

Abstract
Despite increased longevity and resistance to multiple stressors, growth hormone receptor null (GHRKO) mice exhibit severe skeletal impairment. The role of GHR in maintaining osteocyte mitochondrial function is unknown. We found that GHR ablation was detrimental to osteocyte mitochondrial function. In vivo multiphoton microscopy revealed significant reductions of >10% in mitochondrial membrane potential (MMP) in GHRKO osteocytes and reduced mitochondrial volumetric density. Reductions in MMP were accompanied by reductions in glucose transporter-1 levels, steady state ATP, NADH redox index, oxygen consumption rate, and mitochondrial reserve capacity in GHRKO osteocytes. Glycolytic capacity did not differ between control and GHRKO males’ osteocytes. However, osteocytes from aged female GHRKO mice exhibited reductions in glycolytic parameters, indicating impairments in glucose metabolism, which may be sex dependent. GHRKO osteocytes exhibited increased levels of cytoplasmic reactive oxygen species (ROS) (both basal and in response to high glucose), insulin-like growth factor-1 (IGF-1), and insulin. Mitochondrial ROS levels were increased and correlated with reduced glutathione in GHRKO osteocytes. Overall, the compromised osteocyte mitochondrial function and responses to metabolic insults strongly correlated with skeletal impairments, suggesting that despite increased life span of the GHRKO mice, skeletal health span is decreased.
Keywords: OSTEOCYTE, GROWTH HORMONE, RESPIRATION, NADH REDOX, REACTIVE OXYGEN SPECIES
Introduction
Growth hormone (GH) and insulin-like growth factor-1 (IGF-1) are key endocrine factors regulating metabolism,(1) body composition,(2) acquisition of peak bone mass, and maintenance of bone mineral density.(3–5) During postnatal growth, pituitary-derived GH stimulates liver production of IGF-1, which is then delivered to peripheral tissues to stimulate somatic growth. IGF-1 is also produced by all cells and acts in an autocrine/paracrine manner to stimulate tissue growth and function. GH has IGF-1-dependent and –independent actions on tissues, including bone. The two somatic hormones, GH and IGF-1, bind to their cognate receptors, which are found in almost all cells.
In mice, mutations in genes affecting pituitary GH secretion (Ghrhlit/lit) or pituitary development (Prop1df/df and Pit1dw/dw) lead to decreased GH secretion, resulting in decreased hepatic IGF-1 production and overall somatic growth retardation (dwarfism).(6) GH receptor (GHR) null (GHRKO) mice exhibit significantly reduced body and skeletal size, as well as a mechanically inferior skeleton, even after correction for body size.(7,8) Despite their retarded growth, GHRKO mice have a significantly extended life span.(9)
A prolonged life span in association with resistance to oxidative stress and/or increased activity of antioxidant enzymes was initially described in C. elegans bearing mutations in the IGF/insulin receptors or phosphatidylinositol-3 kinase (PI3K).(10–16) The extended life span of GHRKO mice was also attributed to enhanced mitochondrial biogenesis, improved mitochondrial function, and resistance to oxidative stress.(17) Organs and tissues, including muscles,(18) brown adipose tissue,(19) kidneys, and liver,(20–23) of long-lived GHRKO mice are resistant to multiple stressors. However, there has been no previous study of mitochondrial activity in skeletal cells of GHRKO mice, which is severely compromised.(24)
Osteocytes are the most abundant skeletal cells, composing >95% of cells in the adult and aged skeleton. Osteocytes are the longest-lived bone cells and play fundamental roles in maintaining bone-tissue integrity. They reside within the mineralized bone matrix, integrate hormonal and mechanical stimuli, and regulate bone remodeling. Osteocytes communicate via the lacunar canalicular system, a continuous interstitial fluid pathway through which osteocytes obtain nutrients and dispose of wastes. Loss of osteocytes compromises bone quality and is linked to an increased risk of fractures. Apoptosis of osteocytes is linked to aging,(25) absence of mechanical stimuli,(26,27) and hormone withdrawal (estrogen deprivation).(28) A common feature of these physiological changes is impaired mitochondrial function; the consequent inability of cellular detoxifying mechanisms to keep pace with reactive oxygen species (ROS) production eventually leads to apoptosis.(29)
Based on our recent data and the results of previous studies, we hypothesized that GHR regulates skeletal integrity partially via its effects on mitochondrial function and biogenesis in osteocytes. To test this hypothesis, we examined mitochondrial function in vitro in primary osteocytes from adult and aged GHRKO mice, as well as in vivo in bones of intact mice using a multiphoton microscopy approach.(30) In support of our hypothesis, we found that mitochondrial function is compromised in the osteocytes of GHRKO mice.
Materials and Methods
Animals
The generation of the GHRKO model was previously described.(31) All mice were in the C57BL/6J (B6) genetic background. Weaned mice were allocated randomly into cages separated according to their sex. Mice were housed 2 to 5 animals per cage in a facility with 12-hour light/dark cycles and free access to food and water. The different analyses were performed separately in male/female mice at several ages as indicated.
All animal procedures were approved by the Institutional Animal Care and Use Committee of the NYU School of Medicine (assurance number A3435–01, USDA licensed No. 465), and conform to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (http://www.nc3rs.org.uk/arrive-guidelines).
Micro-computed tomography
Micro-computed tomography (μCT) was performed according to published guidelines.(32) The left femora were scanned using a high-resolution SkyScan μCT system (SkyScan 1172, Kontich, Belgium). Images were acquired using a 10 MP digital detector, 10 W energy (100 kV and 100 m A), and a 0.5 mm aluminum filter with a 9.7 μm image voxel size. A fixed global threshold method was used based on the manufacturer’s recommendations and preliminary studies, which showed that mineral variation between groups was not high enough to warrant adaptive thresholds. The cortical region of interest was selected as the 2.0 mm mid-diaphyseal region directly below the third trochanter, which includes the mid-diaphysis and more proximal cortical regions. The trabecular measurements were taken at the femur distal metaphysis 2.5 mm below the growth plate.
Mechanical testing: three-point bending assay
Harvested femurs were stored frozen at −20°C and wrapped in PBS-soaked gauze. At testing time, samples were brought to room temperature in a saline bath. Three-point bending tests to failure were carried out using a BOSE materials testing machine (ElectroForce 3220, Eden Prairie, MN, USA). The upper loading span width was 3.0 mm and the lower support span width was 6.0 mm. Femora were positioned in a saline bath posterior side down on the supports in order to generate bending moments about the medial-lateral axis. Samples were carefully centered on the supports to ensure maximum load at the midpoint of the diaphysis. A preload of 1 N was briefly applied to secure the sample, and then a ramp waveform at a constant displacement rate of 0.10 mm/s was applied until failure. Load-displacement curves were recorded and subsequently analyzed.
Gene expression
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) or by RNeasy Plus kit (Cat# 74134, Qiagen, Valencia, CA, USA), reverse-transcribed to cDNA (Cat# 18080–051, Life Technologies, Carlsbad, CA, USA), and subjected to real-time PCR using SYBR master mix (Life Technologies/Applied Bio-systems, Cat# 4385612) on a BioRad (Hercules, CA, USA) CFX384 real-time machine. Transcript levels were assayed three times in each sample and corrected to 18S. The primer sequences are detailed in Supplemental Table S1.
Primary osteocyte cultures
Cultures were established according to a protocol published by Dr Linda Bonewald.(33) Briefly, upon flushing of the bone marrow, dissected bones were cleaned from remaining soft tissue and the periosteum and underwent sequential digestions with collagenase type-IA (300 active U/mL, Sigma, St. Louis, MO, USA) dissolved in α-minimal essential medium (αMEM; GIBCO#12571–063). Cells were seeded on collagen-coated (Cat# 354236, BD) plates and kept in αMEM medium containing 10% fetal calf serum (Cat# 26140, Thermo Fisher Scientific, Waltham, MA, USA). Osteocyte preparation was verified by determining the expression levels of osteocyte-specific genes (sclerostin, FGF23, MEPE, Phex, DMP1, RANKL, Cox2, Gp38). Note that primary osteocyte cultures are heterogeneous, including osteocytes at different stages of differentiation. Cells were passed 1 to 2 times before vital microscopy analyses and 2 to 3 times before respiration studies (bioanalyzer). Overall, primary cells were held in culture for 21 to 25 days.
For glucose deprivation studies, cells were maintained in XF base medium (Cat# 103193100) supplemented with 1% bovine serum albumin (BSA) for 4 to 6 hours before protein extraction.
Cellular and mitochondrial content of reactive oxygen species (ROS)
Cells were seeded (0.4 × 106 cells/mL) on collagen-coated black-walls 96-well plate incubated for an hour in an assay medium (containing αMEM supplemented with 10 mM HEPES) and incubated for 60 minutes with 10 μg/mL 2′,7′-dichlorofluorescin diacetate (DCFDA) (Cat# ab113851, Abcam, Cambridge, MA, USA). ROS levels were determined at excitation 495 and emission 525 nm (plate reader; Spectra Max5 M5, Molecular Devices [San Jose, CA, USA] with Softmax Pro software). Mitochondrial ROS was determined using MitoSox (5 μM, Cat# M36008, Thermo Fisher Scientific). Images were taken every 10 seconds at excitation 510 and emission 580 nm.
Cellular glutathione levels
Glutathione levels were measured using Monochlorobimane (mBCI/MCB) (50 μM, Cat# M1381MP, Thermo Fisher Scientific). Cells were visualized by 2000E Nikon Microscope Eclipse TE at 20× magnification. Images were taken every 10 seconds at excitation 394 and emission 490 nm.
Cellular ATP levels
Steady-state ATP levels in cell lysates were measured using Luminescent ATP Detection assay kit (Cat# ab113849, Abcam).
NADH auto-fluorescence
NADH auto-fluorescence was measured as described in Weissig and Edeas.(34) Briefly, cells were seeded (0.4 × 106 cells/mL) on collagen-coated glass discs (Cat# 12-545-102, Thermo Fisher Scientific) in assay medium (HBSS: 156 mM NaCl, 3 mM KCl, 2 mM MgSO4, 1.25 mM KH2PO4, 2mM CaCl2, 10 mM glucose, and 10 mM HEPES; pH adjusted to 7.35 with NaOH) and visualized by 2000E Nikon Microscope Eclipse TE at 20× magnification at excitation 351 nm and emission at 375 to 470nm. NADH redox index calculations were done as follows: basal level (taken for 1 to 2 minutes) relative to maximal respiration after 1 μM FCCP (0%) taken for 1 to 2 minutes, and minimal respiration after 1 mM NaCN (100%) taken for 1 to 2 minutes.
Cellular respiration and glycolysis
Primary osteocytes (0.4 × 106 cells/mL, a pool of 2 to 3 mice per group) were seeded on a collagen-coated plate (Cat# 102342–100, Agilent Technologies, Santa Clara, CA, USA) in triplicates. Oxygen consumption rate (OCR) was determined in assay medium (XF base medium Cat# 103193100 supplemented with 1 mM pyruvate, 10 mM glucose, and 2 mM glutamine, Agilent Technologies) using the cellular respiration analyzer (Seahorse Analyzer, Agilent Technologies) at basal, upon oligomycin (1 μM), FCCP (2 μM), and Rotenone/antimycin (0.5 μM) additions. For glycolysis, assay osteocytes were seeded as above using a glucose-free medium (XF Base medium Cat# 103193100, Agilent Technologies) with 2 mM glutamine, and glucose (10 mM) added at the indicated time point according to the manufacturer’s instructions. Data were normalized to cell number determined at the end of the assay and analyzed using the Seahorse Wave software. Assays were repeated >4 times using osteocyte preparations from several mice per group. Data from 3 to 6 assays were combined after normalization of basal OCR to 100%.
