may instead only lead to a reduced viral load set point [111]. Theory suggests that vaccines that completely prevent infection could lead to reduced virulence, either by directly targeting virulence factors (as is the case with the diphtheria vaccines), or by reducing co-infection and hence the strength of within-host competition, which often selects for high replication rates and high virulence [46, 112]. The latter effect is likely to be relevant in certain high-risk populations where infection with multiple strains is common, though in general multiple infections are a rare occurrence [113]. However, “leaky” vaccines that allow some infection but reduce host death rate, like that proposed for HIV, can alter the virulence/infectivity trade-off, making increased virulence less costly (less chance of host death) and more beneficial (to achieve high transmission in face of reduced pathogen titers), and therefore lead to evolution of increased virulence [112]. Virus may also escape vaccine control, similar to escape to naturally occurring immune responses, with unknown consequences for virulence.
HIV evolves resistance to antiretroviral drugs
In the developed world, deaths from AIDS have decreased dramatically since the introduction of highly active antiretroviral therapy (HAART) in the late 1990s [114]. By targeting multiple HIV proteins with drug combinations at high doses, HAART minimizes the likelihood of developing resistance. However, these drugs remain prohibitively expensive and inaccessible to most HIV-infected individuals, and suboptimal adherence and drug resistance remain a problem worldwide. At first, we consider the case of resistance evolving de novo, in an individual initially infected with drug-sensitive HIV; later, we consider the less frequent case where resistance is transmitted.
Like the host immune response, antiretroviral drug treatment provides a strong selective pressure on the virus, and over the course of a single individual’s infection, HIV can evolve drug resistance. Unlike the immune response in a typical individual, drug treatment easily results in a negative growth rate for the virus, limiting ongoing viral replication and slowing the rate of evolution. Resistance mutations can arise from one of three sources [16]. Firstly, if treatment does not completely suppress all viral replication, new mutations may arise during treatment from the residual viral replication. This likelihood of generating resistance depends on the strength of the drug, measured by the fraction of viral replication events prevented by the drug at a given concentration. Secondly, viral loads are generally quite high when treatment starts, and mutations may preexist in the viral population with some low frequency termed mutation–selection balance [115, 116], which is determined by the mutation rate and the fitness cost of the mutation in the absence of treatment. Thirdly, occasionally HIV-infected CD4+ T cells revert to a resting state, bringing with them integrated HIV in their genome. These cells comprise the latent reservoir and may remain in a resting state for many years, unaffected by drug treatment, which only inhibits active viral replication. Cells in the latent reservoir harbor a representative sample of viral genomes that have existed in the plasma over the course of infection. Mutation frequencies in the reservoir are likewise determined by mutation–selection balance, and resting cells may reactivate randomly at any point during drug treatment, reseeding the infection with both wild-type and mutant viruses.
In order for clinical drug resistance to emerge, resistant viral strains must be selected for. Viral fitness is determined by drug concentration and typically follows a sigmoidal dose–response curve with a variable IC50 and slope [117]. Mutant viruses have altered dose–response curves and benefit from having a higher IC50 or lower slope. Resistant mutants also tend to carry a fitness cost, meaning that in the absence of the drug, their fitness is lower than the wild-type virus [118]. As a result, there is only a certain range of intermediate drug concentrations, termed the mutant selection window (MSW), where any particular mutant is selected over the wild type (Fig. 3a). At lower concentrations, the wild type is favored, and at higher concentrations, even mutant fitness is so low that the growth rate is negative. No HIV mutants have been characterized that lack susceptibility to any drug concentration, though many may only be controlled with concentrations that are not clinically achievable due to toxicity [118]. Even when concentrations fall within the MSW, the favored mutant may still be lost to random drift, with a probability depending on the population dynamics of the infection. Heterogeneity and fluctuations in the host environment during treatment generally increase drift, making a favored mutant less likely to establish a persistent lineage (see Box 1).


Fig. 3.
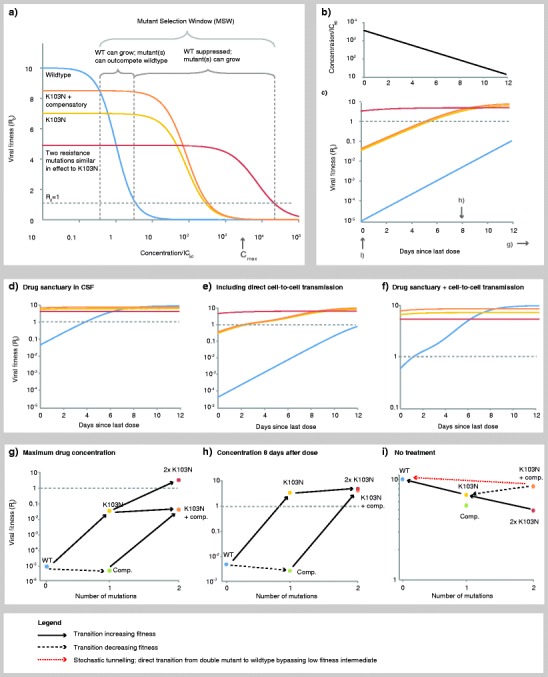
Drug-dependent fitness landscape for the antiretroviral drug efavirenz derived using measured pharamacodynamic and pharmacokinetic parameters for wild-type and the K103N drug-resistant strain. Viral growth rate is positive only when R 0 >1 (above gray dotted line). a Fitness of wild-type and resistant strains follow dose–response curves, resulting in a mutant selection window where resistant strains are selected. We compare a single mutant (K103N) to two hypothetical double mutants by adding either a second equivalent resistant mutation (further increases IC50 and decreases slope) or a compensatory mutation (changes fitness cost only). b Drug concentrations decay over time according to drug half-life. c Relative fitnesses of wild-type and resistant strains consequently change over time as drug decays. d In the CSF, reduced drug penetration results in concentrations reduced to 0.5 %, allowing viral replication at higher systemic drug levels. e Assuming about 10 % of cells are susceptible to direct cell-to-cell transmission, which typically occurs with around 100 virions and is sustainable with a single virion. f Combining the effects of (d) and (e). g At the maximum clinical drug concentration (immediately following a dose), only the doubly resistant mutant can grow. h At intermediate concentrations, the single resistant mutant can grow, facilitating evolution of the compensated mutation or the untreatable doubly resistant mutant. i Without treatment, the wild type is favored. While the doubly resistant mutant can stepwise revert to wild type, the compensated mutant is less likely to do so due to the presence of a fitness valley at either of the intermediate single mutants. See Appendix for methods
Drug concentrations within the MSW may inhibit wild-type growth but select for resistance. Drug concentrations are not constant in a given individual, but fluctuate due to the dose taken, the drug bioavailability and half-life, and the patient’s adherence to medication (Fig. 3b). Consequently, selection pressures also fluctuate (Fig. 3c). Drug concentrations that are high, yet still within the MSW, may slow the rate of emergence of resistance since ongoing viral replication is greatly reduced and resistant mutants can only arise if they are preexisting (either in the actively infected cells or in the latent reservoir).
