

Abstract
We have investigated the effect of the transmembrane domain of three viral ion channel proteins on the lipid bilayer structure by X-ray reflectivity and scattering from oriented planar bilayers. The proteins show a similar effect on the lipid bilayer structural parameters: an increase in the lipid bilayer hydrophobic core, a decrease in the amplitude of the vertical density profile and a systematic change in the ordering of the acyl chains as a function of protein-to-lipid ratio. These results are discussed in a comparative view.
Keywords: Viral ion channel proteins, Lipid bilayer, Acyl chain correlation, X-ray reflectivity
Introduction
Virus-encoded membrane proteins interact with lipid bilayers of their host cells during the infection processes. Virus infection leads to significant alterations in the cell membrane, including morphological changes (e.g., budding) and functional modifications (Gonzalez et al. 2003). Among the most relevant event at the cellular level is the modification of the host’s membrane permeability to ions or other small molecules induced by viral ion channels within the lipid bilayers. A family of viral proteins whose common functional characteristic is their capacity to compromise cell membrane integrity during virus infection are known as viroporins (Gonzalez et al. 2003). Integral membrane proteins are typically comprised of several tens of amino acids (aa) which often contain a predominantly hydrophobic domain which can fold to a trans-membrane (TM) α-helix. The insertion of these proteins into the membrane followed by their oligomerization can lead to a typical hydrophilic pore. For amphiphilic helices these homo-oligomeric pores require hydrophobic amino acid residues aa facing the phospholipid bilayer and the hydrophilic residues forming the pore lumen. The hydrophobic barrier of the membrane which prevents the flow of molecules across the membrane is thus corrupted. However, to allow such passage of ions or foreign molecules through the cell membrane, a particular protein must be able to span the hydrophobic core.
Structural analysis of protein–lipid interactions is an extremely difficult task. Not only conformational degrees of freedom of the protein, but also structural changes of the bilayers must be addressed. Lipid–peptide interactions may lead to significant structural changes both of the polypeptides and the lipid bilayer after partitioning of membrane proteins and peptides (May 2000; Salditt 2003). A suitable method to study structural changes in model membranes is X-ray reflectivity and X-ray scattering from highly aligned planar bilayers on solid support, as reviewed in Salditt (2003, 2005).
In this paper, we compare the structural changes in highly aligned stacks of fluid phospholipid membranes imposed by three different viral membrane proteins. The first two are well-known viral ion channels: the M2 protein from influenza A (M2/A), and the Vpu protein of the human immunodeficiency virus type 1 (HIV-1). The last example is the more recently discovered envelope (E) protein from the coronavirus responsible for the severe acute respiratory syndrome (SARS-CoV) (Poutanen et al. 2003). SARS-CoV E proteins have been identified as viral membrane proteins with possible functions in the viral budding process and possibly also as an ion channel. Motivated by the assumed similarity in function between these viroporins in forming cation-selective channels in planner lipid bilayer, we have investigated the effect of their trans-membrane domain (TMD) on the structure of dimyristoyl-phosphocholine (DMPC) bilayers by X-ray reflectivity, as a function of peptide-to-lipid molar ratio P/L and the hydration level of the headgroups.
M2 is a type I integral protein membrane made of 97 aa, which can be divided into three domains: an N-terminal extracellular domain of 24 aa, a single hydrophobic domain of 19 aa which acts as a transmembrane domain(TMD) which forms the pore of the channel, and finally a 54 aa cytoplasmic tail (Holsinger et al. 1994; Duff and Ashley 1992; Duff et al. 1994). The M2 protein forms homo-oligomers whose active state is a tetramer (Sugrue et al. 1991; Pinto et al. 1997). The tetramer is a 3D model which spans the TM region of M2. In its simple picture, this structure consists of four α-helices arranged with approximate fourfold symmetry about a central channel (i.e., pore). The M2 is thought to function as an ion channel that permits protons to enter virus particles during uncoating of virions in endosomes (Pinto et al. 1992). In addition, in influenza virus-infected cells, the M2 protein channel activity might lead to an equilibration of pH between the acidic lumen of the trans-Golgi network and the cytoplasm. Despite the small size of the active M2 oligomer, evidences indicate that ion channel activity is an intrinsic feature to the M2 protein. In fact, ion channel activity has been observed in systems such as oocytes, mammalian cells and yeast. The activity has also been observed in artificial lipid bilayers from purified M2 protein (Vijayvergiya et al. 2004). Finally, the use of the antiviral compound amantadine, was found to block the channel pore, thereby inhibiting the current associated with the channel activity (Hay et al. 1985; Duff and Ashley 1992). The latter process would hinder the proper budding of virus particles.
Vpu is an oligomeric, type 1 TM phosphor-protein containing 81 aa distributed along an N-terminal hydrophobic region of 31 aa and a C-terminal hydrophilic region, connected by a short stretch of basic amino acid residues (Ewart et al. 1996; Schubert et al. 1996a, b; Park et al. 2003). In the HIV-1 life cycle, Vpu has at least two independent physiological activities that correspond to two domains (Schubert and Strebel 1994; Schubert et al. 1996a). The C-terminal cytoplasmic domain is responsible for the degradation of one of the HIV-1 coreceptor molecules, CD4, allowing the env glycoprotein to be transported to the host cell surface. The N-terminal TM segment is critical for Vpu enhancement of virus particle release from infected cells, but the molecular basis of these actions is unknown (Schubert and Strebel 1994; Schubert et al. 1996a). Vpu exerts its effect indirectly, for example by altering the intracellular milieu near membranes of the endoplasmic reticulum or the exocytic pathway. In analogy to M2 ion channels, it has been shown that reconstitution of Vpu in lipid bilayers forms cation channels (Ewart et al. 1996; Schubert et al. 1996a, b).
