

Abstract
Real-time quantitative reverse transcriptase polymerase chain reaction (rRT-PCR) is now widely used to detect viral pathogens in various human specimens. The application of internal controls to validate the entire process of these assays is necessary to prevent false-negative results caused by unexpected inhibition or inefficient extraction. In the present study, we describe a strategy to produce a stable internal control for rRT-PCR by packaging foreign RNA into influenza virions using plasmid-based reverse genetics technology. The envelope structure of influenza virus can effectively protect RNA segments from RNase digestion, which provides an advantage for its routine use as an internal control. Utilizing this approach, we successfully generated a recombinant influenza virus (rPR8-HCV) containing the 5′ untranslated region (5′ UTR) of the hepatitis C virus (HCV) RNA genome. After inactivation and purification, the rPR8-HCV particles were demonstrated to be RNase resistant and stable at 4 °C for at least 252 days in human plasma, with no degradation even after being frozen and thawed multiple times. These results were reproducible in the COBAS TaqMan HCV test for 164 days. Moreover, the chimeric influenza virus particles could be easily produced in embryonated eggs and were noninfectious after inactivation treatment. Additionally, this strategy could also be adapted for real-time clinical applications of other RNA targets, providing a universal approach with broad clinical applications in rRT-PCR assays.
Keywords: rRT-PCR, Internal control, Influenza virus, Hepatitis C virus, Reverse genetics technology
Introduction
Real-time quantitative reverse transcriptase polymerase chain reaction (rRT-PCR) assays are widely used as a diagnostic tool for the detection and quantification of viral RNA in clinical samples (Espy et al. 2006). However, an inherent problem is frequently encountered with false-negative PCR results, caused mainly by the degradation of target RNA and the presence of PCR inhibitors. Therefore, internal controls are incorporated into these assays to ensure reliable PCR results, in which true-negative results can be distinguished from false-negative results that are due to human error or inhibition (Hoorfar et al. 2004).
An ideal internal control should closely mimic the natural virus in order to monitor all the stages of rRT-PCR, including nucleic acid extraction, RNA reverse transcription, and amplification (Niesters 2004; Pasloske et al. 1998). Additionally, an internal control should be easy to prepare, stable, and safe for routine use. Currently, many diagnostic assays in which viral RNA is detected and quantified use a naked RNA control (Chang et al. 2012; Scholtes et al. 2012; Wernike et al. 2012). However, naked RNA is unstable and susceptible to degradation by RNase (Dreier et al. 2005; Felder and Wolfel 2014). Furthermore, when using naked RNA as a control, the lysis process is not monitored during RNA extraction, and therefore only partial quality assurance is achieved. Thus, developing an improved internal control for viral nucleic acid amplification tests is of the utmost importance.
Armored RNA is a noninfectious synthetic pseudoviral particle produced by packaging the exogenous target RNA sequences into the MS2 coat protein (Pasloske et al. 1998). To date, it is the most appropriate strategy for RNA control preparation. It can serve as a surrogate for the primary target virus owing to its safety, RNase resistance, and stability after prolonged incubation in plasma (Huang et al. 2006). This technology has been successfully adapted to prepare internal controls for various rRT-PCR assays, such as those for human immunodeficiency virus (HIV), hepatitis C virus (HCV), enterovirus, and H7N9 influenza A viruses (Okello et al. 2010; Pasloske et al. 1998; Song et al. 2011; Sun et al. 2013; WalkerPeach et al. 1999). However, commercially available armored RNA preparations are expensive owing to their patent protection (US patent number 5,677,124, http://asuragen.com/), which has greatly limited the widespread application in most routine clinical laboratories.
Influenza virus, which belongs to the family of Orthomyxoviridae, is an enveloped, negative-sense RNA virus with a segmented, single-stranded genome. Reverse genetics technology can be used to generate chimeric influenza viruses containing a foreign RNA fragment based on the modification of an influenza virus internal gene (De Baets et al. 2013; Reece et al. 2013; Wu et al. 2010). Previous studies have shown that influenza virus carrying a reporter gene, such as GFP, can replicate normally and stably express the reporter gene in infected cells, which makes these viruses feasible for extensive propagation (Li et al. 2010; Manicassamy et al. 2010). Furthermore, the virion morphology of chimeric viruses is indistinguishable from those of wild-type viruses when observed by electron microscopy, indicating that chimeric viruses may be RNase resistant with lipid envelope protection (Maloney et al. 2015; Wu et al. 2010). Therefore, we conclude that a chimeric influenza virus carrying the target RNA fragment could potentially be used as internal control for rRT-PCR.
In the present study, based on the manipulation of influenza virus, we developed a novel artificial RNA control for HCV rRT-PCR assays using reverse genetics technology. This was accomplished by inserting the 5′ untranslated region (5′ UTR) of HCV into the nonstructural protein (NS) segment of PR8 influenza virus, inactivating and purifying the chimeric viruses, and then adding to appropriate clinical sample matrices for HCV. We also demonstrated that influenza virus had good potential for use as vector for a stable and low-cost internal control preparation.
Materials and methods
Cells and plasmid
293T cells (ATCC, CRL-3216), purchased from ATCC, were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10 % fetal bovine serum (FBS). The pHW2000 plasmid was kindly provided by E. Hoffmann and R. G. Webster (Hoffmann et al. 2000).