Keeping drug levels above the MSW is required to guarantee the resistant mutant will not be selected for. If these concentrations are clinically possible, then this particular mutant alone is not an insurmountable threat since if it arose, it could be eradicated by ensuring that drug concentrations exceed the MSW for sufficient time. However, clinically relevant resistance may require more than a single mutation. If a single resistance mutation arises, it may facilitate the emergence of stronger, untreatable strains with multiple mutations (Fig. 3g), or with compensatory mutations, which reduce the fitness cost of the first mutation. It is possible that these strains will then be untreatable at maximum clinical drug concentrations, in which case they will lead to treatment failure.
Most current first-line antiretroviral regimens include a drug that can reduce viral replication by at least five orders of magnitude, even at minimum clinical plasma concentrations [117, 129], and so it is likely that multiple factors acting in concert are typically required to provoke treatment failure. Three main environmental factors can conspire to enable viral replication despite ongoing treatment. First, drug concentrations may temporarily drop, either when the concentration reaches a minimum prior to a scheduled dose or when the patient fails to adhere to the regimen. Certain drugs, notably the protease inhibitors, have very sharp dose–response curves, meaning that despite having the benefit of requiring lower concentrations to achieve the same inhibition, have the disadvantage that a small decline in drug concentration can lead to a large drop in viral suppression [117]. Secondly, replication rates are increased in tissues such as lymph nodes that are densely populated with target cells. In particular, multiple infections per cell (for example, by local spread of virus or direct cell-to-cell transmission via virological synapses) may occur in such tissues [130, 131], although the evidence for a high multiplicity of infection in vivo is limited [57, 58]. The presence of even a small collection of cells vulnerable to this mode of transmission may suffice to dampen a drug regimen’s inhibitory effect by an order of magnitude [132] (Fig. 3e, f).
Finally, drug concentrations in certain anatomical compartments may be a small fraction of plasma levels. The concentration of efavirenz in the CNS, for instance, is typically only 0.5 % of the plasma level [133] (Fig. 3d, f). Infected monocytes may introduce the virus to the CNS or other drug refuge, thereby initiating a locally blooming infection, despite successful viral suppression elsewhere in the host [133–135]. For example, on the basis of a simple calculation using dose–response curves measured in vitro, we estimate that wild-type (drug-susceptible) virus can generate a self-sustaining infection in the CNS if target cells are also capable of direct cell-to-cell transmission [132], in the presence of efavirenz therapy just below the clinical minimum drug concentration (Fig. 3d). A single missed or delayed dose would allow concentrations to dip below this minimum level, allowing the infection to grow locally. If a resistance mutation occurs every 104 replication events, then a transient growth in viral load to detectable levels (~50 copies/ml, or 25 virions/ml) confined to the CNS (~200 ml), experiencing daily turnover [124], should generate a resistant mutant every other day. This explains why resistant strains may evolve in vulnerable anatomical compartments even while plasma viral load is suppressed, potentially leading to therapy failure [136, 137]. Equations and parameters for generating Fig. 3 are given in the Appendix.
Drugs that inhibit binding of HIV to host cells (entry or fusion inhibitors) are currently in various stages of development. Like antiretrovirals that act at other phases of the viral lifecycle, these drugs come with a risk for the development of resistance, but they may also have other consequences for disease progression. Models have shown that drugs that inhibit the CCR5 co-receptor (for example, FDA-approved Maraviroc) could facilitate the switch to more pathogenic X4 variants, while anti-CXCR4 drugs may have the added benefit of decreasing selection pressure for X4 strains [138, 139].
The risk of resistance can be reduced with higher drug concentrations and by ensuring that drugs penetrate all tissue compartments; combination therapy plays a crucial role. When multiple drugs with different sites of action are used, the virus may need mutations that reduce susceptibility to all drugs in order for treatment failure to occur. Since multiple independent mutation events are relatively unlikely to occur on the same viral genome, it is generally more difficult for the virus to become fully resistant to combination therapy than to single therapy.
Predicting drug resistance should be an important consideration when designing dosing regimes for antiretroviral drugs, and would ideally be done as a part of the drug design process. Laboratory assays can characterize costs and benefits of mutant strains in the presence of drugs [118]; however, mutations are typically chosen for study only after they are observed in patients failing therapy. Bioinformatic techniques are needed to predict sites where resistance-conferring mutations may occur and how multiple mutations interact [140–142]. High-throughput implementations of known techniques for creating and testing mutant strains could help prevent the emergence of resistance and aid in design of "resistance-proof" drug regimens.
There are a few novel ideas for drug treatments that act in a very different way from current antiretrovirals, which reduce viral infectivity; instead, they aim to alter HIV’s intrinsic mutation rate. Drugs have been proposed which increase the mutation rate beyond the error catastrophe discussed previously, besetting the viral genome with unsustainable levels of deleterious mutational load (reviewed in Section 4.3 of [11]). An example is inhibitors of the viral protein Vif, which prevents the cellular APOBEC3G enzyme from hypermutating HIV cDNA during reverse transcription. A mutagenic drug (ribavirin) that acts with a different mechanism already exists for hepatitis C, foot and mouth disease, and respiratory syncytial virus [143]. It is also possible that drugs that decrease the mutation rate may be useful since much of HIV’s proposed pathogenesis is due to its high mutation rate. It has also been hypothesized that drugs could be used in concert with natural immune control to create an evolutionary trap for HIV: if drugs are designed such that any potential resistance mutations are potent CTL-eliciting epitopes, then the virus would be unable to escape both methods of control simultaneously [144].
HIV exhibits a lurker strategy, allowing the infection to “wait out” drug treatment
A range of cells harbor HIV in different forms. The virus can exist as a productive retroviral infection in active CD4+ T cells, macrophages, and monocytes; as integrated yet unexpressed viral DNA in resting CD4+ T cells; or as opsonized viral particles trapped on the surface of B cells and follicular dendritic cells. Though this last case does not involve infection per se, the envelope proteins of these trapped particles appear to be protected from decay, prolonging the particles’ infectivity until they can be transferred to a CD4+ T cell or other target [145]. The longevity of these cell types varies by several orders of magnitude—with productively infected T cells lasting no more than a few days, resting T cells lasting years, and the other cell types existing for an intermediate period of time [134]. Transmission of virus between these different cell types results in complex viral dynamics and allows HIV to enact a strategy of drug evasion in treated individuals: though an infected cell may initially inhabit a drug-rich environment hostile to viral replication, longer-lived cells can store the virus and later transmit it to short-lived cells in a replication-permissive environment, allowing the infection to persist and ultimately grow. We term this general pattern of turnover between long-lived and short-lived target cells a lurker strategy as it relies on a population of unexpressed virus that persists in the presence of antiretroviral drugs. A similar strategy observed in Escherichia coli involves alternation between a normal and a slow-growing phenotype, allowing a clone to survive as it periodically confronts antibiotic-rich or other stressful environments [146]. The most prominent example attesting to HIV’s ability to lurk is the persistence of the latent reservoir of resting CD4+ T cells, which, along with the inability of the immune response to control even low-level viremia, prevents antiretroviral therapy from eradicating the infection [147]. Treatment interruption presents a “window of opportunity” for a strain residing in long-lived cells to transmit to cells with faster turnover and grow in abundance. The ability of HIV to take advantage of this lurker strategy may constitute of form of cryptic drug resistance that cannot be detected in phenotypic assays that measure viral replication under a constant drug concentration.
