

Abstract
Objective
To investigate to what extent low-frequency genetic variants (with minor allele frequencies <5%) affect the risk of intracranial aneurysms (IAs).
Methods
One thousand fifty-six patients with IA and 2,097 population-based controls from the Netherlands were genotyped with the Illumina HumanExome BeadChip. After quality control (QC) of samples and single nucleotide variants (SNVs), we conducted a single variant analysis using the Fisher exact test. We also performed the variable threshold (VT) test and the sequence kernel association test (SKAT) at different minor allele count (MAC) thresholds of >5 and >0 to test the hypothesis that multiple variants within the same gene are associated with IA risk. Significant results were tested in a replication cohort of 425 patients with IA and 311 controls, and results of the 2 cohorts were combined in a meta-analysis.
Results
After QC, 995 patients with IA and 2,080 controls remained for further analysis. The single variant analysis comprising 46,534 SNVs did not identify significant loci at the genome-wide level. The gene-based tests showed a statistically significant association for fibulin 2 (FBLN2) (best p = 1 × 10−6 for the VT test, MAC >5). Associations were not statistically significant in the independent but smaller replication cohort (p > 0.57) but became slightly stronger in a meta-analysis of the 2 cohorts (best p = 4.8 × 10−7 for the SKAT, MAC ≥1).
Conclusion
Gene-based tests indicated an association for FBLN2, a gene encoding an extracellular matrix protein implicated in vascular wall remodeling, but independent validation in larger cohorts is warranted. We did not identify any significant associations for single low-frequency genetic variants.
Intracranial aneurysms (IAs) have a strong familial component.1 To date, genome-wide association studies (GWAS) have detected several risk loci for IA: common genetic variants (defined as a minor allele frequency [MAF] ≥5%) near the genes STARD13-KL, RBBP8, SOX17, CDKN2BAS, CNNM2, EDNRA, and HDAC92–5 and low-frequency variants (MAF <5%) near FSTL1 and EPM2A found to be associated with IA.6 However, these variants explain only a small part of the estimated heritability of IA,4,6 suggesting that additional genetic variation can contribute to IA.
Whole-genome and whole-exome sequencing studies have shown that low-frequency genetic variants play a role in complex diseases.7 Because these studies are expensive to perform on a large scale, the exome chip has been designed as an affordable alternative to detect low-frequency genetic variants within the coding regions of the genome.8,9 With this method, low-frequency variants have recently been detected for several common diseases and traits, including insulin processing and secretion,10 macular degeneration,11 cholesterol levels and myocardial infarction,12 and esophageal carcinoma.13
We aimed to assess to what extent low-frequency variants in the exome affect the susceptibility to IA by performing an exome-wide association analysis using the exome chip in patients with IA and controls. In addition, we looked up the association of low-frequency variants at established GWAS IA loci to search for additional variants that could be responsible for the associations of common variants with IA.14 Finally, we applied gene-based association analyses to test whether multiple variants within the same gene may be collectively associated with IA.
Methods
Study populations
In the discovery cohort, we included Dutch patients with IA (n = 1,056) treated at the University Medical Center Utrecht, the Netherlands, between 1997 and 2011. IAs were identified by conventional angiogram, CT angiogram, or magnetic resonance angiogram. Patients with fusiform IA, possible traumatic subarachnoid hemorrhage, arteriovenous malformations, polycystic kidney disease, or other connective tissue diseases were excluded. As controls, we retrieved a subgroup of 2,097 participants from the Utrecht Health Project (UHP). The UHP (lrgp.nl/research/) is an ongoing dynamic general population-based cohort that includes residents of the Leidsche Rijn area within the city of Utrecht. An extensive overview of the study has been published previously.15 All new inhabitants were invited by their general practitioner to participate in the UHP. Written informed consent was obtained, and an individual health profile was made by dedicated research nurses. All controls were >18 years of age, were from European descent, and were not genetically related to other people in the cohort.
