

Abstract
The use of disulfide-rich backbone cyclized polypeptides as molecular scaffolds to design a new generation of bioimaging tools and drugs that are potent and specific, and thus might have fewer side effects than traditional small molecule drugs is gaining increasing interest among the scientific but also in the pharmaceutical industry. Highly constrained macrocyclic polypeptides are exceptionally more stable to chemical, thermal and biological degradation and show better biological activity when compared to their linear counterparts. Many of these relatively new scaffolds have been also found to be highly tolerant to sequence variability, aside from the conserved residues forming the disulfide bonds, able to cross cellular membranes and modulate intracellular protein-protein interactions both in vitro and in vivo. These properties make them ideal tools for many biotechnological applications. This article provides and overview of the new developments on the use of several disulfide-rich backbone cyclized polypeptides including cyclotides, θ-defensins and sunflower trypsin inhibitor peptides in the development of novel bioimaging reagents and therapeutic leads.
Keywords: cyclotides, CCK, cystine-knot, drug design, backbone cyclized polypeptides, protein-protein interactions, cyclic peptides
Introduction
The genomic and proteomic revolutions have provided us with an ever-increasing number of mechanistic insights into human diseases. Mutated genes and pathologic protein products are emerging as the basis for the development of novel therapeutic agents to treat human diseases such as cancer or autoimmune diseases. However, the selective disruption of protein-protein interactions (PPIs) still remains a very challenging task, as the interacting surfaces are relatively large and relatively flat. In fact, among the most intractable of targets are those involving intracellular protein-protein interactions, which require the therapeutic agent to efficiently cross the cell membrane.
Broadly speaking, there are only two major structural classes of approved drugs, small molecules and protein therapeutics (also known as biologics). Small molecules typically show good stability and good pharmacological properties, but their intrinsic small size (≤100 atoms) endows them with only a modest overall surface area available to contact a protein target. This limits their ability to effectively target large surfaces involved in protein-protein interactions. Protein-based therapeutics, on the other hand, possess high specificity/selectivity and high affinity for protein targets (1). The use of therapeutic monoclonal antibodies to target extracellular protein receptors is just one example (2, 3). Antibodies, however, suffer from clear limitations: they are expensive to produce, cannot be delivered orally, show low tissue penetration and are unable to reach intracellular targets (4). The potential problems associated with the use of antibody fragments have led to the exploration of alternative protein scaffolds as a source for novel protein-based therapeutics (5–12). However, the utility of protein-based therapeutics has been typically hampered by their generally poor stability and limited bioavailability (13). In order to overcome these limitations, special attention has been recently given to the use of highly constrained peptides, also known as micro-proteins, for the modulation of protein-protein interactions (14–16).
In this review, we will present the recent developments in the use of Cys-rich backbone-cyclized polypeptides for targeting protein-protein interactions. Circular Cys-rich peptides are widely distributed natural product polypeptides among different species including animals and plants (Scheme 1). They have attractive features including thermal, chemical and biological stability against proteases. Peptides in this group include cyclotides, mammalian θ-defensins, and sunflower protease inhibitors.
Scheme 1.
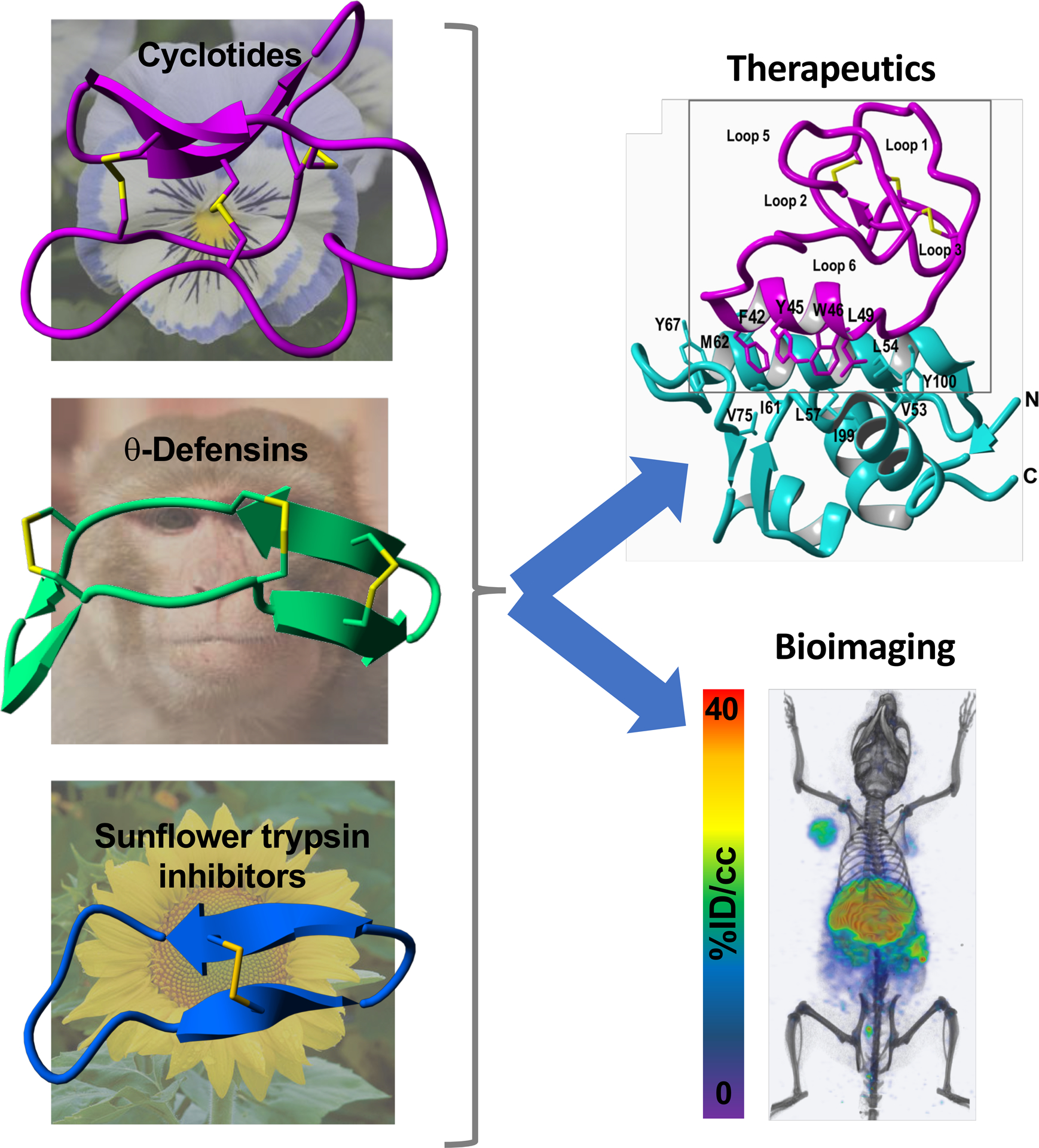
Schematic representation of the structural complexity of the naturally occurring disulfide-rich backbone cyclized peptides that are commonly used as tools for the development of novel bioimaging agents (left, depicting a cyclotide able to visualize CXCR4-expressing tumors in vivo) and therapeutic leads (right, depicting a cyclotide able to activate the p53 pathway in cancer cells in vivo). These include θ-defensins (RTD-1), cyclotides (cycloviolacin O2) and sunflower peptide trypsin inhibitors (SFTI-1).
Cyclotides
Cyclotides are fascinating micro-proteins (≈30 residues long) present in plants from the Violaceae, Rubiaceae, Cucurbitaceae, and more recently Fabaceae families (17). They display various biological properties such as protease inhibitory, anti-microbial, insecticidal, cytotoxic, anti-HIV, and hormone-like activities (see references (14, 15) for recent reviews of the biotechnological applications of cyclotides). They share a unique head-to-tail circular Cys-knotted (CCK) topology of three disulfide bridges, with one disulfide penetrating through a macrocycle formed by the two other disulfides and inter-connecting peptide backbones, forming what is called a cystine knot topology (Fig. 1). Cyclotides can be considered as natural combinatorial peptide libraries structurally constrained by the cystine-knot scaffold and head-to-tail cyclization but in which hypermutation of essentially all residues is permitted with the exception of the strictly conserved cysteines that comprise the knot (18–20). The main features of cyclotides are a remarkable stability due to the cystine knot, a small size making them readily accessible to chemical synthesis, and an excellent tolerance to sequence variations. For example, the first cyclotide to be discovered, kalata B1, is an orally effective uterotonic (21), and other cyclotides have been also shown to be orally bioavailable and capable of crossing cell membranes (22, 23) to efficiently target extracellular (24–26) and intracellular PPIs in vivo (27). In addition, cyclotides have been shown to be poorly immunogenic due to their highly constrained nature (7, 28). Cyclotides thus appear as highly promising leads or frameworks for peptide drug design (15, 29–31).
Figure 1.