It is unclear whether the various mechanisms prolonging the viral life cycle can be considered adaptations that were selected for the specific purpose of using a lurker strategy to cope with a dynamic host environment. In other words, a lurker strategy may be a mere side effect of selection for CD4 tropism, resulting from the heterogeneity of cell types expressing the CD4 receptor and the ability of CD4+ T cells to establish a long-lived memory phenotype. Given the short timeline of HIV evolution in the human species and the observation that HIV phylogenies reveal very little positive selection at the global population level [148], any selection for lurking would need to have taken place over the history of SIV evolution, prior to the modern human zoonosis. This strategy would therefore not have evolved in response to fluctuating drug pressures, but rather in response to the ancestral simian host/immune environment. The dynamic environment presented by the immune system, including shifting immunodominance [63], may have provided the necessary alternation between replication-permissive and harsh environments to select for mechanisms that enable lurking.
One such mechanism involves HIV’s ability to probabilistically postpone viral production [149]. The regulatory circuit controlled by the viral protein Tat (Trans-Activator of Transcription) is a stochastic switch that controls viral production, and so it is a promising candidate mechanism in which to investigate selection for lurking. Tat is a small protein (101 amino acids) that has existed for at least 7 million years in ancestral lentivirus strains [30], and it is expressed early at low abundance in infected cells. When acetylated, Tat enhances elongation of the 5′-LTR viral promoter. As a consequence, Tat also enhances its own transcription, triggering a transient feedback loop that generates a “pulse” of transcriptional activity. The duration of this pulse is determined stochastically by the initial cellular concentration of Tat. Starting from a high concentration, the pulse may last several days, exhausting the productive lifetime of an infected CD4+ T cell [150]. If the initial Tat concentration is too low, however, the duration may not support complete viral production, meaning that the proviral genome remains essentially inactive until a subsequent stochastic fluctuation in Tat expression triggers self-sustaining viral expression [151]. This unreliable feedback loop creates a variable delay before the start of productive infection, during which time the host cell avoids much of the cytopathic effect of viral proteins. If an infected CD4+ T cell can enter a resting state with constant probability per unit time, then a long unproductive state would increase the chance that the cell enters the latent reservoir. Stochastic fluctuations in Tat therefore mediate a probabilistic lurking strategy.
That the virus can take advantage of this stochastic machinery to enter latency does not, by itself, imply that the machinery evolved for the purpose of enabling a lurker strategy. Increasing the number of acetylation sites in Tat would increase the rate at which Tat becomes active, possibly to the extent of kicking the feedback loop into an “always on” state [150]. The fact that Tat contains two of these sites, and no more, may be because continual transactivation precludes a beneficial lurker strategy, or it could merely be because the potential benefit of increased Tat activation does not outweigh the fitness cost involved in lengthening the genome to add an additional site. Explicit modeling of the evolutionary trade-offs involved in modifying the parameters of the Tat feedback loop may clarify the range of dynamic environments under which lurking would be selected.
Could HIV become a drug-resistant epidemic?
It is generally assumed that individuals are infected with drug-sensitive virus, and that suboptimal treatment (i.e., monotherapy) or poor adherence leads to the de novo generation and selection of resistant mutants within that individual, as described earlier. Recent data, however, suggest a significant minority of cases where individuals are infected with a resistant strain. Worldwide, the detection of transmitted drug resistance in treatment-naive individuals is becoming increasingly worrisome. Between 10 and 25 % of individuals in high-risk groups in the USA and Europe are infected with virus harboring mutations associated with resistance to one or more antiretroviral drug [152, 153]. Surprisingly, drug resistance is transmitted not only from treated individuals, but may be transmitted directly between untreated individuals [154]. Transmitted drug resistance occurs much more frequently in regions where antiretroviral use is more prevalent. As drugs become more readily available globally, will the HIV epidemic become untreatable?
Widespread transmission of drug resistance requires both the generation of resistance within infected individuals as well as the maintenance of these mutations upon transmission to uninfected individuals. Traditionally, it was believed that the fitness cost of resistance mutations, resulting in them having a lower fitness that wild-type strains in untreated individuals, made persistence of resistance unlikely (Fig. 3a, i). Often, reversion of resistance mutations is observed when studying transmission pairs. However, reversion to wild type is observed to occur much more slowly than the initial take-over by a resistant mutant [155], sometimes taking years due to smaller relative difference in fitness in the absence of drugs to that in their presence (see Fig. 3g–i). In other cases, resistance may persist. This occurs when the fitness of resistance mutations is offset by the accumulation of compensatory mutations, which have been identified for many resistance mutations. Even if these compensatory mutations are unable to fully offset the cost, reversion may be very unlikely to occur if intermediate mutational steps leading back to the wild type are less fit than the original (multi-step) mutant [152, 156, 157] (Fig. 3i). When this occurs, resistance is no longer reversible, and persistence of resistant strains in untreated individuals will occur, compromising their likelihood of treatment success when they begin antiretroviral therapy.
Transmitted drug resistance is just beginning to be studied in the context of HIV. The finding that overall, lower viral loads are not observed with transmitted drug resistance [158] suggests that the mutations are not costly, and it was also shown that transmitted resistance impairs treatment responses [159]. A recent modeling study for the spread of drug resistance in San Francisco, where prevalence of transmitted resistance is among the highest in the world, suggested that the most important determinants of spread are the relative fitnesses of the wild-type and resistant strain, especially during the asymptomatic (i.e., early, untreated) stage of infection [160].
Many other diseases have already established widespread, costly, and often deadly drug-resistant epidemics: examining them may help assess the threat of spread of drug-resistant HIV (reviewed in [6, 161] (Chapter 10 of [6]). Antibiotic-resistant bacterial epidemics, such as methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus, are facilitated in hospital settings by widespread antibiotic use and easy transfer between high densities of often immunocompromised patients. Bacteria may acquire drug-resistant genes on plasmids through horizontal gene transfer with unrelated bacterial species. Since many antibiotics were derived from naturally occurring compounds, resistance genes may preexist in certain environmental bacteria [162]. Widespread use of antibiotics in livestock exacerbates this problem. Multiple factors contribute to the persistence of resistance genes even when bacteria infect untreated individuals. When carried on small plasmids or linked (on plasmids) to beneficial genes, there may be little fitness cost incurred for carrying the resistance gene. Plasmids may potentially carry resistance genes to multiple drugs. Drug-resistant infections may be more or less virulent than susceptible ones, due to either linked virulence factors or fitness costs to resistance, and studies have found that both scenarios occur [163]. The multidrug-resistant tuberculosis epidemic in the developed world first emerged in immunocompromised individuals, particularly AIDS patients [164], and has been suggested though not consistently demonstrated to be less virulent and transmissible [165]. It is clear that persistence of HIV drug resistance is not influenced by many of these factors facilitating antibiotic resistance.