For the replication cohort, we used the Familial Intracranial Aneurysm (FIA) study.5 Samples in this study were recruited from 26 clinical sites (41 centers) in North America, New Zealand, and Australia. Only families with at least 2 members who had IAs were included. Samples with the following conditions were excluded: (1) a fusiform-shaped unruptured IA of a major intracranial trunk artery; (2) an IA that is part of an arteriovenous malformation; (3) a family or personal history of polycystic kidney disease, Ehlers-Danlos syndrome, Marfan syndrome, fibromuscular dysplasia, or Moya-Moya disease; or (4) failure to obtain informed consent from the patient or family members. A Verification Committee reviewed all medical records and related data. For the present analysis, a set of unrelated cases was created by selecting 1 affected individual from each FIA family. Controls were individuals without IA from the Genetic and Environmental Risk Factors for Hemorrhagic Stroke Study (GERFHS; NS036695) and were identified through random digit dialing to match cases of hemorrhagic stroke by age (±5 years), race, and sex. The Cincinnati Control Cohort was a supplemental control cohort using an identical interview from the GERFHS study. The Cincinnati Control Cohort used a frequency-based matching algorithm to a stroke population and were also identified through random digit dialing. Individuals with spontaneous brain hemorrhage or intracerebral hemorrhage were excluded, but prior stroke was permitted.
Standard protocol approvals, registrations, and patient consents
For the discovery cohort, the study has been approved by the Medical Ethics Committee of the University Medical Center Utrecht. All participants provided written informed consent. The design of the UHP has been discussed with representatives of the Dutch Patient and Healthcare Consumer Platform and has been approved by the Dutch Health Care Inspectorate. The masking of all personal data for researchers and for other possible users of LRGP has been regulated in a legal document. For the replication cohort, the FIA study was approved by the Institutional Review Boards/Ethics Committees at all clinical and analytical centers and recruitment sites.
Genotyping
DNA was isolated from peripheral blood drawn during study inclusion. Cases and controls of the discovery cohort were genotyped together, as part of the Netherlands ExomeChip Project (bbmri.nl), on the Illumina HumanExome Beadchip version 1.1 (Illumina, San Diego, CA). In total, 242,901 single nucleotide variants (SNVs) were genotyped. Genotypes were subsequently called with the GenomeStudio software from Illumina. We used zCall16 to call genotypes of variants that could not be called by GenomeStudio.
Cases and controls of the replication cohort were genotyped on the IlluminaExome-12 version 1.1 (Illumina). Illumina Genome Studio (version 2010.3, Genotyping Module version 1.8.4) was used to call genotypes with the default clustering algorithm (GenCall). Of the 242,901 markers on the array, 218,523 were successfully genotyped.
Quality control
For the discovery cohort, we performed quality control (QC) using PLINK version 1.0717 in the cases and controls separately and then again after merging cases and controls. Samples with a call rate <95% were removed. We included common, independent, high-quality SNVs using the following criteria: SNVs without deviation from Hardy-Weinberg equilibrium (HWE) (p > 0.001), with MAF >5%, rate of missing genotypes <1%, and linkage disequilibrium r2 < 0.05. Using this subset of SNVs, we removed participants on the basis of the following criteria: discordant sex, heterozygosity (participants were excluded if the inbreeding coefficient deviated >4 SDs from the mean), and cryptic relatedness (by calculating identity by descent [IBD] for each pair of individuals). In each pair with an IBD proportion of >20%, an individual was excluded, if that individual exhibited distant relatedness with >1 individual. For case-control pairs, we removed the controls. In case-case or control-control pairs, the individual with the lowest call rate was excluded.
Next, we performed principal component (PC) analysis using EIGENSTRAT18 on the remaining study participants and HapMap-CEU participants. We created PC plots with the first 4 PCs using R version 2.11.19 On the basis of visual inspection of these plots, we excluded individuals who appeared to be outliers with respect to the CEU or the study population. A PC plot of the first 2 PCs after outlier removal is shown in supplementary figure 1 available from Dryad (doi.org/10.5061/dryad.099bk53). PCs were recalculated for only the study participants after outlier removal and tested for association with case-control status (logistic regression) to be used in the single variant analysis (see below).