Construction of the chimeric plasmid pHW-NS-HCV
The reverse complement sequence of highly conserved HCV 5′ UTR (nt 1 to 339, GenBank accession no. AF176573) was inserted into the influenza (A/Puerto Rico/8/34) NS gene (GenBank accession no. AF389122) by using overlap extension PCR. During the first round of PCR, three separate fragments were amplified as follows. NS1, including the 5′ genomic noncoding region but lacking the 3′ stop codon, and NEP, including the 3′ genomic noncoding region, were amplified from a pHW-NS plasmid (constructed by our laboratory) using primer pairs NS-F/NS1-OL-R (HCV) and NEP-OL-F (HCV)/NS-R. The HCV 5′ UTR with the 3′ PTV-1 2A peptide-coding sequence was amplified from a pMD18-HCV plasmid (constructed by our laboratory) using primers HCV-OL-F (NS1) and HCV-OL-R (NEP). After cleanup, the HCV plus NEP fragment was constructed by subsequent rounds of PCR using primers HCV-OL-F (NS1) and NS-R. PCR products were gel purified. Then, the final round of PCR fused NS1 to the N terminal of HCV-NEP via a short GSG linker using primers NS-F and NS-R. To introduce silent mutations at the splice acceptor site in NS1, nucleotides 524 to 527 (5′-TCCA-3′) were replaced with 5′-CCCG-3′ using primers NS1-M-F and NS1-M-R. All of the primer sequences that were used in this study are shown in Table 1. The engineered NS-HCV segment was cloned into the BsmBI site of the pHW2000 vector to generate the chimeric plasmid pHW-NS-HCV. The construct was sequenced across the entire NS gene to confirm insertion. Chimeric plasmids carrying the other seven gene segments of the PR8 virus (pHW-PB2, -PB1, -PA, -HA, -NP, NA and -M) were already constructed in our previous studies (Wang et al. 2012).
Table 1.
Primers for PCR amplification
| Primer name | Primer sequence (5′ to 3′)a |
|---|---|
| NS-F | TATTCGTCTCAGGGAGCAAAAGCAGGGTGACAAA |
| NS-R | ATATCGTCTCGTATTAGTAGAAACAAGGGTGTTTT |
| NS1-OL-R (HCV) | TGCACCGCCGGACCCAACTTCTGACCTAATTGTTCCCGC |
| HCV-OL-F (NS1) | GAAGTTGGGTCCGGCGGTGCACGGTCTACGAGACCTCCC |
| HCV-OL-R (NEP) | TTCTTCCACATCGCCCGCCTGTTTCAGCAGGCTAAAGTTGGTCGCGCCGCTGCCCAGCCCCCGATTGGGGGCGACA |
| NEP-OL-F (HCV) | ACCAACTTTAGCCTGCTGAAACAGGCGGGCGATGTGGAAGAAAACCCGGGCCCGATGGATTCAAACACTGTGTCAAGCT |
| NS1-M-F | CACCATTGCCTTCTCTCCCGGGACATACTGCTGAGG |
| NS1-M-R | CCTCAGCAGTATGTCCCGGGAGAGAAGGCAATGGTG |
| NP-F | AGCAAAAGCAGGGTAGATAATCACT |
| NP-R | AGTAGAAACAAGGGTATTTTTCTTT |
a BsmB I restriction site is indicated by underscoring; silent mutations are indicated in boldface type
Production of chimeric rPR8-HCV virus as internal control
The method for generating chimeric influenza viruses was performed as described previously (Hoffmann et al. 2000; Wang et al. 2012). Briefly, 293T cells were transfected with 1 μg each of eight expression plasmids for viral genes (pHW-PB2 (GenBank accession no. AF389115), -PB1 (GenBank accession no. AF389116), -PA (GenBank accession no. AF389117), -HA (GenBank accession no. AF389118), -NP (GenBank accession no. AF389119), -NA (GenBank accession no. AF389120), -M (GenBank accession no. AF389121), and NS-HCV) using Lipofectamine 2000 (2 μl/μg of plasmid, Invitrogen/Life Technologies, USA). Forty-eight hours after transfection, supernatants and cells were collected and subsequently inoculated into 10-day-old specific pathogen-free chicken embryos for virus propagation. At 72 h post-inoculation, allantoic fluids were harvested and centrifuged at 5000×g for 20 min to remove cell debris. The harvested allantoic fluids were subjected to hemagglutination (HA) assay to determine the presence of viruses. To explore the stability of rPR8-HCV, the chimeric viruses were subcultured on chicken embryos for eight passages. RNA was extracted from the serial passages for RT-PCR amplification of the partial NS-HCV gene fragment using primers HCV-OL-F (NS1) and NS-R.
For inactivation, the virus stocks were mixed with 0.1 % formalin at 4 °C for 7 days. Inactivation of viruses was confirmed by the absence of detectable HA activity after three passages of the treated materials into embryonated eggs. Inactivated viruses were purified by sucrose gradient centrifugation at 30,000×g for 3 h at 4 °C. The purified viruses were then suspended in phosphate-buffered saline and stored at 4 °C. RT-PCR and sequencing were used to confirm the identity of the NS-HCV gene in prepared virus stocks.