Like the above-mentioned viroporins, it has been shown recently that also SARS-CoV E protein has the ability to form an ion channels in a lipid bilayer (Wilson et al. 2004). In general, E proteins are well conserved within each of the different groups of coronaviruses (Godet et al. 1992). SARS-CoV E protein is a small protein (∼76 residues) with an unusually long hydrophobic stretch (25–30 aa), placed in between a hydrophilic N- and C-terminus, ∼8 and ∼40 residues long (Marra et al. 2003; Rota et al. 2003). Note that the length of the hydrophobic segment of E protein is significantly larger than the average length of a transmembrane α-helix which is only about 21 residues (Arkin et al. 1998; Arbely et al. 2004). We have recently studied SARS-CoV E protein in model membranes by infrared spectroscopy, and X-ray reflectivity (Arbely et al. 2004; Khattari et al. 2006).
For comparison, and to obtain more complete experimental evidence, we have extended our previous study to the two other viral proteins in this work. The paper is organized as follows: Materials and methods, Results and finally Summary and conclusions.
Materials and methods
Materials
Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was purchased from Avanti Polar lipids (Alabama, AL). The purity of DMPC is claimed to be 99%. Therefore the lipid was used without further purifications. Chloroform (Chl) and 1,1,1,3,3,3-Hexaflour-2-propanal (HFI) (purity: 99.8%) were purchased from Sigma (Germany).
The three peptides were synthesized and purified by standard solid-phase N-(9-fluorenyl) methoxycarbonyl (Fmoc) chemistry as described in Arbely et al. (2004). All the peptides were made in a length encompassing the TMD. The influenza A M2 protein was made of the residues Pro25-Leu43, HIV-1 Vpu protein was made of the residues Met1-Lys31 corresponding to the N-terminal domain, and the SARS-CoV E protein was made of the residues Glu7-Arg38. Below the TMD is indicated in bold within the full protein sequences.
Influenza A virus protein M2
MSLLTEVETPIRNEWGCRCNDSSDPLVVAASIIGILHLILWILDRLF FKCIYRFFEHGLKRGPSTEGVPESMREEYRKEQQSAVDADDSHFVSI ELE
-
(b)
Human Immunodeficiency virus type 1 protein Vpu
MQPIQIAIVALVVAIIIAIVVWSIVIIEYRKILRQRKIDRLIDRIERA EDSGNESEGEISALVEMGVEMGHHAPWDVDDL
-
(c)
Severe Acute Respiratory Syndrome E protein
MYSFVSEETGTLIVNSVLLFLAFVVFLLVTLAILTALRLCAYCCN IVNVSLVKPTVYVYSRVKNLNSSEGVPDLLV
Sample preparation
Lipids were used as purchased to prepare multilamellar stacks of protein/DMPC bilayers following the procedure described by Seul and Sammon (1990). The lipids were first dissolved in a solution of Chl:HFI (1:1 v/v) at a concentration of 20 mg/ml, while the proteins were dissolved in a solution of Chl:HFI (40%:60% v/v) at 2 mg/ml since the protein is more soluble at higher HFI concentration. Varied amounts of the protein stock solution was then mixed with the DMPC stock solution at a final concentration of 5 mg/ml stock solution which yield the desired P/L ratio. Pure solvents were added in order to yield identical final lipid concentration.
The protein to lipid ratio P/L ranged from 1/500-1/7.5. The mixed solutions were spread on Silicon substrates cleaned by two 15-min cycles of ultrasonic bath in methanol, followed by two 15-min cycles in ultra pure water (18 MΩ cm, Millipore, Bedford, MA), and finally dried under nitrogen stream. A droplet of 200 μl was then spread on the Si-wafer of typically 15 × 25 mm2 positioned in an exactly horizontal plane. The spread solution was allowed to dry very slowly to prevent film rupture and dewetting. The samples were then exposed to high vacuum for 12 h in order to remove completely all solvent traces. Afterwards the samples were rehydrated, yielding film thicknesses in the range of D≃2–5 μm. Such a procedure produces well-aligned multilamellar stacks with respect to the substrate with a typical mosaicity (orientational distribution) less than the instrumental resolution (i.e., 0.01°) (Salditt et al. 1999). A very low mosaicity is a prerequisite in applying interface-sensitive X-ray scattering techniques for structural studies of solid-supported bilayers. For inspection by light microscopy, the samples have been kept in the fluid state at the same temperature as for the X-ray experiments. A significant effect on the domain size and texture of the DMPC bilayer stacks was observed by varying the protein type. Images recorded in the L α state at temperature T = 45° and RH = 98% are shown in Fig. 1. The samples were kept in a sealed temperature-controlled chamber (Julabo, Germany) with a water reservoir at the bottom for hydration. Note that multilamellar films are known to exhibit a pronounced domain structure with a large variation of local film thickness. At the same time the orientation of the bilayers is almost perfect, despite the limited lateral extension of the domains, leading to a patch like morphology, see Fig. 1a for the case of pure DMPC. A transition to a smooth interface is caused by adding M2 protein to the DMPC lipid where small empty holes (diameter ≤ 5μm) are formed in the film (Fig. 1b). The formation of small irregularly shaped domains, which comprise a significant fraction of the bilayer surface area are observed instead of the relatively large domain structures in pure DMPC films in the case of Vpu protein, see Fig. 1c. A star like morphology with relatively large smooth areas evolves on the top of the lipid film in the case of SARS-CoV E protein, see Fig. 1d. These changes are very reproducible for different samples and upon translation of the illuminated spot on the sample. The results show that in the L α phase, the viral ion channel proteins drastically affects the multilayers morphology, possibly by changing the line tension between the domains.
Fig. 1.