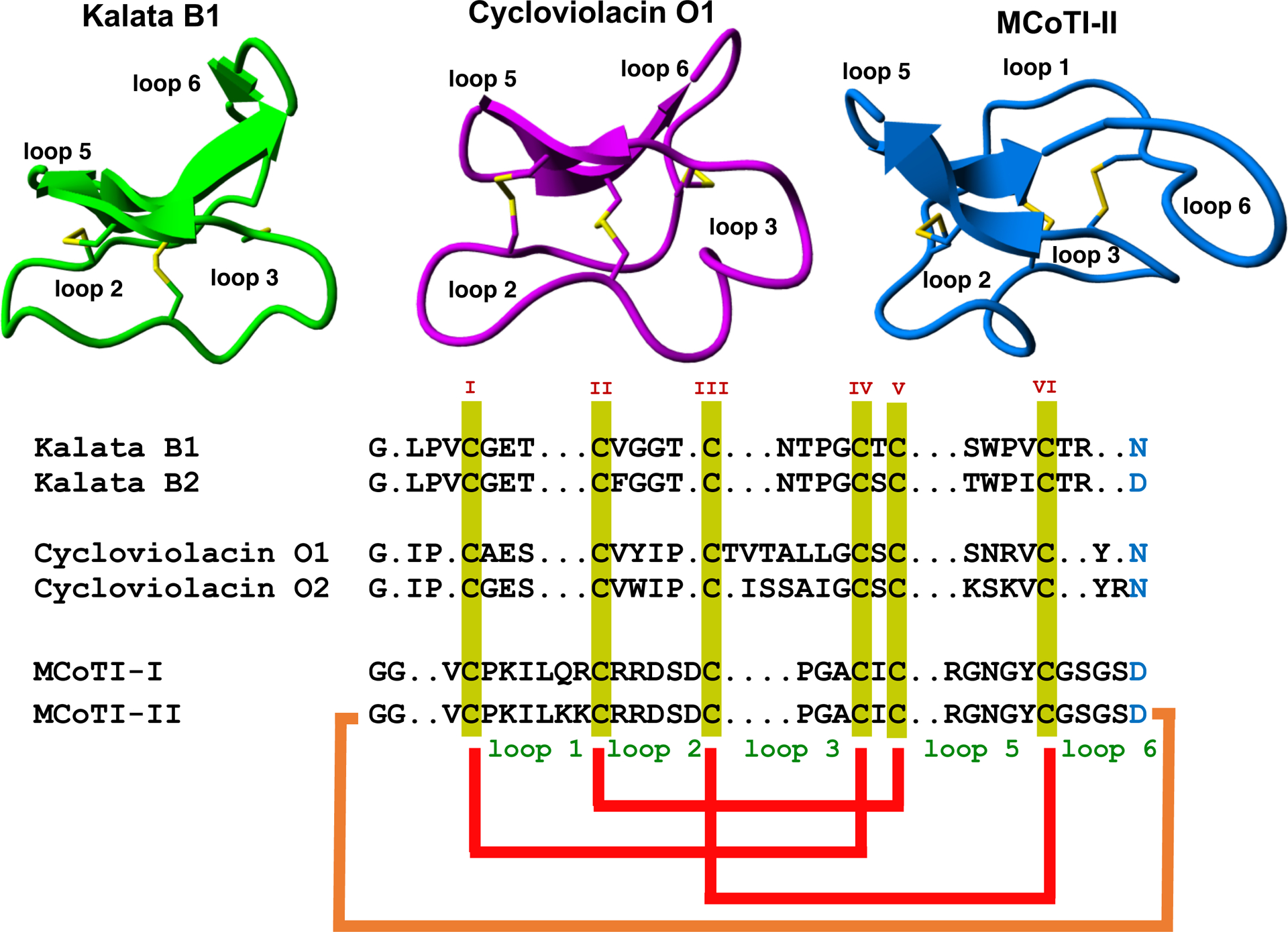
Sequence alignment and structures of different cyclotides belonging to the Möbius (kalata B1, pdb: 1NB1), bracelet (cycloviolacin O1, pdb: 1NBJ) and trypsin inhibitor (MCoTI-II, pdb: 1IB9) subfamilies. Disulfide connectivities and backbone-cyclization are shown in red and orange, respectively. The six Cys residues are labeled with roman numerals whereas loops connecting the different Cys residues are designated with arabic numerals. Conserved Cys and Asp/Asn (required for backbone cyclization in nature) residues are marked in yellow and light blue, respectively. Molecular graphics were created using Yasara (www.yasara.org).
Cyclotides are classified into three subfamilies known as the Möbius, bracelet, and trypsin inhibitor cyclotide subfamilies. All the subfamilies have the same CCK topology although the composition of the loops is slightly different (Fig. 1). Bracelet cyclotides are usually larger and more structurally diverse than Möbius cyclotides, and the make up around 60% of all the cyclotides known so far (32). Bracelet cyclotides are more difficult to obtain by chemical synthesis than either Möbius or trypsin inhibitor cyclotides due to the difficulties associated to fold them correctly in vitro. Due to this, bracelet cyclotides have been less used as molecular scaffolds to target PPIs.
Cyclotides from the trypsin inhibitor subfamily are found in the seeds from several plants of Momordica genus (33, 34) and as their name indicates are potent trypsin inhibitors (35). This is the cyclotide subfamily with fewer members identified thus far showing little sequence homology with the other cyclotides beyond the presence of the CCK fold. In fact, these cyclotides seem related to linear cystine-knot squash trypsin inhibitors and sometimes are also referred to as cyclic knottins (36). Cyclotides from this family possess a longer sequence in loop 1, making the Cys-knot slightly less rigid than in cyclotides from the other two subfamilies.
Naturally occurring cyclotides are produced in plants from dedicated genes that can encode multiple copies of the same cyclotide or even mixtures of different cyclotide sequences (37). Recent studies indicate that an asparaginyl endopeptidase (AEP)-like ligase is responsible for the backbone-cyclization of cyclotides, however, the complete mechanism of how the precursor cyclotide protein is processed has not completely elucidated yet (29). Cyclotides can also be chemically synthesized using solid-phase peptide synthesis as they are relatively small polypeptides containing typically around 30–40 residues (38). More recently, the use of protein splicing in cis and trans has also allowed the production of fully folded cyclotides inside bacterial and yeast cells using heterologous expression systems (39, 40).
Table 1 summarizes recent examples on the use of the cyclotide scaffold to target PPIs. The first two examples the demonstrated the pharmacological potential of engineered cyclotides were aimed for the development of novel peptide-based novel anti-cancer (41, 42) and anti-viral peptide-based therapeutics (43). The development of molecules targeting angiogenesis has been shown to potential therapeutic avenues in cancer treatment as tumor growth is usually associated with unregulated angiogenesis. The molecular grafting of an Arg-rich peptide antagonist of the vascular endothelial growth factor A (VEGF-A) receptor onto several loops of the Möbius cyclotide kalata B1 yielded antagonists with low μM activity for blocking VEGF activity (41). This was one of the first examples where a bioactive peptide was used to produce a novel cyclotide with a specific biological activity, however, it should be noted that the biological activity would still need to be improved by several orders of magnitude for a potential pharmacological application in vivo. A similar approach using the molecular framework of kalata B1 was recently used in the development of cyclotides able to modulate the bradykinin and melanocortin 4 receptors for pain and obesity management, respectively (44, 45). It is worth mentioning, that one of the designed kalata B1-based bradykinin antagonists in this work was shown to be orally bioavailable (45). In this context, a recent study using A point mutated kalata B1 cyclotide was also reported to have oral bioavailability and in a mouse model of multiple sclerosis (46). These findings highlight the potential of the cyclotide molecular framework for the development of novel orally-bioavailable peptide-based therapeutics.
Table 1.
Summary of work published in engineered cyclotides with novel biological activities leading to therapeutic and bioimaging applications.
| Cyclotide | Biological activity | Loop Mlodified | Application | Ref. |
|---|---|---|---|---|
| Möbius subfamily | ||||
| Kalata B1 | VEGF-A antagonist 2 | 2, 3, 5 & 6 | Anti-angiogenic, potential anti-cancer activity | (41) |
| Kalata B1 | Dengue NS2B-NS3 Protease inhibitor | 2 & 5 | Anti-viral for Dengue virus infections | (108) |
| Kalata B1 | Bradikynin B1 receptor antagonist | 6 | Chronic and inflammatory pain | (45) |
| Kalata B1 | Melanocortin 4 receptor Agonist | 6 | Obesity | (44) |
| Kalata B1 | Neuropilin-1/2 antagonist | 5 & 6 | Inhibition of endothelial cell migration and angiogenesis | (109) |
| Kalata B1 | Immunomodulator | 5 & 6 | Protecting against multiple sclerosis | (110) |
| Kalata B1 | Immunomodulator | 4 | Protecting against multiple Sclerosis | (111) |
| Trypsin inhibitor subfamily | ||||
| MCoTI-I | CXCR4 antagonist | 6 | Anti-metastatic and anti-HIV PET-CT imaging | (24–26) |
| MCoTI-I | p53-Hdm2/HdmX Antagonist | 6 | Anti-tumor by activation of p53 pathway | (27) |
| MCoTI-II | FMDV 3C protease Inhibitor | 1 | Antiviral for foot-and-mouth disease | (43) |
| MCoTI-II | β-Tryptase inhibitor | 3, 5 & 6 | Inflammation disorders | (49) |
| MCoTI-II | β-Tryptase inhibitor Human elastase inhibitor | 1 | Inflammation disorders | (42) |
| MCoTI-II | CTLA-4 antagonist | 1,3 & 6 | Immunotherapy for cancer | (54) |
| MCoTI-II | Tryptase inhibitor | 1 | Anticancer | (81) |
| MCoTI-II | VEGF receptor agonist | 6 | Wound healing and cardiovascular damage | (87) |
| MCoTI-I | α-Synuclein-induced cytotoxicity inhibitor | 6 | Parkinson’s disease Validate phenotypic screening of genetically-encoded cyclotide libraries | (40) |
| MCoTI-II | BCR-Abl kinase Inhibitor | 1 & 6 | Chronic myeloid leukemia Attempt to graft both a cell penetrating peptide and kinase inhibitor | (112) |
| MCoTI-I | MAS1 receptor agonist | 6 | Lung cancer and myocardial infarction | (113) |
| MCoTI-II | SET antagonist | 6 | Potential anticancer | (50) |
| MCoTI-II | FXIIa and FXa inhibitors | 1 & 6 | Antithrombotic and cardiovascular disease | (114) |
| MCoTI-II | Thrombospondin-1 (TSP-1) agonist | 6 | Microvascular endothelial cell migration inhibition Anti-angiogenesis | (85) |
| MCoTI-II | Antiangiogenic | 5 & 6 | Anti-cancer | (103) |
The cyclotides from the trypsin inhibitory subfamily have also used as templates to engineer cyclotides with novel biological activities by means of molecular grafting. For example, the cyclotide MCoTI-I has been recently used for the design of a potent CXCR4 antagonist (47). Overexpression of the CXCR4 receptor has been observed in multiple cancers where it is believed it promotes metastasis, angiogenesis, and tumor growth and/or survival (48).