After sample QC, we excluded SNVs that were monomorphic, had >5% missing genotypes, had a missing genotype rate higher than MAF, deviated from HWE (p < 0.001), or had a differential degree of missing genotypes between cases and controls (p < 1 × 10−5, χ2 test).
For the replication cohort, SNVs with call rates <95% or evidence of deviation from HWE (p < 1.0 × 10−10) were excluded. Pairwise IBD calculated from PLINK was used to detect cryptic relatedness. PCs were calculated with EIGENSTRAT.18 Only samples clustering with HapMap-CEU were included in the analysis. Samples with discrepancies between genetically identified sex and reported sex were excluded.
Single variant association analysis
For this analysis, we selected all SNVs with a minor allele count (MAC) > 5 in either of the 2 cohorts. Association testing was carried out in PLINK with the Fisher exact test. We calculated a genomic inflation factor (λGC) for the single variant analysis.20 After Bonferroni correction for the number of SNVs tested, we considered SNV associations with p < 1.07 × 10−6 (= 0.05/46,534 SNVs) as statistically significant, but only after visual inspection of the cluster plots. If the cluster assignment of GenomeStudio did not match the visual cluster assignment, the SNV was removed from further analyses.
We compared the allele frequencies of the significantly associated variants between our study samples and the European population reported by the Exome Aggregation Consortium database.21
Gene-based association analysis
We tested the collective effect of multiple low-frequency variants within the same gene on IA risk using SCORE-Seq.22 All SNVs that passed QC with an MAF <0.05 were included in this analysis. We performed 2 separate association analyses in which we included only the subset of SNVs with a minor allele observed at least 6 times (MAC >5) or at least once (MAC >0).
For both analyses, with MAC thresholds of 5 and 0, we applied 2 different gene-based tests. The first test is the variable threshold (VT) test,23 which includes SNVs at different MAF thresholds <0.05. The second test is the sequence kernel association test (SKAT),24 a weighted sum of individual score statistics. This test includes the association evidence across all SNVs considered within a gene but weighted inversely proportionally to their MAF.
In the first analysis including SNVs with MAC >5, genes with a value of p < 7 × 10−6 (after Bonferroni correction for the number of tested genes: 0.05/7,110) were defined as statistically significant. In the second analysis with MAC >0, the significance level was set at p < 3 × 10−6 after adjustment for 14,648 tested genes.
Replication
Significant results from the single variant analyses were tested in the replication cohort and the 2 cohorts combined using methods similar to those described above. Significant results from the gene-based analyses were also tested in the replication cohort with Rvtest.25 Then both cohorts were meta-analyzed with RAREMETAL (genome.sph.umich.edu/wiki/RAREMETAL).
Look-up of known IA loci
We selected SNVs and genes in 11 known IA risk loci (supplementary table 3 available from Dryad, doi.org/10.5061/dryad.099bk53)2–6 and looked up their association results in our single variant and gene-based analyses. The selection of IA risk loci was based on genome-wide significance (p < 5 × 10−8) in previous GWAS in European populations. Genotyped SNVs within 500 kb from the selected index SNV were included. We were particularly interested in low-frequent SNVs (MAF <0.05) associated with IA with a value of p < 8 × 10−4 (after Bonferroni correction for the number of low-frequent SNVs: 0.05/64) to detect low-frequency causal variants responsible for the associations of these risk loci.
Power calculation
Given our sample size of 995 cases and 2,080 controls in the discovery cohort (after QC), we have >80% power to detect an association of individual genetic variants with MAF = 1% with an effect size of 3.3 (p < 5 × 10−8).26
Data availability
The authors agree to share summary statistics of any unpublished data on request. For some cohorts, individual-level genetic data are also available on request.