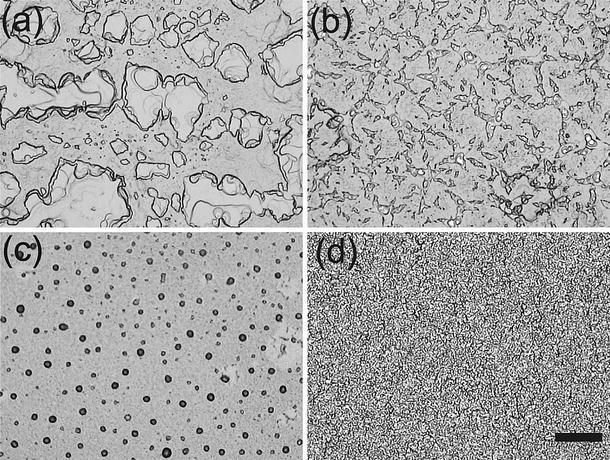
Protein/lipid films imaged in the fluid state (i.e., T = 45°C, RH = 98%) by bright field contrast for a the pure DMPC, b M2, c Vpu and d SARS-CoV E protein at a concentration of P/L = 1/10. The structure of the lipid film changes with the protein type. The scale bar corresponds to 100 μm
X-ray reflectivity
The reflectivity experiments were carried out at two instruments. For SARS-CoV E protein samples, the measurements were performed at the bending magnet beamline D4 of the DORIS storage ring at the synchrotron radiation laboratory HASYLAB/DESY (Hamburg, Germany) using a photon energy of 11 keV (i.e., λ = 1.13 Å), set by a Si(111) monochromator. The reflectivity curves were measured with a fast scintillation counter (Cyberstar, Oxford instruments), using motorized collimating slits on both incident and reflected beam paths. The reflectivity curves were corrected for ring current, sample illumination, and diffuse background (offset-scan). For the other two proteins (i.e., M2 and Vpu) samples, the measurements were preformed at a home built reflectometer with a sealed tube of CuK α (i.e., λ = 1.54 Å) radiation, equipped with collimating X-ray multilayer mirror, motorized slits and a fast scintillation counter. The setup is described in details elsewhere (Li 2005). The reflectivity curves were corrected for sample illumination, and diffuse background (offset-scan). At both instruments, the chamber was mounted on the z-axis diffractometer with the samples oriented vertically.
Prior to X-ray reflectivity measurements, the resulting multilamellar stacks were inserted in a closed temperature and humidity controlled chamber. The chamber consists of two concentric stainless steel cylinders with kapton windows. The chamber temperature was maintained by a flow of water/glycol mixture connected to a temperature-controlled reservoir (Julabo, Germany). The temperature was measured close to the sample holder by a Pt100 sensor with thermal stability in the range of 0.02 K over several hours (Münster et al. 2000). The average temperature of the samples were kept at T = 45°C, well above the chain melting transition. The samples were mounted in the inner cylinder of the chamber facing a humid atmosphere controlled by adding a water or salt–water mixture to a reservoir placed at the bottom of the cylinder (Brotons et al. 2003). While more dehydrated conditions have also been used, here we will concentrate on relative humidity (RH) values of ≈98%. During the measurements, M2 and Vpu samples were facing a humid atmosphere produced by salt free-reservoir, while in the case of E protein, a reservoir of K2SO4 solution was used to achieve the desired relative humidity. The pure DMPC bilayers were swollen up to a repeat distance of typically d≃ 50 Å indicative of partially hydrated samples.
Results
The reflectivity curves of the three proteins at various P/L are shown in Fig. 2, shifted vertically for clarity. The reflectivity is plotted as a function of the vertical momentum transfer q z after subtraction of the diffuse scattering (offset scan), and after illumination correction. The curves show the typical features of highly oriented multilamellar films: the plateau of total reflection at small q z, and a set of sharp and intense, equidistant Bragg peaks. The intensity and number of Bragg peaks decreases with increasing P/L, indicative of peptide-induced lamellar disorder. The complete P/L series has been measured at constant temperature T = 45°C and RH≈0.98% (i.e., the sample are in the L α phase). In the fluid state, the reflectivity curves depend in a systematic way on the peptide concentration. This dependency and the corresponding d-spacing depend on the specific protein. For example at P/L = 1/20, Vpu has the largest effect on the membrane repeat distance d while the effect of the E protein effect was the smallest. The SARS-CoV E protein samples were measured at DESY/Hamburg (see Fig. 2a) and the other samples were measured at the home built reflectometer. Therefore, the peaks are sharper in the SCoV E series than in the other two series. For M2 and vpu, it is becoming difficult to analyze the curves above a certain P/L ratio due to the lower number of Bragg peaks in the reflectivity curves.
Fig. 2.
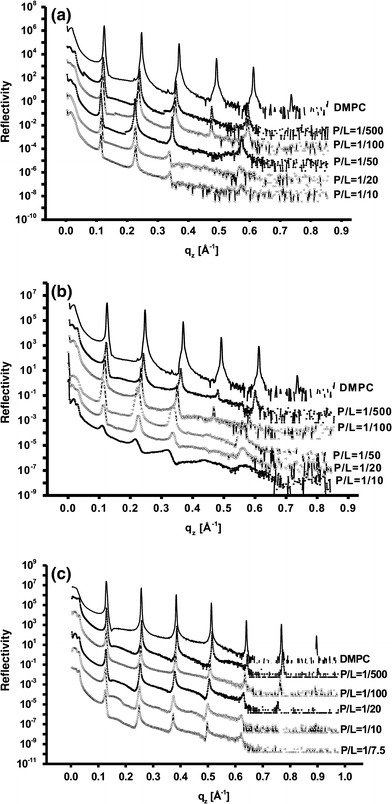
Reflectivity curves of multilamellar protein/DMPC membranes at RH≈98% and constant T = 45°C. Different P/L ratios are presented for a M2, b Vpu and c SARS-CoV E protein. The curves are shifted vertically for clarity. The curves exhibit the typical pattern of a lamellar structure with well-defined periodicities d. The smaller peak heights for P/L = 1/10 reflect the increase of fluctuations and/or static disorder with P/L
One of our goals is to achieve full q
z range fits of each curve to determine the electron density profile (i.e., ρ(z)). However, thermal fluctuations, lamellar disorder and resolution effects make this task difficult (Salditt et al. 2002). While such work is in progress, we apply here an empirical Fourier synthesis (FS) scheme, exploiting only the area under Bragg peak intensities, as it is often used for such multilamellar lipid membranes, e.g., see Li et al. (2004). In simple terms the one-dimensional electron density profile ρ(z) normal to the interface is computed by N
0 Fourier coefficients f
n (up to a normalization constant) by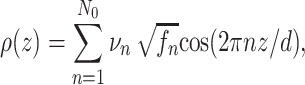 where the phases νn are reduced to positive/negative signs due the mirror plane symmetry of the bilayer. Note that the maximum Bragg peak intensities I
n have been corrected by an empirical Lorentz-like correction factor to calculate the nth Fourier coefficient as f
n = I
n
q