Cyclotides from the trypsin inhibitor subfamily have been also used for the development of protease inhibitors with pharmacological relevance. The cyclotide MCoTI-II was transformed into a potent and selective foot-and-mouth-disease (FMDV) 3C protease inhibitor by introducing mutations onto loops 1 and 6 (43). A similar approach was used by the same authors to generate β-tryptase and human leukocyte elastase inhibitors with low nM Ki values (42, 49).
The most exciting feature of the cyclotide scaffold is that some members of the trypsin inhibitor subfamily can cross the cellular membrane of mammalian cells, therefore, making possible the intracellular delivery of biologically active cyclotides to target intracellular PPIs (22, 23). Our group was recently able to generate a potent activator of p53 function by inhibiting the interaction between p53 and the proteins Hdm2/HdmX using the cyclotide MCoTI-I as molecular scaffold (Fig. 2) (27). The resulting cyclotide MCo-PMI was able to bind with high affinity the p53-binding domains of both Hdm2 and HdmX, showed high ex-vivo stability in serum and was cytotoxic to wild-type p53 cancer cell lines by activating the p53 tumor suppressor pathway both in vitro and in vivo (Fig. 2) (27). This work represents the first example showing an engineered cyclotide able to target an intracellular protein-protein interaction in an animal model of prostate cancer, therefore, highlighting the therapeutic potential of MCoTI-cyclotides for targeting intracellular protein-protein interactions. Exactly the same approach but using cyclotide MCoTI-II instead has been also employed to obtain an antagonist for the SET protein that is overexpressed in some human cancers (50).
Figure 2.
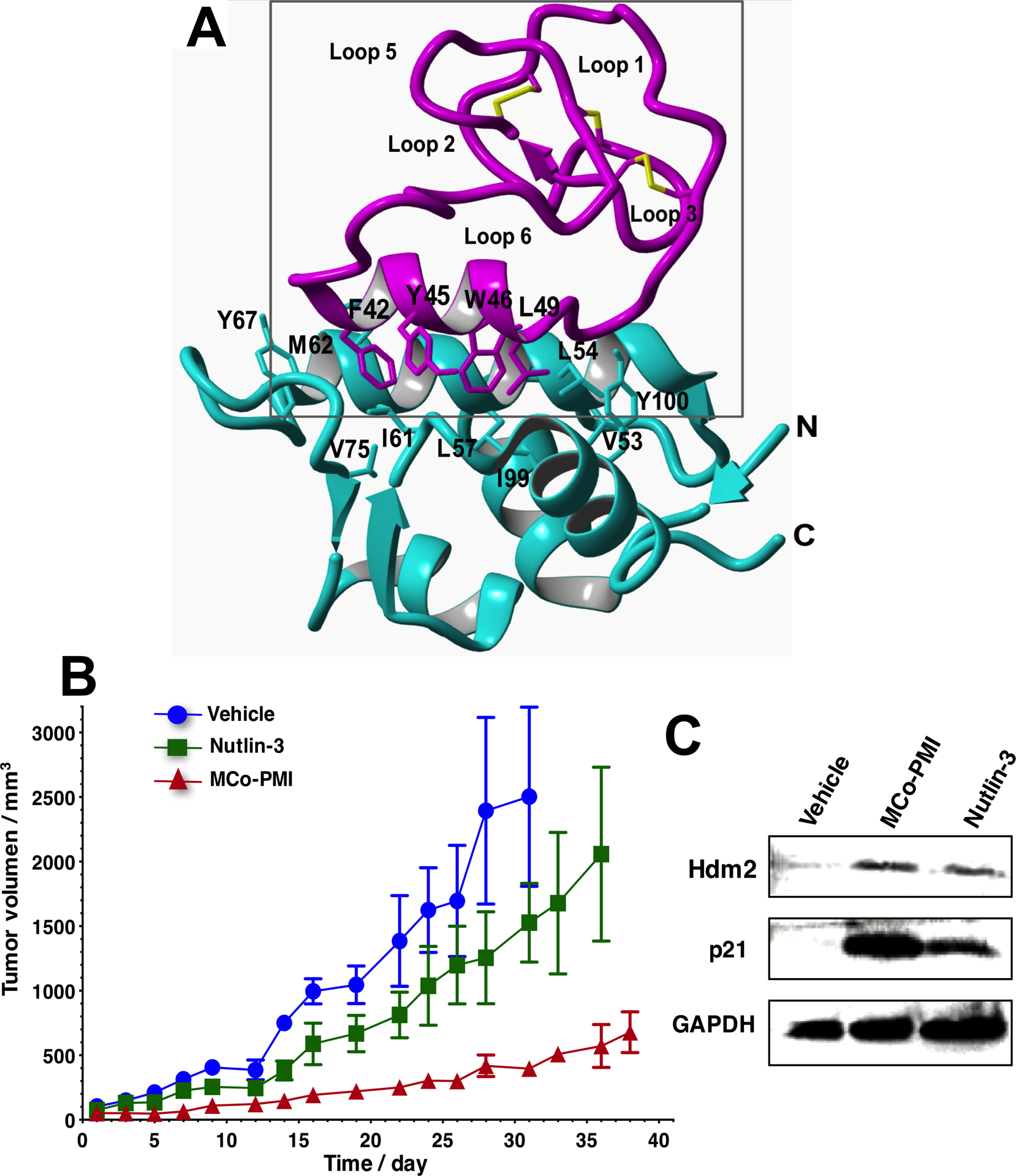
Structure and in vivo activity of the first cyclotide designed to antagonize an intracellular protein-protein interaction in vivo (27). A. Solution structure of the engineered cyclotide MCo-PMI (magenta) and its intracellular molecular target, the p53 binding domain of oncogene Hdm2 (blue) (pdb: 2M86). The cyclotide binds with low nM affinity to both the p53-binding domains of Hdm2 and HdmX. B. Cyclotide MCo-PMI activates the p53 tumor suppressor pathway and blocks tumor growth in a human colorectal carcinoma xenograft mouse model. C. Tumors samples were subjected to SDS-PAGE and analyzed by western blotting for p53, Hdm2 and p21, indicating activation of p53 on tumor tissue.
Our group has also recently shown that cyclotides can be obtained by heterologous expression using both prokaryotic and eukaryotic expression systems (39, 40). For example, a novel MCoTI-grafted cyclotide (MCoCP4) was able to inhibit α-synuclein-induced cytotoxicity in yeast Saccharomyces cerevisiae (40). α-Synuclein is a small lipid-binding protein that is prone to misfolding and aggregation and has been linked to Parkinson’s disease making it a validated therapeutic target for Parkinson’s disease.
The ability to produce natively folded cyclotides in the cell (40, 51, 52) as described earlier makes possible the generation and rapid screening of large libraries of cyclotides, potentially containing billions of members, that are genetically-encoded cyclotides. The generation of such tremendous molecular diversity permits the development of selection of strategies that mimic the evolutionary processes found in nature for the selection of novel cyclotide sequences able to target specific molecular targets. For example as proof of principle, our group used in cell expression of a small library based in cyclotide MCoTI-II in E. coli where every residue in loops 1, 2, 3, 4 and 5 was mutated to explore the effects on folding and trypsin binding activity of the resulting mutants (52). This early study revealed that most of the mutations did not affect folding on the resulting cyclotides, therefore, emphasizing the high plasticity and sequence tolerance of MCoTI-based cyclotides (52).
The use of an acyclic version of cyclotide kalata B1 was employed for the screening and selection of novel cyclotides specific for the VEGFA binding site on neuropilin-1 (53). This study used bacterial display libraries and the authors were able to obtain kalata-based cyclotides with high affinity (Kd ≈50 nM), increased protease resistance, and conferred improved potency for inhibiting endothelial cell migration in vitro (EC50 ≈100 nM) (53).
A yeast surface display approach of an acyclic version of cyclotide MCoTI-II was also employed for the screening of a linearized cyclotide library to select strong binders cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), an inhibitory receptor expressed by T lymphocytes, that has emerged as a target for the treatment of metastatic melanoma (54).
More recently, a cyclotide-based library was employed for phenotypic screening in eukaryotic cells (40). In this work, an engineered cyclotide (MCoCP4) that was designed to reduce the toxicity of human α-synuclein in live yeast cells was selected by phenotypic screening from cells transformed with a mixture of plasmids encoding MCoCP4 and inactive cyclotide MCoTI-I in a ratio of 1 to 50,000. These results show the potential for performing rapid phenotypic screening of genetically encoded cyclotide-based libraries in eukaryotic cells for the selection of bioactive compounds. These exciting results demonstrate the potential to perform phenotypic screening of genetically encoded cyclotide-based libraries in eukaryotic cells for the rapid selection of novel bioactive cyclotides. Moreover, expression in eukaryotic systems should allow the production of cyclotides with different post-translational modifications not available in bacterial expression systems.
The development of efficient methods for the chemical synthesis, cyclization and folding of cyclotides has also made possible to perform high throughput screening on chemically-generated libraries of cyclotides (26). A small library of MCoTI-based CXCR4 cyclotide antagonists was chemically obtained using a ‘tea-bag’ approach in combination with high efficiency folding protocols (26). The approach described in this work also included an efficient purification procedure to rapidly remove non-folded or partially folded cyclotides from the cyclization-folding crude. This approach can be also employed for the purification of cyclotide mixtures thereby allowing the synthesis of amino acid and positional scanning libraries to perform efficient screening of large chemical-generated libraries (55).
The potential of bioactive cyclotide to be employed as bioimaging agents has also been recently reported (24). An MCoTI-based CXCR4 antagonist cyclotide (MCo-CVX-6D) was shown to be an excellent bioimaging tool to visualize CXCR4-overexpressing cancer cells in a mouse model. A [64Cu]-DOTA-labeled version of cyclotide MCo-CVX-6D was used for the efficient detection of tumors containing CXCR4-expressing cells in mice using positron emission tomography-computed tomography (PET-CT) (Fig. 3).(24)
Figure 3.