Examining the potential for transmitted drug resistance from an evolutionary point of view suggests that it will likely be easier to prevent such an epidemic from occurring than to control it once it emerges. Worldwide, there have been multiple population level attempts to reduce the prevalence of drug-resistant infections by lowering subtherapeutic use of antibiotics (for example, in agriculture, or in mild infections of humans): they have shown very little success. Predicting resistance during drug development will be an important step. Improving patient adherence to drugs is extremely important. Most currently HAART regimes involve drug combinations that suppress viral replication to such an extent that clinical resistance is extremely unlikely, and the strongest predictor of resistance is patient adherence, which currently averages around 70 % [166]. While using antiretroviral treatment as a preventative measure against HIV infection in high-risk groups (called preexposure prophylaxis, or PrEP) may reduce the number of new infections, in some cases it could increase the percent of infections that are drug resistant [167]. A similar effect could potentially occur for “test-and-treat” strategies, which aim to scale-up diagnostic tests in underserved areas and to start antiretroviral treatment as soon as individuals are diagnosed with HIV [168].
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOC 145 kb)
Acknowledgments
We are grateful for the support from the National Science Foundation/National Institutes of Health joint program in mathematical biology (M.A.N., A.L.H.), the Bill & Melinda Gates Foundation (M.A.N., A.L.H.), a National Science Foundation Graduate Research Fellowship (D.I.S.R.), the John Templeton Foundation (M.A.N.), and J. Epstein (M.A.N.). We thank Pleuni Pennings, Alal Eran, Alireza Rabi, and two anonymous reviewers for helpful comments with the manuscript.
References
- 1.Fauci AS. Infectious diseases: considerations for the 21st century. Clin Infect Dis. 2001;32:675–685. doi: 10.1086/319235. [DOI] [PubMed] [Google Scholar]
- 2.Pier GB. On the greatly exaggerated reports of the death of infectious diseases. Clin Infect Dis. 2008;47:1113–1114. doi: 10.1086/592123. [DOI] [PubMed] [Google Scholar]
- 3.Spellberg B. Dr. William H. Stewart: mistaken or maligned? Clin Infect Dis. 2008;47:294. doi: 10.1086/589579. [DOI] [PubMed] [Google Scholar]
- 4.Joint United Nations Programme on HIV/AIDS and World Health Organization (2009) AIDS epidemic update. Available: http://www.who.int/hiv/data/en/. Accessed 2 Jan 2012
- 5.CDC HIV surveillance—United States, 1981–2008. MMWR. 2011;60:689–693. [PubMed] [Google Scholar]
- 6.Stearns SC, Koella JC. Evolution in health and disease. 2. New York: Oxford University Press; 2008. [Google Scholar]
- 7.Trevathan WR, Smith EO, McKenna J. Evolutionary medicine and health: new perspectives. 1. New York: Oxford University Press; 2007. [Google Scholar]
- 8.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bull JJ, Sanjuán R, Wilke CO. Theory of lethal mutagenesis for viruses. J Virol. 2007;81:2930–2939. doi: 10.1128/JVI.01624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eigen M. Error catastrophe and antiviral strategy. Proc Natl Acad Sci USA. 2002;99:13374–13376. doi: 10.1073/pnas.212514799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmes EC. Evolution and emergence of RNA viruses. New York: Oxford University Press; 2009. [Google Scholar]
- 12.Martin AN. What is a quasispecies? Trends Ecol Evol. 1992;7:118–121. doi: 10.1016/0169-5347(92)90145-2. [DOI] [PubMed] [Google Scholar]
- 13.Kamp C, Wilke CO, Adami C, Bornholdt S. Viral evolution under the pressure of an adaptive immune system: optimal mutation rates for viral escape. Complexity. 2002;8:28–33. doi: 10.1002/cplx.10067. [DOI] [Google Scholar]
- 14.M’Gonigle LK, Shen JJ, Otto SP. Mutating away from your enemies: the evolution of mutation rate in a host–parasite system. Theor Popul Biol. 2009;75:301–311. doi: 10.1016/j.tpb.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J Virol. 2010;84:9864–9878. doi: 10.1128/JVI.00915-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenbloom DIS, Hill AL, Rabi SA, Siliciano RF, Nowak MA (2012) Antiretroviral dynamics determines HIV evolution and predicts therapy outcome (in press) [DOI] [PMC free article] [PubMed]
- 17.Ariën KK, Vanham G, Arts EJ. Is HIV-1 evolving to a less virulent form in humans? Nat Rev Micro. 2007;5:141–151. doi: 10.1038/nrmicro1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/S0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 19.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 20.Chiu Y-L, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 21.Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci USA. 2004;101:4204–4209. doi: 10.1073/pnas.0306764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomson MM, Nájera R. Molecular epidemiology of HIV-1 variants in the global AIDS pandemic: an update. AIDS Rev. 2005;7:210–224. [PubMed] [Google Scholar]
- 23.Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santiago ML, Range F, Keele BF, Li Y, Bailes E, et al. Simian Immunodeficiency virus infection in free-ranging sooty mangabeys (Cercocebus atys atys) from the Taï Forest, Côte d’Ivoire: implications for the origin of epidemic human immunodeficiency virus type 2. J Virol. 2005;79:12515–12527. doi: 10.1128/JVI.79.19.12515-12527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Worobey M, Gemmel M, Teuwen DE, Haselkorn T, Kunstman K, et al. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature. 2008;455:661–664. doi: 10.1038/nature07390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolfe ND, Switzer WM, Carr JK, Bhullar VB, Shanmugam V, et al. Naturally acquired simian retrovirus infections in central African hunters. Lancet. 2004;363:932–937. doi: 10.1016/S0140-6736(04)15787-5. [DOI] [PubMed] [Google Scholar]
- 27.May RM, Anderson RM. Transmission dynamics of HIV infection. Nature. 1987;326:137–142. doi: 10.1038/326137a0. [DOI] [PubMed] [Google Scholar]
- 28.Anderson RM, May RM. Infectious diseases of humans: dynamics and control. New York: Oxford University Press; 1991. [Google Scholar]
- 29.de Sousa JD, Müller V, Lemey P, Vandamme A-M. High GUD incidence in the early 20th century created a particularly permissive time window for the origin and initial spread of epidemic HIV strains. PLoS One. 2010;5:e9936. doi: 10.1371/journal.pone.0009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katzourakis A, Tristem M, Pybus OG, Gifford RJ. Discovery and analysis of the first endogenous lentivirus. Proc Natl Acad Sci USA. 2007;104:6261–6265. doi: 10.1073/pnas.0700471104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity. 2003;18:441–452. doi: 10.1016/S1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 32.Estes JD, Gordon SN, Zeng M, Chahroudi AM, Dunham RM, et al. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J Immunol. 2008;180:6798–6807. doi: 10.4049/jimmunol.180.10.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goulder PJR, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 35.Wertheim JO, Worobey M. Dating the age of the SIV lineages that gave rise to HIV-1 and HIV-2. PLoS Comput Biol. 2009;5:e1000377. doi: 10.1371/journal.pcbi.1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmes EC. Evolutionary history and phylogeography of human viruses. Annu Rev Microbiol. 2008;62:307–328. doi: 10.1146/annurev.micro.62.081307.162912. [DOI] [PubMed] [Google Scholar]