RNA isolation
RNA was extracted from purified rPR8-HCV particles using TRIzol reagent according to the manufacturer’s instructions (Life Technologies, USA). Briefly, 250 μl of purified viruses was mixed with 750 μl of TRIzol reagent. After 5-min incubation, RNA was separated into the aqueous phase with the addition of 200 μl of chloroform. RNA was then precipitated with isopropanol, washed with 75 % ethanol, and resuspended in 50 μl of RNase-free water. All RNA samples were kept on ice until use.
RNA electrophoresis
To confirm if the generated rPR8-HCV virus contained the chimeric NS-HCV segment, RNA electrophoresis was performed. Briefly, the extracted RNA was run on a 2.8 % polyacrylamide gel containing 7 M urea at 60 V for 3 h, which was then stained with a silver staining kit (Bio-Rad, Hercules, CA, USA).
Identification of purified rPR8-HCV particles
Reverse transcription (RT) reactions were performed in 25 μl reaction mixtures containing 5 μl of purified RNA, 0.2 μl of 25 μM unit 12 primer (5′-AGCAAAAGCAGG-3′), 5 μl of 5× RT buffer, 2 μl of 10 mM deoxynucleoside triphosphate, 1 μl of 40 U/μl RNase inhibitor, and 1 μl of 10 U/μl AMV reverse transcriptase (Promega, Madison, WI, USA). RT reactions were performed using the GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA, USA) at 42 °C for 60 min, followed by 5 min at 95 °C for reverse transcriptase inactivation.
To assess the incorporation of the chimeric NS-HCV gene segments into the rescued virions, PCR was performed in a 50 μl final reaction mixture containing 5 μl of the cDNA obtained from the RT reaction, 0.5 μl of 25 μM primers for amplification of different genes (NS, HCV, and NP gene), 5 μl of 10× PCR buffer, 2 μl of 10 mM deoxynucleoside triphosphate, and 0.5 μl of 5 U/μl High Fidelity PCR enzyme mix (Thermo Fisher Scientific, Rockford, IL, USA). After an initial denaturation at 94 °C for 4 min, 30 amplification cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1.5 min were performed followed by a final extension of 72 °C for 10 min. The PCR products were analyzed by electrophoresis on a 1 % agarose gel and visualized by ethidium bromide staining. Then, PCR products were purified and subcloned into the pM18-T vector (Takara, Dalian, China) and verified by sequencing.
Nuclease resistance of rPR8-HCV particles
To examine whether the purified rPR8-HCV particles were nuclease resistant, rPR8-HCV particles and RNA isolated from rPR8-HCV particles were incubated with 1 μg/μl RNase A (Qiagen, Hilden, Germany) at 37 °C for 1 h. After treatment, the samples were tested using a commercial HCV RNA PCR fluorescence quantitative diagnostic kit (Shanghai Kehua Bio-engineering, Shanghai, China).
Stability of rPR8-HCV particles in plasma
The stability of rPR8-HCV particles was examined in human plasma, seronegative for HCV RNA and anti-HCV. An HCV RNA PCR kit (Sansure Biotech, Hunan, China) was used for sample quantification. Samples containing approximately 104 and 106 IU/ml of the quantified rPR8-HCV particles were prepared with normal human EDTA-preserved plasma. For each stability study, a single batch of rPR8-HCV particles was separated aliquots in individual time point samples of 200 μl, the volume used in real time PCR assay. Samples were then incubated at −20 °C, 4 °C, 37 °C, and room temperature for set periods of time. rPR8-HCV samples were quantified in duplicate at each time point. The data were then analyzed using ABI7500 software (Applied Biosystems, Foster City, CA, USA). The rPR8-HCV particles were stored at −20 °C and thawed to room temperature five times. They were quantified in duplicate.
Chimeric rPR8-HCV as positive controls in clinical diagnosis
In order to investigate whether the chimeric rPR8-HCV can serve as a positive control in clinical diagnosis, the Roche COBAS Ampliprep/COBAS TaqMan system (CAP/CTM) was used. This system is the most widely used real-time PCR assay for HCV RNA quantification. CAP/CTM combines automated extraction of HCV RNA on the COBAS AmpliPrep instrument with real-time PCR on the COBAS TaqMan analyzer, thus greatly reducing time needed for sample preparation, amplification, and detection. Previous studies have shown that CAP/CTM is sensitive, specific, precise, and reproducible with a broad dynamic range of quantification (Bossler et al. 2011; Sarrazin et al. 2008). First, the quantified rPR8-HCV was diluted with normal human plasma to a final HCV RNA concentration of approximately 104 IU/ml in a total volume of 1 ml for each. Aliquoted samples were stored at 4 °C until further use. rPR8-HCV samples were assayed in parallel with the COBAS TaqMan HCV test positive control in regular clinical runs for the determination of HCV viral loads. The rPR8-HCV control and the positive control in the COBAS TaqMan HCV test kit were assayed once a week for approximately 5 months.
Linear analysis of rPR8-HCV in rRT-PCR assays for HCV
The rPR8-HCV particles were serially diluted 10-fold in normal human plasma to obtain concentrations of 2 × 107, 2 × 106, 2 × 105, 2 × 104, and 2 × 103 IU/ml. Three replications of each concentration were tested in the same assay run using the HCV RNA PCR kit (Sansure Biotech, Hunan, China), and the resulting quantification values were averaged.