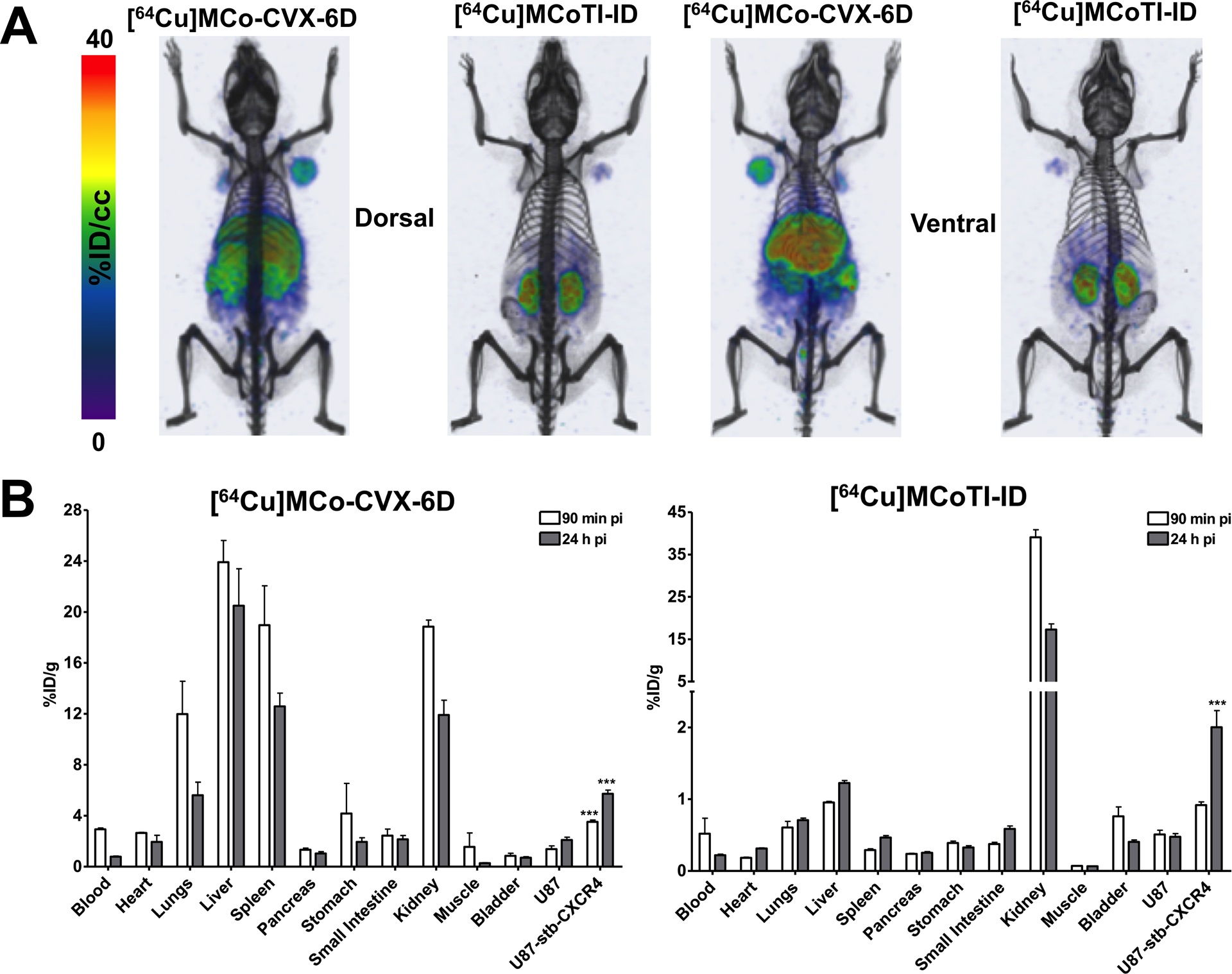
Use of a CXCR4-targeting cyclotide as a bioimaging tool for detecting CXCR4-overexpressing tumor cells in animal models (24). Distribution of [64Cu]MCo-CVX-6D (CXCR4-targetign cyclotide) and [64Cu]MCoTI-ID (a DOTA-labeled variant of native trypsin inhibitor cyclotide MCoTI-I) in NOD/SCID mice bearing U87 and U87-stb-CXCR4 tumors with PET-CT. B. Ex vivo evaluation of [64Cu]MCo-CVX-6D and [64Cu]MCoTI-ID distribution and specificity in NOD/SCID mice bearing U87 and U87-stb-CXCR4 tumors. Ex vivo biodistribution analysis was performed at 90 min and 24 h after post tracers injection.
Given the good pharmacological properties of some bioactive cyclotides, their biodistribution of has been recently studied (24, 56). These studies indicate that MCoTI-cyclotides are distributed predominantly to the serum and kidneys, confirming that they are stable in serum and suggesting that they are eliminated from the blood through renal clearance (Fig. 3B) (24, 56). However, no significant uptake into the brain was observed for cyclotide MCoTI-II (56).
Mammalian θ-defensins
Defensins are cysteine-rich antimicrobial peptides (AMP) that play a critical role in the innate immune defense of mammals (57–59). Although they are classically known for their antimicrobial activities they have been also shown to be involved in other defense mechanisms including immune modulation, neutralization of endotoxins, and anti-cancer properties (59, 60). Mammalian defensins are peptides containing mostly β-sheet structures, six Cys residues forming three intramolecular disulfides and a high content on positively charged residues. They have been classified into α-, β- and θ-defensins depending on their overall structure.
In contrast with α- and β-defensins, θ-defensins are backbone cyclized peptides formed by the head-to-tail covalent assembly of two nonapeptides derived from α-defensin related precursors (Fig. 4A) (57, 61). θ-Defensins are to date the only known cyclic polypeptides expressed in animals (57).
Figure 4.
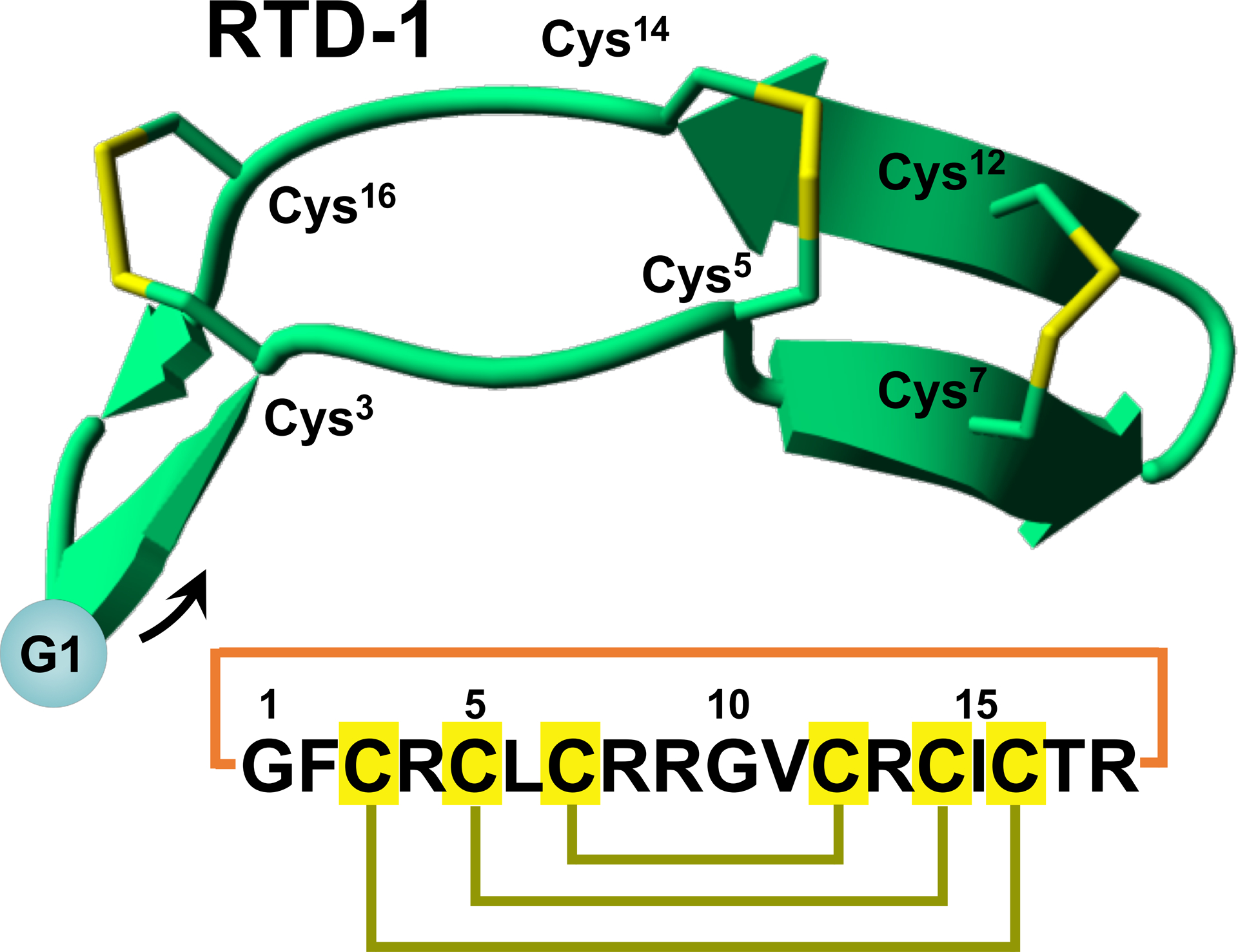
Sequences and structures of naturally-occurring disulfide-rich backbone cyclized peptides θ-defensin (RTD-1) (A) and sunflower trypsin inhibitor 1 (SFTI-1) (B). The backbone cyclized peptide (connecting bond shown in blue) is stabilized by the three disulfide bonds in a ladder formation (disulfide bonds shown in yellow). Molecular graphics were created using Yasara (www.yasara.org).