- 37.Worobey M, Telfer P, Souquière S, Hunter M, Coleman CA, et al. Island biogeography reveals the deep history of SIV. Science. 2010;329:1487. doi: 10.1126/science.1193550. [DOI] [PubMed] [Google Scholar]
- 38.Wertheim JO, Kosakovsky Pond SL. Purifying selection can obscure the ancient age of viral lineages. Mol Biol Evol. 2011;28:3355–3365. doi: 10.1093/molbev/msr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abecasis AB, Lemey P, Vidal N, de Oliveira T, Peeters M, et al. Recombination confounds the early evolutionary history of human immunodeficiency virus type 1: subtype G is a circulating recombinant form. J Virol. 2007;81:8543–8551. doi: 10.1128/JVI.00463-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cvitkovitch DG, Li Y-H, Ellen RP. Quorum sensing and biofilm formation in Streptococcal infections. J Clin Invest. 2003;112:1626–1632. doi: 10.1172/JCI20430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards SV. Is a new and general theory of molecular systematics emerging? Evolution. 2009;63:1–19. doi: 10.1111/j.1558-5646.2008.00549.x. [DOI] [PubMed] [Google Scholar]
- 42.Chung Y, Ané C. Comparing two Bayesian methods for gene tree/species tree reconstruction: simulations with incomplete lineage sorting and horizontal gene transfer. Syst Biol. 2011;60:261. doi: 10.1093/sysbio/syr003. [DOI] [PubMed] [Google Scholar]
- 43.Valen V. A new evolutionary law. Evol Theory. 1973;1:1–30. [Google Scholar]
- 44.May RM, Anderson RM. Epidemiology and genetics in the coevolution of parasites and hosts. Proc R Soc B. 1983;219:281–313. doi: 10.1098/rspb.1983.0075. [DOI] [PubMed] [Google Scholar]
- 45.May RM, Nowak MA. Coinfection and the evolution of parasite virulence. Proc R Soc B. 1995;261:209–215. doi: 10.1098/rspb.1995.0138. [DOI] [PubMed] [Google Scholar]
- 46.Nowak MA, May RM. Superinfection and the evolution of parasite virulence. Proc R Soc B. 1994;255(1342):81–89. doi: 10.1098/rspb.1994.0012. [DOI] [PubMed] [Google Scholar]
- 47.Müller V, Fraser C, Herbeck JT. A strong case for viral genetic factors in HIV virulence. Viruses. 2011;3:204–216. doi: 10.3390/v3030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shirreff G, Pellis L, Laeyendecker O, Fraser C. Transmission selects for HIV-1 strains of intermediate virulence: a modelling approach. PLoS Comput Biol. 2011;7:e1002185. doi: 10.1371/journal.pcbi.1002185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herbeck JT, Müller V, Maust BS, Ledergerber B, Torti C, et al. Is the virulence of HIV changing? A meta-analysis of trends in prognostic markers of HIV disease progression and transmission. AIDS. 2012;26:193–205. doi: 10.1097/QAD.0b013e32834db418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dixit NM, Perelson AS. HIV dynamics with multiple infections of target cells. Proc Natl Acad Sci USA. 2005;102:8198–8203. doi: 10.1073/pnas.0407498102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wodarz D, Levy DN. Effect of multiple infection of cells on the evolutionary dynamics of HIV in vivo: implications for host adaptation mechanisms. Exp Biol Med. 2011;236:926–937. doi: 10.1258/ebm.2011.011062. [DOI] [PubMed] [Google Scholar]
- 52.Wilke CO, Novella IS. Phenotypic mixing and hiding may contribute to memory in viral quasispecies. BMC Microbiol. 2003;3:11. doi: 10.1186/1471-2180-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Novick A, Szilard L. Virus strains of identical phenotype but different genotype. Science. 1951;113:34–35. doi: 10.1126/science.113.2924.34. [DOI] [PubMed] [Google Scholar]
- 54.Boerlijst MC, Bonhoeffer S, Nowak MA. Viral quasi-species and recombination. Proc R Soc B. 1996;263:1577–1584. doi: 10.1098/rspb.1996.0231. [DOI] [Google Scholar]
- 55.Mostowy R, Kouyos RD, Fouchet D, Bonhoeffer S. The role of recombination for the coevolutionary dynamics of HIV and the immune response. PLoS One. 2011;6:e16052. doi: 10.1371/journal.pone.0016052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bretscher MT, Althaus CL, Müller V, Bonhoeffer S. Recombination in HIV and the evolution of drug resistance: for better or for worse? Bioessays. 2004;26:180–188. doi: 10.1002/bies.10386. [DOI] [PubMed] [Google Scholar]
- 57.Jung A, Maier R, Vartanian J-P, Bocharov G, Jung V, et al. Recombination: multiply infected spleen cells in HIV patients. Nature. 2002;418:144. doi: 10.1038/418144a. [DOI] [PubMed] [Google Scholar]
- 58.Josefsson L, King MS, Makitalo B, Brännström J, Shao W, et al. Majority of CD4+ T cells from peripheral blood of HIV-1–infected individuals contain only one HIV DNA molecule. Proc Natl Acad Sci. 2011;108:11199–11204. doi: 10.1073/pnas.1107729108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Batorsky R, Kearney MF, Palmer SE, Maldarelli F, Rouzine IM et al (2011) Estimate of effective recombination rate and average selection coefficient for HIV in chronic infection. Proc Natl Acad Sci USA. Available: http://www.ncbi.nlm.nih.gov/pubmed/21436045 [DOI] [PMC free article] [PubMed]
- 60.Neher RA, Leitner T. Recombination rate and selection strength in HIV intra-patient evolution. PLoS Comput Biol. 2010;6:e1000660. doi: 10.1371/journal.pcbi.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nowak MA, Anderson RM, McLean AR, Wolfs TF, Goudsmit J, et al. Antigenic diversity thresholds and the development of AIDS. Science. 1991;254:963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- 62.Schenzle D. A model for AIDS pathogenesis. Stat Med. 1994;13:2067–2079. doi: 10.1002/sim.4780131916. [DOI] [PubMed] [Google Scholar]
- 63.Nowak MA, May RM, Phillips RE, Rowland-Jones S, Lalloo DG, et al. Antigenic oscillations and shifting immunodominance in HIV-1 infections. Nature. 1995;375:606–611. doi: 10.1038/375606a0. [DOI] [PubMed] [Google Scholar]
- 64.Nowak MA, May RMC. Virus dynamics: mathematical principles of immunology and virology. New York: Oxford University Press; 2000. [Google Scholar]
- 65.Phillips AN. Reduction of HIV concentration during acute infection: independence from a specific immune response. Science. 1996;271:497. doi: 10.1126/science.271.5248.497. [DOI] [PubMed] [Google Scholar]
