

A rationally designed bi-adjuvant nanovaccine enhances neoantigen immunogenicity and enables combination immunotherapy.
Abstract
Neoantigen vaccines have been enthusiastically pursued for personalized cancer immunotherapy while vast majority of neoantigens have no or low immunogenicity. Here, a bi-adjuvant neoantigen nanovaccine (banNV) that codelivered a peptide neoantigen (Adpgk) with two adjuvants [Toll-like receptor (TLR) 7/8 agonist R848 and TLR9 agonist CpG] was developed for potent cancer immunotherapy. Specifically, banNVs were prepared by a nanotemplated synthesis of concatemer CpG, nanocondensation with cationic polypeptides, and then physical loading with hydrophobic R848 and Adpgk. The immunogenicity of the neoantigen was profoundly potentiated by efficient codelivery of neoantigen and dual synergistic adjuvants, which is accompanied by reduced acute systemic toxicity. BanNVs sensitized immune checkpoint programmed death receptor 1 (PD-1) on T cells, therefore, a combination of banNVs with aPD-1 conspicuously induced the therapy response and led to complete regression of 70% neoantigen-specific tumors without recurrence. We conclude that banNVs are promising to optimize personalized therapeutic neoantigen vaccines for cancer immunotherapy.
INTRODUCTION
Cancer immunotherapy enables patients’ own immune system to eradicate tumor cells (1–3). By intervening in different steps in the cancer-immunity cycle, multiple approaches to cancer immunotherapy have been clinically tested, with notable examples of immune checkpoint blockade, chimeric antigen receptor T (CAR-T) cell therapy, and therapeutic cancer vaccines (4–8). Therapeutic tumor vaccines represent an approach to eliciting antigen-specific antitumor immunity. However, conventional tumor therapeutic vaccines, which use overexpressed or re-expressed proteins in tumor cells as antigens, have shown limited therapeutic efficacy largely due to central immune tolerance as well as suboptimal delivery to lymphoid organs where immunomodulation can be coordinated.
Recent advancement in immune-oncology, oncogenomics, and bioinformatics has sparked the identification of a class of tumor antigens called neoantigens, which are the products of events such as tumor somatic mutations and frameshifting mutations that occur only in tumor cells but not in healthy cells. As such, neoantigens are exclusively present in cancer cells, making neoantigens free from central immune tolerance. These unique properties endow neoantigens as excellent candidates for therapeutic cancer vaccines, which has been supported by multiple recent clinical investigations for cancer immunotherapy (9–11). In addition to vaccine-based therapy, neoantigen-specific immune responses have been observed to directly correlate with many other types of immunotherapies, including immune checkpoint blockade and CAR-T cell therapy. These observations collectively indicated a “final pathway” of somatic cancer mutations in cancer immunotherapy (12, 13). Effective cancer therapeutic vaccines rely on potent and durable neoantigen-specific antitumor immune responses. However, most cancer neoantigens have undetectable immunogenicity; this is further complicated by the fact that most cancers, including notable examples of pancreatic cancer and glioblastoma, have extremely low tumor mutation burdens. It is thus highly desired to develop an approach that can potentiate the immunogenicity of neoantigens toward potent and durable cancer immunotherapy.
The efficacy of subunit vaccines can be substantially potentiated by (i) codelivery of adjuvants that stimulate distinct innate immunity and cancer-specific antigens that can guide the elicitation of antigen-specific adaptive immunity such as T cell responses (14, 15), (ii) synergistic modulation of multiple immune signaling pathways, and (iii) multiepitope antigens that elicit a broad spectrum of immune responses. Microbial pathogens set an excellent example that leverages all these approaches. As a current clinical benchmark of peptide neoantigen vaccines, adjuvants such as polyinosinic-polycytidylic acid-poly-l-lysine carboxymethylcellulose (poly-ICLC) and/or anti-CD40 have been mixed together with peptide neoantigens. Yet, coadministered adjuvants and antigens may not guarantee robust immune responses, as soluble formulations of vaccines will result in chaotic dissemination in the body that could restrict vaccine immunogenicity against tumors (16, 17). Pathogen-mimicking nanovaccines hold great potential to potentiate the immunogenicity of neoantigens for cancer immunotherapy and address the above challenges. First, nanovaccines can be efficiently delivered to secondary lymphoid tissues such as lymph nodes (LNs) and antigen-presenting cells (APCs) because medium-sized (5 to 100 nm) nanovaccine could be drained and retained in LNs for a relatively long time. Second, nanovaccines can further codeliver multiple synergistic adjuvants and antigens into LNs (4, 18–20) and APCs (14, 21–25), which is a prerequisite for optimal tumor immunotherapy. Third, by coencapsulating adjuvants and antigens, nanovaccine can improve the pharmacokinetic properties of their drug payloads to further potentiate the resulting immunomodulation.