Statistical analysis
All assay data were naturally log transformed prior to analysis, and the changes in the HCV RNA level over time were evaluated by linear regression as described previously (Jose et al. 2005; Krajden et al. 1999). A p value of <0.05 was considered statistically significant.
Results
Generation and purification of a chimeric influenza virus
To generate an influenza virus containing the HCV 5′ UTR segment, the NS gene of PR8 virus was modified to express NS1-HCV and NEP monocistronically with a PTV-1 2A autoproteolytic cleavage site between them. This cleavage site allowed NEP to be translated independently of the NS1-HCV fusion protein. The splice acceptor site was removed to prevent splicing of NS mRNA. Additionally, the original NS1 translation stop codon TGA was deleted by site-directed mutagenesis so that the HCV gene could be fused to the NS1 protein product. The resultant cDNA was then inserted into the bidirectional expression pHW2000 plasmid to obtain the pHW-NS-HCV construct (Fig. 1a). The recombinant influenza virus, named rPR8-HCV, was successfully rescued by co-transfection with plasmids containing the other seven influenza genes into 293T cells, as described in “Materials and methods” (Fig. 1b). The successful incorporation of NS-HCV into rPR8-HCV virions was examined by silver staining. As expected, an absence of the NS segment within the rPR8-HCV virus was observed. The rPR8-HCV virus had a chimeric NS-HCV segment along with the NP segment, which migrated to nearly the same position (Fig. 1c). Specific NS-HCV chimeric DNA could be readily detected from passages 1 to 8 for chimeric viruses, demonstrating that the rPR8-HCV virus was genetically stable (Fig. 1d).
Fig. 1.
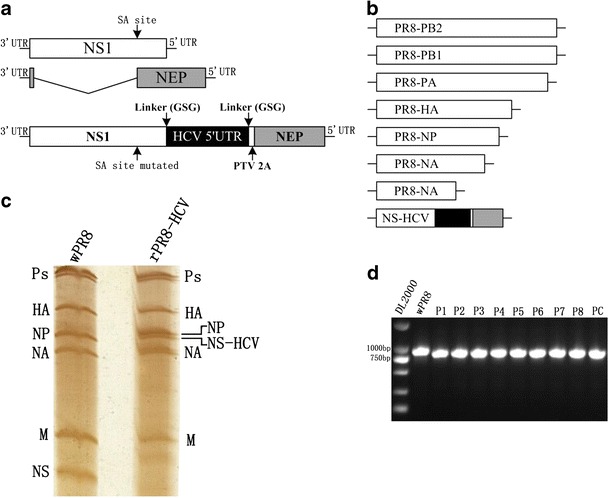
Generation of recombinant influenza virus rPR8-HCV. a Schematic representation of the NS segment of wild-type PR8 virus (wPR8) (top) and of the rPR8-HCV virus (bottom). To prevent mRNA splicing, silent mutations were introduced into the splice acceptor site of the NS1 (nucleotides 524 to 527: 5′-TCCA-3′ to 5′-CCCG-3′). An HCV 5′ UTR sequence (dark) was inserted between NS1 and the “autocleave” PTV 2A peptide-coding sequence via a short GSG linker, and the resulting fragment was fused to the NEP open reading frame (gray). b Eight-plasmid virus rescue system. Seven A/PR/8/34 plasmids (pHW-PB1, pHW-PB2, pHW-PA, pHW-HA, pHW-NP, pHW-NA, and pHW-M) and one NS-based construct (pHW-NS-HCV) were used to generate the rPR8-HCV virus by reverse genetics. c RNA electrophoresis. RNA was isolated from purified rPR8-HCV viruses and wPR8, run on a 2.8 % acrylamide gel, and visualized by silver staining. The positions of the polymerase (Ps), HA, NP, NA, M, NS (wPR8), and NS-HCV (rPR8-HCV) segments are indicated. d Viral genome stability analysis. Chimeric influenza viruses were consecutively inoculated into 10-day-old chicken embryos for eight passages. NS and HCV-NEP genes were detected using RT-PCR. pHW-NS-HCV plasmid were used as positive control (PC)
To ensure the safety of the chimeric virus for use as a positive control, rPR8-HCV viruses were treated with formalin and then their HA activity was examined. The results show that HA activity was not observed after three passages of the virus into embryonated eggs (data not shown), indicating that the treatment resulted in a complete loss of viral infectivity. After being inactivated and purified through sucrose gradient centrifugation, the total RNA was isolated and RT-PCR was performed followed by sequencing. The size of the generated cDNAs confirmed that the full-length chimeric NS-HCV gene was packaged into the chimeric virus (Fig. 2). DNA sequencing revealed neither loss of gene segments nor change in the nucleotide acid sequence after inactivation by formalin (data not shown). These results indicate that the chimeric influenza viruses were successfully obtained. They also demonstrate that the formalin treatment did not destroy the structure of the rPR8-HCV virus.
Fig. 2.
RT-PCR detection of NS, HCV, and NP genes from purified influenza viruses. The NS (upper), HCV (middle), and NP (lower) genes from purified rPR8-HCV and wPR8 were detected using RT-PCR
Durability of chimeric rPR8-HCV particles
The durability of chimeric rPR8-HCV particles was tested by subjecting them to RNase treatment. The results show that rPR8-HCV particles were completely resistant to RNase treatment, while the naked RNA was degraded rapidly after RNase digestion (Fig. 3). This suggests that the purified rPR8-HCV particles containing intact virions could protect encapsulated RNA from RNase degradation.