Calculation of mitochondrial volume and mitochondrial membrane potential (MMP)
Mitochondrial volume and potential were determined in primary osteocytes seeded (0.4 × 106 cells/mL) on collagen-coated glass plates in αMEM medium and visualized by 2000E Nikon Microscope Eclipse TE at 60× magnification Osteocyte mitochondrial volume/cell volume was determined from 3D images reconstructed using the Amira FEI software (Thermo Fisher Scientific). Cell volume was calculated from live cells labeled with Calcein AM (Invitrogen, excitation 495 emission 516), and mitochondria labeled with TMRE (loaded with 60 nM TMRE for 30 minutes, washed, and imaged at 15 nM TMRE; excitation 548 nm emission 573 nm) (n = 20 cells/genotype) of cells seeded in αMEM medium. MMP in osteocytes was estimated from peak TMRE intensity (software NIS ElementsAR) of several mitochondria per cell of >20 cells per culture. At the end of each experiment, FCCP (1 μM) was added to validate mitochondrial (depolarization) integrity.
In vivo multiphoton microscopy
In vivo experiments were performed with TMRM as previously described.(30) TMRM was injected intravenously (2.5 μg/g) to isoflurane-anesthetized mice. Animals were then allowed normal cage activity for 1 hour. Ambulating during this time helps the convective transport of TMRM in the bone tissue fluid and ensures a similar concentration through the cortical bone cortex. Note that during this time mice are allowed to drink; thus they also urinate TMRM. The dorsal metatarsal surfaces were incised, and bones with intact periosteum were exposed. The mouse was placed on a warming blanket and the foot submerged in Hanks Balanced Salt Solution at 37°C. The entire apparatus was set onto the stage of an MPM (Ultima, Bruker Instruments, Inc., Middleton, WI, USA). Olympus LUMPLFLN 40XW water immersion, NA = 0.8; working distance = 3.3 mm at excitation 965 nm and emissions at 565 to 615 nm for TMRM. Z-stack images were analyzed using ImageJ software with “adaptive 3D threshold” plugin. Osteocytes at a distance of 20 to 50 μm from the periosteal surface were selected and analyzed individually.
Western immunoblotting
Proteins were extracted in modified CHAPS buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1.25% CHAPS, 2 mM NaF, 10 mM Na-pyrophosphate, 8 mM β-glycerophosphate, 1 mM Na-orthovanadate, and complete protease inhibitor cocktail (Roche, Mannheim, Germany, Cat# 04693132001). Lysates (20 to 30 μg) were separated using 4% to 20% gradient SDS-PAGE (Life Technologies, Cat# NP0335) and transferred to nitrocellulose membranes (Bio-Rad, Cat# 170–4158). Proteins were detected using primary antibodies detailed in Supplemental Table S2.
Statistical analyses
Data are presented as means ± SEM. Differences between groups were tested using t test or ANOVA and post hoc Tukey’s test, as appropriate. Significance accepted at p < 0.05. Scientists were blinded to sample IDs in all experimental procedures.
Results
Compromised skeletal morphology and mechanical properties of GHRKO mice
It is well established that GHRKO mice are growth retarded, exhibit reduced body weight and length throughout life, have increased body adiposity, and, in laboratory settings, live longer than control mice.(9) We used μCT to follow the skeletal morphology of GHRKO and control male mice through 4, 12, and 24 months of age. GHRKO mice exhibited reduced bone size at all ages, as reflected by reduced total cross-sectional area (Tt.Ar) (Fig. 1A), bone area (B.Ar) (Fig. 1B), cortical bone thickness (Cs.Th) (Fig. 1C), and marrow area (Ma.Ar) (Fig. 1D). Whereas the relative cortical bone area (RCA) was similar between control and GHRKO mice (Fig. 1E), bone robustness (Fig. 1F), which reflects radial versus linear bone growth, was reduced in GHRKO mice at all ages. These data suggest that GHRKO bones are insufficient to support the body weight of these animals and are not simply proportionally smaller in size. Indeed, controls showed a linear relationship between Tt.Ar and body weight throughout life, whereas GHRKO mice did not exhibit this correlation (Fig. 1G). Reduced polar moment of inertia (J0) in GHRKO bones (Fig. 1H) suggested that GHRKO bones were mechanically inferior. Three-point bending assay revealed reductions in strength at all ages (Fig. 1I), reduced toughness, and decreased post-yield diffraction at 24 months of age (Fig. 1K, L). This skeletal characterization confirmed the results of previous studies of GHRKO mice(7,8) at three different ages: young adult, middle-aged, and aged mice.
Fig. 1.
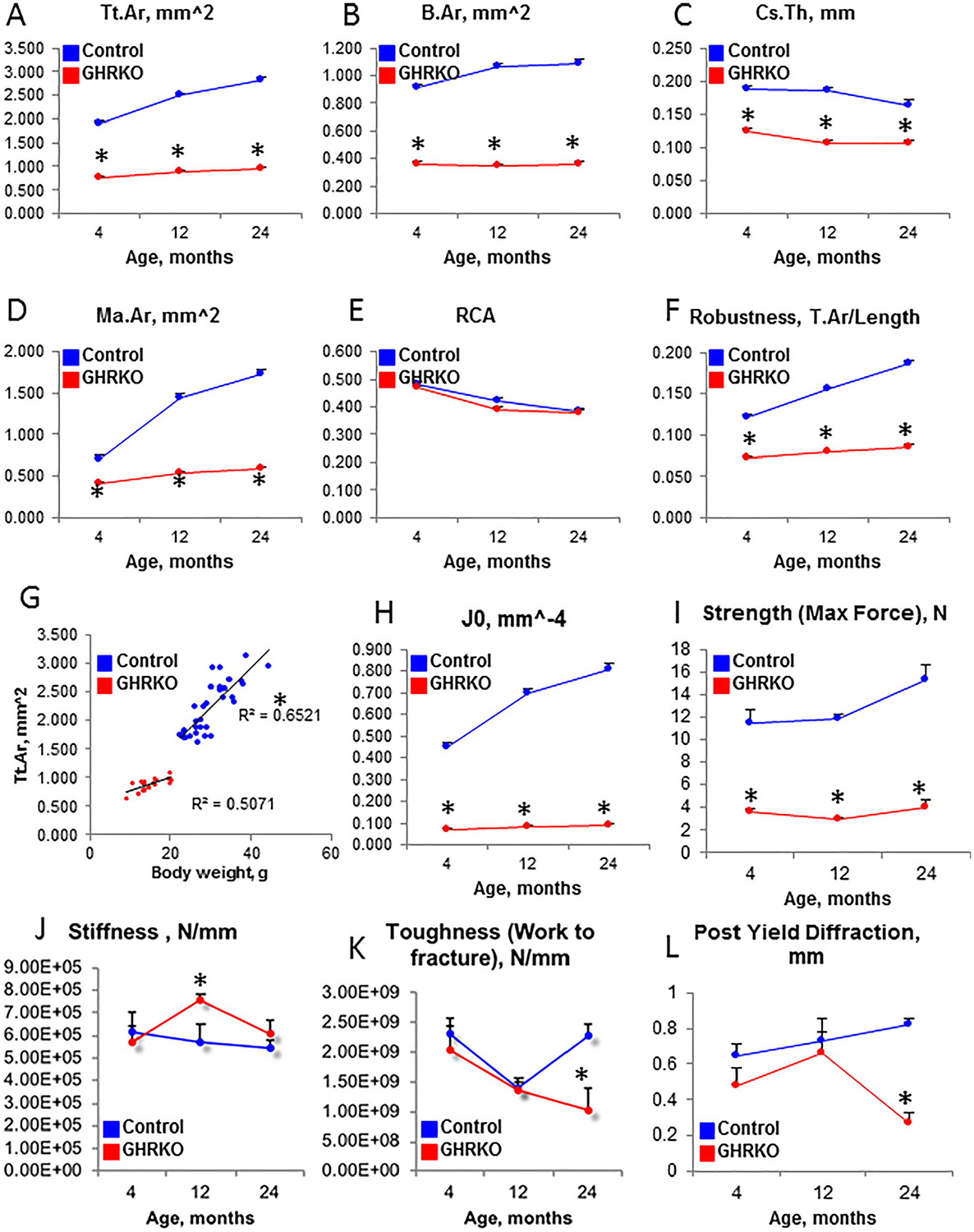
Morphologic and mechanical analyses of GHRKO femurs. Femurs from male GHRKO and control mice (n > 8 per genotype per age) were dissected at 4, 12, and 24 months of age. They were subjected to micro-computed tomography (μCT), followed by the 3-point bending assay. Cortical bone morphology was determined at the femur mid-diaphysis, where (A) total cross-sectional area (Tt.Ar), (B) bone area (B.Ar), (C) cortical bone thickness (Cs.Th), and (D) marrow area (Ma.Ar) were measured. (E) Relative cortical area (RCA) did not differ between controls and GHRKO bones at all ages, whereas (F) bone robustness (calculated as Tt.Ar/length) decreased significantly. There was a strong linear relationship between Tt.Ar and body weight in control mice (G), suggesting bone adaptation to increased body weight. (H) Polar moment of inertia (J0), measured by μCT, suggested that GHRKO bones have reduced mechanical strength. This was evident by the 3-point bending assay, in which (I) bone strength, (J) stiffness, (K) toughness, and (L) post-yield diffraction were measured. Data are presented as mean ± SEM, and significance was accepted at p < 0.05.
Establishment of primary osteocyte cultures
Our next studies focused on osteocytes from the skeletons of mature and aged GHRKO mice. Primary osteocyte cultures were established according to a protocol published by Dr Linda Bonewald.(33) Osteocyte preparation was validated by the expression of osteocyte-specific genes, as compared with the gene expression of the differentiated osteocyte-like cell line IDGSW3.(35) As expected, preparations from bones of young mice were a mixed population of early and late differentiated osteocytes. Accordingly, we found high levels of dentin matrix protein-1 (DMP-1) expression, a marker of mineralizing osteocytes, as well as phosphate regulating endopeptidase homolog X-linked (Phex) and matrix extracellular glycoprotein (Mepe), markers of osteoid osteocytes. Podoplanin (GP38/E11), a marker of early osteocytes, did not differ between control and GHRKO osteocytes (Fig. 2). In accordance with our previous publication(36) using osteocyte-specific GHRKO mice (DMP-1-GHRKO), we found that expression of sclerostin (an endogenous Wnt inhibitor) and fibroblast growth factor 23 (FGF23; a phosphate modulator) were higher in young GHRKO-osteocytes than in control osteocytes (Fig. 2).
Fig. 2.