Results
Study population and QC
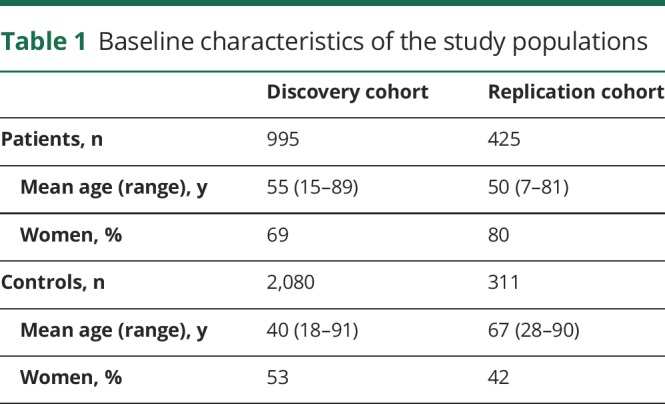
After QC, the discovery sample included 995 cases of IA and 2,080 controls, while the replication sample included 425 cases of IA and 311 controls. The baseline characteristics of the patients and controls of the discovery cohort and the replication cohort are shown in table 1. The number of samples and SNVs removed at each QC step in the discovery cohort is shown in supplementary table 1 available from Dryad (doi.org/10.5061/dryad.099bk53). PC plots after removal of outliers showed well-matched cases and controls (supplementary figure 1 available from Dryad). None of the first 10 PCs were significantly associated with case-control status (p ≥ 0.09 in logistic regression). The final discovery sample analysis included 46,534 SNVs.
Table 1.
Baseline characteristics of the study populations
We performed visual inspection of genotype cluster plots for all SNVs that showed statistically significant associations with IA in the single variant association analysis. In total, we identified 20 SNVs associated with p values ranging from 6.5 × 10−7 to 2.8 × 10−56 (supplementary table 2 available from Dryad, doi.org/10.5061/dryad.099bk53), but on close inspection of the genotype cluster plots, we concluded that they were incorrectly clustered.9 These were all rare SNVs in controls (MAF <0.02) with higher frequencies in cases (MAF up to 0.08). Supplementary figure 2 available from Dryad shows an example of one of these cluster plots. After appropriate manual adjustment of the clusters, none of the associations were statistically significant. Consequently, we interpreted all these associations as false-positive results and excluded these SNVs from further analyses. This illustrates the difficulty and importance of high-quality genotype calling for lower-frequency variants.
Single variant association analysis
In total, 46,514 SNVs were tested for association with IA in the discovery cohort. We did not observe inflation of the test statistic distribution to the expected median (λGC = 0.92; supplementary figure 3 available from Dryad, doi.org/10.5061/dryad.099bk53), and we did not observe any statistically significant associations (other than the false-positive associations mentioned above). Supplementary figure 5 available from Dryad shows the results of the association analysis.
Gene-based association analysis
The results of the gene-based association analyses are presented in table 2. In the first analysis including SNVs with MAC >5, we found 1 statistically significant association of the fibulin 2 gene (FBLN2) on chromosome 3, with p values of 1.0 × 10−6 (VT test) and 6.0 × 10−6 (SKAT) in the discovery cohort. Only 2 SNVs in this gene were analyzed in the gene-based tests: exm292082/rs111389908 and exm292166/rs201160150. In a second analysis, which also included SNVs with an MAC between 1 and 5, the associations became slightly weaker, with p values of 8.2 × 10−6 (SKAT) and 0.26 (VT test), based on a total number of 13 SNVs in this region. After visual inspection of the cluster plots of all 13 SNVs, we did not detect incorrect cluster assignment by GenomeStudio (supplementary figure 4 available from Dryad, doi.org/10.5061/dryad.099bk53). No other statistically significant gene-based associations were found in this second analysis.
Table 2.