Herein, this study presented bi-adjuvant neoantigen nanovaccines (banNVs), which codelivered synergistic Toll-like receptor 7/8 (TLR7/8) agonist (TLR7/8a) R848 and TLR9 agonist CpG oligodeoxynucleotides (ODNs), as well as a peptide neoantigen to potentiate the immunogenicity of neoantigen for potent and durable cancer immunotherapy, especially when combined with anti–PD-1 (aPD-1), which has been indicated to invigorate CD8+ T cells during the immune induction phase (26). TLR agonists are a class of immune adjuvants that have been used or investigated for cancer treatment in clinic (27). Specifically, TLR7/8a imiquimod and resiquimod (R848) (28) have been approved by the U.S. Food and Drug Administration to treat skin lesions topically; CpG ODNs have been widely studied in clinic for cancer treatment. The combination of multiple agonists against TLR7/8 and TLR9 exhibited synergistic therapeutic efficacy mainly via triggering MyD88 and cascade signaling pathways (29). However, the poor aqueous solubility and/or unfavorable pharmacokinetics of these drugs have refrained them from wide applications in cancer immunotherapy (17, 30, 31). Pharmaceutical engineering of these drugs using nanocarriers has shown the potential to efficiently codeliver multiple adjuvants as well as antigens and eventually potentiate immune responses. Nevertheless, most current vaccine nanotechnologies are based on either admixing adjuvants attached by polymer scaffolds with peptide antigens or encapsulating adjuvants and antigens within nanoparticles separately. However, the discrepancy of pharmacokinetic and physicochemical properties of small-molecule adjuvants, ODN adjuvants, and macromolecular peptide antigens remains a major obstacle in vaccine-based cancer immunotherapy (32, 33). Herein, this study synthesized banNVs without complicated chemical appendages and demonstrated that banNVs elicited potent and durable antitumor immunity for tumor immunotherapy in a colorectal cancer model in syngeneic mice (Fig. 1A). By strategically loading three vaccine components in one nanoparticle, banNVs coordinated to elicit both innate and adaptive immunity for cancer immunotherapy with minimal systemic toxicity. BanNVs promoted the codelivery of adjuvants and neoantigens to LNs and LN-residing APCs, mediated efficient antigen uptake and presentation on APCs, and augmented neoantigen-specific cytotoxic T cell responses that eventually eradicated most neoantigen-specific tumors when combined with immune checkpoint blockade (Fig. 1B). Together, banNVs represent a potent cancer therapeutic vaccine for combination cancer immunotherapy.
Fig. 1. Schematic depiction of the development of banNVs to potentiate neoantigen immunogenicity for personalized cancer immunotherapy.
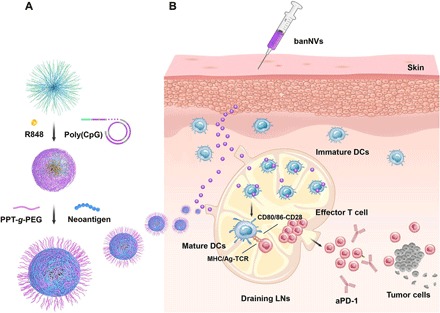
(A) BanNVs were self-assembled in three steps: First, via a nano-templated RCR reaction using DNA primer-coated amphiphilic PEG-PLA micelles and CpG-encoding template DNA, microparticles were self-assembled with a core of PEG-PLA micelle and a shell of polymeric CpG, and the resulting microparticles were then physically loaded with hydrophobic R848; second, the above microparticles were shrunk using cationic PEG-g-PPT to reduce the particle size and promote intranodal and intracellular vaccine delivery, and the hydrophobic PPT additionally enabled efficient loading of hydrophobic peptide neoantigens. (B) BanNVs enable efficient codelivery of adjuvants and antigen peptides to LNs, inducing DC mutation by adjuvants, facilitating antigen cross presentation, and profoundly expanding the repertoires of neoantigen-specific CD8+ effector T cells. Such cancer therapeutic vaccines, especially when combined with aPD-1, markedly inhibited tumor progression and led to complete regression of most MC38 tumors without tumor relapse.