Fig. 3.
Resistance of purified rPR8-HCV to RNase. Equal amounts of rPR8-HCV and isolated RNA from rPR8-HCV were incubated with 1 μg/μl RNase A at 37 °C for 1 h. After digestion, the samples were tested by real-time PCR
Stability of chimeric rPR8-HCV particles in human plasma
The stability of purified rPR8-HCV preparations was examined in human plasma. Two hundred microliters of the rPR8-HCV dilutions (106 or 104 IU/ml) was incubated at 4 °C for 252 days, 25 °C for 120 days, or 37 °C for 21 days, followed by RNA extraction and HCV real-time PCR analysis. As shown in Fig. 4, there was no significant difference in the stabilities between the high and low concentration samples stored at the same temperature (p > 0.05). No loss of signal was observed when samples were maintained at 4 °C, indicating that this temperature could ensure rPR8-HCV stability for at least 8 months in plasma. The mean for low concentration samples at 4 °C was 17,243 IU/ml (4.24 log10; range 10,160–24,691 IU/ml), and the coefficient of variation was 24.7 %. The mean for high concentration samples at 4 °C was 1,758,462 IU/ml (6.25 log10; range 1,076,027–2,343,720 IU/ml), and the coefficient of variation was 19.9 %. However, the rPR8-HCV samples in plasma were not stable for more than 45 days at 25 °C or for more than 7 days at 37 °C, which suggests that samples kept at temperatures above 4 °C were not completely stable for a long time (p < 0.05). Moreover, samples of purified rPR8-HCV in human plasma that underwent long-term storage (over 12 months) at −20 °C showed no decline in their viral load compared with their original concentrations (data not shown). Additionally, rPR8-HCV preparations at high and low concentrations in human plasma were all stable after five freeze–thaw cycles (data not shown).
Fig. 4.
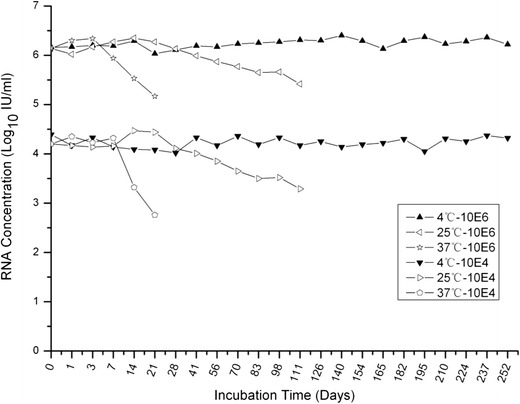
Stability evaluation of rPR8-HCV particles. rPR8-HCV was added to human plasma to a final concentration of 104 and 106 IU/ml. Samples were incubated at 4 °C, 37 °C, or room temperature for set periods of time. Samples were qualified in duplicate at each time point using a HCV RNA PCR fluorescence quantitative diagnostic kit, and the detection results were averaged. The changes in HCV RNA levels in samples were evaluated by linear regression analysis
Chimeric rPR8-HCV particles as positive controls in a clinical assay
To assess the performance of rPR8-HCV particles as positive controls in clinical viral load testing, the samples were assayed for comparison with the positive controls from the COBAS TaqMan HCV test kit, which were non-infectious armored RNA constructs containing a HCV gene fragment (Colucci et al. 2007). For the rPR8-HCV controls, the mean was 18,120 IU/ml (4.26 log10), with a range of 12,400 to 25,600 IU/ml and a coefficient of variation of 19.7 %. The positive control provided within the commercial kit had a mean of 961 IU/ml (2.98 log10), with a range of 693 to 1290 IU/ml and a coefficient of variation of 17.9 %. These results show that the coefficients of variation for the rPR8-HCV controls and for the commercial kit controls were comparable, which demonstrates that the rPR8-HCV control performed as reliably as the controls provided by the commercial kit (Fig. 5).
Fig. 5.
Comparison of the rPR8-HCV positive controls with the COBAS TaqMan HCV test positive controls in a clinical setting. rPR8-HCV was diluted with human plasma to a final concentration of 104 IU/ml and was stored at 4 °C in aliquots of 1.0 ml. The RNA concentrations of samples were assayed once per week for 24 weeks with the COBAS TaqMan HCV test
Linear analysis of rPR8-HCV in the rRT-PCR assays for HCV
The performance of rPR8-HCV as a calibrator for real-time PCR assays was evaluated. A 10-fold dilution of rPR8-HCV particles was individually added into human plasma and quantified using the HCV RNA PCR kit. The correlation between the expected concentration and the observed concentration was determined, and an R 2 value of 0.999 was calculated by linear regression analysis (Fig. 6). The results show that the rPR8-HCV control could function in HCV RNA fluorescence quantitative assays as a calibration standard for the quantification of HCV RNA.
Fig. 6.