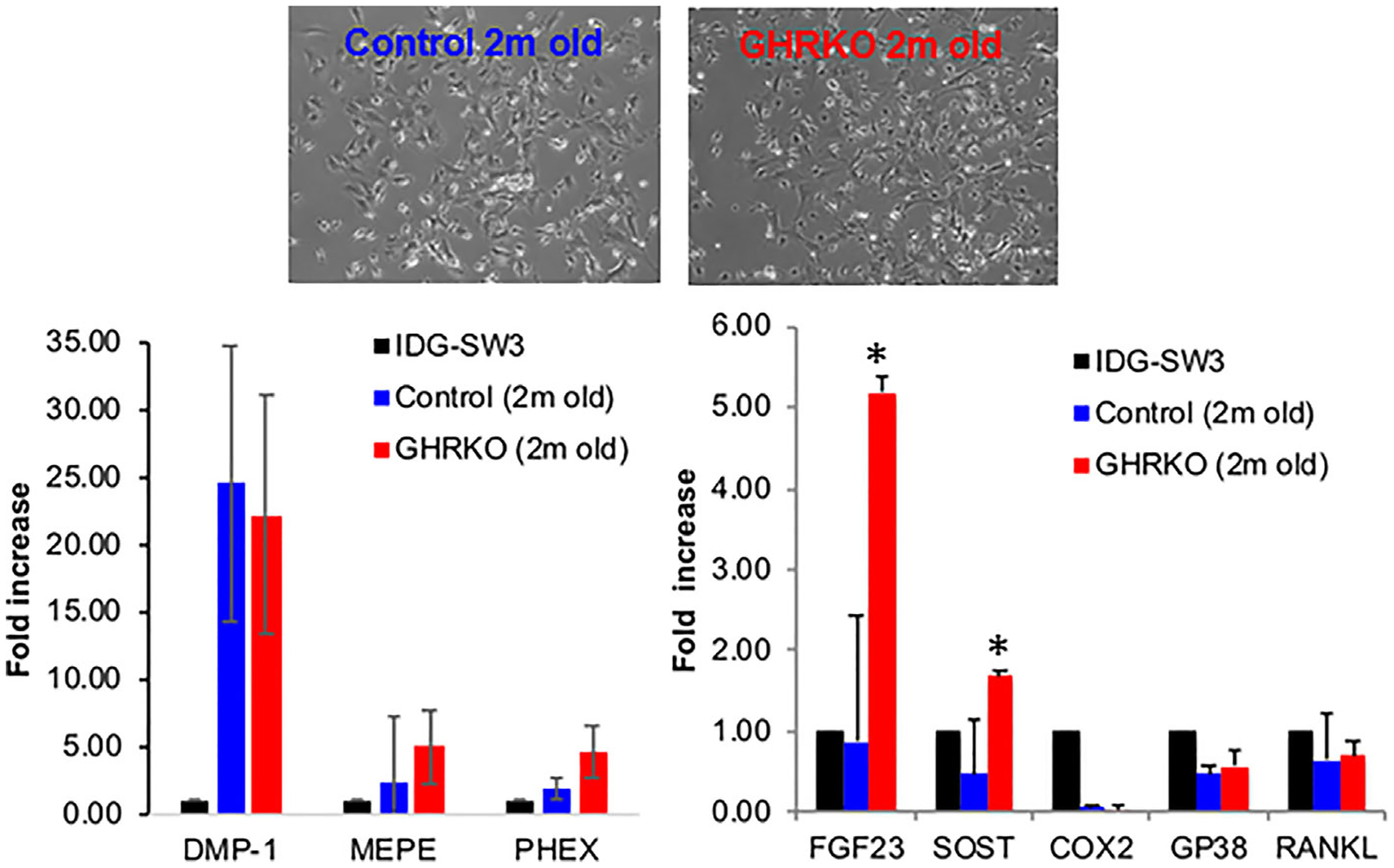
Establishment of primary osteocyte cultures from GHRKO and control mice. Long bones (femur, tibia, and humerus) were used to establish primary osteocyte cultures. The purity of each preparation was verified by expression of osteocyte-specific genes using real-time polymerase chain reaction. RNA from differentiated osteocyte-like IDG-SW3 cell line was used as a reference. Shown is a representative experiment with primary osteocyte cultures derived from 2-month-old control (n = 4) and GHRKO male mice (n = 6). Data are presented as mean ± SEM, and significance was accepted at p < 0.05.
Mitochondrial biogenesis in GHRKO osteocytes
Studies of long-lived GHRKO mice have shown increased gene expression of key regulators of mitochondrial biogenesis in the kidneys and heart.(37–39) Further, long-lived calorie-restricted mice with diminished GH/IGF-1 signaling exhibited increased mitochondrial biogenesis in several tissues.(40) To test whether these observations also apply to osteocytes in bone, we assessed mitochondrial biogenesis by measuring total mitochondrial volume, mitochondrial protein levels, and mitochondrial gene expression. Cell and mitochondrial volumes were calculated from live cells labeled with Calcein AM and TMRE, respectively (n = 20 cells/genotype). Mitochondrial to cell volume was calculated from 3-dimensional images reconstructed using Amira FEI software. Unlike previous findings for the kidneys and heart, we found that in vitro mitochondrial volume in GHRKO osteocytes did not differ from that of controls and was approximately 20% of the cell volume (Fig. 3A). These findings were verified by Western immunoblotting showing similar levels of electron transport chain (ETC) proteins in osteocytes from control and GHRKO mice (Fig. 3B) and by gene expression studies of mitochondria-specific markers (Fig. 3C). No significant differences were found between control and GHRKO primary osteocytes (by all assays used), suggesting no differences in mitochondrial biogenesis, at least in vitro. To address how excess GH, on the other hand, affect mitochondrial volume and function, we used young (2 to 4 months old) bovine-GH transgenic (bGH) mice. bGH mice showed significant increases in body weight and a robust skeletal growth (which was detailed in our recent publication(41)). Despite increases in osteocyte cell volume in bGH mice, no significant difference in relative mitochondrial volume was found between osteocytes from bGH and control mice (Supplemental Fig. S1A). This was supported by Western blot analyses of mitochondrial ETC protein levels in osteocyte cultures (Supplemental Fig. S1C) and by gene expression studies of mitochondria-specific markers (Supplemental Fig. S1D).
Fig. 3.
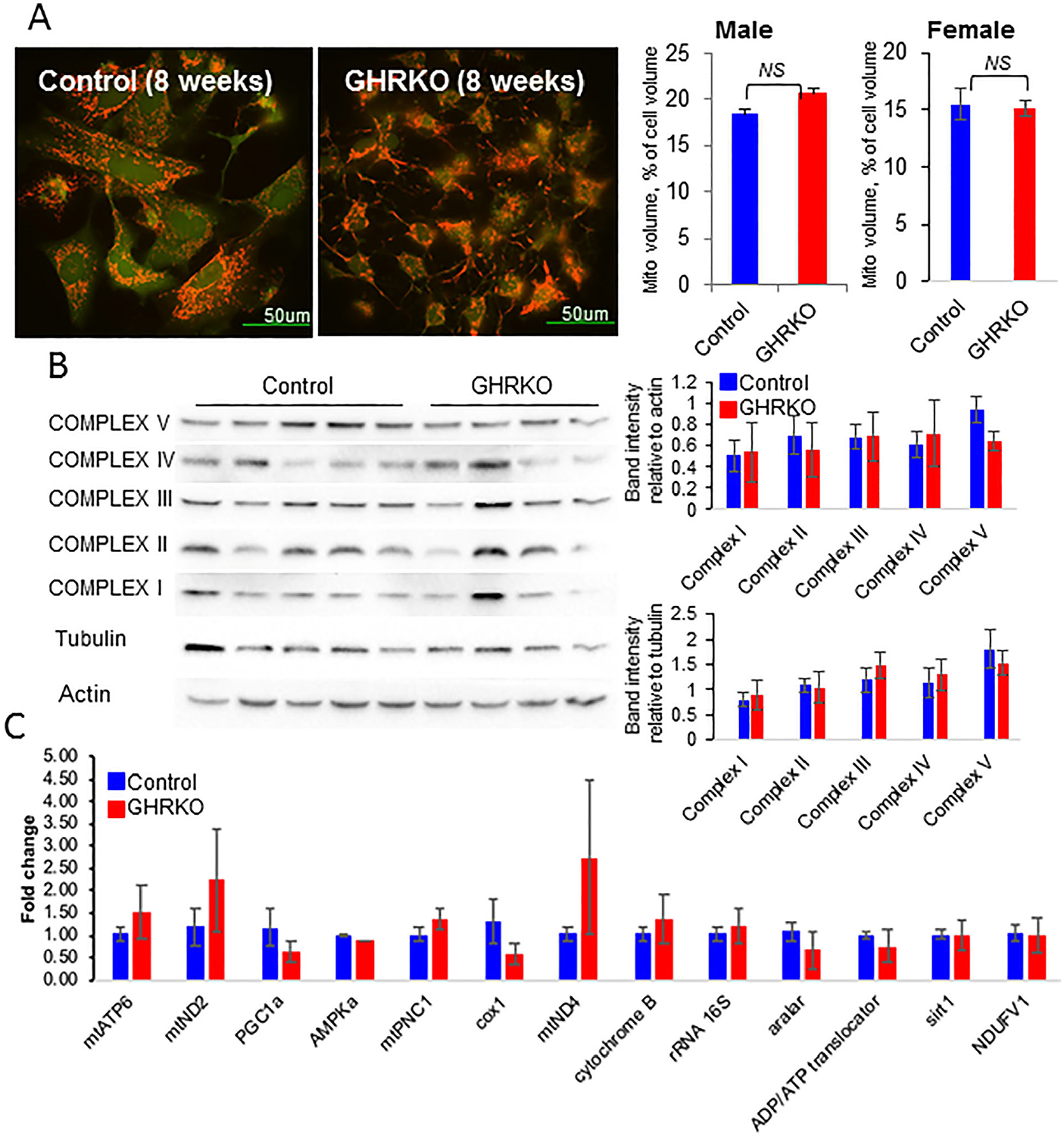
Mitochondria biogenesis in primary osteocyte cultures from control and GHRKO mice. (A) Primary osteocytes were seeded (0.4 × 106 cells/mL) on collagen-coated glass plates and visualized using the 2000E Nikon Microscope Eclipse TE microscope at 60× magnification. Osteocyte mitochondrial volume/cell volume was determined from 3-dimensional images of z-stack volumes reconstructed using the Amira FEI software. Live cells were labeled with Calcein AM, and mitochondria were labeled with tetramethylrhodamine, ethyl ester (TMRE) (n = 20 cells/genotype/age). (B) Protein levels of electron transport chain complexes (I–V) were determined from cell lysates of primary osteocytes by Western immunoblotting using the Total OXPHOS cocktail. (C) Mitochondria-specific gene expression in primary osteocytes was determined using real-time polymerase chain reaction. Shown is a representative experiment with primary osteocyte cultures from 4-month-old control (n = 7) and GHRKO (n = 5) male mice. Data are presented as mean ± SEM, and significance was accepted at p < 0.05.
Mitochondrial membrane potential (MMP) in GHRKO osteocytes
Central to mitochondrial function is the maintenance of proton transport across the inner mitochondrial membrane to generate an electrochemical potential (ΔΨm) that drives ATP synthesis by ATP-synthase.
Changes in ΔΨm reflect alterations in substrate or oxygen availability, activity of the ETC, and permeability of the mitochondrial membrane. To measure ΔΨm, cells were incubated with the fluorescent probe TMRE (60 nM) in αMEM for 30 minutes, washed once with αMEM, and subsequently visualized in αMEM containing 15 nM TMRE using confocal microscopy. ΔΨm was estimated from peak TMRE intensity (software NIS ElementsAR) of >10 mitochondria/cell in 25 to 30 cells/culture from several mice. We found a 10% decrease in TMRE fluorescence intensity in GHRKO osteocytes, compared with control cells, which was age- and sex-independent (Fig. 4A), indicating decreased mitochondrial membrane potential. In osteocytes from young (2 to 4 months old) bGH mice, on the other hand, we did not detect changes in MMP (Supplemental Fig. 1A). Data from GHRKO osteocytes suggest that GHR is implicated in the regulation of mitochondrial function in osteocytes.
Fig. 4.