- 66.Simon V, Ho DD, Abdool Karim Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet. 2006;368:489–504. doi: 10.1016/S0140-6736(06)69157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker C, Moody D, Stites D, Levy J. CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science. 1986;234:1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- 68.Lifson JD, Rossio JL, Piatak M, Parks T, Li L, et al. Role of CD8+ lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J Virol. 2001;75:10187–10199. doi: 10.1128/JVI.75.21.10187-10199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wodarz D, Page KM, Arnaout RA, Thomsen AR, Lifson JD, et al. A new theory of cytotoxic T-lymphocyte memory: implications for HIV treatment. Phil Trans R Soc B. 2000;355:329–343. doi: 10.1098/rstb.2000.0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei X, Decker JM, Wang S, Hui H, Kappes JC, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 71.Chang JJ, Altfeld M. Innate immune activation in primary HIV-1 infection. J Infect Dis. 2010;202:S297–S301. doi: 10.1086/655657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boutwell CL, Rolland MM, Herbeck JT, Mullins JI, Allen TM. Viral evolution and escape during acute HIV-1 infection. J Infect Dis. 2010;202:S309–S314. doi: 10.1086/655653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kouyos RD, von Wyl V, Hinkley T, Petropoulos CJ, Haddad M, et al. Assessing predicted HIV-1 replicative capacity in a clinical setting. PLoS Pathog. 2011;7:e1002321. doi: 10.1371/journal.ppat.1002321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goulder PJR, Watkins DI. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol. 2004;4:630–640. doi: 10.1038/nri1417. [DOI] [PubMed] [Google Scholar]
- 75.Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature. 1991;354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 76.Althaus CL, De Boer RJ. Dynamics of immune escape during HIV/SIV infection. PLoS Comput Biol. 2008;4:e1000103. doi: 10.1371/journal.pcbi.1000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Allen TM, Altfeld M, Geer SC, Kalife ET, Moore C, et al. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J Virol. 2005;79:13239–13249. doi: 10.1128/JVI.79.21.13239-13249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carlson JM, Brumme ZL, Rousseau CM, Brumme CJ, Matthews P, et al. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput Biol. 2008;4:e1000225. doi: 10.1371/journal.pcbi.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, et al. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One. 2009;4:e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Regoes RR, Bonhoeffer S. The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol. 2005;13:269–277. doi: 10.1016/j.tim.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 81.Levy JA. HIV pathogenesis: 25 years of progress and persistent challenges. AIDS. 2009;23:147–160. doi: 10.1097/QAD.0b013e3283217f9f. [DOI] [PubMed] [Google Scholar]
- 82.Sheppard HW, Celum C, Michael NL, O’Brien S, Dean M, et al. HIV-1 infection in individuals with the CCR5-[Delta] 32/[Delta] 32 genotype: acquisition of syncytium-inducing virus at seroconversion. J Acquir Immune Defic Syndr. 2002;29:307. doi: 10.1097/00126334-200203010-00013. [DOI] [PubMed] [Google Scholar]
- 83.Lehmann C, Däumer M, Boussaad I, Sing T, Beerenwinkel N, et al. Stable coreceptor usage of HIV in patients with ongoing treatment failure on HAART. J Clin Virol. 2006;37:300–304. doi: 10.1016/j.jcv.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 84.Regoes RR, Wodarz D, Nowak MA. Virus dynamics: the effect of target cell limitation and immune responses on virus evolution. J Theor Biol. 1998;191:451–462. doi: 10.1006/jtbi.1997.0617. [DOI] [PubMed] [Google Scholar]
- 85.Kamp C, Bornholdt S. From HIV infection to AIDS: a dynamically induced percolation transition? Proc R Soc B. 2002;269:2035–2040. doi: 10.1098/rspb.2002.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Asquith B. The evolutionary selective advantage of HIV-1 escape variants and the contribution of escape to the HLA-associated risk of AIDS progression. PLoS One. 2008;3:e3486. doi: 10.1371/journal.pone.0003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cummins NW, Badley AD. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death and Dis. 2010;1:e99. doi: 10.1038/cddis.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol. 2003;84:1649–1661. doi: 10.1099/vir.0.19110-0. [DOI] [PubMed] [Google Scholar]
- 89.Yates A, Stark J, Klein N, Antia R, Callard R. Understanding the slow depletion of memory CD4+ T cells in HIV infection. PLoS Med. 2007;4:e177. doi: 10.1371/journal.pmed.0040177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Galvani AP. The role of mutation accumulation in HIV progression. Proc R Soc B. 2005;272:1851–1858. doi: 10.1098/rspb.2005.3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bartha I, Simon P, Müller V. Has HIV evolved to induce immune pathogenesis? Trends Immunol. 2008;29:322–328. doi: 10.1016/j.it.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 92.Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27:406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 93.Koup RA, Graham BS, Douek DC. The quest for a T cell-based immune correlate of protection against HIV: a story of trials and errors. Nat Rev Immunol. 2011;11:65–70. doi: 10.1038/nri2890. [DOI] [PubMed] [Google Scholar]
- 94.O’Connell KA, Brennan TP, Bailey JR, Ray SC, Siliciano RF, et al. Control of HIV-1 in elite suppressors despite ongoing replication and evolution in plasma virus. J Virol. 2010;84(14):7018–7028. doi: 10.1128/JVI.00548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wright JK, Brumme ZL, Carlson JM, Heckerman D, Kadie CM, et al. Gag-protease-mediated replication capacity in HIV-1 subtype C chronic infection: associations with HLA type and clinical parameters. J Virol. 2010;84:10820–10831. doi: 10.1128/JVI.01084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miura T, Brockman MA, Brumme ZL, Brumme CJ, Pereyra F, et al. HLA associated alterations in replication capacity of chimeric NL4-3 viruses encoding gag-protease from HIV-1 elite controllers. J Virol. 2008;83:140–149. doi: 10.1128/JVI.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miura T, Brumme CJ, Brockman MA, Brumme ZL, Pereyra F, et al. HLA associated viral mutations are common in human immunodeficiency virus type 1 elite controllers. J Virol. 2009;83:3407–3412. doi: 10.1128/JVI.02459-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nowak MA, May RM. Mathematical biology of HIV infections: antigenic variation and diversity threshold. Math Biosci. 1991;106:1–21. doi: 10.1016/0025-5564(91)90037-J. [DOI] [PubMed] [Google Scholar]
- 99.Pantaleo G, Koup RA. Correlates of immune protection in HIV-1 infection: what we know, what we don’t know, what we should know. Nat Med. 2004;10:806–810. doi: 10.1038/nm0804-806. [DOI] [PubMed] [Google Scholar]
- 100.Korber B, Gnanakaran S. Converging on an HIV vaccine. Science. 2011;333:1589–1590. doi: 10.1126/science.1211919. [DOI] [PubMed] [Google Scholar]
- 101.Blankson JN. The study of elite controllers: a pure academic exercise or a potential pathway to an HIV-1 vaccine? Curr Opin HIV AIDS. 2011;6:147–150. doi: 10.1097/COH.0b013e3283457868. [DOI] [PubMed] [Google Scholar]
- 102.Johnson WE, Desrosiers RC. Viral persistence: HIV’s strategies of immune system evasion. Annu Rev Med. 2002;53:499–518. doi: 10.1146/annurev.med.53.082901.104053. [DOI] [PubMed] [Google Scholar]
- 103.Stearns SC. The evolution of life history traits: a critique of the theory and a review of the data. Annu Rev Ecol Syst. 1977;8:145–171. doi: 10.1146/annurev.es.08.110177.001045. [DOI] [Google Scholar]
