

TRPC1 channel facilitates virus infection.
Abstract
Mammalian transient receptor potential (TRP) channels are major components of Ca2+ signaling pathways and control a diversity of physiological functions. Here, we report a specific role for TRPC1 in the entry of herpes simplex virus type 1 (HSV-1) into cells. HSV-1–induced Ca2+ release and entry were dependent on Orai1, STIM1, and TRPC1. Inhibition of Ca2+ entry or knockdown of these proteins attenuated viral entry and infection. HSV-1 glycoprotein D interacted with the third ectodomain of TRPC1, and this interaction facilitated viral entry. Knockout of TRPC1 attenuated HSV-1–induced ocular abnormality and morbidity in vivo in TRPC1−/− mice. There was a strong correlation between HSV-1 infection and plasma membrane localization of TRPC1 in epithelial cells within oral lesions in buccal biopsies from HSV-1–infected patients. Together, our findings demonstrate a critical role for TRPC1 in HSV-1 infection and suggest the channel as a potential target for anti-HSV therapy.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a ubiquitous and contagious human virus that remains a tremendous worldwide health care burden and has the potential to cause substantial morbidity (1). Initial infection with HSV-1 starts with binding to and then entry into host cells. The entry of HSV-1 is essential for subsequent infection and occurs by a complicated mechanism that involves interactions between viral components and host cell surface receptors (2, 3). For example, viral glycoprotein D (gD) interacts with cell surface receptors, including HVEM (herpesvirus entry mediator), nectin-1 (4), and nectin-2 (5) in the early step of entry (6, 7).
HSV-1 entry into cells has been shown to induce the release of Ca2+ from endoplasmic reticulum and cause intracellular Ca2+ ([Ca2+]i) elevation, which contributes to HSV-1 infection (8). However, there is little information regarding the role of Ca2+ influx in HSV-1 infection. Notably, Ca2+ entry is important in infection by several types of virus, such as Sindbis virus, West Nile (9), human immunodeficiency virus, filovirus, and arenavirus (10, 11).
In this study, we aimed to investigate the involvement and relevance of host cell Ca2+ influx pathways during HSV-1 entry and infection using the HEp-2 cell line, mouse models, and buccal biopsies from healthy and HSV-1–infected patients. Our data suggest that during HSV-1 entry, Ca2+-entry mediated by Orai1/STIM1 (Ca2+ release–activated Ca2+ channel protein 1/stromal interaction molecule 1) causes translocation of TRPC1 (transient receptor potential canonical 1). We further demonstrated a specific role for TRPC1 in binding HSV-1 and facilitating its entry into cells. These findings reveal a previously unidentified mechanism that contributes to HSV-1 infection and pathology.
RESULTS AND DISCUSSION
Involvement of store-operated Ca2+ entry during HSV-1 infection
HSV-1 infection of HEp-2 cells elicited intracellular Ca2+ release, measured as an increase in [Ca2+]i in Ca2+-free external medium, as well as Ca2+ influx, seen as a [Ca2+]i increase when extracellular Ca2+ was provided (Fig. 1A). The Ca2+ influx component was blocked by the well-established store-operated Ca2+ entry (SOCE) inhibitor GSK-7975A and 2-aminoethoxydiphenyl borate (2-APB) (Fig. 1A) (12–16). GSK-7975A did not inhibit either thapsigargin (TG)– or HSV-1–induced Ca2+ release (fig. S1A). Furthermore, GSK-7975A did not affect 1-oleoyl-2-acetyl-sn-glycerol (OAG)–induced Ca2+ influx, which is not regulated by endoplasmic reticulum Ca2+ store depletion (fig. S1B) (17). The HSV-1–induced intracellular Ca2+ release was abolished by pretreating cells with TG (fig. S1C), suggesting that HSV-1 induced the release of Ca2+ from a TG-sensitive Ca2+ store. Together, these findings demonstrate that HSV-1 induces SOCE in HEp-2 cells. When HEp-2 cells were preinfected with HSV-1 for 15 min at 37°C, the TG treatment induced a threefold greater Ca2+ influx than in mock-infected control cells (Fig. 1B), suggesting that HSV-1 infection enhances Ca2+ influx in host cells.
Fig. 1. SOCE is activated during HSV-1 infection of HEp-2 cells.
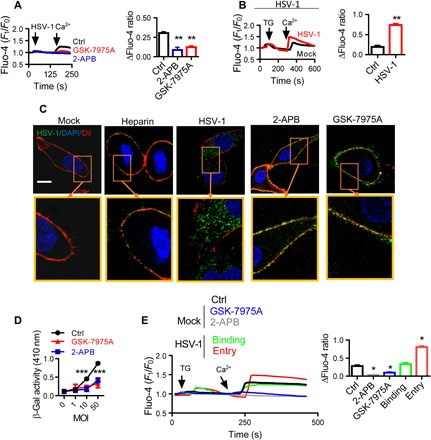
(A) HSV-1 infection elicits SOCE. Fluo-4–loaded HEp-2 cells were infected with 0.5 MOI of HSV-1 in the absence of extracellular Ca2+ (first arrow) and then treated with 50 μM 2-APB or 10 μM GSK-7975A before Ca2+ was added back (second arrow). Traces show relative fluorescence intensity. Bar graph to the right shows statistical analysis of peak fluorescence increase due to Ca2+ influx over the resting levels. For each condition, data were obtained from three replications, each of which included 10 cells, meaning a total of 30 cells per condition [same for (B), (C), and (E)]. (B) SOCE is increased in HSV-1–infected cells. HEp-2 cells were infected with HSV-1 at 0.5 MOI for 15 min and then treated with TG (left arrow). Ca2+ was then returned to the cells (right arrow). (C and D) SOCE modulates HSV-1 entry. HEp-2 cells were treated with 50 μM 2-APB or 10 μM GSK-7975A and then infected with HSV-1. (C) Representative immunofluorescence images showing the entry of HSV-1. DiI (red) indicates the PM; soluble heparin (10 μg/ml) was used to block binding as an additional negative control. Scale bar, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole. (D) Viral entry analyzed with β-Gal assays. (E) HSV-1 entry is associated with SOCE modulation. SOCE was analyzed during HSV-1 binding or entry into HEp-2 cells as described in (B). Alternatively, uninfected cells were treated with 2-APB or GSK-7975A as negative controls. *P < 0.05, **P < 0.01, and ***P < 0.001 by unpaired t test (B), one-way analysis of variance (ANOVA) (A and E), and two-way ANOVA (D). Graphs show the mean ± SD.
We assessed the contribution of Ca2+ entry to HSV-1 infection by determining the HSV-1 localization in control cells and those treated with the SOCE inhibitors 2-APB and GSK-7975A. Blocking SOCE significantly inhibited the cytoplasmic presence of HSV-1 antigen, as shown by the strong peripheral signal of HSV-1 in cells treated with 2-APB or GSK-7975A, indicating that viral entry into host cells was decreased. In contrast, control cells showed evident HSV-1 antigen in the cytosol in a discrete vesicular pattern, an indicator of viral entry (Fig. 1C). Note that 2-APB and GSK-7975A did not affect HSV-1 binding (fig. S1D). The viral entry was also quantitatively assessed by infecting cells with HSV-1 (KOS) gL86 (18–20). GL86 is a recombinant virus whose gL gene has been replaced by the lacZ gene encoding β-galactosidase (β-Gal). Production of β-Gal in host cells indicates HSV-1 entry into the host cell and transport to the nucleus where gene expression is elicited. The results showed that both 2-APB and GSK-7975A significantly suppressed the production of β-Gal (Fig. 1D). Together, these data show that SOCE plays a role in HSV-1 entry.