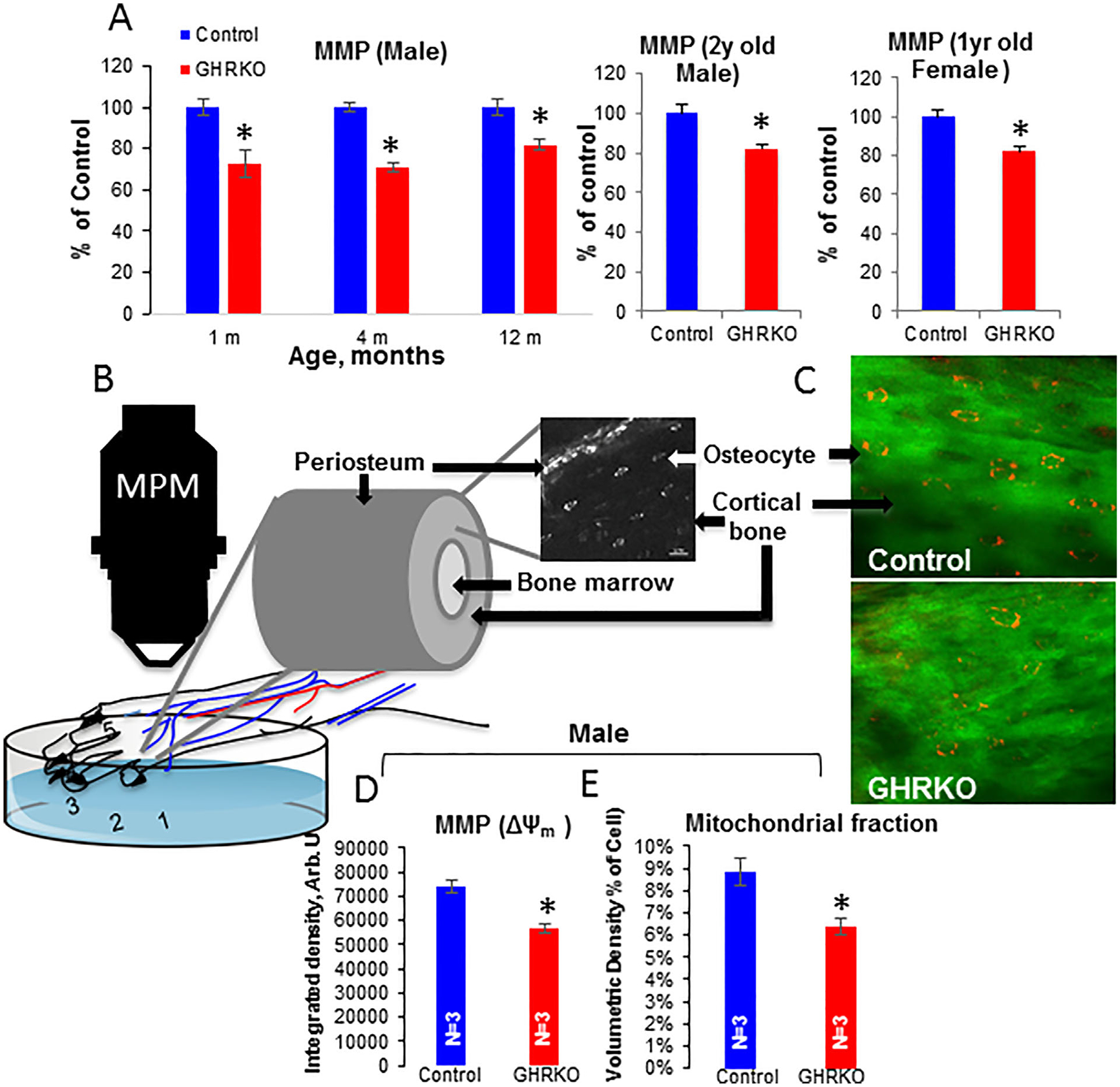
Mitochondrial membrane potential (MMP) in GHRKO osteocytes. (A) Primary osteocytes were seeded (0.4 × 106 cells/mL) on collagen-coated glass plates and visualized using the 2000E Nikon Microscope Eclipse TE microscope at 60× magnification. Osteocyte MMP was calculated from peak tetramethylrhodamine, ethyl ester (TMRE) intensity (n = 20 cells/genotype/age). Data are presented as mean ± SEM, and significance was accepted at p < 0.05. (B) Schematic representation of measurements of MMP in vivo in cortical bone osteocytes. (C) Multiphoton microscopy images from the metatarsals of control and GHRKO mice injected with TMRM. (D) ΔΨm was estimated from the integrated intensity density of TMRM-injected male mice for >20 osteocytes per mouse. (E) “Active mitochondrial fraction” was calculated from the volumetric density of TMRM-injected male mice for >20 osteocytes per mouse. Data are presented as mean ± SEM, and significance was accepted at p < 0.05.
Decreases in ΔΨm in GHRKO osteocytes were verified in vivo. Mice were injected intravenously with TMRM (2.5 μg/g body weight), and cortical osteocytes in the 2nd and 3rd metatarsals were visualized by multiphoton microscopy (Fig. 4B, C). ΔΨm was calculated for cortical osteocytes at 5 to 25 mm depth from z-stack images taken at several locations along the metatarsal cortex. ΔΨm was estimated from the integrated intensity density of TMRM for >20 osteocytes per mouse (Fig. 4D) and “active mitochondrial fraction” was calculated from volumetric density for >20 osteocytes per mouse (Fig. 4E). We found that intensity density of TMRM (reflecting ΔΨm) was decreased by approximately 15% in GHRKO mice in vivo, and volumetric density (reflecting mitochondrial fraction) was also reduced. In accordance with the in vitro data, cortical osteocytes in bGH mice did not reveal significant differences in ΔΨm or mitochondrial fraction in vivo (Supplemental Fig. S1B).
NADH redox state in GHRKO osteocytes
Decreases in ΔΨm in osteocytes from GHRKO mice could result from decreases in the availability of NADH, the first electron donor in the ETC, or from compromised ETC function. The reduced form of NAD (NADH) is produced in the tricarboxylic acid (TCA) cycle. Consequently, NADH donates a proton and an electron to the respiratory chain, producing the oxidized form of NAD (NAD+), which can be reused in the TCA cycle. Thus, the NAD+/NADH ratio is a key parameter reflecting TCA and respiratory chain function. Using fluorescent microscopy, we measured NADH autofluorescence in primary osteocytes. Application of the mitochondrial uncoupler FCCP to cells maximized cell respiration and depleted the mitochondrial NADH pool (min, 0% NADH). In contrast, application of NaCN, a complex IV inhibitor, suppressed mitochondrial respiration, allowing NADH to fully accumulate (max, 100% NADH) (Fig. 5A). The redox index was calculated as the ratio of basal to max NADH autofluorescence. We observed a decreased NADH redox index in osteocytes from young (2-month-old) or aged (2-year-old) male GHRKO mice, as compared with their respective control mice, indicating that NADH levels were lower in GHRKO mice (Fig. 5B, C). Similar data were obtained from osteocytes of young (3-month-old) and aged (1-year-old and 2-year-old) female GHRKO mice (Fig. 5D, E). In contrast, NADH redox in osteocytes from 4- and 12-month-old bGH mice increased as compared with controls (Supplemental Fig. S1E). These data suggest that ablation of GHR in osteocytes results in decreased TCA cycle activity or increased NADH consumption by the respiratory chain due to reduced chain efficiency.
Fig. 5.
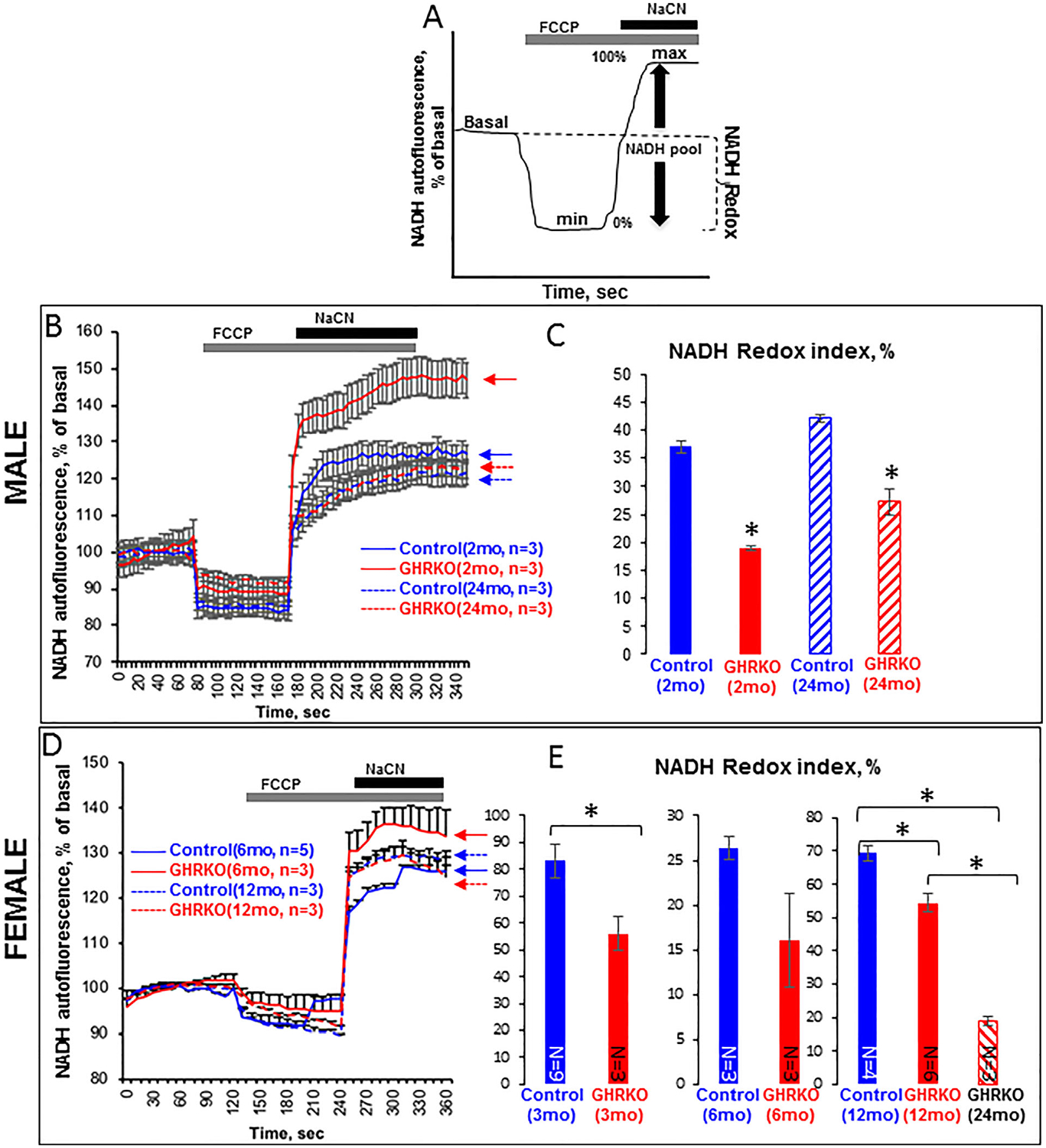
NADH redox state in primary osteocyte cultures from control and GHRKO mice. Cells were seeded (0.4 × 106 cells/mL) on collagen-coated glass plates and visualized using the 2000E Nikon Microscope Eclipse TE microscope at 20× magnification. (A) Schematic representation of redox index calculations: basal level relative to maximal respiration after carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 1 μM) (0%) and minimal respiration after NaCN (1 mM) (100%). (B) Time lapse of NADH autofluorescence in osteocytes from male mice, and (C) NADH redox index in osteocytes from male mice calculated according to (A). (D) Time lapse of NADH autofluorescence in osteocytes from female mice, and (E) NADH redox index in osteocytes from female mice calculated according to (A). Data are presented as mean ± SEM of 20 cells per time point.