- 104.Stearns SC. Life-history tactics: a review of the ideas. Q Rev Biol. 1976;51(1):3–47. doi: 10.1086/409052. [DOI] [PubMed] [Google Scholar]
- 105.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 106.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature. 2009;458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fryer HR, Scherer A, Oxenius A, Phillips R, McLean AR (2009) No evidence for competition between cytotoxic T-lymphocyte responses in HIV-1 infection. Proc R Soc B. doi:10.1098/rspb.2009.1232 [DOI] [PMC free article] [PubMed]
- 108.Heeney JL, Dalgleish AG, Weiss RA. Origins of HIV and the evolution of resistance to AIDS. Science. 2006;313:462–466. doi: 10.1126/science.1123016. [DOI] [PubMed] [Google Scholar]
- 109.Moir S, Chun T-W, Fauci AS. Pathogenic mechanisms of HIV disease. Annu Rev Pathol. 2011;6:223–248. doi: 10.1146/annurev-pathol-011110-130254. [DOI] [PubMed] [Google Scholar]
- 110.Fellay J, Shianna KV, Telenti A, Goldstein DB. Host genetics and HIV-1: the final phase? PLoS Pathog. 2010;6:e1001033. doi: 10.1371/journal.ppat.1001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barouch DH. Challenges in the development of an HIV-1 vaccine. Nature. 2008;455:613–619. doi: 10.1038/nature07352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gandon S, Mackinnon MJ, Nee S, Read AF. Imperfect vaccines and the evolution of pathogen virulence. Nature. 2001;414:751–756. doi: 10.1038/414751a. [DOI] [PubMed] [Google Scholar]
- 113.van der Kuyl AC, Cornelissen M. Identifying HIV-1 dual infections. Retrovirology. 2007;4:67. doi: 10.1186/1742-4690-4-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 115.Bonhoeffer S, Nowak MA. Pre-existence and emergence of drug resistance in HIV-1 infection. Proc R Soc B. 1997;264:631–637. doi: 10.1098/rspb.1997.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ribeiro RM, Bonhoeffer S, Nowak MA. The frequency of resistant mutant virus before antiviral therapy. AIDS. 1998;12:461. doi: 10.1097/00002030-199805000-00006. [DOI] [PubMed] [Google Scholar]
- 117.Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, et al. Dose–response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat Med. 2008;14:762–766. doi: 10.1038/nm1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sampah MES, Shen L, Jilek BL, Siliciano RF. Dose–response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc Natl Acad Sci USA. 2011;108:7613–7618. doi: 10.1073/pnas.1018360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hartl DL, Clark AG. Principles of population genetics. Sunderland: Sinauer Associates; 2007. [Google Scholar]
- 120.Pennings PS (2011) Standing genetic variation and the evolution of drug resistance in HIV. Arxiv preprint arXiv:11116164 [DOI] [PMC free article] [PubMed]
- 121.Kouyos RD, Althaus CL, Bonhoeffer S. Stochastic or deterministic: what is the effective population size of HIV-1? Trends Microbiol. 2006;14:507–511. doi: 10.1016/j.tim.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 122.Dinoso JB, Kim SY, Wiegand AM, Palmer SE, Gange SJ, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci. 2009;106:9403. doi: 10.1073/pnas.0903107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hockett RD, Michael Kilby J, Derdeyn CA, Saag MS, Sillers M, et al. Constant mean viral copy number per infected cell in tissues regardless of high, low, or undetectable plasma HIV RNA. J Exp Med. 1999;189:1545–1554. doi: 10.1084/jem.189.10.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Markowitz M, Louie M, Hurley A, Sun E, Di Mascio M, et al. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J Virol. 2003;77:5037–5038. doi: 10.1128/JVI.77.8.5037-5038.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ewens WJ. Mathematical population genetics: theoretical introduction. Berlin: Springer; 2004. 1.4.3 The stochastic theory; pp. 27–28. [Google Scholar]
- 126.Rouzine I, Coffin J. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc Natl Acad Sci USA. 1999;96:10758. doi: 10.1073/pnas.96.19.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brown AJL. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc Natl Acad Sci USA. 1997;94:1862. doi: 10.1073/pnas.94.5.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Margot N, Lu B, Cheng A, Miller M. Resistance development over 144 weeks in treatment-naive patients receiving tenofovir disoproxil fumarate or stavudine with lamivudine and efavirenz in Study 903*. HIV Med. 2006;7:442–450. doi: 10.1111/j.1468-1293.2006.00404.x. [DOI] [PubMed] [Google Scholar]
- 129.Shen L, Rabi SA, Siliciano RF. A novel method for determining the inhibitory potential of anti-HIV drugs. Trends Pharmacol Sci. 2009;30:610–616. doi: 10.1016/j.tips.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J Exp Med. 2004;199:283. doi: 10.1084/jem.20030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Groot F, Welsch S, Sattentau QJ. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood. 2008;111:4660. doi: 10.1182/blood-2007-12-130070. [DOI] [PubMed] [Google Scholar]
- 132.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, et al. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature. 2011;477:95–98. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 133.Best BM, Koopmans PP, Letendre SL, Capparelli EV, Rossi SS, et al. Efavirenz concentrations in CSF exceed IC50 for wild-type HIV. J Antimicrob Chemother. 2011;66:354. doi: 10.1093/jac/dkq434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med. 2002;53:557–593. doi: 10.1146/annurev.med.53.082901.104024. [DOI] [PubMed] [Google Scholar]
- 135.Crowe S, Zhu T, Muller WA. The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J Leukocyt Biol. 2003;74:635–641. doi: 10.1189/jlb.0503204. [DOI] [PubMed] [Google Scholar]
- 136.Smit TK, Brew BJ, Tourtellotte W, Morgello S, Gelman BB, et al. Independent evolution of human immunodeficiency virus (HIV) drug resistance mutations in diverse areas of the brain in HIV-infected patients, with and without dementia, on antiretroviral treatment. J Virol. 2004;78:10133. doi: 10.1128/JVI.78.18.10133-10148.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Edén A, Fuchs D, Hagberg L, Nilsson S, Spudich S, et al. HIV-1 viral escape in cerebrospinal fluid of subjects on suppressive antiretroviral treatment. J Infect Dis. 2010;202:1819. doi: 10.1086/657342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Regoes R, Bonhoeffer S. HIV coreceptor usage and drug treatment. J Theor Biol. 2002;217:443–457. doi: 10.1006/jtbi.2002.3049. [DOI] [PubMed] [Google Scholar]
- 139.Weinberger AD, Perelson AS, Ribeiro RM, Weinberger LS. Accelerated immunodeficiency by anti-CCR5 treatment in HIV infection. PLoS Comput Biol. 2009;5:e1000467. doi: 10.1371/journal.pcbi.1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Beerenwinkel N, Schmidt B, Walter H, Kaiser R, Lengauer T, et al. Diversity and complexity of HIV-1 drug resistance: a bioinformatics approach to predicting phenotype from genotype. Proc Natl Acad Sci. 2002;99:8271–8276. doi: 10.1073/pnas.112177799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Beerenwinkel N, Lengauer T, Daumer M, Kaiser R, Walter H, et al. Methods for optimizing antiviral combination therapies. Bioinformatics. 2003;19:i16. doi: 10.1093/bioinformatics/btg1001. [DOI] [PubMed] [Google Scholar]