z. The choice of this factor can be debated (Li et al. 2004). Note that in this simplified approach, changes in the peak intensities with P/L are always attributed to the form factor, since the structure factor is tacitely assumed to be that of the ideal lattice. This assumption is an idealization, which is justified if thermal fluctuations are weak. Conditions of partial hydration and the presence of a flat boundary help to maintain in the validity range. However, the decrease of peak intensities with P/L is probably primarily a disordering effect. Nevertheless the ratios of the peak intensities can be exploited to compute the functional profile ρ(z), which is to be regarded as the intrinsic profile convoluted with a positional distribution function (smearing) due to disorder. For example, more than seven lamellar reflectivity reflections have been observed for the peptide-free bilayers, while only five or four orders persist in the presence of peptide sequence. This phenomenon is typical for many membrane active peptides or proteins, and leads to a smoothing of the deduced bilayer profile. Of course the local profile is not necessarily flatter for high P/L. Instead this can be regarded as a consequence of the increased lamellar disorder, since the determined profiles have to be regarded not as the intrinsic profile, but rather to the convolution of the intrinsic profile with the distribution function of the bilayer position, which broadens with increasing lamellar disorder. However, five orders of reflectivity data are clearly enough for calculating the electron distribution of the peptide/bilayer with a sufficiently high resolution to determine structural quantities, such as the distance between the headgroups, e.g., the distance between the two maxima corresponding to the phosphorous atoms d
pp.
where the phases νn are reduced to positive/negative signs due the mirror plane symmetry of the bilayer. Note that the maximum Bragg peak intensities I
n have been corrected by an empirical Lorentz-like correction factor to calculate the nth Fourier coefficient as f
n = I
n
q
z. The choice of this factor can be debated (Li et al. 2004). Note that in this simplified approach, changes in the peak intensities with P/L are always attributed to the form factor, since the structure factor is tacitely assumed to be that of the ideal lattice. This assumption is an idealization, which is justified if thermal fluctuations are weak. Conditions of partial hydration and the presence of a flat boundary help to maintain in the validity range. However, the decrease of peak intensities with P/L is probably primarily a disordering effect. Nevertheless the ratios of the peak intensities can be exploited to compute the functional profile ρ(z), which is to be regarded as the intrinsic profile convoluted with a positional distribution function (smearing) due to disorder. For example, more than seven lamellar reflectivity reflections have been observed for the peptide-free bilayers, while only five or four orders persist in the presence of peptide sequence. This phenomenon is typical for many membrane active peptides or proteins, and leads to a smoothing of the deduced bilayer profile. Of course the local profile is not necessarily flatter for high P/L. Instead this can be regarded as a consequence of the increased lamellar disorder, since the determined profiles have to be regarded not as the intrinsic profile, but rather to the convolution of the intrinsic profile with the distribution function of the bilayer position, which broadens with increasing lamellar disorder. However, five orders of reflectivity data are clearly enough for calculating the electron distribution of the peptide/bilayer with a sufficiently high resolution to determine structural quantities, such as the distance between the headgroups, e.g., the distance between the two maxima corresponding to the phosphorous atoms d
pp.
The centrosymmetric electron density profiles of the bilayer-containing proteins are shown in Fig. 3, on arbitrary scale. The profiles were calculated using Eq. 1 with appropriate choice of phases (− , − , + , − , + , − , −). The curves have been normalized such that the area under the first Bragg peak is set to be one, while the higher order Bragg peaks were properly scaled with respective to the first peak intensity. Typical electron density profiles have been obtained for such model systems. The simple way to interpreted the profiles is the following: the two mean peaks of ρ(z) on either sides of the figure correspond to phospholipid headgroups, the two sides minima to the water layer and the central minimum is associated with terminal methyl moiety of the hydrocarbon chains.
Fig. 3.
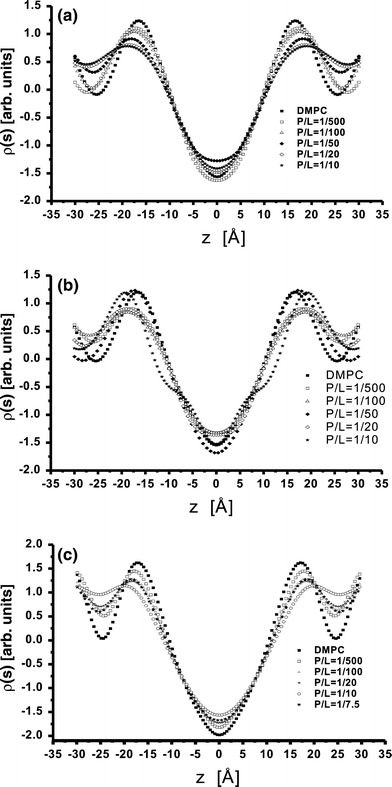
The electron density profiles of the pure DMPC bilayer and different P/L ratios of a M2, b Vpu and c SARS-CoV E protein. The curves have been computed from the integrated peak intensities shown in Fig. 3 by FS method. The normalization procedure is described in the text
The multilamellar d-spacing and the bilayer thickness d pp are shown in Fig. 4 as a function of P/L for all DMPC–protein complexes in the fluid state. For all P/L samples, d was obtained from the reflectivity curves by fitting the q z values at each Bragg peak as a function of Bragg order (i.e., n) to a straight line. The presence of peptides in the lipid induces a shift in Bragg peaks toward smaller q z values as compared to the pure lipid. At fixed P/L ratio, this shift varies with the protein type. The solid lines in the curves serve as guide to the eye (empirical fits) and do not represent any theoretical model. The d-spacing increase is strongest in the case of the Vpu-DMPC samples. The E protein shows the smallest swelling effect. Finally, above a critical P/L ratio, the swelling for the M2 and vpu saturates to a uniform value.