Maximal respiration capacity in GHRKO osteocytes
To further investigate the mechanism underlying mitochondrial dysfunction, we measured maximal respiration capacity (Seahorse Analyzer, Agilent Technologies). Primary osteocytes were seeded on a collagen-coated plate (0.4 × 106 cells/mL) in triplicate. OCR was determined at basal state and upon the addition of oligomycin (1 μM), FCCP (2 μM), and rotenone/antimycin (0.5 μM). The FCCP concentration required for maximal respiration was established by titration (data not shown) using osteocytes from control mice. Data were normalized to cell number determined at the end of the assay. We found that osteocytes from young (2-month-old) (Fig. 6A, B) and aged (2-year-old) (Fig. 6C, D) male GHRKO mice had comparable basal and ATP-linked OCR but exhibited reduced maximal OCR (after addition FCCP), and loss of mitochondrial reserve capacity as compared with age-matched controls. In osteocytes from young (2-month-old) GHRKO females, OCR was normal (Fig. 6E, F). However, osteocytes from 1-year-old GHRKO mice showed significant reduction in maximum respiration capacity (Fig. 6G, H). The origin of sexual dimorphism in mitochondrial respiration between GHRKO male and female mice is not clear yet and requires further investigation. Interestingly, osteocytes from young (4-month-old) bGH mice showed increases in maximal OCR and significantly enhanced ATP production rate as compared with controls (Supplemental Fig. S1F).
Fig. 6.
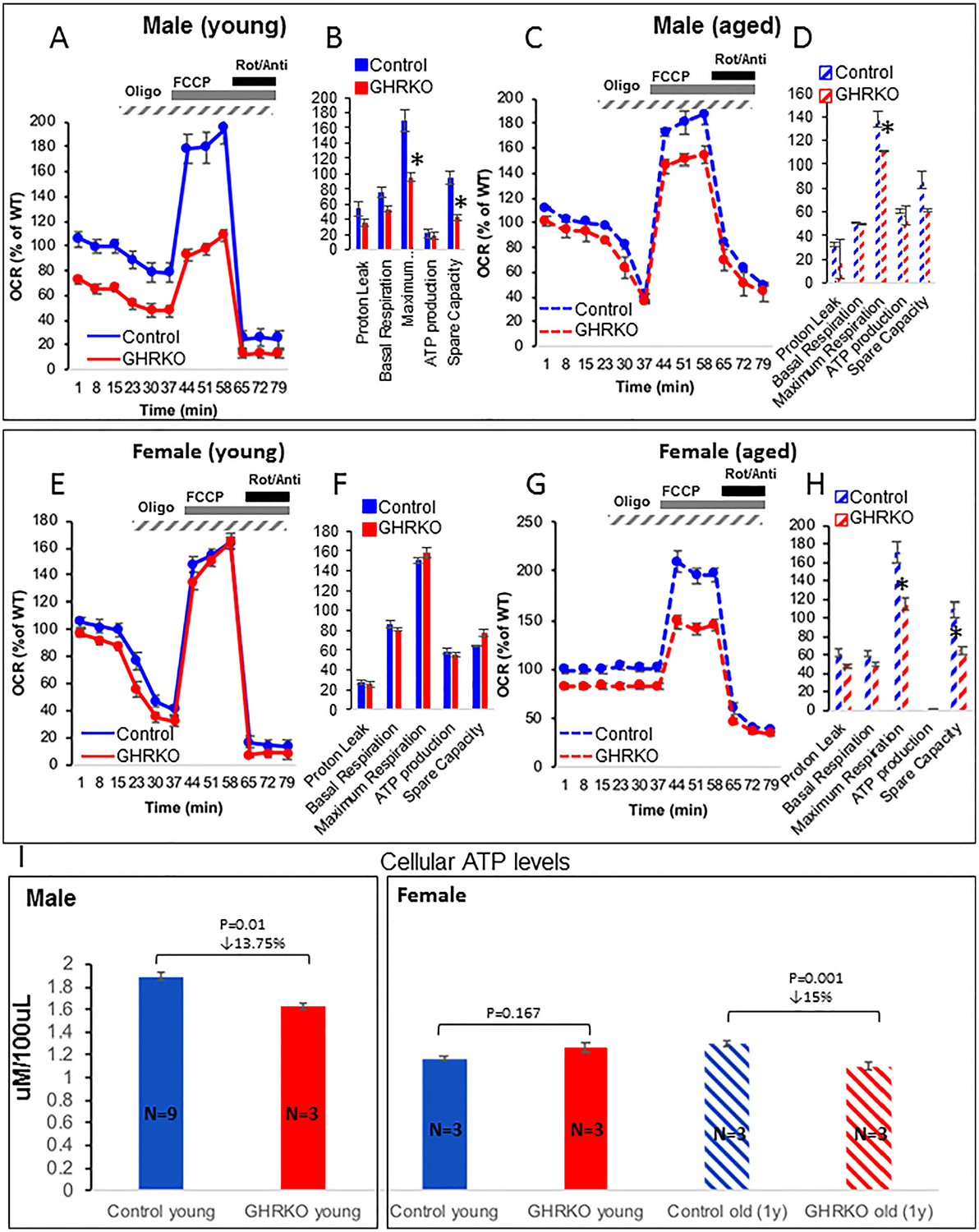
Mitochondrial respiration in primary osteocyte cultures from control and GHRKO mice. Primary osteocytes were seeded on collagen-coated plates (0.4 × 106 cells/mL) in triplicate. Oxygen consumption rate (OCR) was determined in the basal state and upon the addition of oligomycin (1 μM), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 2 μM), and rotenone/antimycin (Roti/Anti; 0.5 μM). Data were normalized to cell number (determined at the end of the assay). (A, B) Mitochondrial respiration in primary osteocytes from 2-month-old (young) and (C, D) 2-year-old (aged) male control and GHRKO mice. (E, F) Mitochondrial respiration in primary osteocytes from 2-month-old (young) and (G, H) 1-year-old (aged) female control and GHRKO mice. Data were calculated according to the manufacturer’s algorithm. (I) Steady-state ATP levels were measured in primary osteocyte cultures from male and female mice using Luminescent ATP Detection assay. Data are presented as mean ± SEM of triplicates in multiple assays. Significance was accepted at p < 0.05.
The dynamic measurement of OCR revealed no differences in the rate of ATP production between control and GHRKO osteocytes. Thus, we set to measure steady-state levels of ATP using luminescence-based assay (Fig. 6I). In males, GHRKO osteocytes showed significant but modest decrease in steady-state ATP levels. In females however, we found significant decreases in steady-state ATP only in osteocytes from aged GHRKO mice. In contrast, ATP levels in bGH osteocytes increased significantly (Supplemental Fig. S1G). Altogether, these data indicate that mitochondrial function is compromised in the absence of GHR. Of note, fibroblasts from GHRKO mice also exhibited reduced maximal OCR in the cellular respiration assay (Supplemental Fig. S2).
Osteocyte ROS production in response to metabolic insults in GHRKO cells
Based on our findings showing compromised mitochondrial function in osteocytes from GHRKO mice, we hypothesized that these cells would be more sensitive to ROS-inducing insults. ROS were detected using the cell membrane permeable dye 2′,7′-dichlorofluorescin diacetate (DCFDA), which is deacetylated by cellular esterases to a nonfluorescent compound and oxidized by ROS into the fluorescent 2′,7′-dichlorofluorescein (DCF). We tested the response of osteocytes from young adult (4-month-old) and aged (2-year-old) male control and GHRKO mice to increasing concentrations of glucose, as well as physiological concentrations of insulin (1 nM) and IGF-1 (10 nM). Control osteocytes exhibited increased ROS production with age under normal glucose levels (5 mM), but ROS did not increase significantly with age when glucose levels rose to 25 mM (Fig. 7A). These data suggest that basal ROS production in osteocytes is age-dependent only under normal glucose levels. Interestingly, osteocytes from young male GHRKO mice showed higher basal ROS production under normal glucose levels as compared with controls, but the difference between GHRKO and control mice was no longer significant when glucose levels rose to 25 mM (Fig. 7A). In osteocytes from young females, we did not find differences in cellular ROS between control and GHRKO under normal glucose levels. However, cellular ROS increased in both aged control and GHRKO female and was significantly higher in aged GHRKO osteocytes as compared with aged control osteocytes. These data suggest that in males, basal ROS in osteocytes is GHR-dependent under normal glucose levels, even in young mice. In females, however, basal ROS under normal glucose is GHR-dependent only in aged osteocytes. ROS levels in response to insulin and IGF-1 was elevated compared with untreated cells, but the increase was greater in GHRKO than in control osteocytes (Fig. 7B). These data suggest that ablation of GHR in osteocytes renders these cells more sensitive to metabolic insults in an age- and sex-dependent manner.
Fig. 7.
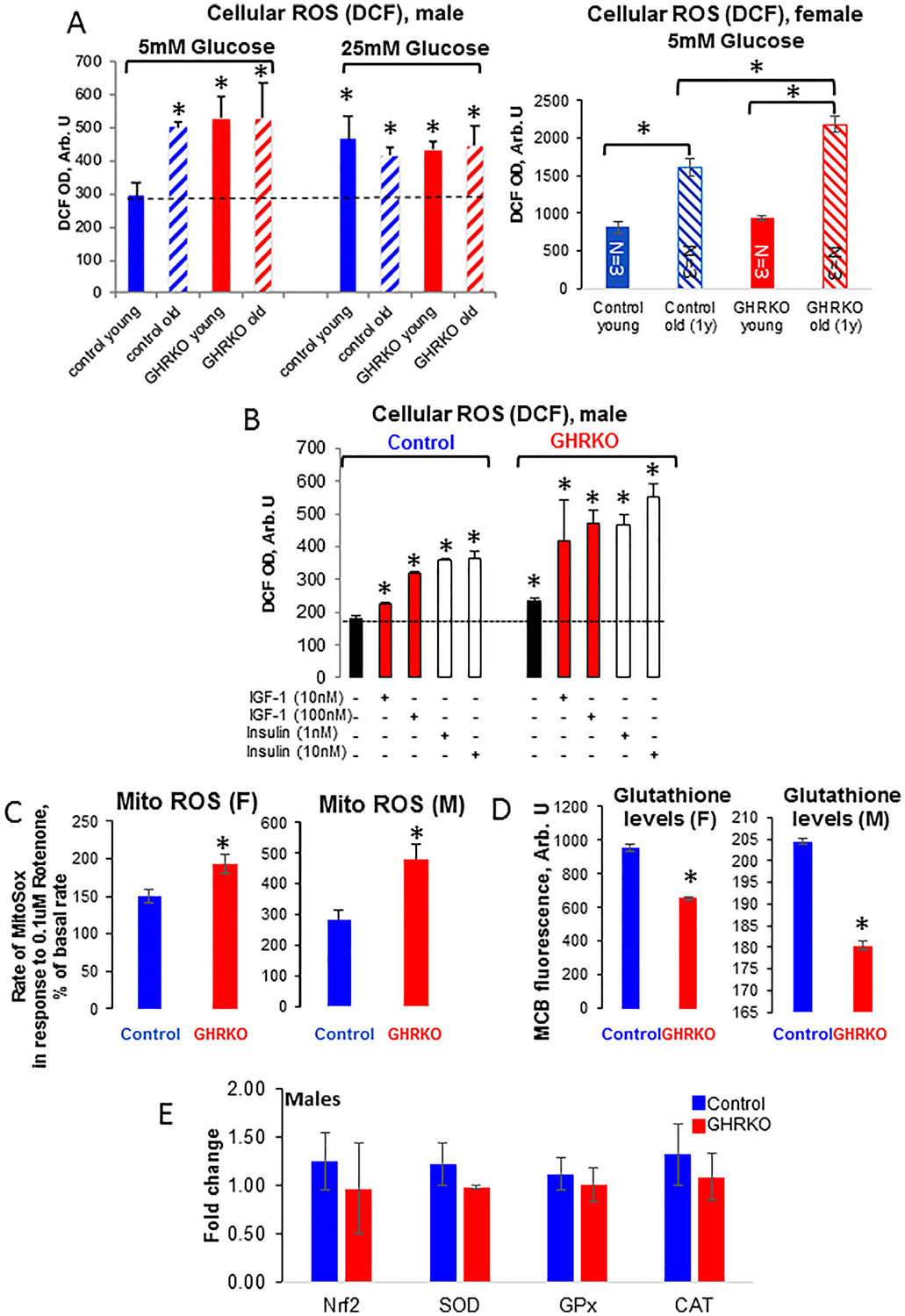
Reactive oxygen species (ROS) production in primary osteocytes from GHRKO and control mice. Cells were seeded (0.4 × 106 cells/mL) on collagen-coated black-walls 96-well plates and (A) incubated for 30 minutes with 2′,7′-dichlorofluorescin diacetate (DCFDA; 10 μg/mL). ROS was determined at excitation 495 nm and emission 525 nm (plate reader; Spectra Max5 M5, Molecular Devices with Softmax Pro software). Presented are ROS levels in the presence of 5 or 25 mM glucose in osteocytes from male and female mice, or (B) in the presence of insulin or insulin growth factor (IGF)-1 in osteocytes from male mice. (C) Cells were seeded as in (A) and incubated for 15 minutes with MitoSox (5 μM). Mitochondrial (mito) ROS levels in response to 0.1 μM rotenone were determined at excitation 510 nm and emission 580 nm. (D) Glutathione levels in osteocytes from young female and male mice in response to 0.1 μM rotenone were determined at excitation 394 nm and emission 490 nm. Data are presented as mean ± SEM of triplicates from three young (2-month-old) and three aged (2-year-old) control and GHRKO mice in each group. (E) Expression levels of oxidative stress defense enzymes (nuclear factor erythroid 2–related factor 2-Nrf2, superoxide dismutase-SOD, glutathione peroxidase-Gpx, catalase-CAT) in osteocytes from control and GHRKO mice. Significance (based on analysis of variance) as compared with young controls was accepted at p < 0.05. DCF = dichlorofluorescein; OD = optical density.