To determine whether SOCE is also involved in HSV-1 binding, cells were maintained at 4°C for 1 hour, conditions under which HSV-1 entry is inhibited. Alternatively, HSV-1 entry was assessed by incubating HEp-2 cells with the virus at 37°C for 15 min (see Materials and Methods). TG-stimulated SOCE was increased by HSV-1 entry into HEp-2 cells but not by binding (Fig. 1E). Consistent with this, a mutant HSV-1 that does not express gD (21) did not enter HEp-2 cells and failed to enhance SOCE (fig. S1E). Together, the data shown in Fig. 1 suggest that (i) SOCE is activated as a consequence of HSV-1–induced intracellular Ca2+ release, (ii) SOCE is enhanced by HSV-1 entry, and (iii) SOCE is required for HSV-1 entry.
Involvement of TRPC1 during HSV-1 infection
To further elucidate the mechanism of HSV-1–induced SOCE, major cellular components of SOCE, STIM1, Orai1, and TRPC1 (22, 23) were examined for their role in HSV-1 infection. We then found that HSV-1–induced SOCE, but not intracellular Ca2+ release, was abrogated by knockdown of STIM1, Orai1, and TRPC1 using small interfering RNAs (siRNAs) (siSTIM1, siOrai1, and siTRPC1; Fig. 2A and fig. S1F). Unlike siOrai1 and siSTIM1, siTRPC1 only caused a partial reduction of Ca2+ influx, consistent with previous findings (22, 23). On the other hand, knockdown of TRPC3, TRPC4, TRPC5, and TRPC6 did not significantly affect HSV-1–stimulated Ca2+ entry in HEp-2 cells (fig. S1G).
Fig. 2. Involvement of TRPC1 during HSV-1 infection.
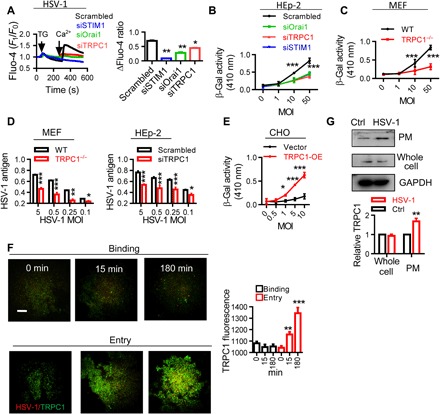
(A) STIM1, Orai1, and TRPC1 are involved in HSV-1–induced SOCE. HEp-2 cells treated with siOrai1, siTRPC1, or siSTIM1 were infected with HSV-1 at 0.5 MOI for 15 min, and SOCE was analyzed as described in Fig. 1B. (B) STIM1, Orai1, and TRPC1 are involved in HSV-1 entry. Entry of HSV-1 into HEp-2 cells treated with siOrai1, siTRPC1, or siSTIM1 was analyzed with β-Gal assays. n = 6 for each treatment, same in (C) to (E). (C) TRPC1 modulates HSV-1 entry. HSV-1 entry into TRPC1−/− MEFs assessed with β-Gal assays. (D) TRPC1 modulates HSV-1 replication. HSV-1 replication in TRPC1−/− MEFs (left) or siTRPC1-treated HEp-2 cells (right) was analyzed by ELISA. (E) TRPC1 enables HSV-1 entry into CHO cells. CHO cells were transfected with TRPC1 overexpression vector (TRPC1-OE), and HSV-1 entry was assessed with β-Gal assays. (F) HSV-1 entry triggers TRPC1 translocation. Increased TRPC1 (green) in the PM during HSV-1 binding or entry into HEp-2 cells was visualized by TIRF microscope. Scale bar, 10 μm. For each condition, data were obtained from three replications, each of which included 5 cells, meaning a total of 15 cells per condition. (G) HSV-1 entry triggers TRPC1 surface expression. Left, representative blots; right, quantitation of TRPC1 surface expression. n = 3 blots for each treatment. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA (A and F), two-way ANOVA (B to E), or unpaired t test (G). Graphs show the mean ± SD.
Unlike the results for SOCE, siOrai1, siSTIM1, and siTRPC1 equally inhibited virus entry (Fig. 2B), suggesting that the presence of TRPC1 protein is crucial for HSV-1 entry. To further investigate the crucial role of TRPC1 in HSV-1 infection, we assessed the HSV-1 infection in mouse embryonic fibroblasts (MEFs) derived from TRPC1−/− mice (24). HSV-1 entry was significantly decreased in these cells following HSV-1 infection (Fig. 2C), and furthermore, the cells displayed a reduced level of HSV-1 antigen after 18 hours of viral replication (Fig. 2D), similar to that seen in siTRPC1-treated HEp-2 cells (Fig. 2D). In contrast, overexpression of TRPC1 enhanced TG-induced Ca2+ entry in HSV-1–infected HEp-2 cells (fig. S2A) together with an increase in HSV-1 entry and replication (fig. S2B). Knockdown or overexpression of TRPC1 did not affect the amount of HSV-1 antigen in cells under conditions of binding (fig. S2C).
In addition, we followed the “gain of function” protocol demonstrated by Montgomery et al. (18). Here, Chinese hamster ovary (CHO) cells, which lack the known entry receptor for HSV-1 and are resistant to HSV-1 entry (18, 25), were transfected with TRPC1 overexpression vector and then inoculated with HSV-1 to determine whether the overexpressed TRPC1 could serve as a bona fide receptor for HSV-1 entry. Figure 2E shows that HSV-1 infection was significantly increased in TRPC1-expressing CHO cells, thus suggesting that TRPC1 is an entry receptor for HSV-1.
The localization of TRPC1 in the plasma membrane (PM) is a dynamic process and can affect the total Ca2+ influx following store depletion (15, 26). We examined TRPC1 localization in the PM by using total internal reflection fluorescence (TIRF) microscopy. First, TG treatment induced marked TRPC1 translocation in HEp-2 cells (fig. S3A), suggesting that SOCE is able to trigger TRPC1 translocation in HEp-2 cells. Then, the TRPC1 translocation was assessed in HSV-1–infected HEp-2 cells. Viral entry increased the TRPC1 signal within the TIRF microscopy plane (Fig. 2F), and this was not seen during viral binding. Consistent with this, gD-negative HSV-1 did not cause significant translocation of TRPC1 (fig. S3A). Viral entry also caused an increase in the surface expression of TRPC1 (Fig. 2G). Whole-cell expression of TRPC1, STIM1, and Orai1 was not affected under these conditions (Fig. 2G for TRPC1, and fig. S3B for STIM1 and Orai1).
The TRPC1 mutant TRPC1D581K, which carries a mutation at the ion permeation pore region of TRPC1 (27), was used to transfect HEp-2 cells, and SOCE, TRPC1 translocation, and viral entry were monitored. Consistent with previous studies and the notion that this pore-dead mutant is dominant negative to TRPC1-mediated SOCE (15, 27), the HSV-1–induced increase in SOCE as seen in Fig. 1B was diminished in TRPC1D581K-transfected HEp-2 cells (fig. S3C). However, the HSV-1–induced PM translocation of TRPC1D581K protein was comparable to that of wild-type (WT) TRPC1 (fig. S3D). The entry of HSV-1 remained unchanged and comparable to that of WT TRPC1 (fig. S3E). Therefore, the results suggest that translocation of TRPC1 to the PM is an important factor for HSV-1 entry. Notably, this experiment does not rule out the importance of TRPC1-mediated SOCE during HSV-1 entry. This SOCE elicits downstream pathways, i.e., nuclear factor κB pathways (28, 29), which are closely associated with viral replication processes after entry (30, 31).