Fig. 4.
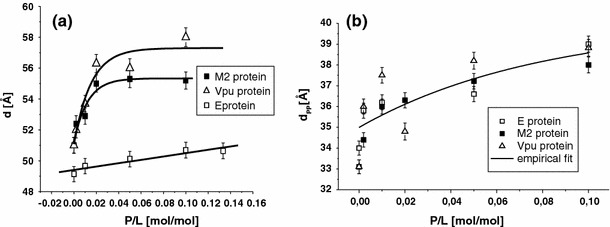
a The membrane repeat distance d and b the bilayer thickness d pp defined as the peak-to-peak distance in the electron density profiles plotted as a function of P/L at T = 45°C and RH≈0.98%. All three systems show a bilayer thickening. Solid lines are empirical fits to illustrate the trend in the data
The bilayer thickness defined as the distance between the two maxima associated with the phosphorous group d pp as a function of P/L was determined from ρ(z). Note that, d pp is unaffected by the normalization procedure of the electron density profiles (Wu et al. 1995). The effect of the three viral proteins on d pp in the fluid state is presented in Fig. 4b.
An increase in d pp is observed for all three viral protein systems. The exact functional form is difficult to extract due to the statistical scatter in the data. A linear fit would describe the increase fairly well with a total change of up to 4Å at high P/L. However, at small P/L the changes seem to be accentuated, and saturation may occur at high P/L. These data reveal a remarkable effect on the bilayer structure depending on the protein concentration. All three proteins show a thickening effect as a function of P/L which is in contrast to the bilayer thinning observed for many α-helical antibiotic peptides in the surface state (parallel state) with the helical axis in the horizontal plane. For this class of peptides, bilayer thinning drives the transmembrane insertion. Above a critical concentration (P/L)*, there is a transition from a parallel to a perpendicular (i.e., transmembrane) conformation with respect to the membrane plane (Huang and Wu 1991; Ludtke et al. 1995; Chen et al. 2003). For the present class of peptides, it is known from FTIR and X-ray reflectivity measurements that the E protein (e.g., at least at high P/L concentration) is oriented perpendicular to the membrane plane (Arbely et al. 2004), and it has also been shown that the other two viral ion channel proteins are oriented perpendicular to the bilayer (Kukol et al. 1999; Kukol and Arkin 1999).
It would be interesting to connect the thickening in d pp to the hydrophobic length and hydrophobic mismatch with DMPC of the different proteins. A simplistic assumption of a TM α-helix with each amino acid contributing an axial length of 1.5 Å (Killian et al. 1996), would result in 28.5 Å based on the 19 aa of M2. Contrarily, this simplistic argument would give 46.5 Å based on the 31 aa for Vpu, and 48 Å based on the 32 aa for SCoV E protein. Indeed, the helical domain of Vpu is known to extend beyond the hydrophobic core from solid-state NMR studies (Sharp et al. 1999). Furthermore, the helical axis is tilted to reduce the mismatch. In the case of SCoV E protein, the enormous mismatch prohibits the formation of a single TM helix. Instead, FTIR and X-ray data support the conclusion of a hairpin conformation (Arbely et al. 2004). Finally, M2 should not exhibit a large mismatch. A tetrameric pore structure has been reported (Tian et al. 2003; Sakaguchi et al. 1997). Here, we see that despite very different structures and length of the TMD, all three proteins lead to a bilayer thickening.
Having quantified the bilayer thickness d pp, we can also briefly address the increase of water spacing d w = d − d pp and associated changes in the interaction potentials with P/L. All measurements have been carried out at partial hydration (corresponding to an osmotic pressure of about 2-3 × 106 Pa, optimized for structural investigations and reduced thermal fluctuations. Thus, at a hydration high enough so that the bilayer structure is already indistinguishable from that at full hydration, but small enough so that about six lamellar diffraction orders can be observed at P/L = 0. The swelling behavior (corrected for bilayer thickness variation) is as follows: for M2, d w is approximately constant, for Vpu an increase, and for SARS E a slight decrease with P/L is observed. Electrostatic forces and interaction with the headgroup dipoles may also be important for this swelling or and deswelling behavior. An estimated effective charge of the proteins − 0.8, +1, and − 1, respectively, is calculated at neutral pH, from the sequence only by single amino acid titration curves (Protein Calculator v3.3, http://www.cdputnam@scripps.edu). For Vpu, the swelling could also be an effect of steric repulsion, since lamellar fluctuations seem to increase strongly with P/L. Concerning lamellar disordering, the general picture is the following: With increasing P/L the lamellar orders decay, until at P/L = 0.1 only 3–5 orders remain, depending on the protein. This is is the result of either increased (dynamic) fluctuation amplitudes or static disordering of the lamellae by the proteins. In the case of Vpu the effect is particularly strong, in the case of SARS E protein it is rather small. In addition, a broadening of the lamellar peaks is observed for Vpu which cannot be explained in the framework of standard smectic elasticity theory, e.g., by an increase of the Caille parameter η with P/L. Instead, domain size distribution and/or the functional form of the positional correlation functions must change. Rather than by changes in the bending rigidity and associated steric (Helfrich) repulsion, the observed effects may be explained by perturbations or static disordering of the lamellar stack at high protein concentration.
The lateral structure of the membranes on molecular length scales can be probed by X-ray scattering in the geometry of grazing incidence diffraction (GID) (Salditt 2003). Here, we probe the lateral ordering of acyl chains and any changes of this ordering induced by the viral membrane proteins. The same sample series (function of P/L) as for the reflectivity have been investigated. The experiments have been carried out at the D4 station of the Doris III storage ring of HASYLAB/DESY Hamburg, using a photon energy of 20 keV. The samples were placed horizontally at the bottom of the chamber, and the temperature was set to T = 45°C and RH = 75% to insure that the DMPC bilayers are in the L α phase. We have measured the scattering distribution in GID geometry for all P/L of the three proteins as a function of lateral momentum transfer q ∥. All three sample series showed a strong decrease in the intensity of the acyl chain correlation peak. This seems to be a general finding for lipid–protein mixtures and indicates a disordering effect, or more precisely a loss of scattering volume (area).