RESULTS
Synthesis of banNV
Briefly, the banNVs were prepared in three steps: First, thiolated DNA primer was conjugated to maleimide-functionalized poly(ethylene oxide)-block-poly(d,l-lactic acid) (MAL-PEG-b-PLA) micelles; then, a circularized CpG template (table S1) was immobilized on primer-PEG-PLA nanoparticle surface through hybridization, followed by in situ rolling circle amplification (RCA) to self-assemble core-shell CpG microparticles (CpG MPs), the hydrophobic PLA core of which was exploited for encapsulating hydrophobic adjuvant molecules; last, the resulting MPs were condensed into banNVs by cationic PEG-grafted polypeptide (PPT-g-PEG) via electrostatic interaction between PPT-g-PEG and negatively charged CpG; meanwhile, hydrophobic PPTs were used for physical loading of peptide neoantigens (Fig. 1A). In particular, amphiphilic PEG-b-PLA was self-assembled into micelles (d = 30.43 ± 3.04 nm) (Fig. 2A), as shown by transmission electron microscopy (TEM). Then, DNA primer for RCA was conjugated on the surface of PEG-PLA micelles, as verified by particle size increasing from 44.72 to 57.09 nm using dynamic light scattering (DLS) (fig. S1A and table S1), as well as mobility shift of DNA versus DNA-polymer conjugates in agarose gel electrophoresis (fig. S1B). The conjugates were purified by high-performance liquid chromatography (HPLC) to remove free DNA and bare MAL-PEG-b-PLA (fig. S1C), followed by verification using agarose gel electrophoresis (Fig. 2B). Next, a linear phosphorylated CpG DNA template (table S1) was used for in situ RCA to generate tandem CpG on the surface of micelles, resulting in formation of MPs with uniform diameters, good dispersity, and sponge-like surface porosity as validated by scanning electron microscopy (SEM) and TEM (Fig. 2C and fig. S2) and agarose gel electrophoresis (Fig. 2D). The process of CpG MPs formation was monitored by SEM at a series of time points during RCA. CpG MPs were produced after 12 hours with wrinkled surface morphological structure, and the sheet-like structures on CpG MP surfaces were reorganized until 24 hours after RCA (Fig. 2E).
Fig. 2. Characteristics of banNVs.

(A) Transmission electron microscopy (TEM) images of PLA-PEG micelles. (B) An image of agarose gel electrophoresis showing the retarded migration of DNA-PEG-PLA conjugates. bp, base pairs. (C) Scanning electron microscopy (SEM) images of CpG MPs self-assembled via in situ RCA. (D) An image of agarose gel electrophoresis of CpG MPs. The smeared band represents unreacted DNA. (E) SEM images of in situ RCA products during a series of time points. (F) Fluorescence images of CpG MPs labeled with Cy5-dUTP. (G) STEM-based EDS mapping of CpG MPs. (H) TEM images of banNVs after condensation. (I) A DLS graph showing the hydrodynamic diameters of products during the process of banNV synthesis. (J) Image of agarose gel electrophoresis showing the retardation of banNVs (the concentrations of nucleic acids were varied in different groups). The definitions of the abbreviations are listed in table S2.