Because Orai1-mediated Ca2+ entry has been shown to control the PM expression of TRPC1, we studied the effect of HSV-1 entry on Orai1. We found that Orai1 was diffusely localized in control cells but showed punctate signals under conditions of HSV-1 entry (fig. S3F). Furthermore, TRPC1 was colocalized with Orai1 in response to HSV-1 entry, and a close association between the two channels was shown by fluorescence resonance energy transfer (FRET) measurements (fig. S3F) (32, 33). In addition, we studied HSV-1–induced TRPC1 translocation using TIRF microscope in HEp-2 cells loaded with the intracellular Ca2+ chelator BAPTA-AM [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl ester)]. BAPTA-AM did not inhibit the TRPC1 TIRF signal after HSV-1 entry (fig. S3G). Furthermore, BAPTA, which chelates the extracellular Ca2+, decreased HSV-1–induced TRPC1 translocation (fig. S3G). These results indicates that Ca2+ entry from extracellular space is critical for the translocation of TRPC1, and the site(s) of the Ca2+ action must be very close to the passage of the Ca2+ influx. Otherwise, the calcium ions that entered the cell would have been captured by the intracellular BAPTA before they are able to exert the effect on TRPC1.
In Fig. 1, we have shown that HSV-1 entry enhances SOCE and, conversely, SOCE promotes HSV-1 entry in HEp-2 cells. Together with the data in Fig. 2, we further demonstrated that HSV-1 increased the surface localization of TRPC1, which likely accounts for the increase in SOCE.
Interaction between TRPC1 and HSV-1 gD
We next determined whether HSV-1 directly interacts with TRPC1. FRET measuring TRPC1 and HSV-1 gD interaction on the PM showed a significant enhancement of the FRET signal during HSV-1 entry (Fig. 3A). Other viral glycoproteins, such as gB, did not interact with TRPC1 (Fig. 3A). Consistently, after infection with HSV-1 for 15 min, host cell TRPC1 coimmunoprecipitated with HSV-1 gD (Fig. 3B) but not with gB and gH (fig. S4A). Nectin-1, a known gD receptor (4), was present in the immunoprecipitated fraction together with gD and TRPC1, with a time course similar to the gD-TRPC1 interaction (fig. S4A), indicating that gD associates with both TRPC1 and nectin-1. In addition, TRPC1 coimmunoprecipitated with gD in CHO cells that lack nectin-1 (Fig. 3B) (18, 34). These data suggest that the gD-TRPC1 interaction does not require nectin-1. Whether the presence of nectin-1 interferes with the efficacy of the gD-TRPC1 interaction, and whether there is competition or synergy between TRPC1 and nectin-1 in binding gD, deserves further evaluation.
Fig. 3. Interaction between TRPC1 and HSV-1 gD.
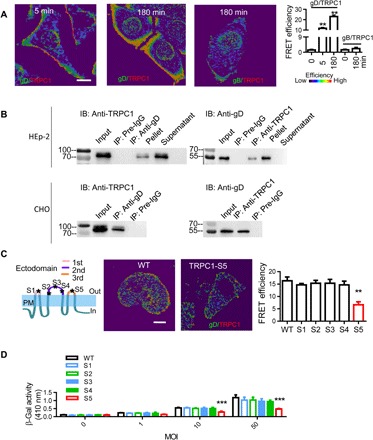
(A) Representative images and statistics of interaction between TRPC1 and gD. Colocalization of TRPC1 with gD or gB was analyzed by FRET at different time points of infection. For each condition, data were obtained from three replications, each of which included 5 cells, meaning a total of 15 cells per condition [same for (C)]. (B) TRPC1 coimmunoprecipitates with gD. HEp-2 cells (upper panels) or TRPV1-OE–transfected CHO cells (lower panels) were infected with HSV-1, and the interaction between TRPC1 and HSV-1 gD was analyzed by coimmunoprecipitation. Preimmune IgG served as a control. A converse coimmunoprecipitation was also performed for each treatment. Input was from the whole-cell extract as a positive control; supernatant and pellet were obtained after immunoprecipitation (n = 3 blots for each condition). (C) Effects of TRPC1 mutants on the gD-TRPC1 interaction. Five mutations were introduced on the three ectodomains of TRPC1 as shown in the cartoon. Middle and right panels show representative images and statistics for gD-TRPC1 interactions measured by FRET in HEp-2 cells transfected with vectors overexpressing mutant TRPC1 (S1 to S5). (D) Effect of TRPC1 mutants on HSV-1 entry. HEp-2 cells were transfected with mutant TRPC1 (S1 to S5) vectors, and viral entry was analyzed with β-Gal assays. Scale bars, 10 μm. **P < 0.01 and ***P < 0.001 by one-way ANOVA (A and C) or two-way ANOVA (D). Graphs show the mean ± SD.
Five sites (S1 to S5) were mutated in the three ectodomains of TRPC1 (see cartoons in Fig. 3C and fig. S4B). These mutants were expressed individually in HEp-2 cells along with HSV-1 infection. Similar TG-stimulated Ca2+ entry was seen in cells expressing the different TRPC1 mutants (fig. S4C). FRET was then used to measure interaction between viral gD and different mutant TRPC1s in HEp-2 cells. Those expressing TRPC1 with a mutation in S5 (third ectodomain) displayed a lower gD-TRPC1 interaction than cells expressing the other TRPC1 mutants (Fig. 3C). HEp-2 cells expressing S5-mutated TRPC1 also showed less β-Gal enzyme activity (Fig. 3D). Furthermore, when CHO cells were transfected with the S1-S5 TRPC1 vectors and then infected with HSV-1 gL86, overexpression of S1-S4 TRPC1 mutants enabled viral entry. However, for the S5 mutant, the HSV-1 entry was comparable to that of control vector–transfected cells (fig. S4D). Therefore, we concluded that the TRPC1 S5 site is important for both the gD-TRPC1 interaction and HSV-1 entry. Together, the findings suggest that HSV-1–induced SOCE leads to enhancement of TRPC1 in the PM and direct interaction between TRPC1 and viral gD at the cell membrane promotes HSV-1 entry. Notably, TRPC1 is present at a low level in the PM before HSV-1 infection (35), so we hypothesized that the initial entry of HSV-1 into cells is mediated by these TRPC1 proteins. SOCE, activated subsequent to HSV-1 entry and intracellular Ca2+ release, further enhances the level of TRPC1 in the PM, which can then bind to extracellular HSV-1 and promote entry of the virus.
Interaction between TRPC1 and HSV-1 gD in mice and human samples
To further examine the role of the TRPC1–HSV-1 interaction under physiological conditions of HSV-1 infection, we established an ocular infection model in WT and TRPC1−/− mice using two HSV-1 strains, 17 syn+ and McKrae. Note that the HSV-1 strain KOS used in in vitro experiments display low virulence in mouse models. In WT mice, both 17 syn+ and McKrae HSV-1 strains induced marked edema in the eyeballs with hypercellularity of the sclera and detachment of the retina. In contrast, TRPC1−/− mice displayed mild ocular abnormality, with relatively little retinal detachment (Fig. 4A). Quantitatively, TRPC1−/− mice had significantly lower herpes stromal keratitis (HSK) scores, which were measured on day 5 after infection (Fig. 4B). HSK development (i.e., the time taken to induce 50% corneal opacity) was significantly delayed in TRPC1−/− mice (Fig. 4C). TRPC1−/− mice had significantly higher survival rates after HSV-1 infection than infected WT mice (Fig. 4D).
Fig. 4. TRPC1 modulates HSV-1 infection in mice and patients with oral herpes lesion.