Figure 5 illustrates the effect of the viral ion channel proteins on the ordering of the acyl tails. Lorentzian fits to the scattering distribution have been performed for each P/L (i.e., see solid line in Fig. 5a at P/L = 1/10) yielding the peak position q 0(P/L), and consequently the nearest neighbor distance of acyl chains d xy (i.e., 2π/q 0), as well as the correlation length ξr(P/L) = 1/HWHM, determined from the width (half width at half maximum, HWHM) of the correlation peak. The fluid correlation peak moves to slightly to higher q ∥ as P/L is increased, corresponding to a decrease in the nearest neighbor spacing. The corresponding shift is quantified in Fig. 5b. In all of the samples, the measured HWHM values correspond to correlation lengths of the exponentially decaying positional correlations between 4.5 and 7 Å. These values depend strongly on the hydration, and the dependence on P/L is difficult to extract.
Fig. 5.
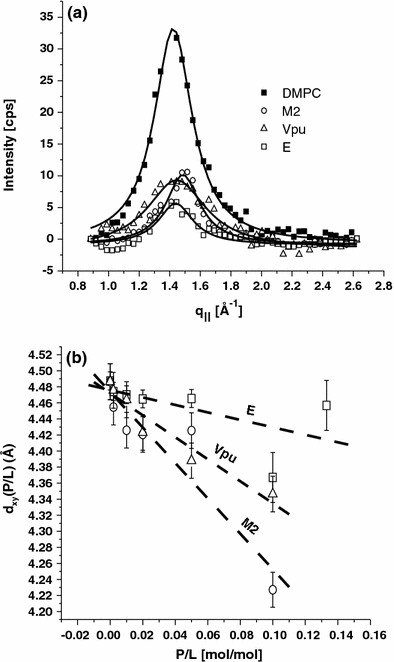
a Lipid chain correlation peak of pure DMPC, along with the samples containing the three viral ion channel proteins at P/L = 1/10. The curves are shown after background and ring current correction of the raw data. b The nearest neighbor distance between acyl chains if found to decrease with P/L
Summary and conclusions
In summary, incorporation of viral membrane proteins in fluid phospholipid membranes induces significant changes in the bilayer structure depending on the protein concentration. These changes start already at the lowest peptide concentration P/L = 0.002. Firstly, the reflectivity curves change. In particular, the lamellar diffraction intensities decrease, indicative of increased disorder in inter-bilayer positions. A small shift of the peak position reflects a swelling of the water layer as well as a slight thickening of the bilayer with P/L. The relative peak intensities change resulting in a flattening of the density profile. This is probably also primarily a disordering effect. At the same time, the average bilayer thickness increases with P/L. Secondly, the chain correlation peak decreases and slightly shifts its position. The intensity decrease reflects increasing lateral disordering of the chains on a molecular scale. The shift of the peak corresponds to a decrease of the average distance between acyl chains. This could be dominated by the contribution of lipids next to the protein which are forced to elongate to minimize the hydrophobic mismatch.
The changes with P/L can only be explained if a local insertion of a protein changes bilayer properties in a non-local collective manner. A local insertion thus results in a medium range perturbation. This result is particularly pronounced for M2 and Vpu. Indeed, a pronounced change in scattering intensity even at relatively small concentrations is particularly evident for M2 and Vpu. This implies that the viral proteins change the state of the bilayer in its vicinity over some range, and not only locally. In general terms, a hydrophobic mismatch of the TM α-helix or more generally the perturbation of the bilayer at the protein surface can be the local source of the such collective elastic effect. If the protein has a large hydrophobic surface, it will tend to elongate the adjacent chains. This leads to a thickening of the bilayer (on the average). This is in contrast to the bilayer thinning effect which is observed for amphiphilic membrane proteins which are oriented horizontally at the hydrophobic/hydrophilic interface of the bilayer. Since the bilayer is an elastic medium it will react on chain elongation by lateral compression, as quantified by bilayer elasticity and packing theories (Ben-Shaul and Gelbart 1994; May 2000). In a simple physical view, the protein acts as an inclusion which imposes a boundary condition on the elastic medium of the acyl chain fluid. At the same time, the total scattering intensity of the acyl chain peak decreases strongly indicative of a disordering effect, probably associated with a laterally non-constant thickness profile. This variation of bilayer thickness may also explain the broadening of the vertical density profile. The observed effects on lateral distance d xy and bilayer thickness d pp are consistent. All observed effects are qualitatively the same for all three viral ion channels, but quantitatively different. For example, Vpu has the largest effect on the bilayer thickness, whereas M2 has the largest effect on d xy.
Let us briefly address the molecular conformations of the proteins in the lipid bilayer. For SARS-CoV E protein a hydrophobic sequence of 26 aa and a corresponding total transmembrane length of about 39 Å would lead to an impossibly high hydrophobic mismatch for a transmembrane helix. However, FTIR results show that SARS-CoV E protein helices are oriented perpendicular (Arbely et al. 2004). At the same time, we know from the electron density profiles of site-specific iodination labels [The phenylalanine (i.e., residue 23)] that this label is located at the hydrophilic/hydrophobic interface of the bilayer (Khattari et al. 2005). Along with the palindromic sequence, this has led us to conclude that the protein forms a small α-helical hairpin. For Vpu and M2, published NMR structures provide an atomistic view of these structures. From the present X-ray reflectivity and scattering study detailed conclusions on the conformation of these proteins cannot be made, in part due to the present lack of labeled proteins and respective site-labeled density profiles. As for SCoV E protein, simple counting of hydrophobic aa in Vpu would also give a large hydrophobic mismatch. Namely, the TMD of Vpu protein is composed of 31 aa of which about 26 ± 2 aa are probably associated with the transmembrane domain, leading to a hypothetical length of 39 ± 3 Å) (Kukol and Arkin 1999). In the case of the M2 protein, the axial length is about 28 ± 3 Å, noticeably different from the other two proteins (Kukol et al. 1999).