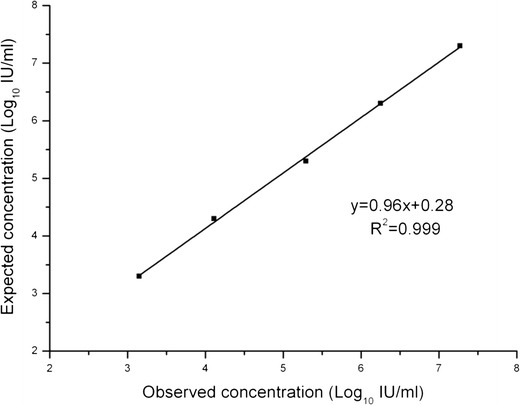
Linearity of the rPR8-HCV control in the HCV RNA fluorescence quantitative assay. rPR8-HCV particles were diluted by 10-fold serial dilutions in normal human plasma to the following concentrations: 2 × 107, 2 × 106, 2 × 105, 2 × 104, and 2 × 103 IU/ml. Each run was performed in triplicate and the mean values were calculated
Discussion
Influenza viruses have been developed as viral vectors for vaccine development and gene therapy, using plasmid-based reverse genetics by inserting a desired RNA sequence into the influenza virus (De Baets et al. 2013; Reece et al. 2013; Wu et al. 2010). Similarly, we hypothesized that chimeric influenza virus could also be used as internal control for nucleic acid test, because the lipid envelope structure of influenza virus would protect the RNA of interest from adverse effect. To date, chimeric influenza virus carrying a long foreign gene have centered on modifying the neuraminidase (NA), matrix protein (M), or NS genes of the influenza viral genome (Kittel et al. 2005; Li et al. 2010; Manicassamy et al. 2010; Wu et al. 2010). Thus, we tried to insert the HCV 5′ UTR into one of these three genes and only the chimeric influenza virus carrying the HCV 5′ UTR was successfully generated by utilizing the smallest NS segment. The insertion of HCV 5′ UTR into NS segment is analogous to the NS1-GFP virus, which was previously reported by Manicassamy et al. (Manicassamy et al. 2010). After that, we evaluated the inactivated and purified chimeric virus particles as an internal control for routine clinical diagnostic rRT-PCR assay. The results show that our obtained chimeric influenza virus is resistant to RNase and is stable, even in plasma, for up to 252 days. Moreover, we also show that our novel HCV positive control is comparable with a commercial armored RNA control when they are used in the real-time PCR detection of HCV.
We developed the chimeric viral particles to overcome the limitations of naked RNA control, mainly their instability. Using influenza reverse genetics, specifically designed RNA was packaged into virions, thereby protecting the target RNA from RNase degradation within the viral envelope. Thus, the chimeric viruses used as internal control could be more stable than the naked RNA. As expected, rPR8-HCV particles were found to be resistant to RNase digestion and exhibited improved storage and handling properties, which will ensure the reliability of qRT-PCR diagnostic assays. Although HCV RNA within the viral particles was not completely stable at temperatures over 4 °C, its stability was preserved in plasma at 4 °C for at least 8 months. It could also be stored for 12 months at −20 °C and endured five freeze–thaw cycles in plasma. Additionally, the chimeric viral particles can be used as control to monitor the entire process of rRT-PCR assays, including the RNA extraction step, which could not be achieved when using naked RNA transcripts.
As an alternative approach to armored RNA technology, the strategy presented here could bypass the patent-related costs of using armored RNA controls. The lower cost of our method makes it more practical for use in routine clinical applications. Moreover, the chimeric viral particles could more authentically mimic target virus particles than armored RNA preparations, because they have a similar genomic organization and surface structure with HCV and other RNA viruses.
Nevertheless, the manipulation of the internal gene faces some potential problems that must be overcome both theoretically and operationally. First, the influenza virus RNA genome is relatively small, and this limits the length of the incorporated target RNA (Garcia-Sastre and Palese 1995). The foreign genes introduced into influenza virus are usually restricted in several hundred nucleotides, which is suitable for most rRT-PCR assays because the RNA length in these assays is usually less than 500 bases. Indeed, the results presented here indicate that a 330-nucleotide HCV RNA sequence could be successfully inserted into virions. A previous report has shown that a GFP gene up to 720 nucleotides could be packaged and propagated using this same strategy, indicating that it is possible to carry a target gene of more than 700 nucleotides (Manicassamy et al. 2010). Further investigation is required to determine if this technique will work for inserting longer fragments, which are particularly important for assays based on signal amplification (e.g., bDNA hybridization) instead of sequence amplification (e.g., rRT-PCR). Second, the high mutation rate of the influenza RNA genome may lead to the loss of inserted target genes (Luytjes et al. 1989). However, the chimeric rPR8-HCV appeared to be completely genetically stable and was able to replicate to high titers in embryonated eggs. Interestingly, even after eight passages in embryonated eggs, the rPR8-HCV virus still showed no loss of the foreign HCV gene. Finally, infectivity may be a potential disadvantage for the routine use of chimeric influenza viruses as internal controls (Falchieri et al. 2012). The PR8 influenza virus bears infectivity, but it has strong host specificity for mice. It is noninfectious to humans, and thus there is little danger of accidental infection during manufacture. Furthermore, embryonated egg cultures confirmed that all chimeric viruses had lost their infectivity after inactivation by formaldehyde. Therefore, the chimeric virus particles can be developed as internal controls that present no infection risks, allowing them to be shipped and used safely. Nonetheless, proper clinical laboratory safety precautions should be followed because the chimeric virus particles represent genetically modified organisms.