- 142.Lawyer G, Altmann A, Thielen A, Zazzi M, Sönnerborg A, et al. HIV-1 mutational pathways under multidrug therapy. AIDS Res Ther. 2011;8:26. doi: 10.1186/1742-6405-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Crotty S, Cameron C, Andino R. Ribavirin’s antiviral mechanism of action: lethal mutagenesis? J Mol Med. 2002;80:86–95. doi: 10.1007/s00109-001-0308-0. [DOI] [PubMed] [Google Scholar]
- 144.Ávila-Ríos S, Reyes-Terán G, Espinosa E. Cornering HIV: taking advantage of interactions between selective pressures. Med Hypotheses. 2007;69:422–431. doi: 10.1016/j.mehy.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 145.Smith-Franklin BA, Keele BF, Tew JG, Gartner S, Szakal AK, et al. Follicular dendritic cells and the persistence of HIV infectivity: the role of antibodies and Fc-gamma receptors. J Immunol. 2002;168:2408. doi: 10.4049/jimmunol.168.5.2408. [DOI] [PubMed] [Google Scholar]
- 146.Kussell E, Kishony R, Balaban NQ, Leibler S. Bacterial persistence: a model of survival in changing environments. Genetics. 2005;169:1807–1814. doi: 10.1534/genetics.104.035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Siliciano JD, Siliciano RF. The latent reservoir for HIV-1 in resting CD4+ T cells: a barrier to cure. Curr Opin HIV AIDS. 2006;1:121–128. doi: 10.1097/01.COH.0000209582.82328.b8. [DOI] [PubMed] [Google Scholar]
- 148.Rambaut A, Posada D, Crandall KA, Holmes EC. The causes and consequences of HIV evolution. Nat Rev Genet. 2004;5:52–61. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- 149.Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 150.Weinberger LS, Shenk T. An HIV feedback resistor: auto-regulatory circuit deactivator and noise buffer. PLoS Biol. 2006;5:e9. doi: 10.1371/journal.pbio.0050009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Singh A, Weinberger LS. Stochastic gene expression as a molecular switch for viral latency. Curr Opin Microbiol. 2009;12:460–466. doi: 10.1016/j.mib.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pingen M, Nijhuis M, de Bruijn JA, Boucher CAB, Wensing AMJ. Evolutionary pathways of transmitted drug-resistant HIV-1. J Antimicrob Chemother. 2011;66(7):1467–1480. doi: 10.1093/jac/dkr157. [DOI] [PubMed] [Google Scholar]
- 153.van de Vijver D, Wensing AMJ, Boucher CAB. The epidemiology of transmission of drug resistant HIV-1. In: Leitner T, editor. HIV Sequence Compendium 2007. Los Alamos: Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; 2007. [Google Scholar]
- 154.Hué S, Gifford RJ, Dunn D, Fernhill E, Pillay D. Demonstration of sustained drug-resistant human immunodeficiency virus type 1 lineages circulating among treatment-naïve individuals. J Virol. 2009;83:2645–2654. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Little SJ, Frost SDW, Wong JK, Smith DM, Pond SLK, et al. Persistence of transmitted drug resistance among subjects with primary human immunodeficiency virus infection. J Virol. 2008;82:5510–5518. doi: 10.1128/JVI.02579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Handel A, Regoes RR, Antia R. The role of compensatory mutations in the emergence of drug resistance. PLoS Comput Biol. 2006;2:e137. doi: 10.1371/journal.pcbi.0020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schulz zur Wiesch P, Engelstadter J, Bonhoeffer S. Compensation of fitness costs and reversibility of antibiotic resistance mutations. Antimicrob Agents Chemother. 2010;54:2085–2095. doi: 10.1128/AAC.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Harrison L, Castro H, Cane P, Pillay D, Booth C, et al. The effect of transmitted HIV-1 drug resistance on pre-therapy viral load. AIDS. 2010;24:1917–1922. doi: 10.1097/QAD.0b013e32833c1d93. [DOI] [PubMed] [Google Scholar]
- 159.Wittkop L, Günthard HF, de Wolf F, Dunn D, Cozzi-Lepri A, et al. Effect of transmitted drug resistance on virological and immunological response to initial combination antiretroviral therapy for HIV (EuroCoord-CHAIN joint project): a European multicohort study. Lancet Infect Dis. 2011;11:363–371. doi: 10.1016/S1473-3099(11)70032-9. [DOI] [PubMed] [Google Scholar]
- 160.Smith RJ, Okano JT, Kahn JS, Bodine EN, Blower S. Evolutionary dynamics of complex networks of HIV drug-resistant strains: the case of San Francisco. Science. 2010;327(5966):697–701. doi: 10.1126/science.1180556. [DOI] [PubMed] [Google Scholar]
- 161.zur Wiesch PA, Kouyos R, Engelstädter J, Regoes RR, Bonhoeffer S. Population biological principles of drug-resistance evolution in infectious diseases. Infect Dis. 2011;11:236–247. doi: 10.1016/S1473-3099(10)70264-4. [DOI] [PubMed] [Google Scholar]
- 162.Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, et al. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Micro. 2010;8:251–259. doi: 10.1038/nrmicro2312. [DOI] [PubMed] [Google Scholar]
- 163.Cohen ML. Epidemiology of drug resistance: implications for a post-antimicrobial era. Science. 1992;257:1050–1055. doi: 10.1126/science.257.5073.1050. [DOI] [PubMed] [Google Scholar]
- 164.Edlin BR, Tokars JI, Grieco MH, Crawford JT, Williams J, et al. An outbreak of multidrug-resistant tuberculosis among hospitalized patients with the acquired immunodeficiency syndrome. N Engl J Med. 1992;326:1514–1521. doi: 10.1056/NEJM199206043262302. [DOI] [PubMed] [Google Scholar]
- 165.Cohen T, Murray M. Modeling epidemics of multidrug-resistant M. tuberculosis of heterogeneous fitness. Nat Med. 2004;10:1117–1121. doi: 10.1038/nm1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bangsberg DR, Acosta EP, Gupta R, Guzman D, Riley ED, et al. Adherence-resistance relationships for protease and non-nucleoside reverse transcriptase inhibitors explained by virological fitness. AIDS. 2006;20:223–231. doi: 10.1097/01.aids.0000199825.34241.49. [DOI] [PubMed] [Google Scholar]
- 167.Supervie V, García-Lerma JG, Heneine W, Blower S. HIV, transmitted drug resistance, and the paradox of preexposure prophylaxis. Proc Natl Acad Sci USA. 2010;107:12381–12386. doi: 10.1073/pnas.1006061107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Blower S, Bodine E, Kahn J, McFarland W. The antiretroviral rollout and drug-resistant HIV in Africa: insights from empirical data and theoretical models. AIDS. 2005;19:1–14. doi: 10.1097/00002030-200501030-00001. [DOI] [PubMed] [Google Scholar]
- 169.Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73:10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lee HY, Perelson AS, Park S-C, Leitner T. Dynamic correlation between intrahost HIV-1 quasispecies evolution and disease progression. PLoS Comput Biol. 2008;4:e1000240. doi: 10.1371/journal.pcbi.1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nowak MA. Evolutionary dynamics: exploring the equations of life. Cambridge: Belknap Press of Harvard University Press; 2006. [Google Scholar]
- 173.Galvani AP. The role of mutation accumulation in HIV progression. Proc R Soc B. 2005;272:1851–1858. doi: 10.1098/rspb.2005.3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 145 kb)