θ-Defensins have shown to possess antimicrobial activity against both Gram-positive and Gram-negative bacteria (57), as well as anti-fungal (57) and anti-HIV (62, 63) activities.
Chemically-produced human θ-defensin, derived from human pseudogene sequences, have also shown to protect human cells from infection by HIV-1 (62) and have been even evaluated as a topical anti-HIV agent (64–66). θ-defensins have been shown to have moderate activity against several bacterial toxins and proteases (67), as well as human metalloproteases like TNFα converting enzyme (TACE) (68). θ-defensins have also been shown to possess anti-inflammatory properties in animal models (69). In addition, θ-defensins present high resistance to proteolytic degradation in serum and plasma due to the network of disulfide bonds and backbone-cyclized topology (68, 69). Altogether, these unique features have made θ-defensin an ideal molecular framework for the development of novel peptide-based therapeutics (69, 70).
Our group has developed efficient approaches for the chemical and recombinant production of θ-defensins (61, 68, 71–73). Using a ‘tea-bag’ approach in combination with a one-pot cyclization and oxidative folding method our group has obtained potent θ-defensins analog able to inhibit anthrax lethal factor (LF) and TACE with Ki values ≈40 nM and ≈157 nM, respectively (68). It is worth mentioning that these analogs showed also significant activity in the presence of 0.1% bovine serum albumin (BSA) (68).
The θ-defensin scaffold has also recently employed for designing integrin antagonists (74). This was accomplished by grafting the integrin-binding Arg-Gly-Asp (RGD) peptide motif into the θ-defensin RTD-1 molecular framework. The most active compound had an IC50 of ≈18 nM for the αvβ3 integrin and presented high serum stability. These results clearly show the high robustness of the θ-defensin scaffold for the generation of molecular diversity and can provide a stable and conformationally restrained scaffold for bioactive epitopes in a β-strand or turn conformation.
Sunflower trypsin inhibitor 1 (SFTI-1)
SFTI-1 is a 14 amino acid backbone-cyclized peptide containing a single disulfide bond that is naturally found in the seeds of sunflower (Helianthus annuus) (75). SFTI-1 belongs to the Bowman-Birk inhibitor (BBI) family, whose members are found in many plants and are potent serine protease inhibitors (76). Structural analysis of SFTI-1 shows a well-defined double β-hairpin loop linked by two short antiparallel β-strands (Fig. 4B) (32, 75, 77). SFTI-1 is a potent trypsin inhibitor that belongs to the Bowman-Birk protease inhibitor (BBI) family. The backbone-cyclized SFTI-1 is the smallest and the most potent protease member of the family with a Ki against trypsin in the low nM range (75, 76).
The relatively rigid backbone of SFTI-1 makes its protease-binding loop well-defined, which can serve as a general scaffold for serine protease inhibitors. For example, introduction of several mutations within this loop has produced SFTI-based analogs able to inhibit a variety of serine proteases (78–82). The structural features of SFTI-1 characterized by the presence of a backbone cyclization combined with an internal disulfide bond and an extensive hydrogen binding network make it exceptional stable to thermal or enzymatic degradation (81, 83). SFTI-1 has been also shown to be non-toxic and be able to cross cellular membranes (22). Moreover, as shown with other backbone cyclized disulphide-rich scaffolds, the molecular framework provided by SFTI-1 can also be readily re-engineered by grafting foreign biological active peptide sequences into one the loops producing SFTI-analogs with novel biological activities (84–87).
SFTI-1 has been found to be a potent inhibitor of matriptase (Ki ≈ 1 nM) (88). Matriptase is a type II transmembrane serine protease found in most cancer cells, where it has been shown that can activate key pro-metastatic substrates to trigger cell migration, cancer invasion, and metastasis; therefore providing a target of therapeutic intervention (89). Amino acid scanning of the position P2′ residue (Ile) in SFTI-1 has been also accomplished to produce a potent mesotrypsin inhibitor (90). Replacement of this residue by aromatic residues (Tyr, Phe, Trp or the non-canonical amino acid 4,4′-biphenyl-L-alanine (Bip)) yielded an SFTI-I analog that maintained a similar structure to SFTI-1 and showed marked improvements in activity against mesotrypsin (exceeding 100-fold) (90).
SFTI-1 has also been re-engineered to produce potent human kallikrein-related peptidase 4 (KLK4) inhibitor (82). Human KLK4 is a potential target for prostate cancer treatment because of its proteolytic ability to activate many tumorigenic and metastatic pathways including the protease activated receptors (PARs) (91, 92). In this work, SFTI-1 was modified by introducing several mutations (K5R, T4Q and R2F), identified by using a combination of molecular modeling and sparse matrix substrate screening, to produce the analog SFTI-FCQR that was potent and selective KLK4 inhibitor (Ki ≈3 nM) (82). Further optimization of the KLK4 inhibitor SFTI-FCQR by the same authors using molecular dynamic algorithms produced a significantly improved inhibitor (SFTI-FCQR Asn14) with a Ki value of ≈64 pM (93). The same authors have also employed a similar approach to improve the weaker activity of SFTI-I against cathepsin G (Ki ≈ 570 nM) through optimization of the binding loop to generate molecules with higher potency (Ki = 1.6 nM) and higher selectivity (over 360-fold) for other related proteases (94).
Development of SFTI-based inhibitors of kallikrein‐related peptidases 7 (KLK7) and 5 (KLK5) was also recently accomplished by grafting of the reactive‐center loop (RCL) of several serpins reported to inhibit KLK5 and KLK7 (95). KLK7 and KLK5 are expressed in human skin, and their dysregulation is associated with skin diseases such as Netherton syndrome, atopic dermatitis, and psoriasis (96); and different types of human cancer (97). The best SFTI-derived inhibitors against KLK7 and KLK5 provide Ki values ranging from 0.6 to 0.9 μM values.
The SFTI-1 scaffold has been recently used in combination with phage display techniques to screen and select a novel SFTI-based ITGαvβ6-binding peptide (SFITGv6) (98). The ITGαvβ6 receptor is highly expressed on head and neck squamous cell carcinoma (HNSCC) and is the target of the antiangiogenic RGD-peptide cilengitide (99). In this work, a linearized version of SFITGv6 peptide labelled with the radiotracer 177Lu-DOTA was employed as bioimaging agent for ITGαvβ6-positive carcinomas (98, 100). SFTI-1 has also been employed to graft the binding loop of micro-protein Min-23 selected to bind angiogenesis marker delta‐like ligand4 (Dll4) by using a phage-display approach (101). The micro-protein Min-23 is a two disulfide‐bridge stabilized scaffold, which was rationally designed by miniaturization of its parent knottin Ecballium elaterium trypsin inhibitor II (EETI‐II) (102). In this work, the resulting grafter SFTI-derived peptide was able to preserve the Dll4-binding specificity and the tumor-targeting capability of the original micro-protein (101). A similar grafting approach using anti-angiogenic peptides was also able to yield potent anti-angiogenic SFTI-derived peptides (103). The grafted peptide SFTI-PEDF displayed an almost similar degree of activity as the linear grafted peptide with a better proteolytic stability profile. SFTI-1 has also been transformed into a furin inhibitor by replacing the trypsin-binding loop of SFTI-1 by a natural furin substrate (104). The resulting grafted SFTI-based peptide was further optimized by using computer-based structural modelling to generate a sub-nanomolar furin inhibitor (Ki ≈ 0.5 nM) that showed very good selectivity over trypsin (>10,000) and matriptase (>1,000) (104).
The SFTI-1 molecular framework was recently employed to produce a cyclic peptide for for reducing inflammation in models of inflammatory bowel diseases (IBDs) (105). In this work, the authors grafted a small bioactive peptide from the annexin A1 protein into trypsin binding loop of SFTI-1. The resulting SFTI-based peptide (cyc-MC12) maintained the overall fold of the naturally occurring cyclic peptide as more effective at reducing inflammation in a mouse model of acute colitis than the bioactive peptide alone showing enhanced ex vivo stability in human serum.
The SFTI-1 scaffold was also recently employed for the production of a novel subtype of selective melanocortin receptor (MCR) agonists (106). This was accomplished by grafting the α/β/γ-melanocyte stimulating hormone (MSH)-derived HFRW tetrapeptide into the different loops of SFTI-1 in combination with systematic N-methylation of the grafted pharmacophore. One of the double N-methylated SFTI-derived peptides was able to show low nM activity for human MC1R being about 100 times more selective for this receptor than for MC3R. The nuclear magnetic resonance structural analysis of this grafted peptide revealed the key role of peptide bond N-methylation in shaping the conformation of the grafted peptide pharmacophore. This work highlights the potential of cyclic peptide scaffolds for epitope grafting in combination with N-methylation to introduce receptor subtype selectivity in the context of peptide-based drug discovery (106).
An interesting application of bioactive grafted SFTI-based peptides was recently reported as a scavenger for anticitrullinated protein/peptide autoantibodies (ACPA) for potential diagnostic and/or treatment of rheumatoid arthritis (107). In this interesting work, the SFTI-1 scaffold was engineered to display a citrulline-containing ACPA binding epitope identified from the α-chain of human fibrinogen. The resulting cyclic peptide scavenger showed high apparent affinity and subtype-specific binding for ACPA, as well as superior serum stability.
All these examples show the great potential of SFTI-1 as molecular framework for grafting or engineering new activities. Its small size and high resistance to proteolytic degradation are making it increasingly recognized as an excellent template for engineering studies as it does not require structural optimisation and the inhibitor’s inherent activity can be re-directed to other serine proteases by substituting residues that form major binding contacts.