The morphological characteristics of the CpG MPs were further studied by confocal microscopy to localize CpG molecules in MPs. To do this, Cy5-labeled deoxyuridine triphosphate (dUTP) was added during RCA, which yielded MPs with red fluorescence throughout the particles (Fig. 2F and fig. S3), indicating that the CpG was distributed evenly throughout the particles. MPs were expected to be composed of CpG and magnesium pyrophosphate (Mg2P5O7). By STEM-based energy dispersive x-ray spectroscopy (EDS) mapping (Fig. 2G) (34), crystalline Mg2P5O7 was visualized in the sheet structures of MPs. The map of carbon suggested a homogeneous distribution of CpG throughout the MPs. The porous substructures in CpG MPs, together with the hydrophobic PLA cores in the micelle, were expected to encapsulate R848 and release the drugs in response to acidic environment. Last, MPs were condensed by cationic PPT-g-PEG into banNVs. Twelve hours after incubating with PPT-g-PEG, MPs were purified by centrifugation to remove free peptides. TEM revealed that MPs were condensed into uniform nanoparticles (Fig. 2H), with diameters reduced from 2222 ± 145 nm for MPs to 171 ± 22 nm for NVs (Fig. 2I). Gel electrophoresis confirmed that CpG molecules in MPs were completely complexed with PPT-g-PEG, as supported by the nearly complete retardation of CpG in banNVs (Fig. 2J). Because of the intertwining hydrophobic PPT, a macromolecular peptide neoantigen, namely, Adpgk, was generated from mutations (ASMTNRELM → ASMTNMELM) in mouse MC38 tumor cells that were physically loaded into the final nanoparticles (35), thereby generating a previously unidentified nanovaccine with bi-adjuvants and neoantigens. The molar ratio of drug loaded in banNVs was determined to be approximately 10:10:1 of Adpgk/R848/CpG (the definitions of the abbreviations are listed in table S2).
Dendritic cells (DCs) immunostimulation of banNVs
Many TLR ligands stimulate innate immune responses in APCs characterized by the secretion of a series of cytokines and the up-regulated expression of proinflammatory factors, which eventually promote both innate immunity and adaptive immunity. To test the in vitro immunostimulation of DCs by our nanovaccines, DCs were treated with the equivalent doses of CpG and CpG nanoparticles (NPs). Forty-eight hours later, comparable levels of interleukin-12 (IL-12), IL-6, and tumor necrosis factor–α (TNF-α) were detected (Fig. 3, A to C). This confirms the immunostimulatory activity of CpG assembled in CpG NPs. As a control, GpC NPs, which were assembled from DNA where the CpG dinucleotides were mutated to GpC control, did not detectably activate DCs (36). Moreover, treatment of DCs with CpG NPs/R848 further promoted the levels of IL-12p40 and IL-6, relative to CpG NPs or R848 alone (Fig. 3, A and B), which was attributed to not only the additive but also the synergistic immunoactivation via both TLR7/8 and TLR9 pathways (29). Besides, CpG NPs/R848 outperformed mixed CpG and R848, likely due to the sustained release of R848 and CpG from banNVs. Consistently, compared to CpG, CpG NPs, or R848 alone, CpG NPs/R848 promoted the expression of costimulatory factor CD80 and CD86 on DCs (fig. S4, A to D), which indicated strong immunostimulation induced by bi-adjuvant CpG NPs/R848.
Fig. 3. Immune stimulatory and sustained antigen presentation on DCs.
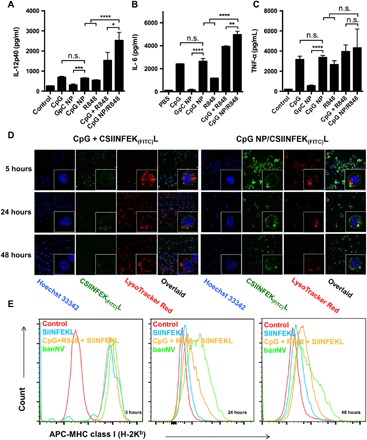
ELISA results of IL-12p40 (A), IL-6 (B), and TNF-α (C) secretion from DC2.4 cells after banNVs treatment (CpG: 200 nM; R848: 500 nM). (D) Confocal microscopy images of CSIINFEK(FITC)L presentation by DC2.4 cells after treatment with banNVs for 5, 24, and 48 hours (CpG: 200 nM and CSIINFEK(FITC)L: 2 μM). (E) Flow cytometric analysis of SIINFEKL/H-2Kb complex expression on DC2.4 cells after pretreatment with banNVs for 5, 24, and 48 hours (CpG: 200 nM and CSIINFEK(FITC)L: 2 μM). Data are represented as means ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (Student’s t test). n.s., not significant. The definitions of the abbreviations are listed in table S2.