Association results of gene-based tests in the discovery cohort, replication cohort, and meta-analysis of the 2 cohorts

In the replication cohort, 11 SNVs in the FBLN2 region were available for further analyses. HWE p values of these SNVs were between 0.005 and 1. Gene-based tests of FBLN2 did not reveal a significant association, with p values of 0.57 (SKAT, MAC >0), 0.68 (SKAT, MAC >5), and 0.78 (VT, MAC >0). However, the directions of effect were the same as those in the discovery cohort. A VT test with an MAC >5 was not applied in the replication cohort due to the presence of only 1 SNV. The association of FBLN2 became slightly stronger in the meta-analysis of the 2 cohorts, with p values of 4.8 × 10−7 (SKAT, MAC >0), 5.5 × 10−6 (SKAT, MAC >5), and 0.17 (VT, MAC >0).
In the single variant analysis, 3 of 13 FBLN2 SNVs showed a nominal level of association (p < 0.05) with IA in the discovery cohort: exm292082/rs111389908 (p = 2.0 × 10−3, odds ratio [OR] 2.6, 95% confidence interval [CI] 1.40–5.01 for A allele), exm292166/rs201160150 (p = 6.3 × 10−4, OR 3.4, 95% CI 1.62–7.41 for T allele), and exm292103/rs113265853 (p = 1.8 × 10−3, OR 4.2, 95% CI 0.06–0.67 for allele C). Two of these SNVs (exm292082 and exm292166) were also present in the replication cohort, with similar directions of effect (table 2). After data from the 2 cohorts were combined, the association of exm292082 became slightly weaker (p = 3.5 × 10−3, OR 2.31, 95% CI 1.29–4.21), while the association of exm292166 became stronger (p = 7.4 × 10−5, OR 3.39, 95% CI 1.76–6.80).
According to the Exome Aggregation Consortium database (exac.broadinstitute.org),27 exm292082 (p.Ala311Thr) is a missense variant with an MAF of 0.011 in the European population. The variant exm292103 (p.Pro409Leu; MAF 0.005) is also a missense variant, and exm292166 (c.2433C>T, MAF 0.004) is located at the splicing region. Further details about the associations of all tested SNVs in FBLN2 are shown in tables 2 and 3.
Table 3.
Association results of 13 SNVs in FBLN2 gene on chromosome 3 in the Dutch study cohort of 995 cases and 2,080 controls, replication cohort of 425 cases and 311 controls, and the 2 cohorts combined

Look-up of known IA loci
The association results in the discovery cohort for SNVs and genes in 11 established IA GWAS risk loci are shown in supplementary table 3 available from Dryad (doi.org/10.5061/dryad.099bk53). We observed no statistically significant associations of low-frequent SNVs in these loci. We observed statistically significant associations (p < 3.3 × 10−4 with Bonferroni correction) with only 2 previously reported common risk SNVs: rs10958409 at 8q11 (MAF 0.18 in cases, p = 1.7 × 10−4) and rs10931779 (MAF 0.42 in cases, proxy of known risk SNV rs919433 with r2 = 0.92) at 2q33 (p = 6.9 × 10−5).
Discussion
In our exome-wide association analysis, we found no statistically significant associations of individual low-frequency genetic variants with IA. A gene-based test indicated that variants in FBLN2 may be associated with IA, but further validation is necessary. We did not find evidence of low-frequency variants within established IA GWAS loci2–4 that contribute to the IA associations due to common variants.
The FBLN2 gene may be an interesting candidate gene for IA because it encodes an extracellular matrix protein that belongs to the fibulin family28 and plays a role in the formation of the elastic lamina of blood vessels.29
A principal limitation of this study is the sample size and therefore the statistical power to detect modest effects due to genetic variants (either on their own or collectively within genes). The lack of significant findings in our study is in line with other genome-wide studies of low-frequency variants in common diseases. Indeed, a published study of intracerebral hemorrhage with a similar sample size (n = 1,553) also did not reveal any novel associations.30 In contrast, larger studies (n > 8,000) of insulin secretion,10 myocardial infarction and cholesterol levels,12 and blood pressure31 reported statistically significant associations of single low-frequency variants.