Next, we measured basal and rotenone-induced (0.1 μM) mitochondrial ROS using MitoSOX, a mitochondrial-specific ROS probe that measures superoxide. Basal MitoSOX fluorescence rate did not differ significantly between control and GHRKO osteocytes. However, the rotenone-stimulated mitochondrial ROS production rate increased significantly in osteocytes from both male and female GHRKO mice (Fig. 7C). To test whether a reduction in glutathione, which is a cellular defense mechanism to reduce ROS products, may explain the increased rate of rotenone-stimulated mitochondrial ROS production, we used an MCB probe. MCB is a nonfluorescent probe that emits fluorescent light when bound to reduced/oxidized glutathione. We found that MCB levels were significantly lower in osteocytes from GHRKO mice than in those from control mice (Fig. 7D), possibly contributing to the increased ROS in those cells. Lastly, despite increases in cellular and mitochondrial ROS in GHRKO cells, the expression levels of genes involved in oxidative stress defense did not differ between control and GHRKO osteocytes (Fig. 7E).
Cellular energy sensing in osteocytes from control and GHRKO mice
Cellular energy sensing plays pivotal roles in the function of many organs. Recent studies suggest that osteoblasts, the precursors of osteocytes, take up glucose via the glucose transporter-1 (GLUT-1) and utilize aerobic glycolysis for ATP production.(42) Our initial gene expression studies have shown that GLUT-1 and GLUT-4 could be detected by real-time PCR (though this does not exclude the possibility that other GLUTs may be expressed). We found that only GLUT-1 expression was significantly decreased (Fig. 8A) in osteocytes from male GHRKO mice as compared with controls. However, Western blot analyses revealed that both GLUT-1 and GLUT-4 proteins reduced in GHRKO osteocytes (Fig. 8B). Next, we tested how glucose deprivation affects the AMP-activated protein kinase (AMPK), which senses the overall cellular energy change and is a master regulator of cellular energy homeostasis. We found that glucose deprivation (4 to 6 hours) induced osteocyte apoptosis. Under this condition of glucose deprivation, the total protein levels of AMPKa (catalytic subunit) were not affected, but its phosphorylation significantly decreased in osteocytes from both control and GHRKO male mice (Fig. 8C). In contrast, protein levels of the AMPKb (regulatory subunit) reduced after glucose deprivation, whereas its phosphorylation levels were not affected in either control or GHRKO osteocytes. Reductions in AMPKa phosphorylation associated with reductions in the levels of the autophagy marker LC3II and significant increases in cleaved caspase-3 (CC3), an apoptotic marker (Fig. 8D) in both GHRKO and control osteocytes. Overall, these data suggest that osteocytes are very sensitive to glucose deprivation in vitro.
Fig. 8.
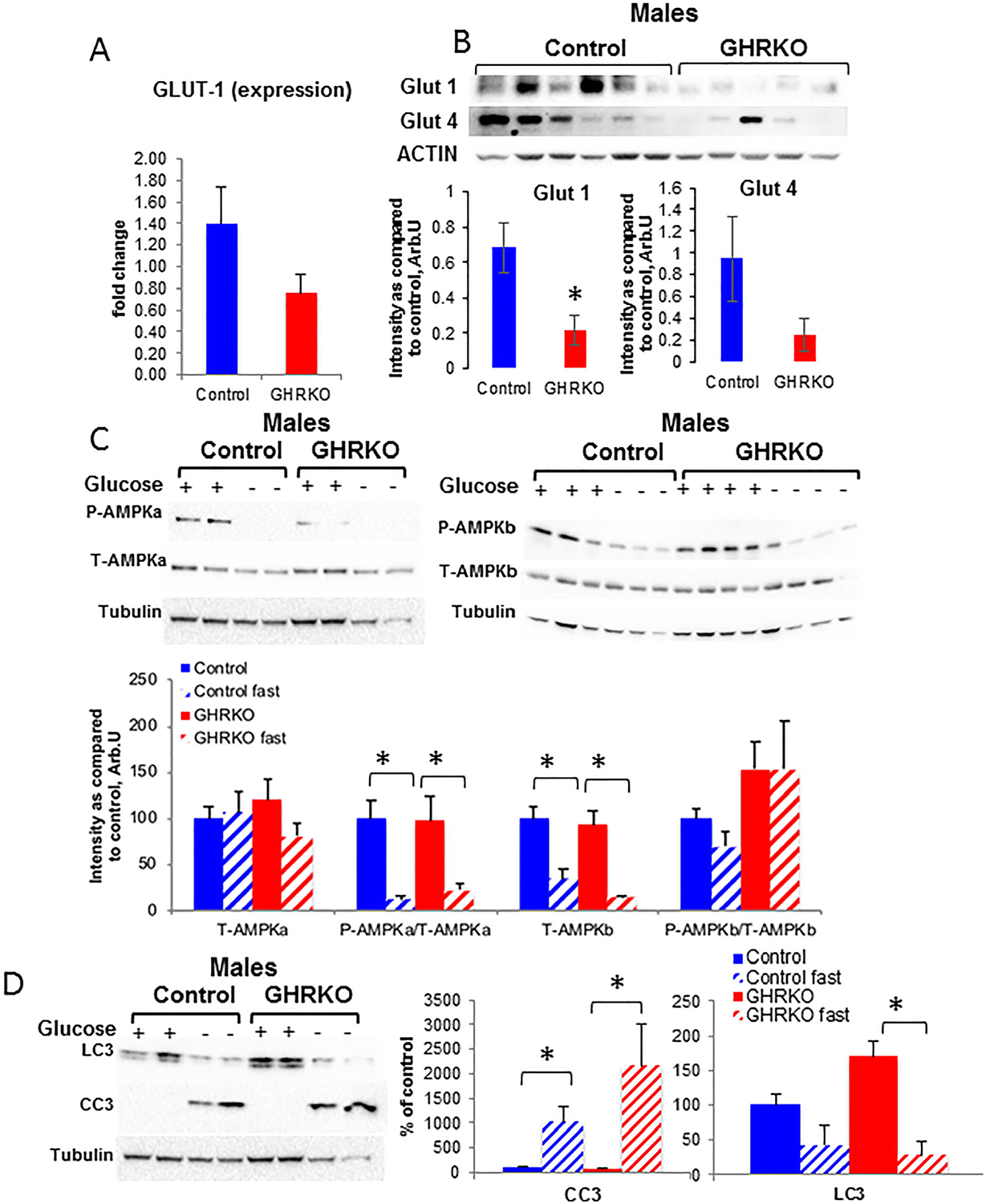
Osteocyte response to glucose deprivation. Primary osteocytes from male mice were seeded (0.4 × 106 cells/mL) on collagen-coated plates for 10 to 15 days. (A) Expression levels of GLUT-1 were determined by real-time PCR (control n = 10 samples from >20 mice; GHRKO n = 5 samples from 12 mice). (B) GLUT-1 and GLUT-4 protein levels were determined from cell lysates of primary osteocytes by Western immunoblotting (control n = 8 samples from >12 mice; GHRKO n = 6 samples from 10 mice). (C) Total and phosphorylated AMPKa and b were determined from cell lysates of primary osteocytes with or without (4 to 6 hours) glucose by Western immunoblotting (control n = 4 mice; GHRKO n = 4 mice). (D) LC3II and cleaved caspase 3 (CC3) were determined from cell lysates of primary osteocytes by Western immunoblotting (control n = 4 mice; GHRKO n = 4 mice). Data are presented as mean ± SEM of multiple assays. Significance was accepted at p < 0.05.
Next, we applied a glucose stress assay (Seahorse Analyzer, Agilent Technologies) to osteocytes from male (Fig. 9A–C) and female mice (Fig. 9D–F). In osteocytes from 6-month-old male mice, provision of glucose increased the mitochondrial supply of NADH through the TCA cycle, whereas increasing the extracellular acidification rate (ECAR) (Fig. 9A) and decreasing OCR (Fig. 9B), a phenomenon known as the “Crabtree effect.” Addition of oligomycin further increased ECAR (due to lactate accumulation) and significantly reduced OCR. Addition of a 2-deoxy-glucose (2DG) analogue blocked the changes in both ECAR and OCR. There was no difference between male control and GHRKO osteocytes, although glycolysis and glycolytic capacity tended to be reduced in GHRKO osteocytes (Fig. 9C). Similar data were obtained with osteocytes from 6-month-old females (Fig. 9D–F). However, differences were apparent in osteocytes from 1-year-old females, in whom glycolytic capacity, glycolytic reserve, and non-glycolytic acidification were reduced in GHRKO osteocytes (Fig. 9F). The glucose stress assay does not measure glucose uptake, but it indicates that glucose metabolism is impaired in aged female GHRKO mice, which requires further studies to unravel the molecular mechanism involved.
Fig. 9.