Concluding remarks
It is fair to say that the use of highly constrained disulphide-rich backbone cyclized polypeptides is becoming to gain acceptance as molecular scaffolds for the potential design of novel peptide-based therapeutics and diagnostic tools. This is in part to their unique properties which include their small size that allows chemical synthesis, the ability to be expressed using standard expression systems, high resistance to chemical, physical and biological degradation and in some case the ability to cross cellular membranes to target intracellular PPIs.
Of the three types of scaffolds reviewed here, the cyclotide scaffold without question is one of the more exciting. The cyclotide unique knotted arrangement of three disulfide bonds and exceptional tolerance sequence variation in all their hypervariable loops provides an exceptional molecular platform to design novel cyclotides with new biological activities by rational design using molecular grafting techniques or by employing molecular evolution techniques making use of the multiple loops found on the cyclotide scaffold. Cyclotides have also been shown to cross mammalian cellular membranes to target protein-protein interactions in vitro but also and more importantly in animal models. This highlights the high stability of the Cys-knot to be degraded/oxidized under complex biological conditions.
As mentioned earlier, the relative small size of these scaffolds facilitates their chemical synthesis allowing the introduction of chemical modifications such as non-natural amino acids and PEGylation to improve their pharmacological properties.
None of the scaffolds reviewed here have reached human clinical trials yet, however, the results obtained with several bioactive compounds in animal models may hint that this could occur in a not too distant future. One of the main challenges that affect this type of constrained polypeptides is their oral bioavailability if they want to be competitive with small-molecule therapeutics. Although we have seen that some cyclotides have been shown to be orally active, there is still little information about their oral bioavailability. It is anticipated, however, that more studies on the biopharmaceutical properties of these exciting new peptide-based molecular scaffolds will be available very soon.
Acknowledgements
This work was supported by National Institutes of Health Research Grant R01-GM113363, Department of Defense Congressionally Directed Medical Research Programs in Lung Cancer Grant LC150051, BROAD Medical Research Program-Crohn’s & Colitis Foundation of America Grant #483566, Lupus Research Institute and Whittier Foundation.
References
- 1.Stumpp MT, Binz HK, Amstutz P. DARPins: a new generation of protein therapeutics. Drug Discov Today. 2008;13(15–16):695–701. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391–400. [DOI] [PubMed] [Google Scholar]
- 3.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126–36. [DOI] [PubMed] [Google Scholar]
- 4.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craik DJ, Simonsen S, Daly NL. The cyclotides: novel macrocyclic peptides as scaffolds in drug design. Curr Opin Drug Discov Devel. 2002;5(2):251–60. [PubMed] [Google Scholar]
- 6.Craik DJ, Cemazar M, Daly NL. The cyclotides and related macrocyclic peptides as scaffolds in drug design. Curr Opin Drug Discov Devel. 2006;9(2):251–60. [PubMed] [Google Scholar]
- 7.Craik DJ, Clark RJ, Daly NL. Potential therapeutic applications of the cyclotides and related cystine knot mini-proteins. Expert Opin Investig Drugs. 2007;16(5):595–604. [DOI] [PubMed] [Google Scholar]
- 8.Kolmar H, Skerra A. Alternative binding proteins get mature: rivalling antibodies. FEBS J. 2008;275(11):2667. [DOI] [PubMed] [Google Scholar]
- 9.Skerra A Alternative binding proteins: anticalins - harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities. FEBS J. 2008;275(11):2677–83. [DOI] [PubMed] [Google Scholar]
- 10.Sancheti H, Camarero JA. “Splicing up” drug discovery. Cell-based expression and screening of genetically-encoded libraries of backbone-cyclized polypeptides. Adv Drug Deliv Rev. 2009;61(11):908–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bloom L, Calabro V. FN3: a new protein scaffold reaches the clinic. Drug Discov Today. 2009;14(19–20):949–55. [DOI] [PubMed] [Google Scholar]
- 12.Lewis RJ. Conotoxin venom Peptide therapeutics. Advances in experimental medicine and biology. 2009;655:44–8. [DOI] [PubMed] [Google Scholar]
- 13.McGregor DP. Discovering and improving novel peptide therapeutics. Curr Opin Pharmacol. 2008;8(5):616–9. [DOI] [PubMed] [Google Scholar]
- 14.Wang CK, Craik DJ. Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat Chem Biol. 2018;14(5):417–27. [DOI] [PubMed] [Google Scholar]
- 15.Gould A, Camarero JA. Cyclotides: Overview and biotechnological applications. ChemBioChem. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gongora-Benitez M, Tulla-Puche J, Albericio F. Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem Rev. 2014;114(2):901–26. [DOI] [PubMed] [Google Scholar]
- 17.Poth AG, Colgrave ML, Lyons RE, Daly NL, Craik DJ. Discovery of an unusual biosynthetic origin for circular proteins in legumes. Proc Natl Acad Sci U S A. 2011;108(25):1027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Austin J, Kimura RH, Woo YH, Camarero JA. In vivo biosynthesis of an Ala-scan library based on the cyclic peptide SFTI-1. Amino Acids. 2010;38(5):1313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang YH, Colgrave ML, Clark RJ, Kotze AC, Craik DJ. Lysine-scanning mutagenesis reveals an amendable face of the cyclotide kalata B1 for the optimization of nematocidal activity. J Biol Chem. 2010;285(14):10797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simonsen SM, Sando L, Rosengren KJ, Wang CK, Colgrave ML, Daly NL, et al. Alanine scanning mutagenesis of the prototypic cyclotide reveals a cluster of residues essential for bioactivity. J Biol Chem. 2008;283(15):9805–13. [DOI] [PubMed] [Google Scholar]
- 21.Saether O, Craik DJ, Campbell ID, Sletten K, Juul J, Norman DG. Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata B1. Biochemistry. 1995;34(13):4147–58. [DOI] [PubMed] [Google Scholar]
- 22.Cascales L, Henriques ST, Kerr MC, Huang YH, Sweet MJ, Daly NL, et al. Identification and characterization of a new family of cell-penetrating peptides: cyclic cell-penetrating peptides. J Biol Chem. 2011;286(42):36932–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Contreras J, Elnagar AY, Hamm-Alvarez SF, Camarero JA. Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J Control Release. 2011;155(2):134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lesniak WG, Aboye T, Chatterjee S, Camarero JA, Nimmagadda S. In vivo Evaluation of an Engineered Cyclotide as Specific CXCR4 Imaging Reagent. Chemistry (Easton). 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aboye TL, Ha H, Majumder S, Christ F, Debyser Z, Shekhtman A, et al. Design of a novel cyclotide-based CXCR4 antagonist with anti-human immunodeficiency virus (HIV)-1 activity. J Med Chem. 2012;55(23):10729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aboye T, Kuang Y, Neamati N, Camarero JA. Rapid parallel synthesis of bioactive folded cyclotides by using a tea-bag approach. ChemBioChem. 2015;16(5):827–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji Y, Majumder S, Millard M, Borra R, Bi T, Elnagar AY, et al. In Vivo Activation of the p53 Tumor Suppressor Pathway by an Engineered Cyclotide. J Am Chem Soc. 2013;135(31):11623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slazak B, Kapusta M, Malik S, Bohdanowicz J, Kuta E, Malec P, et al. Immunolocalization of cyclotides in plant cells, tissues and organ supports their role in host defense. Planta. 2016;244(5):1029–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Camarero JA. Cyclotides, a versatile ultrastable micro-protein scaffold for biotechnological applications. Bioorg Med Chem Lett. 2017;27(23):5089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia AE, Camarero JA. Biological activities of natural and engineered cyclotides, a novel molecular scaffold for peptide-based therapeutics. Curr Mol Pharmacol. 2010;3(3):153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henriques ST, Craik DJ. Cyclotides as templates in drug design. Drug Discov Today. 2010;15(1–2):57–64. [DOI] [PubMed] [Google Scholar]
- 32.Wang CK, Kaas Q, Chiche L, Craik DJ. CyBase: a database of cyclic protein sequences and structures, with applications in protein discovery and engineering. Nucleic Acids Res. 2008;36(Database issue):D206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heitz A, Hernandez JF, Gagnon J, Hong TT, Pham TT, Nguyen TM, et al. Solution structure of the squash trypsin inhibitor MCoTI-II. A new family for cyclic knottins. Biochemistry. 2001;40(27):7973–83. [DOI] [PubMed] [Google Scholar]
- 34.Mylne JS, Chan LY, Chanson AH, Daly NL, Schaefer H, Bailey TL, et al. Cyclic peptides arising by evolutionary parallelism via asparaginyl-endopeptidase-mediated biosynthesis. Plant Cell. 2012;24(7):2765–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hernandez JF, Gagnon J, Chiche L, Nguyen TM, Andrieu JP, Heitz A, et al. Squash trypsin inhibitors from Momordica cochinchinensis exhibit an atypical macrocyclic structure. Biochemistry. 2000;39(19):5722–30. [DOI] [PubMed] [Google Scholar]