Importantly, the presented approach has flexibility for inserting different foreign RNA sequences by influenza reverse genetics. Thus, it is an appropriate method for preparing positive controls for other RNA viruses, such as HIV and SARS-CoV. The resulting controls would be simple to produce in high yields, and they could be standardized according to standard operating procedures (SOPs) like those used for the manufacture of inactivated influenza whole-virus vaccines. Moreover, formalin inactivation, which does not destroy the RNA or inhibit the PCR reaction, avoids the potential danger of handling of infectious controls (Jonges et al. 2010).
In conclusion, our results demonstrate that inactivated chimeric influenza virus particles can be used as internal controls for clinical rRT-PCR assays. They more closely mimic true viral particles. The application of these particles may provide a practical solution to the well-known problem coming from established controls. The approach described here is also appropriate for clinical rRT-PCR assays of other RNA viruses with clinical significance, such as HIV, enterovirus, or any other RNA targets.
Acknowledgments
The authors are grateful to Jie Lian for the valuable comments on the manuscript. This study was supported by the Natural Science Foundation of Shanghai (No. 15ZR1436200) and a grant from joint research project for important diseases by Shanghai Municipal Commission of Health and Family Planning (No. 2013ZYJB0010).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interests.
Ethics statement
The use of human plasma was approved by the ethical committee of Shanghai Centre for Clinical Laboratory, China. This article does not contain any studies with animals performed by any of the authors.
References
- Bossler A, Gunsolly C, Pyne MT, Rendo A, Rachel J, Mills R, Miller M, Sipley J, Hillyard D, Jenkins S, Essmyer C, Young S, Lewinski M, Rennert H. Performance of the COBAS® AmpliPrep/COBAS TaqMan® automated system for hepatitis C virus (HCV) quantification in a multi-center comparison. J Clin Virol. 2011;50:100–103. doi: 10.1016/j.jcv.2010.10.020. [DOI] [PubMed] [Google Scholar]
- Chang M, Gottlieb GS, Dragavon JA, Cherne SL, Kenney DL, Hawes SE, Smith RA, Kiviat NB, Sow PS, Coombs RW. Validation for clinical use of a novel HIV-2 plasma RNA viral load assay using the Abbott m2000 platform. J Clin Virol. 2012;55:128–133. doi: 10.1016/j.jcv.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci G, Ferguson J, Harkleroad C, Lee S, Romo D, Soviero S, Thompson J, Velez M, Wang A, Miyahara Y, Young S, Sarrazin C. Improved COBAS TaqMan hepatitis C virus test (version 2.0) for use with the high pure system: enhanced genotype inclusivity and performance characteristics in a multisite study. J Clin Microbiol. 2007;45:3595–3600. doi: 10.1128/JCM.01320-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Baets S, Schepens B, Sedeyn K, Schotsaert M, Roose K, Bogaert P, Fiers W, Saelens X. Recombinant influenza virus carrying the respiratory syncytial virus (RSV) F85-93 CTL epitope reduces RSV replication in mice. J Virol. 2013;87:3314–3323. doi: 10.1128/JVI.03019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier J, Stormer M, Kleesiek K. Use of bacteriophage MS2 as an internal control in viral reverse transcription-PCR assays. J Clin Microbiol. 2005;43:4551–4557. doi: 10.1128/JCM.43.9.4551-4557.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JD, Wengenack NL, Rosenblatt JE, Cockerill FR, 3rd, Smith TF. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19:165–256. doi: 10.1128/CMR.19.1.165-256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchieri M, Brown PA, Catelli E, Naylor CJ. Avian metapneumovirus RT-nested-PCR: a novel false positive reducing inactivated control virus with potential applications to other RNA viruses and real time methods. J Virol Methods. 2012;186:171–175. doi: 10.1016/j.jviromet.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Felder E, Wolfel R. Development of a versatile and stable internal control system for RT-qPCR assays. J Virol Methods. 2014;208:33–40. doi: 10.1016/j.jviromet.2014.07.028. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A, Palese P. Influenza virus vectors. Biologicals. 1995;23:171–178. doi: 10.1006/biol.1995.0028. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A. 2000;97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoorfar J, Malorny B, Abdulmawjood A, Cook N, Wagner M, Fach P. Practical considerations in design of internal amplification controls for diagnostic PCR assays. J Clin Microbiol. 2004;42:1863–1868. doi: 10.1128/JCM.42.5.1863-1868.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Cheng Y, Guo Q, Li Q. Preparation of a chimeric armored RNA as a versatile calibrator for multiple virus assays. Clin Chem. 2006;52:1446–1448. doi: 10.1373/clinchem.2006.069971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonges M, Liu WM, van der Vries E, Jacobi R, Pronk I, Boog C, Koopmans M, Meijer A, Soethout E. Influenza virus inactivation for studies of antigenicity and phenotypic neuraminidase inhibitor resistance profiling. J Clin Microbiol. 2010;48:928–940. doi: 10.1128/JCM.02045-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose M, Gajardo R, Jorquera JI. Stability of HCV, HIV-1 and HBV nucleic acids in plasma samples under long-term storage. Biologicals. 2005;33:9–16. doi: 10.1016/j.biologicals.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Kittel C, Ferko B, Kurz M, Voglauer R, Sereinig S, Romanova J, Stiegler G, Katinger H, Egorov A. Generation of an influenza A virus vector expressing biologically active human interleukin-2 from the NS gene segment. J Virol. 2005;79:10672–10677. doi: 10.1128/JVI.79.16.10672-10677.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajden M, Minor JM, Zhao J, Rifkin O, Comanor L. Assessment of hepatitis C virus RNA stability in serum by the Quantiplex branched DNA assay. J Clin Virol. 1999;14:137–143. doi: 10.1016/S1386-6532(99)00046-3. [DOI] [PubMed] [Google Scholar]