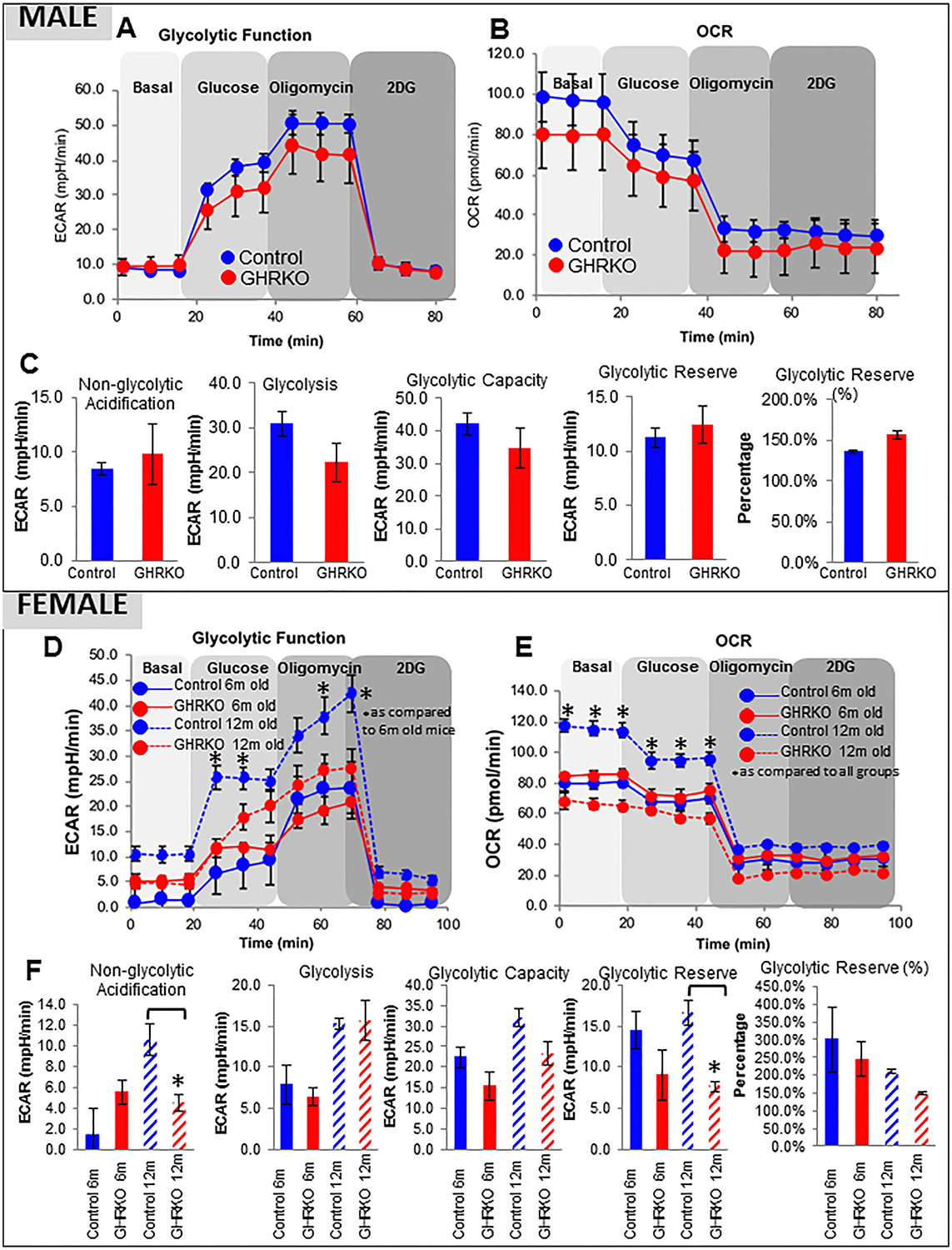
Glycolytic function in osteocytes from young and aged female and male mice. Primary osteocytes were seeded on collagen-coated plates(0.4 × 106 cells/mL) in triplicate. (A) Extracellular acidificance rate (ECAR) and (B) oxygen consumption rate (OCR) in osteocytes from male mice were measured at basal conditions and in response to glucose (10 mM), oligomycin (1 μM), and 2-deoxyglucose (2DG; 50 mM). (C) Non-glycolytic acidification, glycolysis, glycolytic capacity, and glycolytic reserve were calculated according to the manufacturer’s algorithm and presented as mean ± SEM of triplicates in multiple assays. Significance was accepted at p<0.05. (D) ECAR and (E) OCR in osteocytes from female mice. (F) Assay parameters calculated for female osteocytes, as in (C). Data are presented as mean ± SEM of triplicates in multiple assays, and significance was accepted at p < 0.05.
Discussion
4In this study, we show, for the first time, the effects of GHR ablation on mitochondrial function in cortical bone osteocytes in young and aged mice. In vivo studies showed that osteocytes of GHRKO mice exhibited significantly reduced mitochondrial volumetric density (indicative of reduced mitochondrial cellular fraction), as well as significantly reduced mitochondrial intensity density (indicative of reduced ΔΨm). In vitro studies of primary osteocytes, on the other hand, did not show reductions in mitochondrial volume in GHRKO cells. However, we found significantly reduced ΔΨm in primary osteocyte cultures from young and aged GHRKO mice, suggesting that the compromised mitochondrial phenotype persisted in vitro. These data are in conflict with previous reports showing enhanced mitochondrial function in liver, muscle, heart, kidney, and brain of aged GHRKO mice.(17) We posit that this contradiction stems from cell- and organ-specific factors. Specifically, the GHRKO mice show significant changes in body composition and carbohydrate/lipid metabolism, which lead to an overall stress-resistant phenotype. However, osteocytes, which are long-lived cells, buried in the bone matrix, and do not proliferate, show distinct cellular behavior. It is conceivable that mitochondrial stress accumulates throughout life in those cells, considering that the rate of mitochondrial biogenesis in osteocytes is low. Further, the extracellular matrix of the osteocytes is fundamentally different from all other cells in the body in terms of blood supply, nutrient supply and disposal, pH, and growth factors, which likely contribute to their decreased stress resistance.
NADH/NAD+ redox assays demonstrated an increase in basal NAD+ levels in GHRKO osteocytes, indicating impairments in mitochondrial respiration. This was associated with reduced steady-state levels of ATP and reduced ΔΨm. Mitochondria stress test using primary osteocytes (and fibroblasts, shown in Supplemental Fig. S2) confirmed impaired mitochondrial respiration in osteocytes from GHRKO mice. Our data are consistent with several early reports suggesting that reduced GH signaling decreases the basal OCR at the level of the whole organism.(43,44) However, these data contradict other reports showing increased basal oxygen consumption per gram body weight in GHRKO mice.(45,46) This controversy likely stems from in vivo experimental design. In vitro respiration in osteocytes was measured under controlled natural temperature (37°C), whereas in vivo respiration (of mice) was conducted under different temperature settings (not necessarily thermo-neutral conditions), which may have produced opposing results.
Longevity extension due to reduced metabolic rate is relevant to the “oxidative stress hypothesis of aging,” which proposes that increased aerobic respiration is accompanied by enhanced ROS production via enzymes of the ETC, leading to attrition of mitochondrial fitness. The reduced ΔΨm, compromised mitochondrial function, and reduced maximal respiration capacity in GHRKO osteocytes persuaded us to investigate ROS production in these cells. Numerous studies suggest positive correlation between ΔΨm and ROS production.(47,48) However, in cases of mitochondrial disorders associated with dysfunction of the ETC, lower ΔΨm with reduced respiration can be observed with simultaneous increase in ROS production.(49) We show that despite reduced maximal respiration capacity in response to stress (FCCP), we observed increased basal ROS production in osteocytes from young and aged GHRKO mice; these data could be interpreted in two ways: 1) increased ROS in GHRKO osteocytes leads to a secondary increase in stress defense mechanisms, which protect the cells against further damage, or 2) increased ROS increases the sensitivity of GHRKO osteocytes to stress and, thereby, severely compromises their ability to resist further stress. Additional data showing increased mitochondrial ROS and decreased glutathione levels in GHRKO osteocytes favors the latter. Our data are consistent with previous reports showing the protective effects of GH or IGF-1 on mitochondrial function, specifically with regard to ROS metabolism.(50–63)
The effects of GH on the regulation of energy balance, nutrient sensing, and nutrient partitioning are tissue-specific and not fully understood. The GHRKO mice exhibit reduced blood glucose levels and significant reductions in serum insulin levels(64) throughout their life span. This correlated with reduced GLUT-1 levels in osteocytes of GHRKO mice. Because the GHRKO mice show increased insulin sensitivity,(64) it is conceivable that glucose fluctuations significantly affect osteocyte glucose metabolism. Indeed, when stressed and cultured in high-glucose concentrations (25 mM), osteocytes from GHRKO mice exhibited significantly increased ROS production compared with controls, suggesting impaired regulation of ROS homeostasis under stress. Although the glycolytic capacity did not differ between control and GHRKO males, osteocytes from aged female GHRKO mice exhibited reductions in glycolytic parameters, indicating impairments in glucose metabolism, which may be sex dependent. Metabolic alterations in osteocytes lacking the GHR in response to fluctuations in glucose may have clinical implications. Age-induced somatopause, which refers to systemic reductions in GH/IGF-1 signals, correlates with impaired skeletal integrity(65) and with metabolic changes, such as hyperglycemia and hyperinsulinemia that affect stress resistance mechanisms (increases in ROS production) in osteocytes as bones age. It is conceivable that loss of GH signaling in the aging skeleton compromises mitochondrial function in osteocytes, leading eventually to changes in bone matrix composition and bone strength.
Finally, our study has limitations that are inherent in our mouse model. Long-lived GHRKO mice have life-long reductions in GH/IGF-1 signals and may activate adaptive mechanisms to overcome their “congenital somatopause,” specifically in skeletal cells.(66) These mice exhibit increased body adiposity(2) but also show simultaneous enhanced sensitivity to insulin.(67) Further studies are required to investigate whether age-induced reductions in GH/IGF-1 action will compromise the mitochondria of osteocytes. Finally, our metabolic studies were performed using primary cultures, which are enriched in osteocytes but are not pure. We note that these cultures consist of osteoblast lineage cells of various stages. Therefore, our interpretation of the in vitro data suggesting impaired mitochondrial function should be taken with caution until validated in vivo in future studies using more sophisticated assays. Nonetheless, our biochemical, microscopic, and functional data show that osteocytes lacking GHR exhibit compromised mitochondrial function that, at least partially, explains the impaired skeletal phenotype of GHRKO mice.
Supplementary Material
Acknowledgments
All authors concur with the submission. The material submitted for publication has not been previously reported and is not under consideration for publication elsewhere.
This study was supported by funding from the National Institutes of Health (grant #DK100246 to SY) and Bi-national Science Foundation (grant #2013282 to SY and HW).