- 36.Chiche L, Heitz A, Gelly JC, Gracy J, Chau PT, Ha PT, et al. Squash inhibitors: from structural motifs to macrocyclic knottins. Curr Protein Pept Sci. 2004;5(5):341–9. [DOI] [PubMed] [Google Scholar]
- 37.Craik DJ, Malik U. Cyclotide biosynthesis. Curr Opin Chem Biol. 2013;17(4):546–54. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Bi T, Camarero JA. Chemical and biological production of cyclotides. Adv Bot Res. 2015;76:271–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jagadish K, Camarero JA. Recombinant Expression of Cyclotides Using Split Inteins. Methods Mol Biol. 2017;1495:41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jagadish K, Gould A, Borra R, Majumder S, Mushtaq Z, Shekhtman A, et al. Recombinant Expression and Phenotypic Screening of a Bioactive Cyclotide Against alpha-Synuclein-Induced Cytotoxicity in Baker’s Yeast. Angew Chem Int Ed Engl. 2015;54(29):8390–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gunasekera S, Foley FM, Clark RJ, Sando L, Fabri LJ, Craik DJ, et al. Engineering stabilized vascular endothelial growth factor-A antagonists: synthesis, structural characterization, and bioactivity of grafted analogues of cyclotides. J Med Chem. 2008;51(24):7697–704. [DOI] [PubMed] [Google Scholar]
- 42.Thongyoo P, Bonomelli C, Leatherbarrow RJ, Tate EW. Potent inhibitors of beta-tryptase and human leukocyte elastase based on the MCoTI-II scaffold. J Med Chem. 2009;52(20):6197–200. [DOI] [PubMed] [Google Scholar]
- 43.Thongyoo P, Roque-Rosell N, Leatherbarrow RJ, Tate EW. Chemical and biomimetic total syntheses of natural and engineered MCoTI cyclotides. Org Biomol Chem. 2008;6(8):1462–70. [DOI] [PubMed] [Google Scholar]
- 44.Eliasen R, Daly NL, Wulff BS, Andresen TL, Conde-Frieboes KW, Craik DJ. Design, synthesis, structural and functional characterization of novel melanocortin agonists based on the cyclotide kalata B1. J Biol Chem. 2012;287(48):40493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong CT, Rowlands DK, Wong CH, Lo TW, Nguyen GK, Li HY, et al. Orally active peptidic bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew Chem Int Ed Engl. 2012;51(23):5620–4. [DOI] [PubMed] [Google Scholar]
- 46.Thell K, Hellinger R, Schabbauer G, Gruber CW. Immunosuppressive peptides and their therapeutic applications. Drug Discov Today. 2014;19(5):645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aboye TL, Ha H, Majumder S, Christ F, Debyser Z, Shekhtman A, et al. Design of a novel cyclotide-based CXCR4 antagonist with anti-human immunodeficiency virus (HIV)-1 activity. J Med Chem. 2012;55(23):10729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balkwill F The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol. 2004;14(3):171–9. [DOI] [PubMed] [Google Scholar]
- 49.Sommerhoff CP, Avrutina O, Schmoldt HU, Gabrijelcic-Geiger D, Diederichsen U, Kolmar H. Engineered cystine knot miniproteins as potent inhibitors of human mast cell tryptase beta. J Mol Biol. 2010;395(1):167–75. [DOI] [PubMed] [Google Scholar]
- 50.D’Souza C, Henriques ST, Wang CK, Cheneval O, Chan LY, Bokil NJ, et al. Using the MCoTI-II Cyclotide Scaffold To Design a Stable Cyclic Peptide Antagonist of SET, a Protein Overexpressed in Human Cancer. Biochemistry. 2016;55(2):396–405. [DOI] [PubMed] [Google Scholar]
- 51.Camarero JA, Kimura RH, Woo YH, Shekhtman A, Cantor J. Biosynthesis of a fully functional cyclotide inside living bacterial cells. ChemBioChem. 2007;8(12):1363–6. [DOI] [PubMed] [Google Scholar]
- 52.Austin J, Wang W, Puttamadappa S, Shekhtman A, Camarero JA. Biosynthesis and biological screening of a genetically encoded library based on the cyclotide MCoTI-I. ChemBioChem. 2009;10(16):2663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Getz JA, Cheneval O, Craik DJ, Daugherty PS. Design of a Cyclotide Antagonist of Neuropilin-1 and-2 That Potently Inhibits Endothelial Cell Migration. Acs Chemical Biology. 2013;8(6):1147–54. [DOI] [PubMed] [Google Scholar]
- 54.Maass F, Wustehube-Lausch J, Dickgiesser S, Valldorf B, Reinwarth M, Schmoldt HU, et al. Cystine-knot peptides targeting cancer-relevant human cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). J Pept Sci. 2015;21(8):651–60. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Gould A, Aboye T, Bi T, Breindel L, Shekhtman A, et al. Full Sequence Amino Acid Scanning of theta-Defensin RTD-1 Yields a Potent Anthrax Lethal Factor Protease Inhibitor. Journal of medicinal chemistry. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang CK, Stalmans S, De Spiegeleer B, Craik DJ. Biodistribution of the cyclotide MCoTI-II, a cyclic disulfide-rich peptide drug scaffold. J Pept Sci. 2016;22(5):305–10. [DOI] [PubMed] [Google Scholar]
- 57.Tang YQ, Yuan J, Osapay G, Osapay K, Tran D, Miller CJ, et al. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science. 1999;286(5439):498–502. [DOI] [PubMed] [Google Scholar]
- 58.Tanabe H, Yuan J, Zaragoza MM, Dandekar S, Henschen-Edman A, Selsted ME, et al. Paneth cell alpha-defensins from rhesus macaque small intestine. Infect Immun. 2004;72(3):1470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. 2005;6(6):551–7. [DOI] [PubMed] [Google Scholar]
- 60.Lehrer RI. Primate defensins. Nat Rev Microbiol. 2004;2(9):727–38. [DOI] [PubMed] [Google Scholar]
- 61.Conibear AC, Wang CK, Bi T, Rosengren KJ, Camarero JA, Craik DJ. Insights into the Molecular Flexibility of theta-Defensins by NMR Relaxation Analysis. J Phys Chem B. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cole AM, Hong T, Boo LM, Nguyen T, Zhao C, Bristol G, et al. Retrocyclin: a primate peptide that protects cells from infection by T- and M-tropic strains of HIV-1. Proc Natl Acad Sci U S A. 2002;99(4):1813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Penberthy WT, Chari S, Cole AL, Cole AM. Retrocyclins and their activity against HIV-1. Cell Mol Life Sci. 2011;68(13):2231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Munk C, Wei G, Yang OO, Waring AJ, Wang W, Hong T, et al. The theta-defensin, retrocyclin, inhibits HIV-1 entry. AIDS Res Hum Retroviruses. 2003;19(10):875–81. [DOI] [PubMed] [Google Scholar]
- 65.Owen SM, Rudolph D, Wang W, Cole AM, Sherman MA, Waring AJ, et al. A theta-defensin composed exclusively of D-amino acids is active against HIV-1. J Pept Res. 2004;63(6):469–76. [DOI] [PubMed] [Google Scholar]
- 66.Gallo SA, Wang W, Rawat SS, Jung G, Waring AJ, Cole AM, et al. Theta-defensins prevent HIV-1 Env-mediated fusion by binding gp41 and blocking 6-helix bundle formation. J Biol Chem. 2006;281(27):18787–92. [DOI] [PubMed] [Google Scholar]
- 67.Wang W, Mulakala C, Ward SC, Jung G, Luong H, Pham D, et al. Retrocyclins kill bacilli and germinating spores of Bacillus anthracis and inactivate anthrax lethal toxin. J Biol Chem. 2006;281(43):32755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y, Gould A, Aboye T, Bi T, Breindel L, Shekhtman A, et al. Full Sequence Amino Acid Scanning of theta-Defensin RTD-1 Yields a Potent Anthrax Lethal Factor Protease Inhibitor. J Med Chem. 2017;60(5):1916–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schaal JB, Tran D, Tran P, Osapay G, Trinh K, Roberts KD, et al. Rhesus macaque theta defensins suppress inflammatory cytokines and enhance survival in mouse models of bacteremic sepsis. PLoS One. 2012;7(12):e51337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tongaonkar P, Trinh KK, Schaal JB, Tran D, Gulko PS, Ouellette AJ, et al. Rhesus macaque theta-defensin RTD-1 inhibits proinflammatory cytokine secretion and gene expression by inhibiting the activation of NF-kappaB and MAPK pathways. J Leukoc Biol. 2015;98(6):1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bi T, Li YL, Shekhtman A, Camarero JA. In-cell production of a genetically-encoded library based on the theta-defensin RTD-1 using a bacterial expression system. Biorg Med Chem. 2018;26(6):1212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gould A, Li Y, Majumder S, Garcia AE, Carlsson P, Shekhtman A, et al. Recombinant production of rhesus theta-defensin-1 (RTD-1) using a bacterial expression system. Mol Biosyst. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aboye TL, Li Y, Majumder S, Hao J, Shekhtman A, Camarero JA. Efficient one-pot cyclization/folding of Rhesus theta-defensin-1 (RTD-1). Bioorg Med Chem Lett. 2012;22(8):2823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conibear AC, Bochen A, Rosengren KJ, Stupar P, Wang C, Kessler H, et al. The cyclic cystine ladder of theta-defensins as a stable, bifunctional scaffold: A proof-of-concept study using the integrin-binding RGD motif. ChemBioChem. 2014;15(3):451–9. [DOI] [PubMed] [Google Scholar]
- 75.Mogi T, Kita K. Gramicidin S and polymyxins: the revival of cationic cyclic peptide antibiotics. Cell Mol Life Sci. 2009;66(23):3821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Korsinczky ML, Schirra HJ, Craik DJ. Sunflower trypsin inhibitor-1. Curr Protein Pept Sci. 2004;5(5):351–64. [DOI] [PubMed] [Google Scholar]