In general, protein–lipid or peptide–lipid complexes are expected to respond to such an energetically unfavorable mismatch situation in a number of ways, depending on the molecular details of the system. The polypeptides can tilt (Vpu) or kink when their TM hydrophobic length is too long to match the bilayer, thus reducing their effective length. In the opposite situation, a TM helix can adopt a more extended conformation or a non-transmembrane orientation (Zhang et al. 1995). The lipid bilayer can also respond to the mismatch situation by adapting the local thickness and/or curvature, as postulated theoretically a ling time ago (Owicki et al. 1978). In the present work, we have observed such collective bilayer response to the insertion of three different viral ion channels. More structural information on the protein conformation and degrees of freedom could be derived in future from full 2D mappings of reciprocal space, and from site-specific labels in scattering. At the same time, one may speculate that the bilayer degrees of freedom and the indirect alterations which the proteins impose on the bilayer may also be functionally relevant. For instance, lateral strain fields could change important bilayer properties such as ion permeability in the same way as equilibrium thermal fluctuations near the main phase transition (Makarov 2005). To this end, the present work shows how structural changes of the bilayer can be measured experimentally. Ideally, structure of the protein as well as the bilayer should be studied simultaneously with functional properties. To this end, the bilayer lipid composition which has be the same in this study, must also be varied.
Acknowledgment
Financial support by the DFG through the German–Israel–Palestine trilateral project SA 7772/6-1 is gratefully acknowledged.
References
- Arbely A, Khattari Z, Brotons G, Akkawi M, Salditt T, Arkin IT. A highly unusual palindromic transmembrane helical hairpin formed by SARS coronavirus E protein. J Mol Biol. 2004;341:769–779. doi: 10.1016/j.jmb.2004.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin IT, Brunger AT. Statistical analysis of predicted transmembrane alpha-helices. Biochim Biophys Acta. 1998;1429:113–128. doi: 10.1016/s0167-4838(98)00225-8. [DOI] [PubMed] [Google Scholar]
- Ben-Shaul A, Gelbart M (1994) Statistical thermodynamics of amphiphilic self-assembly. In: Gelbart W, Ben-Shaul A, Roux D (eds) Micelles, Membranes, Microemulsions, and Monomers. Springer, Berlin Heidelberg New York
- Brotons G, Salditt T, Dubois M, Zemb Th. Highly oriented, charged multilamellar membranes osmotically stressed by a polyelectrolyte of same sign. Langmuir. 2003;19:8235–8244. doi: 10.1021/la034733j. [DOI] [Google Scholar]
- Chen FY, Lee MT, Huang HW. Evidence for membrane thinning effect as the mechanism for peptide-induced pore formation. Biophys J. 2003;34:3751–3758. doi: 10.1016/S0006-3495(03)75103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff KC, Ashley RH. The transmembrane domain of influenza A M2 protein forms amantadine-sensitive proton channels in planar lipid bilayers. Virology. 1992;190(1):485–489. doi: 10.1016/0042-6822(92)91239-Q. [DOI] [PubMed] [Google Scholar]
- Duff K, Gilchrist P, Saxena A, Brandshaw J. Neutron diffraction reveals the site of amantadine blockade in the influenza A M2 protein ion channel domain. Virology. 1994;202:287–293. doi: 10.1006/viro.1994.1345. [DOI] [PubMed] [Google Scholar]
- Ewart GD, Sutherland T, Gage PW, Cox GB. The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J Virol. 1996;70:7108–7115. doi: 10.1128/jvi.70.10.7108-7115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godet M, L’Haridon R, Vautherot JF, Laude H. TGEV coronavirus ORF4 encodes a membrane protein that is incorporated into virions. Virology. 1992;166:666–675. doi: 10.1016/0042-6822(92)90521-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez ME, Carrasco L. Viroporins. FEBS Lett. 2003;552:28–34. doi: 10.1016/S0014-5793(03)00780-4. [DOI] [PubMed] [Google Scholar]
- Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4(11):3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger L, Nichani D, Pinto L, Lamb R. Influenza A virus M2 ion channel protein: a structure-function analysis. J Virol. 1994;68:1551–1563. doi: 10.1128/jvi.68.3.1551-1563.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HW, Wu Y. Lipid–alamethicin interactions influence alamethicin orientation. Biophys J. 1991;60:1079–1087. doi: 10.1016/S0006-3495(91)82144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattari Z, Brotons G, Arbely E, Arkin IT, Metzger TH, Salditt T. SARS E protein in phospholipid bilayers: an anomalous X-ray reflectivity study. Physica B. 2005;357:34–38. doi: 10.1016/j.physb.2004.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattari Z, Brotons G, Akkawi M, Arbely E, Arkin IT, Salditt T. SARS coronavirus E protein in phospholipid bilayers: a X-ray. Biophys J. 2006;90:2038–2050. doi: 10.1529/biophysj.105.072892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian JA, Salemink I, De Planque MRR, Lindblom G, Koeppe R, Greathouse DV. Induction of nonbilayer structures in diacylphosphatidylcholine model membranes by transmembrane-helical peptides: importance of hydrophobic mismatch and proposed role of tryptophans. Biochemistry. 1996;35:1037–1045. doi: 10.1021/bi9519258. [DOI] [PubMed] [Google Scholar]
- Kukol A, Arkin IT. Vpu transmembrane peptide structure obtained by site-specific Fourier transform infrared dichroism and global molecular dynamics searching. Biophys J. 2003;77:1594–1601. doi: 10.1016/S0006-3495(99)77007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukol A, Adams PD, Rice LM, Brunger AT, Arkin IT. Experimentally based orientational refinement of membrane protein models: a structure for the influenza A M2 H+ channel. J Mol Biol. 1999;286:951–962. doi: 10.1006/jmbi.1998.2512. [DOI] [PubMed] [Google Scholar]