Footnotes
Disclosures
ZL, MES, MBS, DFB, CJR, HW, JJK, EVP, AYA and SY have nothing to declare.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Brown-Borg HM, Bartke A. GH and IGF1: roles in energy metabolism of long-living GH mutant mice. J Gerontol A Biol Sci Med Sci. 2012;67(6):652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.List EO, Berryman DE, Funk K, et al. The role of GH in adipose tissue: lessons from adipose-specific GH receptor gene-disrupted mice. Mol Endocrinol. 2013;27(3):524–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yakar S, Isaksson O. Regulation of skeletal growth and mineral acquisition by the GH/IGF-1 axis: lessons from mouse models. Growth Horm IGF Res. 2016;28:26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langlois JA, Rosen CJ, Visser M, et al. Association between insulin-like growth factor I and bone mineral density in older women and men: the Framingham Heart Study. J Clin Endocrinol Metab. 1998;83(12):4257–62. [DOI] [PubMed] [Google Scholar]
- 5.Ghiron LJ, Thompson JL, Holloway L, et al. Effects of recombinant insulin-like growth factor-I and growth hormone on bone turnover in elderly women. J Bone Miner Res. 1995;10(12):1844–52. [DOI] [PubMed] [Google Scholar]
- 6.Liang H, Masoro EJ, Nelson JF, Strong R, McMahan CA, Richardson A. Genetic mouse models of extended lifespan. Exp Gerontol. 2003;38 (11–12):1353–64. [DOI] [PubMed] [Google Scholar]
- 7.Bonkowski MS, Pamenter RW, Rocha JS, Masternak MM, Panici JA, Bartke A. Long-lived growth hormone receptor knockout mice show a delay in age-related changes of body composition and bone characteristics. J Gerontol A Biol Sci Med Sci. 2006;61(6):562–7. [DOI] [PubMed] [Google Scholar]
- 8.Sjogren K, Bohlooly YM, Olsson B, et al. Disproportional skeletal growth and markedly decreased bone mineral content in growth hormone receptor −/− mice. Biochem Biophys Res Commun. 2000;267(2):603–8. [DOI] [PubMed] [Google Scholar]
- 9.Bartke A, Brown-Borg H. Life extension in the dwarf mouse. Curr Top Dev Biol. 2004;63:189–225. [DOI] [PubMed] [Google Scholar]
- 10.Partridge L, Gems D. Mechanisms of ageing: public or private? Nat Rev Genet. 2002;3(3):165–75. [DOI] [PubMed] [Google Scholar]
- 11.Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408(6809):255–62. [DOI] [PubMed] [Google Scholar]
- 12.Lithgow GJ. Invertebrate gerontology: the age mutations of Caenorhabditis elegans. Bioessays. 1996;18(10):809–15. [DOI] [PubMed] [Google Scholar]
- 13.Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13(11):1385–93. [PubMed] [Google Scholar]
- 14.Larsen PL. Aging and resistance to oxidative damage in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1993;90(19):8905–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lithgow GJ, White TM, Melov S, Johnson TE. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci U S A. 1995;92(16):7540–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanfleteren JR. Oxidative stress and ageing in Caenorhabditis elegans. Biochem J. 1993;292(Pt 2):605–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown-Borg HM, Rakoczy SG, Sharma S, Bartke A. Long-living growth hormone receptor knockout mice: potential mechanisms of altered stress resistance. Exp Gerontol. 2009;44(1–2):10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vescovo G, Ravara B, Gobbo V, Angelini A, Dalla Libera L. Skeletal muscle fibres synthesis in heart failure: role of PGC-1alpha, calcineurin and GH. Int J Cardiol. 2005;104(3):298–306. [DOI] [PubMed] [Google Scholar]
- 19.Stout MB, Swindell WR, Zhi X, et al. Transcriptome profiling reveals divergent expression shifts in brown and white adipose tissue from long-lived GHRKO mice. Oncotarget. 2015;6(29):26702–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rojanathammanee L, Rakoczy S, Brown-Borg HM. Growth hormone alters the glutathione S-transferase and mitochondrial thioredoxin systems in long-living Ames dwarf mice. J Gerontol A Biol Sci Med Sci. 2014;69(10):1199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown-Borg HM, Johnson WT, Rakoczy SG. Expression of oxidative phosphorylation components in mitochondria of long-living Ames dwarf mice. Age. 2012;34(1):43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartke A, Bonkowski M, Masternak M. How diet interacts with longevity genes. Hormones. 2008;7(1):17–23. [DOI] [PubMed] [Google Scholar]
- 23.Brown-Borg HM, Rakoczy SG. Glutathione metabolism in long-living Ames dwarf mice. Exp Gerontol. 2005;40(1–2):115–20. [DOI] [PubMed] [Google Scholar]
- 24.Wu Y, Sun H, Basta-Pljakic J, et al. Serum IGF-1 is insufficient to restore skeletal size in the total absence of the growth hormone receptor. J Bone Miner Res. 2013;28(7):1575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jilka RL, O’Brien CA. The role of osteocytes in age-related bone loss. Curr Osteoporos Rep. 2016;14(1):16–25. [DOI] [PubMed] [Google Scholar]
- 26.Hughes JM, Petit MA. Biological underpinnings of Frost’s mechanostat thresholds: the important role of osteocytes. J Musculoskelet Neuronal Interact. 2010;10(2):128–35. [PubMed] [Google Scholar]
- 27.Mosley JR. Osteoporosis and bone functional adaptation: mechanobiological regulation of bone architecture in growing and adult bone, a review. J Rehabil Res Dev. 2000;37(2):189–99. [PubMed] [Google Scholar]
- 28.Emerton KB, Hu B, Woo AA, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2010;46(3): 577–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin GM, Austad SN, Johnson TE. Genetic analysis of ageing: role of oxidative damage and environmental stresses. Nat Genet. 1996;13(1):25–34. [DOI] [PubMed] [Google Scholar]
- 30.Frikha-Benayed D, Basta-Pljakic J, Majeska RJ, Schaffler MB. Regional differences in oxidative metabolism and mitochondrial activity among cortical bone osteocytes. Bone. 2016;90:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Y, Wang C, Sun H, LeRoith D, Yakar S. High-efficient FLPo deleter mice in C57BL/6J background. PloS One. 2009;4(11):e8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 33.Stern AR, Stern MM, Van Dyke ME, Jahn K, Prideaux M, Bonewald LF. Isolation and culture of primary osteocytes from the long bones of skeletally mature and aged mice. BioTechniques. 2012;52(6):361–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weissig V, Edeas M. Mitochondrial medicine. Preface. Methods Mol Biol 2015;1264:v–xiv. [DOI] [PubMed] [Google Scholar]
- 35.Woo SM, Rosser J, Dusevich V, Kalajzic I, Bonewald LF. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J Bone Miner Res. 2011;26(11):2634–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Z, Kennedy OD, Cardoso L, et al. DMP-1-mediated Ghr gene recombination compromises skeletal development and impairs skeletal response to intermittent PTH. FASEB J. 2016;30(2):635–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gesing A, Masternak MM, Lewinski A, Karbownik-Lewinska M, Kopchick JJ, Bartke A. Decreased levels of proapoptotic factors and increased key regulators of mitochondrial biogenesis constitute new potential beneficial features of long-lived growth hormone receptor gene-disrupted mice. J Gerontol A Biol Sci Med Sci. 2013;68(6):639–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gesing A, Masternak MM, Wang F, et al. Expression of key regulators of mitochondrial biogenesis in growth hormone receptor knockout (GHRKO) mice is enhanced but is not further improved by other potential life-extending interventions. J Gerontol A Biol Sci Med Sci. 2011;66(10):1062–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gesing A, Bartke A, Wang F, Karbownik-Lewinska M, Masternak MM. Key regulators of mitochondrial biogenesis are increased in kidneys of growth hormone receptor knockout (GHRKO) mice. Cell Biochem Funct. 2011;29(6):459–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopez-Lluch G, Navas P. Calorie restriction as an intervention in ageing. J Physiol. 2016;594(8):2043–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Z, Han T, Fishman S, et al. Ablation of hepatic production of the acid-labile subunit in bovine-GH transgenic mice: effects on organ and skeletal growth. Endocrinology. 2017;158(8):2556–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei J, Shimazu J, Makinistoglu MP, et al. Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell. 2015;161(7):1576–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hauck SJ, Hunter WS, Danilovich N, Kopchick JJ, Bartke A. Reduced levels of thyroid hormones, insulin, and glucose, and lower body core temperature in the growth hormone receptor/binding protein knockout mouse. Exp Biol Med. 2001;226(6):552–8. [DOI] [PubMed] [Google Scholar]
- 44.Harman D. Free radical theory of aging. Mutat Res. 1992;275(3–6):257–66. [DOI] [PubMed] [Google Scholar]
- 45.Berryman DE, List EO, Kohn DT, Coschigano KT, Seeley RJ, Kopchick JJ. Effect of growth hormone on susceptibility to diet-induced obesity. Endocrinology. 2006;147(6):2801–8. [DOI] [PubMed] [Google Scholar]
- 46.Westbrook R, Bonkowski MS, Strader AD, Bartke A. Alterations in oxygen consumption, respiratory quotient, and heat production in long-lived GHRKO and Ames dwarf mice, and short-lived bGH transgenic mice. J Gerontol A Biol Sci Med Sci. 2009;64(4):443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552(Pt 2):335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416(1):15–8. [DOI] [PubMed] [Google Scholar]
- 49.Lebiedzinska M, Karkucinska-Wieckowska A, Giorgi C, et al. Oxidative stress-dependent p66Shc phosphorylation in skin fibroblasts of children with mitochondrial disorders. Biochim Biophys Acta. 2010;1797(6–7):952–60. [DOI] [PubMed] [Google Scholar]
- 50.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61(3): 448–60. [DOI] [PubMed] [Google Scholar]
- 51.Torella D, Rota M, Nurzynska D, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94(4):514–24. [DOI] [PubMed] [Google Scholar]
- 52.Gustafsson H, Tamm C, Forsby A. Signalling pathways for insulin-like growth factor type 1-mediated expression of uncoupling protein 3. J Neurochem. 2004;88(2):462–8. [DOI] [PubMed] [Google Scholar]
- 53.Davani EY, Brumme Z, Singhera GK, Cote HC, Harrigan PR, Dorscheid DR. Insulin-like growth factor-1 protects ischemic murine myocardium from ischemia/reperfusion associated injury. Crit Care. 2003;7(6):R176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukunaga K, Kawano T. Akt is a molecular target for signal transduction therapy in brain ischemic insult. J Pharmacol Sci. 2003;92(4):317–27. [DOI] [PubMed] [Google Scholar]
- 55.Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301(5634):839–42. [DOI] [PubMed] [Google Scholar]
- 56.Offen D, Shtaif B, Hadad D, Weizman A, Melamed E, Gil-Ad I. Protective effect of insulin-like-growth-factor-1 against dopamine-induced neurotoxicity in human and rodent neuronal cultures: possible implications for Parkinson’s disease. Neurosci Lett. 2001;316(3):129–32. [DOI] [PubMed] [Google Scholar]
- 57.Kajstura J, Fiordaliso F, Andreoli AM, et al. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50(6): 1414–24. [DOI] [PubMed] [Google Scholar]
- 58.Yamashita K, Kajstura J, Discher DJ, et al. Reperfusion-activated Akt kinase prevents apoptosis in transgenic mouse hearts over-expressing insulin-like growth factor-1. Circ Res. 2001;88(6): 609–14. [DOI] [PubMed] [Google Scholar]
- 59.Cittadini A, Grossman JD, Stromer H, Katz SE, Morgan JP, Douglas PS. Importance of an intact growth hormone/insulin-like growth factor 1 axis for normal post-infarction healing: studies in dwarf rats. Endocrinology. 2001;142(1):332–8. [DOI] [PubMed] [Google Scholar]
- 60.Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis. 1999;6(5):347–63. [DOI] [PubMed] [Google Scholar]
- 61.Heck S, Lezoualc’h F, Engert S, Behl C. Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor kappaB. J Biol Chem. 1999; 274(14):9828–35. [DOI] [PubMed] [Google Scholar]
- 62.Lian JD, al-Jumah M, Cwik V, Brooke MH. Neurotrophic factors decrease the release of creatine kinase and prostaglandin E2 from metabolically stressed muscle. Neuromuscul Disord. 1998;8(1): 7–13. [DOI] [PubMed] [Google Scholar]
- 63.Gluckman P, Klempt N, Guan J, et al. A role for IGF-1 in the rescue of CNS neurons following hypoxic-ischemic injury. Biochem Biophys Res Commun. 1992;182(2):593–9. [DOI] [PubMed] [Google Scholar]
- 64.Basu R, Qian Y, Kopchick JJ. Mechanisms in endocrinology: lessons from growth hormone receptor gene-disrupted mice: are there benefits of endocrine defects? Eur J Endocrinol. 2018;178(5):R155–81. [DOI] [PubMed] [Google Scholar]
- 65.Lombardi G, Tauchmanova L, Di Somma C, et al. Somatopause: dismetabolic and bone effects. J Endocrinol Invest. 2005;28(10 Suppl):36–42. [PubMed] [Google Scholar]
- 66.Bartke A, List EO, Kopchick JJ. The somatotropic axis and aging: benefits of endocrine defects. Growth Horm IGF Res. 2016;27:41–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arum O, Saleh J, Boparai R, et al. Interaction of growth hormone receptor/binding protein gene disruption and caloric restriction for insulin sensitivity and attenuated aging. F1000Res. 2014;3:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.