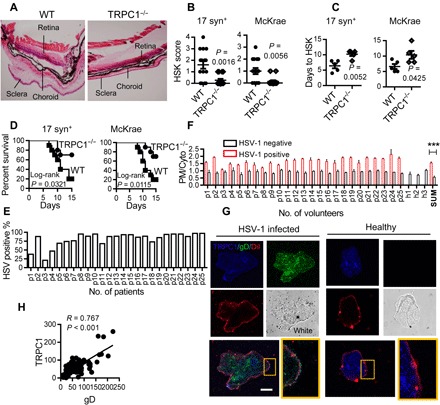
(A) Effect of TRPC1 on in vivo HSV-1 infection. The mice were infected with 104 plaque-forming units (pfu) per mouse of 17 syn+ or 103 pfu per mouse of McKrae HSV-1 [same for (B) to (D)], and the eyeballs were stained with hematoxylin and eosin after 5 days of infection. Figures show representative eyeball morphology caused by McKrae infection; magnification, ×40; n = 15 for each infected group. (B) HSK scores in WT and TRPC1−/− groups on day 5 of infection. n = 15 for each infected group. (C) Days to HSK causing 50% corneal opacity. n = 5 for each infected group. (D) Survival curves of infected mice. Kaplan-Meier curves for TRPC1−/− and WT mice infected with HSV-1 for 15 days (n = 10 for each infected group). (E) Percentage of HSV-1 gD–positive cells in buccal biopsies from each patient. The percentages of HSV-1 gD–positive epithelial cells were calculated for 100 randomly chosen oral epithelial cells in biopsies from three slides for each patient (p1 to p25). (F) Features of TRPC1 fluorescence distribution in human samples. The TRPC1 fluorescence on the PM versus that in the cytoplasm (PM/Cyto; in pixel) in HSV-1–infected or uninfected epithelial cells from buccal biopsies of 25 patients (p1 to p25) and 3 healthy candidates (h1 to h3). SUM, overall PM/Cyto value difference between patients and healthy candidates (t test). (G) Representative images of healthy and HSV-1–infected oral epithelial cells. Scale bar, 10 μm; n = 25 patients, n = 3 healthy individuals; 10 cells from each individual were imaged; DiI (red) indicates PM. (H) Fluorescence intensity of TRPC1 calculated for Pearson correlation with gD on the PM (n = 100 HSV-1–infected cells). ***P < 0.001 by unpaired t test (B, C, and F). Graphs show the mean ± SD.
In humans, the common symptoms of HSV-1 infection are pharyngitis, tonsillitis, and gingivostomatitis (36). Buccal biopsies from patients diagnosed with herpes oral lesions and healthy individuals were used to examine the expression and location of TRPC1 and HSV-1 gD in epithelial cells. In samples from 25 patients, 80.8 ± 3.95% of the oral epithelial cells were infected by HSV-1 (presence of fluorescent HSV-1 gD antigen signals) (Fig. 4E). In healthy uninfected epithelial cells, the fluorescent signal of TRPC1 was dispersed in the cytoplasm. The PM versus cytoplasm (PM/Cyto) value of TRPC1 fluorescence in these uninfected cells was significantly lower in cells positive for HSV-1 gD antigen (Fig. 4, F and G). A few cells from patients with herpes-induced oral lesions were not positive for HSV-1 antigen; they might have been from healthy tissue near the lesion site. These gD-negative cells did not show PM expression of TRPC1 and thus had a lower TRPC1 PM/Cyto ratio (Fig. 4F). There was strong correlation between the fluorescence intensity of TRPC1 and that of gD on the PM in HSV-1–infected cells (Pearson correlation coefficient = 0.767, P < 0.05; Fig. 4H).
Anti–HSV-1 strategies based on gD-TRPC1 interaction
A synthetic peptide, C1E3p [containing the third ectodomain (amino acids 608 to 616) of TRPC1], was tested for its effects on gD-TRPC1 interaction. Note that the mouse and human TRPC1 peptide sequences share great similarity (37). Application of C1E3p to HEp-2 cells reduced the colocalization of TRPC1 and gD compared to untreated cells (Fig. 5A). Significantly, C1E3p reduced HSV-1 replication in HEp-2 cells (Fig. 5B). Symptoms of abnormality in the sclera, choroid, and retina, as well as the occurrence of retinal detachment in mice infected with HSV-1 were decreased (Fig. 5C). The HSK scores for C1E3p-treated mice were lower than for control mice (Fig. 5D), and infection was delayed (Fig. 5E). In addition, C1E3p-treated mice had better survival rates 15 days after HSV infection than untreated mice (Fig. 5F). Together, our data suggest that C1E3p effectively disrupts the gD-TRPC1 interaction and decreases the severity of HSV-1 infection. Notably, C1E3p was preincubated with HSV-1 during these experiments, and further studies are needed to improve the binding efficiency of the competitive peptide to gD by enhancing affinity and stability or devising a new delivery method.
Fig. 5. Targeting gD-TRPC1 interaction and TRPC1 translocation to inhibit HSV-1 infection.
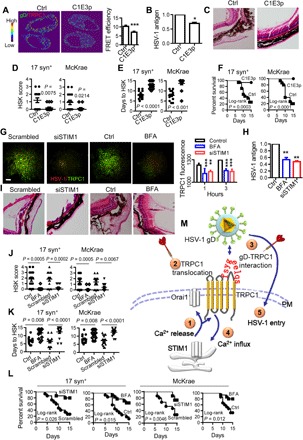
(A) Representative images (left) and statistics (right) of the effect of C1E3p on the gD-TRPC1 interaction. HSV-1 at 0.5 MOI was incubated with 10 μM C1E3p or PBS and then used to infect HEp-2 cells for 15 min. Colocalization of TRPC1 and gD was analyzed by FRET. For each condition, data were obtained from three replications, each of which included 5 cells, meaning a total of 15 cells per condition [same for (G)]. (B) Effect of C1E3p on HSV-1 replication as analyzed by ELISA. HEp-2 cells were infected with HSV-1 as in (A) (n = 6 for each treatment). (C) Effect of C1E3p on HSV-1 infection–induced eye abnormality. 17 syn+ (104 pfu per mouse) or McKrae HSV-1 (103 pfu per mouse) was preincubated with 10 μM C1E3p and then used to infect mice. The C1E3p treatment was repeated every 2 days. Figures show representative eye ball morphology caused by McKrae infection on day 5; magnification, ×40; n = 15 for each infected group. (D to F) Quantitation of effect of C1E3p on in vivo HSV-1 infection. The mice were infected with C1E3p-treated HSV-1 as in (C). HSK scores on day 5 of HSV-1 infection (D), days to HSK causing 50% corneal opacity (E), and survival rates (F) were analyzed. (G) Effect of BFA or STIM1 knockdown on TRPC1 translocation. Representative TIRF microscopy images (left) and statistics (right) for HEp-2 cells incubated with 5 μM BFA or 10 μM siSTIM1 and then infected with HSV-1 at 0.5 MOI for 15 min. Scale bar, 10 μm. (H) Effect of BFA or STIM1 knockdown on HSV-1 replication. HEp-2 cells were treated as in (G), and HSV-1 replication was analyzed by ELISA. PBS or scrambled siRNA served as control, and their values were normalized to 1 (n = 6 for each treatment). (I) Effect of BFA or STIM1 knockdown on HSV-1 infection–induced eye abnormality. The mice were treated with 5 μM BFA or 10 μM siSTIM1 and infected with 104 pfu per mouse of 17 syn+ or 103 pfu per mouse of McKrae HSV-1. Figures show representative eye ball morphology caused by McKrae infection on day 5; magnification, ×40; n = 15 for each infected group. (J to L) Quantitation of the effect of BFA or STIM1 knockdown on in vivo HSV-1 infection. Mice were treated with BFA or siSTIM1 and infected with HSV-1 as described in (I). HSK scores on day 5 of HSV-1 infection (J), days to HSV-1–induced HSK reaching 50% corneal opacity (K), and survival rates for 15 days (L) were calculated (n = 15 for each treatment). (M) Model of the mechanism of TRPC1-mediated HSV-1 infection. ① and ② At an early stage of HSV-1 infection, HSV-1 entry using resting levels of TRPC1 induces intracellular Ca2+ release, which results in activation of STIM1 and Orai1 channels, and consequently TRPC1 translocation to the PM. ③ and ④ TRPC1 and Orai1 also mediate the SOCE; PM TRPC1 serves as receptor for HSV-1 gD. ⑤ TRPC1 facilitates HSV-1 entry into the host cell. Thus, TRPC1 translocation ② and gD-TRPC1 interaction ④ are possible targets (darts) for suppressing HSV-1 infection. *P < 0.05, **P < 0.01, and ***P < 0.001 by unpaired t test (A, B, D, E, J, and K) or one-way ANOVA (G and H). Graphs show the mean ± SD.