Sustained antigen presentation induced by banNVs
To study the cellular uptake and presentation of antigens, lysine with fluorescein isothiocyanate (FITC) was modified in the ε-amino group of model antigen SIINFEKL, an major histocompatibility complex (MHC)–I (H-2Kb)–restricted epitope derived from ovalbumin. The resulting CSIINFEK(FITC)L maintained the binding ability of SIINFEKL to H-2Kb molecules (37). DC2.4 cells were treated with CpG + CSIINFEK(FITC)L and CpG NPs/CSIINFEK(FITC)L (10:1 of antigen/CpG molar equivalent), respectively. Cells were washed after 5 hours before further incubation. In stark contrast, CpG NPs encapsulated with CSIINFEK(FITC)L were highly colocalized in the endolysosome by 5 hours, and robust fluorescence signal of CSIINFEK(FITC)L was observed on DC2.4 membranes at 24 hours and maintained up to 48 hours after incubation (Fig. 3D). The antigen uptake from both formulations declined with time during 5 to 48 hours, likely due to exocytosis of banNVs and dilution effects caused by cell division (38, 39). Intrigued by efficient antigen uptake, the impact of banNVs on the presentation of CSIINFEK(FITC)L on DCs was then assessed by flow cytometric staining using an antibody that binds to SIINFEKL/H-2Kb complexes. Treatment with CpG NPs/SIINFEKL prolonged presentation of CSIINFEK(FITC)L on DCs compared with CSIINFEK(FITC)L + CpG. Note that DCs incubated with soluble CpG + CSIINFEK(FITC)L efficiently presented CSIINFEK(FITC)L after incubating for 5 hours, yet the antigen signal decreased precipitously after 24 hours, implying rapid clearance of antigen due to degradation or disassociation (Fig. 3E). Oppositely, antigen presentation with banNVs gradually increased during the initial period but decreased at a later time, achieving 2.3-fold higher levels of presentation at 24 hours and sustaining approximately 1.6-fold during 48 hours, relative to soluble forms of CpG + CSIINFEK(FITC)L. Together, banNVs facilitated intracellular delivery of antigens and adjuvants and mediated their continuous release within the endosome, thereby eliciting durable antigen presentation and DC maturation, which are expected to promote the downstream cross-priming of CD8+ T cells.
In vivo intranodal and intracellular delivery of banNVs
Subsequently, this study investigated the in vivo delivery of banNVs to LNs. C57BL/6 mice were subcutaneously injected at the tail base with soluble CpG + CSIINFEK(FITC)L versus CpG NP/CSIINFEK(FITC)L. After 12 hours, draining inguinal LNs were harvested for fluorescence imaging. CpG + CSIINFEK(FITC)L showed a marginally increased FITC signal in inguinal LNs compared to tissue autofluorescence (Fig. 4, A and B), likely due to the rapid systemic dissemination of antigen. In contrast, CpG NPs encapsulated with CSIINFEK(FITC)L showed remarkable LN accumulation of antigen peptides (Fig. 4A). Quantification of fluorescence signal intensities of LNs (Fig. 4B) suggested approximately 1.5-fold greater antigen accumulation by CpG NP/CSIINFEK(FITC)L relative to tissue autofluorescence (P < 0.01) and 1.2-fold greater antigen accumulation than soluble CpG + CSIINFEK(FITC)L control group (P < 0.05). The codelivery of adjuvants (labeled with Cy5) and antigen (labeled with FITC) into the same APCs is desired for optimal immunomodulation. The uptake of banNVs in LN-residing APCs was then characterized. C57BL/6 mice were injected with CpG-Cy5 + CSIINFEK(FITC)L and Cy5-CpG NPs/CSIINFEK(FITC)L, respectively. Inguinal LNs were excised and disrupted into single cells for flow cytometric analysis of Cy5 and FITC signals in F4/80+ macrophages and CD11c+ DCs, both of which are APC populations that can internalize exogenous particles and present antigens to naïve T cells. Macrophages (2.3%) and 5.1% DCs exhibited Cy5+FITC+ in banNVs, while only 0.9% macrophages and 1.1% DCs showed Cy5+FITC+ for free vaccines (Fig. 4, C and D), indicating that banNVs promoted the codelivery of adjuvants and antigens in vivo. C57BL/6 mice immunized three times with vaccines showed lymphadenopathy in draining inguinal LNs (fig. S5, A and B), likely due to the buildup of lymphocytes in LNs (fig. S6).
Fig. 4. In vivo delivery of banNVs to LNs and LN-residing APCs.