- Li C (2005) Strukturanalyse von Antibiotischen Peptides in Lipidmembranen mittels Röntgenreflektivität. PhD dissertation, Goerg-August-Universität Göttingen
- Li C, Constantin D, Salditt T. Biomimetic membranes of lipid-peptide model systems prepared on solid support. J Phys Condens Matter. 2004;16:S2439–S2453. doi: 10.1088/0953-8984/16/26/017. [DOI] [Google Scholar]
- Ludtke S, He K, Huang HW. Membrane thinning caused by magainin 2. Biochemistry. 1995;34:16764–16769. doi: 10.1021/bi00051a026. [DOI] [PubMed] [Google Scholar]
- Makarov I (2005) Theoretical and experimental description of permeability of peptide-containing membranes, Ph.D. thesis, University of Göttingen
- Marra MA, et al. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- May S. Theories on structural perturbations of lipid bilayers. Curr Opin Coll Int Sci. 2000;5:244–249. doi: 10.1016/S1359-0294(00)00062-5. [DOI] [Google Scholar]
- Münster C, Lu J, Schinzel S, Bechinger B, Salditt T. Grazing incidence X-ray diffraction of highly aligned phospholipid membranes containing antimicrobial peptides magainin 2. Eur Biophys J. 2000;28:683–688. doi: 10.1007/s002490050008. [DOI] [PubMed] [Google Scholar]
- Owicki JC, Springgate MW, McConnell HM. Theoretical study of protein–lipid interactions in bilayer membranes. Proc Natl Acad Sci USA. 1978;75:1616–1619. doi: 10.1073/pnas.75.4.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Montal MO, Montal M, Opella SJ. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J Mol Biol. 2003;333:409–424. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell. 1992;69(3):517–528. doi: 10.1016/0092-8674(92)90452-I. [DOI] [PubMed] [Google Scholar]
- Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci USA. 1997;94(21):11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poutanen SM, et al. Identification of severe acute respiratory syndrome in Canada. N Engl J Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- Rota PA, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Sakaguchi T, Tu Q, Pinto LH, Lambd RA. The active oligomeric state of the minimalistic influenza virus M2 ion channel is a tetramer. Proc Natl Acad Sci. 1997;94:5000–5005. doi: 10.1073/pnas.94.10.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salditt T. Lipid–peptide interaction in oriented bilayers probed by interface-sensitive scattering methods. Curr Opin Struct Biol. 2003;13:467–478. doi: 10.1016/S0959-440X(03)00113-1. [DOI] [PubMed] [Google Scholar]
- Salditt T. Thermal fluctuations and stability of solid-supported lipid membranes. J Phys Condens Matter. 2005;17:R287–R314. doi: 10.1088/0953-8984/17/6/R02. [DOI] [Google Scholar]
- Salditt T, Münster C, Lu Y, Vogel M, Fenzl W, Souvorov A. Specular and diffuse scattering of highly aligned phospholipids membranes. Phys Rev E. 1999;60:7285–7289. doi: 10.1103/PhysRevE.60.7285. [DOI] [PubMed] [Google Scholar]
- Salditt T, Li C, Spaar A, Mennicke A. X-ray reflectivity of solid-supported multilamellar membranes. Eur Phys J. 2002;7:105–116. [Google Scholar]
- Schubert U, Strebel K. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J Virol. 1994;68(4):2260–2271. doi: 10.1128/jvi.68.4.2260-2271.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Bour S, Ferrer-Montiel AV, Montal M, Maldarell F, Strebel K. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J Virol. 1996a;70(2):809–819. doi: 10.1128/jvi.70.2.809-819.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1 infected cells. FEBS Lett. 1996b;398:12–18. doi: 10.1016/S0014-5793(96)01146-5. [DOI] [PubMed] [Google Scholar]
- Seul M, Sammon MJ. Preparation of surfactant multilayer films on solid substrates by deposition from organic solution. Thin Solid Films. 1990;185:287–305. doi: 10.1016/0040-6090(90)90093-S. [DOI] [Google Scholar]
- Sharpe S, Yau W-M, Tycko R. Structure and dynamics of the HIV-1 Vpu transmembrane domain revealed by solid-state NMR with magic-angle spinning. Biochemistry. 1999;45:918–933. doi: 10.1021/bi051766k. [DOI] [PubMed] [Google Scholar]
- Sugrue RJ, Hay AJ. Structural characteristics of the M2 protein of influenza A viruses: evidence that it forms a tetrameric channel. Virology. 1991;180(2):617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Gao PF, Pinto LH, Lamb RA, Cross TA. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Protein Sci. 2003;12:2597–2605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayvergiya V, Wilson R, Chorak A, Gao PF, Cross TA, Busath DD. Proton conductance of influenza virus M2 protein in planar lipid bilayers. Biophys J. 2004;87(3):1697–1704. doi: 10.1529/biophysj.104.043018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L, Mckinlay C, Gage P, Ewart G. SARS coronavirus E protein forms cation-selective ion channels. Virology. 2004;330:322–331. doi: 10.1016/j.virol.2004.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, He K, Ludtke SJ, Huang HW. X-ray diffraction study of lipid bilayer membrane interacting with amphiphilic helical peptides: diphytanoyl phosphatidylcholine with alamethicin at low concentrations. Biophys J. 1995;68:2361–2369. doi: 10.1016/S0006-3495(95)80418-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YP, Lewis RN, Henry GD, Sykes BD, Hodges RS, Mc Elhaney RN. Peptide models of helical hydrophobic transmembrane segments of membrane proteins. 1. Studies of the conformation, intrabilayer orientation, and amide hydrogen exchangeability of Ac-K2-(LA)12-K2-amide. Biochemistry. 1995;34:2348–2361. doi: 10.1021/bi00007a031. [DOI] [PubMed] [Google Scholar]