In addition, we targeted TRPC1 translocation to modulate the gD-TRPC1 interaction. STIM1 is known to control TRPC1 translocation (38), and we found that knockdown of STIM1 in HEp-2 cells suppressed the HSV-1–induced translocation of TRPC1 (Fig. 5G) and attenuated HSV-1 replication (Fig. 5H). Similarly, blocking intracellular vesicle trafficking with brefeldin A (BFA) (39) suppressed the HSV-1–induced PM recruitment of TRPC1 (Fig. 5G) and decreased HSV-1 replication (Fig. 5H). Key in vivo experiments testing the effects of siSTIM1 and BFA on the ocular abnormality induced by HSV-1 demonstrated that both treatments significantly decreased the ocular symptoms and HSK scores (Fig. 5, I and J), delayed the appearance of HSK (Fig. 5K), and increased survival (Fig. 5L). These data suggest that disrupting TRPC1 translocation decreases HSV-1 infection.
DISCUSSION AND CONCLUSION
TRP channels have been implicated in various pathophysiological processes (40, 41). Here, the data reveal a critical and unidentified role for the Ca2+-permeable cation channel TRPC1 in HSV-1 entry. We propose a model (Fig. 5M) whereby HSV-1 triggers intracellular Ca2+ release that mobilizes STIM1 and Orai1, and this leads to activation of SOCE. SOCE causes recruitment of TRPC1 to the PM, enhancing Ca2+ entry into the cells. The third ectodomain of TRPC1 interacts with HSV-1 gD at the PM. The gD-TRPC1 interaction promotes HSV-1 entry, which we demonstrated in tissue samples from HSV-1–infected patients and mouse models. Last, we showed that the TRPC1-gD interaction and PM recruitment of TRPC1 serve as targets to inhibit HSV-1 entry. Together, these novel findings reveal a strong link between TRPC1 and HSV-1 infection and that TRPC1 is a potential target for anti-HSV therapy.
MATERIALS AND METHODS
Cells, viruses, and reagents
Vero cells expressing gD (VD60 cells) and Vero cells expressing gL (79BV4) were gifts from R. M. Longnecker, Lurie Comprehensive Cancer Center, Northwestern University, Evanston, IL, USA. HEp-2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum at 37°C in a humidified 5% CO2 environment. 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (DiI) cell plasma red dye was from Beyotime (Shanghai, PR China). HSV-1 (KOS) was from the American Type Culture Collection. HSV-1 strains 17 syn+ and McKrae were gifts from J. Zhou, Kunming Institute of Zoology, China. The HSV-1 gD mutant KOS strain and HSV-1 (KOS) GL86 mutant were gifts from R. M. Longnecker. The KOS, 17 syn+, and McKrae strains were propagated in HEp-2 cells. Briefly, parental virus was added to a monolayer of host HEp-2 cells and allowed to propagate until a complete cytopathic effect was observed. Then, the cells and virus were harvested by centrifugation at 2000 rpm for 20 min at 4°C. The pellets combining cells and virus were then sonicated on ice for 20 s to obtain HSV-1 stock, which was titered using HEp-2 cells. The HSV-1 (KOS) gD mutant strain and HSV-1 (KOS) GL86 mutant were propagated and titered using their respective host cells, VD60 and 79BV4. Human TRPC1 complementary DNA (cDNA) (NM_001251845) was cloned into the pcDNA6 vector for overexpression (TRPC1-OE). The plasmid overexpressing gD was a gift from T. Peng, Guangzhou Institutes of Biomedicine and Health, China. STIM1, Orai1, and TRPC1 siRNA was from Santa Cruz Biotechnology (USA).
HSV-1 binding and entry
Assays of HSV-1 binding and entry were carried out as previously reported (42). HSV-1 binding experiments were performed at 4°C for 1 hour. In viral entry assays, the cells were infected with recombinant HSV-1(KOS) gL86 expressing β-Gal at 37°C for 3 hours. Cells were then treated with sodium citrate (pH 3) to inactivate the virus associated with the PM (43). The entry efficiency was assessed with o-nitrophenyl β-d-glucopyranoside [3 mg/ml, in phosphate-buffered saline (PBS) containing 0.5% NP-40] after 7 hours of infection (18). Enzymatic activity was measured as optical density at 410 nm using a spectrophotometer. To measure HSV-1 entry into CHO cells, the cells were seeded on 96-well plates and transfected with 0.6 μg of TRPC1-OE vector for 48 hours. The cells were then exposed to HSV-1 gL86 at the indicated MOIs. Alternatively, polyclonal anti–HSV-1 antibody (Abcam 9533) that detects the glycoproteins and core proteins of HSV-1 was used to detect viral entry after 15 min of infection at 37°C by immunofluorescence. For the imaging process, each condition was replicated for three times. Ten cells were randomly chosen and evaluated in each replication. Statistical results were obtained from a total of 30 cells.
In situ enzyme-linked immunosorbent assay
The binding and replication of HSV-1 was evaluated by enzyme-linked immunosorbent assay (ELISA) following procedures previously described with some modifications (44). HEp-2 cells (1 × 105) were seeded on 96-well microtiter plates, cultured overnight, and then infected with HSV-1 at the required titer and incubation period. In the binding assay, HSV-1 was incubated with HEp-2 cells at 4°C, the cells were washed with PBS, and HSV-1 antigen was detected by ELISA. In the replication assay, HSV-1 was coincubated with HEp-2 cells at 37°C for 18 hours and subsequently assayed via ELISA. In the ELISA assay, the cells were fixed with 0.05% glutaraldehyde for 15 min at 4°C, and HSV-1 was assessed using antibody-detecting HSV-1 antigen (ab9533; Abcam), followed by secondary antibodies and tetramethylbenzidine to develop color.
Measurement of SOCE
Cells were plated on glass coverslips and loaded with 10 μM Fluo-4/AM (Molecular Probes, USA) for 30 min at 37°C and placed in Ca2+-free physiological saline solution (OPSS) containing 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM glucose, 2 mM EGTA, and 5 mM Hepes (pH 7.4). TG (4 μM) or HSV-1 at 0.5 multiplicity of infection (MOI) was used to deplete Ca2+ stores or activate intracellular Ca2+ release. Ca2+ entry was then measured by returning Ca2+ to TG-treated cells. SOCE was inhibited where indicated by addition of 2-APB (50 μM; Sigma, USA) or GSK-7975A (10 μM; a gift from GlaxoSmithKline, UK) before the return of Ca2+. Fluo-4 fluorescence was measured using an Olympus confocal microscope system. In experiments comparing the SOCE induced by HSV-1 binding and entry, HEp-2 cells were first loaded with Fluo-4/AM for 30 min at 37°C, and then HSV-1 was added to the cells and incubated at 4°C for 1 hour (binding) or at 37°C for 15 min (entry). The cells were then treated with sodium citrate (pH 3) to inactivate the virus associated with the PM and washed with OPSS. SOCE was then measured at room temperature in OPSS. In all SOCE experiments, each condition was replicated for three times. Ten cells were randomly chosen and evaluated in each replication. Statistical results were obtained from a total of 30 cells. Fluo-4 values at each time point were normalized to that before TG addition. Each fluorescent trace represents changes in [Ca2+]i, which was calculated as the ratio of fluorescence relative to the fluorescence before Ca2+ addition (F1/F0, for Flou-4 fluorescence).