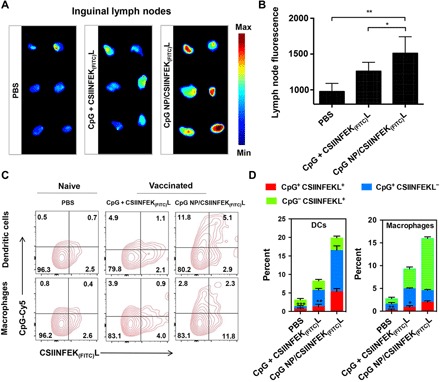
(A) Inguinal LN fluorescence imaging and (B) signal quantification using naïve C57BL/6 mice treated with PBS or C57BL/6 mice immunized with soluble CpG + CSIINFEK(FITC)L and CpG NP encapsulated with CSIINFEK(FITC)L (CpG equivalents: 2 nmol and CSIINFEK(FITC)L: 20 μg) 12 hours after subcutaneous injection at tail base. (C) Flow cytometry plots and (D) quantification showing the codelivery of CpG (modified with Cy5) and CSIINFEK(FITC)L into LN-residing DCs and macrophages 12 hours after subcutaneous injection at tail base in C57BL/6 mice. All error bars in figures show SEM. Data are represented as means ± SEM (n = 3 mice per group). *P < 0.05, **P < 0.01, and ***P < 0.001 (Student’s t test). The definitions of the abbreviations are listed in table S2.
T cell responses elicited by banNVs
Next, the antigen-specific T cell responses elicited by banNVs in vivo were investigated. Adpgk, an MHC-I H-2Db-restricted neoantigen peptide (ASMTNMELM), was used in murine MC38 colorectal cancer. BanNVs were formulated again by encapsulating Adpgk into CpG/R848 NPs. C57BL/6 mice were immunized with phosphate-buffered saline (PBS), CpG + R848 + Adpgk, CpG NPs/Adpgk, GpC NPs/R848 + Adpgk, and banNVs on days 0, 7, and 14. Peripheral blood cells were collected on day 21 to analyze Adpgk–MHC I dextramer+ CD8+ T cells (fig. S7, A and B). CpG + R848 + Adpgk induced 1.1% of Adpgk-specific CD8+ T cells after the boosting immunization, whereas the banNVs treatment elicited remarkable 5.6-fold greater frequencies of Adpgk+ CD8+ T cells (Fig. 5, A and B), indicating that banNVs potentiated systemic T cell responses in vivo. Furthermore, banNVs were more effective than single adjuvant combined with antigen peptide immunization (Fig. 5, A and B), potentially attributed to synergistic activation of two TLR pathways. As a result of banNV stimulation, the expression of PD-1 on Adpgk-specific CD8+ T cells increased by eight times relative to total CD8+ T cells, suggesting exhaustion of these T cells (Fig. 5, C and D, and fig. S8). Next, C57BL/6 mice immunized with different formulations of vaccines were challenged to study antitumor immunity. MC38 tumor cells were inoculated subcutaneously 7 days after the last immunization (fig. S9A). Mice treated with banNVs had no detectable tumor masses during 12 days after inoculation, indicating antitumor efficacy. In contrast, soluble vaccines failed to protect any mice from progression (Fig. 5E and fig. S9B). After immunization with vaccines on days 0, 2, and 4, no signs of splenomegaly or systemic inflammation were observed relative to soluble vaccines (Fig. 5F and fig. S10).
Fig. 5. BanNVs potentiated the neoantigen immunogenicity in mice.