- Li F, Feng L, Pan W, Dong Z, Li C, Sun C, Chen L. Generation of replication-competent recombinant influenza A viruses carrying a reporter gene harbored in the neuraminidase segment. J Virol. 2010;84:12075–12081. doi: 10.1128/JVI.00046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luytjes W, Krystal M, Enami M, Parvin JD, Palese P. Amplification, expression, and packaging of foreign gene by influenza virus. Cell. 1989;59:1107–1113. doi: 10.1016/0092-8674(89)90766-6. [DOI] [PubMed] [Google Scholar]
- Maloney S, Francis F, Bletchly C, Norton R. Evaluation of a commercially available synthetic RNA lipid enveloped control molecule. J Virol Methods. 2015;211:19–21. doi: 10.1016/j.jviromet.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A. 2010;107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesters HG. Molecular and diagnostic clinical virology in real time. Clin Microbiol Infect. 2004;10:5–11. doi: 10.1111/j.1469-0691.2004.00699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello JB, Rodriguez L, Poinar D, Bos K, Okwi AL, Bimenya GS, Sewankambo NK, Henry KR, Kuch M, Poinar HN. Quantitative assessment of the sensitivity of various commercial reverse transcriptases based on armored HIV RNA. PLoS One. 2010;5:e13931. doi: 10.1371/journal.pone.0013931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasloske BL, Walkerpeach CR, Obermoeller RD, Winkler M, DuBois DB. Armored RNA technology for production of ribonuclease-resistant viral RNA controls and standards. J Clin Microbiol. 1998;36:3590–3594. doi: 10.1128/jcm.36.12.3590-3594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece JC, Alcantara S, Gooneratne S, Jegaskanda S, Amaresena T, Fernandez CS, Laurie K, Hurt A, O’Connor SL, Harris M, Petravic J, Martyushev A, Grimm A, Davenport MP, Stambas J, De Rose R, Kent SJ. Trivalent live attenuated influenza-simian immunodeficiency virus vaccines: efficacy and evolution of cytotoxic T lymphocyte escape in macaques. J Virol. 2013;87:4146–4160. doi: 10.1128/JVI.02645-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin C, Dragan A, Gartner BC, Forman MS, Traver S, Zeuzem S, Valsamakis A. Evaluation of an automated, highly sensitive, real-time PCR-based assay (COBAS Ampliprep/COBAS TaqMan) for quantification of HCV RNA. J Clin Virol. 2008;43:162–168. doi: 10.1016/j.jcv.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Scholtes C, Icard V, Amiri M, Chevallier-Queyron P, Trabaud MA, Ramiere C, Zoulim F, Andre P, Deny P. Standardized one-step real-time reverse transcription-PCR assay for universal detection and quantification of hepatitis delta virus from clinical samples in the presence of a heterologous internal-control RNA. J Clin Microbiol. 2012;50:2126–2128. doi: 10.1128/JCM.06829-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Sun S, Li B, Pan Y, Li W, Zhang K, Li J. External quality assessment for enterovirus 71 and coxsackievirus A16 detection by reverse transcription-PCR using armored RNA as a virus surrogate. J Clin Microbiol. 2011;49:3591–3595. doi: 10.1128/JCM.00686-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jia T, Sun Y, Han Y, Wang L, Zhang R, Zhang K, Lin G, Xie J, Li J. External quality assessment for Avian Influenza A (H7N9) Virus detection using armored RNA. J Clin Microbiol. 2013;51:4055–4059. doi: 10.1128/JCM.02018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WalkerPeach CR, Winkler M, DuBois DB, Pasloske BL. Ribonuclease-resistant RNA controls (armored RNA) for reverse transcription-PCR, branched DNA, and genotyping assays for hepatitis C virus. Clin Chem. 1999;45:2079–2085. [PubMed] [Google Scholar]
- Wang X, Zhang W, Liu F, Zheng M, Zheng D, Zhang T, Yi Y, Ding Y, Luo J, Dai C, Wang H, Sun B, Chen Z. Intranasal immunization with live attenuated influenza vaccine plus chitosan as an adjuvant protects mice against homologous and heterologous virus challenge. Arch Virol. 2012;157:1451–1461. doi: 10.1007/s00705-012-1318-7. [DOI] [PubMed] [Google Scholar]
- Wernike K, Hoffmann B, Dauber M, Lange E, Schirrmeier H, Beer M. Detection and typing of highly pathogenic porcine reproductive and respiratory syndrome virus by multiplex real-time rt-PCR. PLoS One. 2012;7:e38251. doi: 10.1371/journal.pone.0038251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Guan Y, Yang Z, Chen J, Wang H, Chen Q, Sui Z, Fang F, Chen Z. A live bivalent influenza vaccine based on a H9N2 virus strain. Vaccine. 2010;28:673–680. doi: 10.1016/j.vaccine.2009.10.102. [DOI] [PubMed] [Google Scholar]