- 77.Korsinczky ML, Clark RJ, Craik DJ. Disulfide bond mutagenesis and the structure and function of the head-to-tail macrocyclic trypsin inhibitor SFTI-1. Biochemistry. 2005;44(4):1145–53. [DOI] [PubMed] [Google Scholar]
- 78.Jendrny C, Beck-Sickinger AG. Inhibitors of kallikrein-related peptidases 7 and 5 by grafting serpin reactive center loop sequences onto sunflower trypsin inhibitor-1 (SFTI-1). Chembiochem. 2015. [DOI] [PubMed] [Google Scholar]
- 79.Gitlin A, Debowski D, Karna N, Legowska A, Stirnberg M, Gutschow M, et al. Inhibitors of Matriptase-2 Based on the Trypsin Inhibitor SFTI-1. Chembiochem. 2015;16(11):1601–7. [DOI] [PubMed] [Google Scholar]
- 80.de Veer SJ, Swedberg JE, Akcan M, Rosengren KJ, Brattsand M, Craik DJ, et al. Engineered protease inhibitors based on sunflower trypsin inhibitor-1 (SFTI-1) provide insights into the role of sequence and conformation in Laskowski mechanism inhibition. The Biochemical journal. 2015;469(2):243–53. [DOI] [PubMed] [Google Scholar]
- 81.Quimbar P, Malik U, Sommerhoff CP, Kaas Q, Chan LY, Huang YH, et al. High-affinity cyclic peptide matriptase inhibitors. J Biol Chem. 2013;288(19):13885–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Swedberg JE, Nigon LV, Reid JC, de Veer SJ, Walpole CM, Stephens CR, et al. Substrate-guided design of a potent and selective kallikrein-related peptidase inhibitor for kallikrein 4. Chem Biol. 2009;16(6):633–43. [DOI] [PubMed] [Google Scholar]
- 83.Daly NL, Chen YK, Foley FM, Bansal PS, Bharathi R, Clark RJ, et al. The absolute structural requirement for a proline in the P3’-position of Bowman-Birk protease inhibitors is surmounted in the minimized SFTI-1 scaffold. J Biol Chem. 2006;281(33):23668–75. [DOI] [PubMed] [Google Scholar]
- 84.Wang CK, Northfield SE, Huang YH, Ramos MC, Craik DJ. Inhibition of tau aggregation using a naturally-occurring cyclic peptide scaffold. European journal of medicinal chemistry. 2016;109:342–9. [DOI] [PubMed] [Google Scholar]
- 85.Chan LY, Craik DJ, Daly NL. Cyclic thrombospondin-1 mimetics: grafting of a thrombospondin sequence into circular disulfide-rich frameworks to inhibit endothelial cell migration. Biosci Rep. 2015;35(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zoller F, Markert A, Barthe P, Zhao W, Weichert W, Askoxylakis V, et al. Combination of phage display and molecular grafting generates highly specific tumor-targeting miniproteins. Angew Chem Int Ed Engl. 2012;51(52):13136–9. [DOI] [PubMed] [Google Scholar]
- 87.Chan LY, Gunasekera S, Henriques ST, Worth NF, Le SJ, Clark RJ, et al. Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood. 2011;118(25):6709–17. [DOI] [PubMed] [Google Scholar]
- 88.Jiang S, Li P, Lee SL, Lin CY, Long YQ, Johnson MD, et al. Design and synthesis of redox stable analogues of sunflower trypsin inhibitors (SFTI-1) on solid support, potent inhibitors of matriptase. Org Lett. 2007;9(1):9–12. [DOI] [PubMed] [Google Scholar]
- 89.Lin CY, Anders J, Johnson M, Dickson RB. Purification and characterization of a complex containing matriptase and a Kunitz-type serine protease inhibitor from human milk. J Biol Chem. 1999;274(26):18237–42. [DOI] [PubMed] [Google Scholar]
- 90.de Veer SJ, Li CY, Swedberg JE, Schroeder CI, Craik DJ. Engineering potent mesotrypsin inhibitors based on the plant-derived cyclic peptide, sunflower trypsin inhibitor-1. Eur J Med Chem. 2018;155:695–704. [DOI] [PubMed] [Google Scholar]
- 91.Veveris-Lowe T, Lawrence M, Collard R, Bui L, Herington A, Nicol D, et al. Kallikrein 4 (hK4) and prostate-specific antigen (PSA) are associated with the loss of E-cadherin and an epithelial-mesenchymal transition (EMT)-like effect in prostate cancer cells. Endocr Relat Cancer. 2005;12(3):631–43. [DOI] [PubMed] [Google Scholar]
- 92.Coughlin SR. How the protease thrombin talks to cells. Proceedings of the National Academy of Sciences. 1999;96(20):11023–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Swedberg JE, De Veer SJ, Sit KC, Reboul CF, Buckle AM, Harris JM. Mastering the canonical loop of serine protease inhibitors: enhancing potency by optimising the internal hydrogen bond network. PLoS One. 2011;6(4):e19302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Swedberg JE, Li CY, de Veer SJ, Wang CK, Craik DJ. Design of Potent and Selective Cathepsin G Inhibitors Based on the Sunflower Trypsin Inhibitor-1 Scaffold. J Med Chem. 2017;60(2):658–67. [DOI] [PubMed] [Google Scholar]
- 95.Jendrny C, Beck-Sickinger AG. Inhibition of Kallikrein-Related Peptidases 7 and 5 by Grafting Serpin Reactive-Center Loop Sequences onto Sunflower Trypsin Inhibitor-1 (SFTI-1). ChemBioChem. 2016;17(8):719–26. [DOI] [PubMed] [Google Scholar]
- 96.Prassas I, Eissa A, Poda G, Diamandis EP. Unleashing the therapeutic potential of human kallikrein-related serine proteases. Nat Rev Drug Discov. 2015;14(3):183–202. [DOI] [PubMed] [Google Scholar]
- 97.Avgeris M, Mavridis K, Scorilas A. Kallikrein-related peptidases in prostate, breast, and ovarian cancers: from pathobiology to clinical relevance. Biol Chem. 2012;393(5):301–17. [DOI] [PubMed] [Google Scholar]
- 98.Altmann A, Sauter M, Roesch S, Mier W, Warta R, Debus J, et al. Identification of a novel ITGαvβ6-binding peptide using protein separation and phage display. Clin Cancer Res. 2017;23(15):4170–80. [DOI] [PubMed] [Google Scholar]
- 99.Raguse J-D, Gath HJ, Bier J, Riess H, Oettle H. Cilengitide (EMD 121974) arrests the growth of a heavily pretreated highly vascularised head and neck tumour. Oral Oncol. 2004;40(2):228–30. [DOI] [PubMed] [Google Scholar]
- 100.Roesch S, Lindner T, Sauter M, Loktev A, Flechsig P, Muller M, et al. Comparative study of the novel RGD motif-containing ITGalphavbeta6 binding peptides SFLAP3 and SFITGv6 for diagnostic application in HNSCC. J Nucl Med. 2018. [DOI] [PubMed] [Google Scholar]
- 101.Zoller F, Markert A, Barthe P, Zhao W, Weichert W, Askoxylakis V, et al. Combination of Phage Display and Molecular Grafting Generates Highly Specific Tumor‐Targeting Miniproteins. Angew Chem Int Ed. 2012;51(52):13136–9. [DOI] [PubMed] [Google Scholar]
- 102.Heitz A, Le-Nguyen D, Chiche L. Min-21 and Min-23, the smallest peptides that fold like a cystine-stabilized beta-sheet motif: Design, solution structure, and thermal stability. Biochemistry. 1999;38(32):10615–25. [DOI] [PubMed] [Google Scholar]
- 103.Chan LY, Craik DJ, Daly NL. Dual-targeting anti-angiogenic cyclic peptides as potential drug leads for cancer therapy. Sci Rep. 2016;6:35347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fittler H, Depp A, Avrutina O, Dahms SO, Than ME, Empting M, et al. Engineering a Constrained Peptidic Scaffold towards Potent and Selective Furin Inhibitors. ChemBioChem. 2015;16(17):2441–4. [DOI] [PubMed] [Google Scholar]
- 105.Cobos Caceres C, Bansal PS, Navarro S, Wilson D, Don L, Giacomin P, et al. An engineered cyclic peptide alleviates symptoms of inflammation in a murine model of inflammatory bowel disease. J Biol Chem. 2017;292(24):10288–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Durek T, Cromm PM, White AM, Schroeder CI, Kaas Q, Weidmann J, et al. Development of Novel Melanocortin Receptor Agonists Based on the Cyclic Peptide Framework of Sunflower Trypsin Inhibitor-1. J Med Chem. 2018;61(8):3674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gunasekera S, Fernandes-Cerqueira C, Wennmalm S, Wahamaa H, Sommarin Y, Catrina AI, et al. Stabilized Cyclic Peptides as Scavengers of Autoantibodies: Neutralization of Anticitrullinated Protein/Peptide Antibodies in Rheumatoid Arthritis. ACS Chem Biol. 2018;13(6):1525–35. [DOI] [PubMed] [Google Scholar]
- 108.Gao Y, Cui T, Lam Y. Synthesis and disulfide bond connectivity-activity studies of a kalata B1-inspired cyclopeptide against dengue NS2B-NS3 protease. Bioorg Med Chem. 2010;18(3):1331–6. [DOI] [PubMed] [Google Scholar]
- 109.Getz JA, Cheneval O, Craik DJ, Daugherty PS. Design of a cyclotide antagonist of neuropilin-1 and −2 that potently inhibits endothelial cell migration. ACS Chem Biol. 2013;8(6):1147–54. [DOI] [PubMed] [Google Scholar]
- 110.Wang CK, Gruber CW, Cemazar M, Siatskas C, Tagore P, Payne N, et al. Molecular grafting onto a stable framework yields novel cyclic peptides for the treatment of multiple sclerosis. ACS Chem Biol. 2014;9(1):156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thell K, Hellinger R, Sahin E, Michenthaler P, Gold-Binder M, Haider T, et al. Oral activity of a nature-derived cyclic peptide for the treatment of multiple sclerosis. Proc Natl Acad Sci U S A. 2016;113(15):3960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang YH, Henriques ST, Wang CK, Thorstholm L, Daly NL, Kaas Q, et al. Design of substrate-based BCR-ABL kinase inhibitors using the cyclotide scaffold. Sci Rep. 2015;5:12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aboye T, Meeks CJ, Majumder S, Shekhtman A, Rodgers K, Camarero JA. Design of a MCoTI-Based Cyclotide with Angiotensin (1–7)-Like Activity. Molecules. 2016;21(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Swedberg JE, Mahatmanto T, Abdul Ghani H, de Veer SJ, Schroeder CI, Harris JM, et al. Substrate-Guided Design of Selective FXIIa Inhibitors Based on the Plant-Derived Momordica cochinchinensis Trypsin Inhibitor-II (MCoTI-II) Scaffold. J Med Chem. 2016;59(15):7287–92. [DOI] [PubMed] [Google Scholar]