It remains an empirical question to what extent the gene burden tests actually help boost the power to find genes compared to the simpler approach of testing all variants, one by one, which has worked so well for GWAS. To date, we are aware of only a single example: rare loss-of-function variants in SETD1A are associated with schizophrenia and development disorders.32 Another recent exome-chip study in meningitis, with a sample size comparable to ours, also reported a gene-based association, although this still requires further validation.33
The exome chip has the advantage that low-frequency variants in coding regions of the genome can be genotyped and tested cheaply for large groups of participants. However, variants in noncoding regions remain untested, although these can also play a role in complex traits. In addition, the exome chip contains only predetermined variants, so novel variants remain undetected. In contrast, sequencing studies can identify novel variants, but these are more expensive to perform on a large scale.
Another possible drawback of this exome-chip study is the genotype calling of low-frequency variants, which can be inefficient because the number of minor alleles is very small.9 Although we used appropriate methods for calling of these variants, we still observed some errors in cluster assignment. We were able to distinguish true-positive from false-positive associations by visual inspection of the cluster plots of the associated SNVs, but theoretically, true associations may have gone undetected due to clustering errors.
This study indicates a possible role of the FBLN2 gene in IA pathology based on the collective association of coding low-frequency variants in this gene. The known role of FBLN2 in blood vessels make this gene an interesting candidate gene for IA, but functional studies are needed to further investigate how this gene is involved in IA pathology. In addition, further replication in larger cohorts and detection of additional associated low-frequency variants in this region are needed to further confirm these findings.
Acknowledgment
The authors thank Jonathan Rosand, MD, MS, and Christina Kourkoulis from Massachusetts General Hospital for performing genotyping of the replication cohort.
Glossary
- CI
confidence interval
- FBLN2
fibulin 2
- FIA
Familial Intracranial Aneurysm
- GERFHS
Genetic and Environmental Risk Factors for Hemorrhagic Stroke Study
- GWAS
genome-wide association studies
- HWE
Hardy-Weinberg equilibrium
- IA
intracranial aneurysm
- IBD
identity by descent
- MAC
minor allele count
- MAF
minor allele frequency
- OR
odds ratio
- PC
principal component
- QC
quality control
- SKAT
sequence kernel association test
- SNV
single nucleotide variant
- UHP
Utrecht Health Project
- VT
variable threshold
Author contributions
Femke N. G. van 't Hof: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis, study supervision. Dongbing Lai: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis. Jessica van Setten: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis. Michiel L. Bots: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Ilonca Vaartjes: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Joseph Broderick: data acquisition, accepts responsibility for conduct of research and will give final approval, acquisition of data, obtaining funding. Daniel Woo: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Tatiana Foroud: drafting/revising the manuscript, study concept or design, accepts responsibility for conduct of research and will give final approval, statistical analysis. Gabriel J.E. Rinkel: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Paul I.W. de Bakker: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, study supervision, obtaining funding. Ynte M. Ruigrok: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data, study supervision, obtaining funding.
Study funding
The exome-chip data were generated in a research project that was financially supported by BBMRI-NL, a Research Infrastructure financed by the Dutch government (NWO 184.021.007). F.N.G.v.H. is supported by a grant from the Dutch Heart Foundation (project 2008B004). Y.M.R. was supported by a clinical fellowship grant by the Netherlands Organization for Health Research and Development (project 90714533). The UHP received grants from the Ministry of Health, Welfare and Sports, the University of Utrecht, the Province of Utrecht, the Dutch Organisation of Care Research, the University Medical Centre of Utrecht, and the Dutch College of Healthcare Insurance Companies. Funding for the studies in the FIA study was provided by NIH R01NS039512 and R03NS083468.