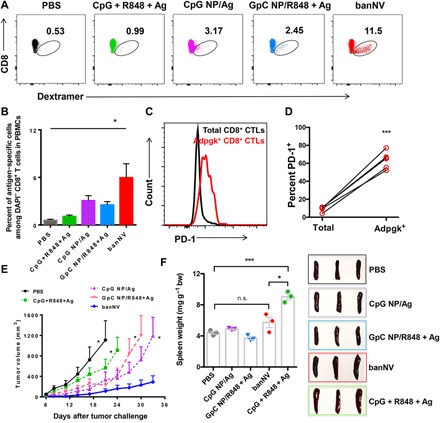
(A) Representative flow cytometric analysis and (B) quantification of the frequency of peripheral Ag (Adpgk)–specific CD8+ T cells from C57BL/6 mice vaccinated with soluble CpG + R848 + Ag, CpG NP/R848, GpC NP/R848 + Ag, and banNVs, respectively, on days 0, 7, and 14. Blood analysis was conducted on day 21 using an H-2Db–ASMTNMELM dextramer (n = 5 mice per group). (C) Representative flow cytometry results and (D) quantification of PD-1 expression on peripheral CD8+ T cells (n = 5 mice per group). (E) Tumor growth curve of C57BL/6 mice subcutaneously challenged with MC38 cells (0.3 × 106) (n = 5 mice per group) 21 days after the first immunization (CpG equivalents: 2 nmol, R848: 8 μg, and Adpgk: 20 μg). (F) Photographs and quantification of spleen/body weight ratios of mice on day 6 after vaccination on days 0, 2, and 4 (n = 3 mice per group). All error bars show SEM. Data are represented as means ± SEM. *P < 0.05 and ***P < 0.001 (one-way ANOVA with Bonferroni post hoc test and Student’s t test). The definitions of the abbreviations are listed in table S2.
Combination of banNVs and aPD-1 for the immunotherapy of colorectal cancer
To validate the therapeutic efficacy of banNVs, C57BL/6 mice were inoculated with Adpgk-specific MC38 cells on day 0. Mice were treated on day 9 with PBS, CpG + R848 + Adpgk, CpG NPs/Adpgk, GpC NPs/R848 + Adpgk, and banNVs. Tumor growth was measured every 3 days until day 39. Soluble vaccines delayed tumor growth relative to the control group. CpG NPs/Adpgk and GpC NPs/R848 + Adpgk showed a similar effect and were superior to soluble vaccines. In contrast, CpG NPs/R848 + Adpgk substantially potentiated the immunotherapeutic efficacy and prolonged mouse survival (Fig. 6A), with approximately 40% of mice survival in 39 days. Although banNVs did not lead to complete regression of tumor, likely due to the immunosuppression mediated by pathways such as PD-1 in CD8+ T cells, combined banNVs with aPD-1 further promoted the tumor therapy efficacy (Fig. 6, B and C). Consistently, the medium tumor progression rate of mice treated with banNV is 3.65 [−1, 6.64], which is superior to that of single adjuvant together with neoantigen (table S3). Tumor growth was suppressed in mice treated with aPD-1 + banNVs, which also increased survival rate to 70% on day 48, compared with a 40% of survival rate by banNVs alone (Fig. 6, A to C). We next evaluated whether immune cell depletion influences the efficacy of banNV (Fig. S11). Our finding showed that depletion of CD8+ T cells leads to nearly complete failure of anticancer ability of banNVs. Instead, depletion of CD4+ T cells and natural killer 1.1 (NK1.1) cells did not alter the therapeutic efficacy of banNVs, suggesting that CD8+ T cell is required for immunotherapy of banNVs against MC38 tumors (Fig. 6D). Similarly, MC38 tumors grew equally well in mice treated with banNVs combined with aPD-1 after CD8+ T cell depletion, demonstrating the central role of CD8+ T cells (Fig. 6E).
Fig. 6. Combination of banNVs with immune checkpoint blockade markedly promoted the response rates and complete regression rates of MC38 tumors in syngeneic mice.
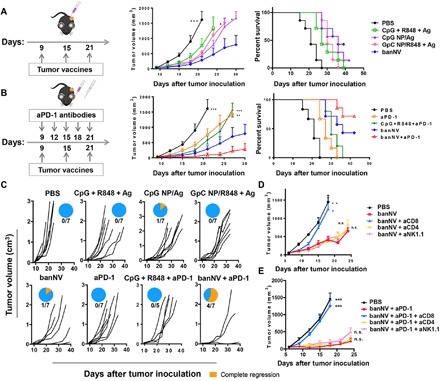
(A) Left: Experimental design for tumor immunotherapy in C57BL/6 mice with indicated formulations of vaccines (CpG: 2 nmol, R848: 8 μg, and Adpgk: 20 μg); middle: tumor growth curve, and right: mouse survival of C57BL/6 mice after subcutaneous inoculation with MC38 tumor cells (0.3 × 106) (n = 6 to 7 mice per group). (B) Left: Experimental design for combination tumor immunotherapy in C57BL/6 mice with banNVs and aPD-1 (CpG: 2 nmol, R848: 8 μg, Adpgk: 20 μg, and aPD-1: 200 μg); middle: tumor growth curve; and right: mouse survival of C57BL/6 mice after subcutaneous inoculation with MC38 tumor cells (0.3 × 106) (n = 5 to 7 mice per group). (C) Individual tumor growth and survival profile of C57BL/6 mice treated with vaccines and/or aPD-1 (CpG: 2 nmol, R848: 8 μg, Adpgk: 20 μg, and aPD-1: 200 μg) during 40 days (n = 5 to 7 mice per group). (D and E) Tumor growth curve after vaccination with banNVs or combination of banNV and aPD-1, together with lymphocyte depletion by anti-CD8, anti-CD4, or anti-NK1.1 (200 μg). All error bars show SEM. Data are represented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 (one-way ANOVA with Bonferroni post hoc test and Student’s t test).