Immunoprecipitation and immunoblots
HEp-2 cells or CHO cells were infected with HSV-1 at 0.5 MOI for 15 min, and then whole-cell lysates were obtained. Immunoprecipitation was carried out using anti–HSV-1 gD protein antibody (sc-21719; Santa Cruz Biotechnology, USA) or anti-TRPC1 antibody (ACC-010; Alomone Labs, Israel); preimmune immunoglobulin G (IgG) was used as a control for anti–HSV-1 gD. The immunoprecipitates were purified with protein A agarose (Roche, USA) and resolved on 12% SDS–polyacrylamide gel electrophoresis (PAGE) for immunoblotting.
In experiments analyzing the membrane distribution of TRPC1 protein, HEp-2 cells were infected with HSV-1 for 15 min and the surface proteins were extracted with the Cell Surface Protein Isolation Kit (Pierce Biotechnology, USA) according to the manufacturer’s instructions. Briefly, the surface proteins of HEp-2 cells with or without HSV-1 infection were biotinylated and labeled proteins were isolated with NeutrAvidin agarose. The bound proteins were subjected to immunoblotting using TRPC1 antibody. Band intensities of total TRPC1 (samples from whole-cell lysates) were first normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from the same gel. Second, TRPC1 band intensity obtained with samples from HSV-1–infected group was normalized to that of the mock-infected control. Furthermore, band intensities of PM TRPC1 were first normalized to that of whole-cell TRPC1 value, and then these values for HSV-1–infected group were normalized to that of the mock-infected control.
For immunoblots, proteins were separated on 12% SDS-PAGE with the primary antibodies at 1:200 dilution. The antibody binding was assessed with an Odyssey imaging system (LI-COR Biosciences, USA). Band intensities were normalized to that of loading control from the same gel, and then the results for siRNA (S1F) or HSV-1–infected group (S3B) were normalized to that of the control (scrambled siRNA for S1F and mock-infected control for S3B) and are shown as relative values.
Site-directed mutagenesis of TRPC1
Single mutations were introduced into the first and third ectodomains of TRPC1 at amino acids 408 to 415 and 608 to 616, respectively, while three sites were mutated in the second ectodomain because this fragment is long, containing 64 amino acids (476 to 539) (35). First, a fragment containing all the designed mutation sites was polymerase chain reaction (PCR)–amplified with primers Trunc-f and Trunc-r, using vectorA harboring the TRPC1 gene as template. The 1.3-kb fragment was inserted into cloning vector pUC19 to yield pUCTruncTRPC1. That plasmid was then amplified by PCR with mutagenic oligonucleotide primers to introduce mutations. The primers are listed in Table 1. PCRs were performed using Pyrobest DNA Polymerase (1.0 μl, 5 U/μl, Takara, China) under the following conditions: 95°C for 300 s; 18 cycles of 95°C for 30 s, 65°C for 30 s, and 72°C for 120 s; then 72°C for 600 s. Dpn I (1 μl; NEB, USA) was then added, and samples were incubated for 3 hours at 37°C, followed by enzyme denaturation for 20 min at 75°C. Next, competent Escherichia coli JM109 cells were transformed with the amplified plasmid and screened on Luria-Bertani plates containing ampicillin (100 μg/ml). The successful introduction of desired mutations was confirmed by sequencing. Plasmids harboring the mutations were digested by Bam HI and Xho I. The 1.3-kb fragments were isolated on agarose gels and ligated to the 7.7 kb fragment recovered from the parent plasmid vectorA, which was digested with the same enzymes, to replace the native sequences. Last, the ligation products were transformed into competent E. coli cells. Trunc-f, 5′-GGTTACCGACGCAAGCCC-3′; Trunc-r, 5′-CTTACTGAGGCTACTAAT-3′.
Table 1. List of mutagenic oligonucleotide primers.
| Primer name | Primer sequence |
| Region1-f | 5′-ggctggccccattgtgtttttcttattattattgtagacaagagagtatagattaa-3′ |
| Region1-r | 5′-ttaatctatactctcttgtctacaataataataagaaaaacacaatggggccagcc-3′ |
| Region2-f | 5′-gtaggatggaatgcatcccaattattccgatcagcaaaatcatgaa-3′ |
| Region2-r | 5′-ttcatgattttgctgatcggaataattgggatgcattccatcctac-3′ |
| Region3-f | 5′-aaaaaagagacgaagataacttagaacattattatttgcaaaaagcccttctgccaccagtgtagg-3′ |
| Region3-r | 5′-cctacactggtggcagaagggctttttgcaaataataatgttctaagttatcttcgtctctttttt-3′ |
| Region4-f | 5′-taacatctgtcccattgaaatctgtaatggattattgatagagctggttgtatacataaaaaagagacg-3′ |
| Region4-r | 5′-cgtctcttttttatgtatacaaccagctctatcaataatccattacagatttcaatgggacagatgtta-3′ |
| Region5-f | 5′-ctcccacaaaggactgcagttcttcattattgctaaatcttgtgacaaagattgcc-3′ |
| Region5-r | 5′-ggcaatctttgtcacaagatttagcaataatgaagaactgcagtcctttgtgggag-3′ |
The aspartic acid of No. 581 of TRPC1 was mutated to lysine. The 50-μl PCR system contained 32 μl of ddH2O, 5 μl of 10*PCR buffer, 5 μl of 2 mM deoxynucleotide triphosphates (dNTPs), 3 μl of 25 mM MgSO4, 1.5 μl of each of the two primers, 2 to 10 ng of template DNA, and 1 μl of KOD-plus-Neo. Each cycle consisted of 98°C for 10 s, 56°C for 30 s, and 68°C for 4.5 min. The PCR cycles ended with an extension step at 72°C for 5 min. The PCR products were treated with 2.5 μl of Dpn I at 37°C for 3 hours, and then 10 μl of each PCR was analyzed by agarose gel electrophoresis. The above PCR products were each transformed into DH5α-competent cells. The transformed cells were spread on a Luria-Bertani plate containing antibiotics and incubated at 37°C overnight. The number of colonies was counted and used as an indirect indication of PCR amplification efficiency. Five colonies from each plate were grown, and the plasmid DNA was isolated. The plasmid was verified using Sanger sequencing to ensure successful mutations. The following primers were used: forward, CTGTGAACAGCAAAGCAATAAGACCTTCCATTCGTTCATTGG; reverse, CCAATGAACGAATGGAAGGTCTTATTGCTTTGCTGTTCACAG.