Disclosure
F. van 't Hof, D. Lai, J. van Setten, M. Bots, I. Vaartjes, J. Broderick, D. Woo, T. Foroud, and G. Rinkel report no disclosures relevant to the manuscript. P. de Bakker is a full-time employee and owns equity of Vertex Pharmaceuticals. Y. Ruigrok reports no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Bor AS, Rinkel GJ, Adami J, et al. Risk of subarachnoid haemorrhage according to number of affected relatives: a population based case-control study. Brain 2008;131:2662–2665. [DOI] [PubMed] [Google Scholar]
- 2.Bilguvar K, Yasuno K, Niemela M, et al. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet 2008;40:1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yasuno K, Bilguvar K, Bijlenga P, et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet 2010;42:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yasuno K, Bakircioglu M, Low SK, et al. Common variant near the endothelin receptor type A (EDNRA) gene is associated with intracranial aneurysm risk. Proc Natl Acad Sci USA 2011;108:19707–19712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foroud T, Lai D, Koller D, et al. Genome-wide association study of intracranial aneurysm identifies a new association on chromosome 7. Stroke 2014;45:3194–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurki MI, Gaal EI, Kettunen J, et al. High risk population isolate reveals low frequency variants predisposing to intracranial aneurysms. PLoS Genet 2014;10:e1004134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ, Richards JB. Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat Rev Genet 2018;19:110–124. [DOI] [PubMed] [Google Scholar]
- 8.Abecasis Lab. Exome Chip Design Wiki Site. Available at: genome.sph.umich.edu/wiki/Exome_Chip_Design. Accessed April 2017. [Google Scholar]
- 9.Guo Y, He J, Zhao S, et al. Illumina human exome genotyping array clustering and quality control. Nat Protoc 2014;9:2643–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huyghe JR, Jackson AU, Fogarty MP, et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet 2013;45:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seddon JM, Yu Y, Miller EC, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet 2013;45:1366–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holmen OL, Zhang H, Fan Y, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet 2014;46:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang J, Zhong R, Tian J, et al. Exome-wide analyses identify low-frequency variant in CYP26B1 and additional coding variants associated with esophageal squamous cell carcinoma. Nat Genet 2018;50:338–343. [DOI] [PubMed] [Google Scholar]
- 14.Service SK, Teslovich TM, Fuchsberger C, et al. Re-sequencing expands our understanding of the phenotypic impact of variants at GWAS loci. PLoS Genet 2014;10:e1004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grobbee DE, Hoes AW, Verheij TJ, Schrijvers AJ, van Ameijden EJ, Numans ME. The Utrecht Health Project: optimization of routine healthcare data for research. Eur J Epidemiol 2005;20:285–287. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein JI, Crenshaw A, Carey J, et al. zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics 2012;28:2543–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 19.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. Available at: R-project.org/. [Google Scholar]
- 20.Devlin B, Roeder K. Genomic control for association studies. Biometrics 1999;55:997–1004. Accessed April 2017. [DOI] [PubMed] [Google Scholar]
- 21.Exome Aggregation Consortium (ExAC). Available at: exac.broadinstitute.org. Accessed September 2015. [Google Scholar]
- 22.Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet 2011;89:354–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price AL, Kryukov GV, de Bakker PI, et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet 2010;86:832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 2011;89:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics (Oxford, England) 2016;32:1423–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003;19:149–150. [DOI] [PubMed] [Google Scholar]
- 27.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pruitt KD, Brown GR, Hiatt SM, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 2014;42:D756–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chapman SL, Sicot FX, Davis EC, et al. Fibulin-2 and fibulin-5 cooperatively function to form the internal elastic lamina and protect from vascular injury. Arterioscler Thromb Vasc Biol 2010;30:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radmanesh F, Falcone GJ, Anderson CD, et al. Rare coding variation and risk of intracerebral hemorrhage. Stroke 2015;46:2299–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu C, Kraja AT, Smith JA, et al. Meta-analysis identifies common and rare variants influencing blood pressure and overlapping with metabolic trait loci. Nat Genet 2016;48:1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh T, Kurki MF, Curtis D, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci 2016;19:571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kloek AT, van Setten J, van der Ende A, et al. Exome array analysis of susceptibility to pneumococcal meningitis. Sci Rep 2016;6:29351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors agree to share summary statistics of any unpublished data on request. For some cohorts, individual-level genetic data are also available on request.