Fluorescence resonance energy transfer
Cells were fixed in 4% paraformaldehyde, washed with PBS, and incubated for 1 hour with 5% bovine serum albumin and 0.1% Triton X-100 in PBS. The cells were incubated with primary antibodies [anti-TRPC1 (sc-20110; Santa Cruz Biotechnology, USA) and anti–HSV-1 gD (sc-21719; Santa Cruz Biotechnology, USA), both at a dilution of 1:100 in PBS containing 5% bovine serum albumin] overnight at 4°C in a humidified chamber. The cells were then labeled with Alexa Fluor 546–conjugated secondary antibodies for TRPC1 and Cy2-conjugated secondary antibodies for HSV-1 gD. After washing and mounting, fluorescence images were acquired using a confocal microscope (Leica TCS SP8). Alexa Fluor 488 and Alexa Fluor 546 were excited at 488 and 546 nm, respectively, with emission at 505 to 530 nm and >560 nm, respectively. First, images of the donor and acceptor distributions were captured. The acceptor dyes were then bleached by repetitive scans with the 514-nm laser at 80% of maximum power. After photobleaching, a donor image was captured again. LSM data acquisition software (Leica TCS SP5 LAS AF version 1.7.0) was used to define regions of interest, and N-FRET was calculated using the method of Xia and Liu (45). For data recording, each condition was replicated for three times. Five cells were randomly chosen and evaluated in each replication. Statistical results were obtained from a total of 15 cells.
TIRF microscopy
HSV-1 was allowed to bind to HEp-2 cells and enter the cells for 0, 15, and 180 min, and then they were fixed in 1% formaldehyde. TRPC1 and gD were detected with anti–HSV-1 gD antibody (sc-21719; Santa Cruz Biotechnology, USA) and anti-TRPC1 primary antibody (sc-20110; Santa Cruz Biotechnology, USA). Secondary antibodies were then used with Donkey anti-Rabbit IgG (H+L) highly cross-adsorbed secondary antibody, AF488 (catalog no. A21206), and Donkey anti-Mouse IgG (H+L) highly cross-adsorbed secondary antibody, AF568 (catalog no. A10037, both from Thermo Fisher Scientific, Shanghai, PR China). TIRF microscopy was performed on a Nikon N-STORM & A1 Cell TIRF system using DU897 EMCCD 100× oil TIRF objective (38). To excite HSV-1 gD and TRPC1, 488- and 561-nm solid-state lasers were used, respectively. The laser alignment was performed individually for each region of interest. Fluorescence image acquisitions were collected with Nis-Elements software, and values were analyzed with ImageJ software. For data recording, each condition was replicated for three times. Five cells were randomly chosen and evaluated in each replication. Statistical results were obtained from a total of 15 cells.
Mice and in vivo studies
All animal experiments were conducted using protocols approved by the Animal Care Committee of Jiangnan University and following Institutional Animal Care and Use Committee guidelines. The parental TRPC1 knockout (TRPC1−/−) mice (46) were from L. Birnbaumer. Mice were bred and maintained under standard conditions at the animal house of Jiangnan University (Wuxi, China). F1 and F2 populations were generated to conduct the genetic studies. Sera from parental TRPC1−/− mice were tested by ELISA to ensure that they were free from HSV-1 infection before being used for experiments. Male WT and TRPC1−/− mice were chosen from the same generation. The cornea of the right eye was scarified, and each mouse was infected with HSV-1 strain 17 syn+ or McKrae, while the left eye was untouched. The phenotypes of the infected mice were scored for HSK (47) as follows: 0, clear; 1, <25%; 2, <50%; 3, <75%; and 4, 75 to 100% corneal opacity and neovascularization. To prepare histology specimens, mice were sacrificed and the eyes were enucleated, fixed, and stained with hematoxylin and eosin.
MEF infection
Primary MEFs were prepared from day 15 embryos of TRPC1−/− or WT mice in DMEM containing 10% fetal bovine serum, and TRPC1 expression was screened by PCR. The MEFs were infected with HSV-1 (KOS) to analyze the nuclear transport of ICP5 by Western blot and infection efficacy by ELISA.
Clinical sample validation
Each patient/healthy individual involved in this study had provided written informed consent. All studies, including patient enrollment and sample analysis, were conducted with approval from the Ethics Committee of Jiangnan University. Patients diagnosed with oral herpes lesions (visible blisters on the lips or around the mouth) were selected. Healthy individuals served as controls. The epithelial cells were collected with a cytobrush from lesion sites in the oral cavity, smeared on glass slides, and fixed in 1% formaldehyde. Anti–HSV-1 gD antibody (sc-21719; Santa Cruz Biotechnology, USA) and anti-TRPC1 antibody (ACC-010; Alomone Labs, Israel) were used to confirm the presence of HSV-1 and TRPC1 by confocal microscopy. The fluorescence intensity (in pixel) of TRPC1 and gD in PM or cytoplasm was analyzed with ImageJ software. Location of PM was indicated by DiI fluorescence.
C1E3p, BFA, and siSTIM1 treatment
Male WT mice were randomized into different treatment groups for each experiment. For C1E3p treatment, the virus was incubated with 10 μM C1E3p or PBS at 37°C for 30 min and then used to infect HEp-2 cells (n = 3) or eyeballs (n = 15 mice). After HSV-1 infection, 2 mM C1E3p was applied to the eyeballs of infected mice every 2 days for 15 days. For BFA and siSTIM1 treatment, HEp-2 cells or mice (n = 15) were infected with HSV-1 and simultaneously treated with BFA (5 μM) or siSTIM1 (10 μM; RiboBio Co. Ltd., PR China). BFA (5 μM) or siSTIM1 (10 μM) was then applied to the eyeballs every 2 days for 15 days.
Statistical analysis
Statistical analyses were performed in GraphPad Prism (version 6). Sample sizes, types of statistical test, and P values for each experiment are indicated in the figures legends.
Supplementary Material
Acknowledgments
We thank I. C. Bruce for critical reading of the manuscript. We thank the Core Facilities in Zhejiang University School of Medicine for assistance in TIRF microscopy experiments. Funding: This work was supported by the National Natural Science Foundation of China (81622007, 81870362, and 91939301). Chang Jiang Scholars Program (Q2015106), Fundamental Research Funds for the Central Universities (JUSRP51704A), and the National First-class Discipline Program of Food Science and Technology (JUFSTR20180101 and JUSRP51615B). The work was also supported, in part, by the Intramural Research Program of the NIH (project Z01-ES-101684) to L.B. and NIDCR-DIR, NIH (Z01-DE00438-31) to I.S.A. Author contributions: X.M. conceived and supervised the project. L.B., I.S.A., Y.Z., X.Y., and S.T. were the chief consultants and helped with the design and writing of the manuscript. A.M., D.H., and Y.L. performed the experiments. D.H. and A.M. analyzed the data. X.M., I.S.A., and D.H. wrote the manuscript. Competing interests: X.M., A.M., D.H., and C.Y. have filed a C1E3p patent to the State Intellectual Property of PR China. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The Trpc1−/− mice can be provided by L.B. pending scientific review and a completed material transfer agreement. Requests for the Trpc1−/− mice should be submitted to birnbau1@gmail.com.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaaz3367/DC1
Fig. S1. Features of SOCE during HSV-1 infection.
Fig. S2. TRPC1 modulates HSV-1 infection.
Fig. S3. Dynamic changes of SOCE-related molecules during HSV-1 infection.
Fig. S4. Determination of gD-TRPC1 interaction.
Fig. S5. Original images of Western blots.
REFERENCES AND NOTES
- 1.Chen A. B., Ben-Porat T., Whitley R. J., Kaplan A. S., Purification and characterization of proteins excreted by cells infected with herpes simplex virus and their use in diagnosis. Virology 91, 234–242 (1978). [DOI] [PubMed] [Google Scholar]
- 2.Spear P. G., Herpes simplex virus: Receptors and ligands for cell entry. Cell. Microbiol. 6, 401–410 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Spear P. G., Longnecker R., Herpesvirus entry: An update. J. Virol. 77, 10179–10185 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geraghty R. J., Krummenacher C., Cohen G. H., Eisenberg R. J., Spear P. G., Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280, 1618–1620 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Warner M. S., Geraghty R. J., Martinez W. M., Montgomery R. I., Whitbeck J. C., Xu R., Eisenberg R. J., Cohen G. H., Spear P. G., A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246, 179–189 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Atanasiu D., Saw W. T., Cohen G. H., Eisenberg R. J., Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84, 12292–12299 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atanasiu D., Saw W. T., Eisenberg R. J., Cohen G. H., Regulation of Herpes Simplex Virus glycoprotein-induced cascade of events governing cell-cell fusion. J. Virol. 90, 10535–10544 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheshenko N., Del Rosario B., Woda C., Marcellino D., Satlin L. M., Herold B. C., Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 163, 283–293 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scherbik S. V., Brinton M. A., Virus-induced Ca2+ influx extends survival of west nile virus-infected cells. J. Virol. 84, 8721–8731 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao H., Duan M., Yang L., Buch S., Platelet-derived growth factor-BB restores human immunodeficiency virus Tat–cocaine-mediated impairment of neurogenesis: Role of TRPC1 channels. J. Neurosci. 32, 9835–9847 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han Z., Madara J. J., Herbert A., Prugar L. I., Ruthel G., Lu J., Liu Y., Liu W., Liu X., Wrobel J. E., Reitz A. B., Dye J. M., Harty R. N., Freedman B. D., Calcium regulation of hemorrhagic fever virus budding: Mechanistic implications for host-oriented therapeutic intervention. PLOS Pathog. 11, e1005220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashmole I., Duffy S. M., Leyland M. L., Morrison V. S., Begg M., Bradding P., CRACM/Orai ion channel expression and function in human lung mast cells. J. Allergy Clin. Immunol. 129, 1628–1635.e2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derler I., Schindl R., Fritsch R., Heftberger P., Riedl M. C., Begg M., House D., Romanin C., The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 53, 139–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bootman M. D., Collins T. J., Mackenzie L., Roderick H. L., Berridge M. J., Peppiatt C. M., 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 16, 1145–1150 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Cheng K. T., Liu X., Ong H. L., Swaim W., Ambudkar I. S., Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLOS Biol. 9, e1001025 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maul-Pavicic A., Chiang S. C., Rensing-Ehl A., Jessen B., Fauriat C., Wood S. M., Sjöqvist S., Hufnagel M., Schulze I., Bass T., Schamel W. W., Fuchs S., Pircher H., McCarl C. A., Mikoshiba K., Schwarz K., Feske S., Bryceson Y. T., Ehl S., ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc. Natl. Acad. Sci. U.S.A. 108, 3324–3329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trebak M., Lemonnier L., Smyth J. T., Vazquez G., Putney J. W. Jr., Phospholipase C-coupled receptors and activation of TRPC channels. Handb. Exp. Pharmacol. 2007, 593–614 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Montgomery R. I., Warner M. S., Lum B. J., Spear P. G., Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87, 427–436 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Burnham L. A., Jaishankar D., Thompson J. M., Jones K. S., Shukla D., Tiwari V., Liposome-mediated herpes simplex virus uptake is glycoprotein-D receptor-independent but requires heparan sulfate. Front. Microbiol. 7, 973 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qie L., Marcellino D., Herold B. C., Herpes simplex virus entry is associated with tyrosine phosphorylation of cellular proteins. Virology 256, 220–227 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Manoj S., Jogger C. R., Myscofski D., Yoon M., Spear P. G., Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc. Natl. Acad. Sci. U.S.A. 101, 12414–12421 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ambudkar I. S., Ong H. L., Organization and function of TRPC channelosomes. Pflugers Arch. 455, 187–200 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Cheng K. T., Ong H. L., Liu X., Ambudkar I. S., Contribution and regulation of TRPC channels in store-operated Ca2+ entry. Curr. Top. Membr. 71, 149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tallóczy Z., Virgin H. W. IV, Levine B., PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2, 24–29 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Nicola A. V., McEvoy A. M., Straus S. E., Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77, 5324–5332 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng K. T., Liu X., Ong H. L., Ambudkar I. S., Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 283, 12935–12940 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X., Singh B. B., Ambudkar I. S., TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the S5-S6 region. J. Biol. Chem. 278, 11337–11343 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Selvaraj S., Watt J. A., Singh B. B., TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP+. Cell Calcium 46, 209–218 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thippegowda P. B., Singh V., Sundivakkam P. C., Xue J., Malik A. B., Tiruppathi C., Ca2+ influx via TRPC channels induces NF-κB-dependent A20 expression to prevent thrombin-induced apoptosis in endothelial cells. Am. J. Physiol. Cell Physiol. 298, C656–C664 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galvan V., Roizman B., Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 95, 3931–3936 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodkin M. L., Morton E. R., Blaho J. A., Herpes simplex virus infection and apoptosis. Int. Rev. Immunol. 23, 141–172 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Marras S. A., Kramer F. R., Tyagi S., Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 30, e122 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagy P., Vereb G., Damjanovich S., Mátyus L., Szöllõsi J., Measuring FRET in flow cytometry and microscopy. Curr. Protoc. Cytom. Chapter 12, Unit12.8 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Shieh M. T., WuDunn D., Montgomery R. I., Esko J. D., Spear P. G., Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J. Cell Biol. 116, 1273–1281 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beech D. J., TRPC1: Store-operated channel and more. Pflugers Arch. 451, 53–60 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Esmann J., The many challenges of facial herpes simplex virus infection. J. Antimicrob. Chemother. 47 (suppl. T1), 17–27 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Xu S.-Z., Beech D. J., TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ. Res. 88, 84–87 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Ma X., Cao J., Luo J., Nilius B., Huang Y., Ambudkar I. S., Yao X., Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4-C1 heteromeric channels to the plasma membrane. Arterioscler. Thromb. Vasc. Biol. 30, 2249–2255 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrandiz-Huertas C., Mathivanan S., Wolf C. J., Devesa I., Ferrer-Montiel A., Trafficking of thermoTRP channels. Membranes 4, 525–564 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nilius B., Szallasi A., Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 66, 676–814 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Ong H. L., de Souza L. B., Cheng K. T., Ambudkar I. S., Physiological functions and regulation of TRPC channels. Handb. Exp. Pharmacol. 223, 1005–1034 (2014). [DOI] [PubMed] [Google Scholar]
- 42.He D. X., Tam S. C., Trichosanthin affects HSV-1 replication in Hep-2 cells. Biochem. Biophys. Res. Commun. 402, 670–675 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Lehman I. R., Boehmer P. E., Replication of herpes simplex virus DNA. J. Biol. Chem. 274, 28059–28062 (1999). [DOI] [PubMed] [Google Scholar]
- 44.McClain D. S., Fuller A. O., Cell-specific kinetics and efficiency of herpes simplex virus type 1 entry are determined by two distinct phases of attachment. Virology 198, 690–702 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Xia Z., Liu Y., Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 81, 2395–2402 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dietrich A., Kalwa H., Storch U., Mederos y Schnitzler M., Salanova B., Pinkenburg O., Dubrovska G., Essin K., Gollasch M., Birnbaumer L., Gudermann T., Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 455, 465–477 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Norose K., Yano A., Zhang X.-M., Blankenhorn E., Heber-Katz E., Mapping of genes involved in murine herpes simplex virus keratitis: Identification of genes and their modifiers. J. Virol. 76, 3502–3510 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaaz3367/DC1
Fig. S1. Features of SOCE during HSV-1 infection.
Fig. S2. TRPC1 modulates HSV-1 infection.
Fig. S3. Dynamic changes of SOCE-related molecules during HSV-1 infection.
Fig. S4. Determination of gD-TRPC1 interaction.
Fig. S5. Original images of Western blots.