DISCUSSION
Cancer immunotherapy has made remarkable breakthrough in a growing number of cancers. However, there are many notable exceptions of cancers that respond poorly to current immunotherapies. Therapeutic tumor vaccine has emerged as a powerful therapy strategy via codelivery of adjuvants with cancer-specific antigens to stimulate distinct innate immune signaling pathways and activate adaptive immunity such as T cell responses (10). Cancer somatic mutations or frameshifting mutations and the resulting cancer neoantigens have emerged as central role players in cancer immunotherapy, yet the overwhelming majority of neoantigens lack effective immunogenicity. Water-in-oil adjuvant systems have been conventionally used to improve immunogenicity in clinic, while cumulative evidence revealed T cell sequestration at the vaccination sites, leading to T cell exhaustion and deletion. Also, current delivery approaches based on direct administration of neoantigens along with adjuvants are often restricted by discrepant physicochemical properties and unfavorable pharmacokinetics, which altogether result in insufficient elicitation of antitumor immunity and potential safety concerns. To overcome these drawbacks, new means of therapeutic interventions to improve T cell immunity are sought. Pathogen-mimicking nanovaccines can address these problems through rational design of adjuvants formulated with defined antigen peptides, which can efficiently accumulate in draining LNs (18–20) and be further internalized by distinct APCs to elicit immune responses (21–25). This study established an alternative, delicate, and scalable strategy to develop banNVs, enabling programmable encapsulation and construction of bi-adjuvants (R848, CpG) and a hydrophobic peptide neoantigen (Adpgk), which were prepared by in situ RCA of tandem CpG on PEG-PLA micelles, followed by nanocondensation of CpG MPs by cationic PPT-g-PEG.
Advances in nanoengineering make it possible to codeliver multiple adjuvants and peptide antigens, which can also improve drug solubility and formulation, promote the interactions between immune cells, and elicit durable and potent T cell responses (17, 30). In these banNVs, the innermost core of PLA and the outer shell of hydrophobic PPT were used for physical loading of R848 and Adpgk, respectively, enabling programmable synthesis of multilayer structure-based nanovaccines. In terms of other vaccine delivery approaches, molecular vaccine components were usually linked to albumin tail and polymer scaffold or were encapsulated into nanocarriers independently in preclinical studies. However, disparate pharmaceutical properties and redundant chemical appendages restricted their further clinical application. BanNVs provide new insights into overcoming these challenges of pharmaceutical discrepancies between different forms of tumor specific vaccine agents and increased safety concerns, which may protract the time window of medication required for cancer treatment. Furthermore, banNVs presented a framework for future immunotherapy with prolonged retention time in LNs, tailor-designed codelivery of adjuvants and peptide neoantigens, tunable drug release, and notable antitumor efficacy especially when combined with immune checkpoint blockade. We anticipate that this platform will influence a broad range of efforts in heterogeneous nanovaccine delivery, with tremendous prospect to play a substantial role in the ongoing revolution of next-generation cancer immunotherapy.
TLRs play a vital role in cancer immunotherapy especially when combined with other therapeutic agents (i.e., tumor antigens, antibodies, and chemotherapeutic drugs) (40). TLR agonists activate immune response by stimulating distinct populations of APCs including all major DC subsets to up-regulate costimulatory factors (CD80, CD86, and CD40), secrete cytokines (TNF, IL-6, IL-12, etc.), and present antigens, thus enhancing the innate immune response and driving the subsequent adaptive immune response (17). TLR9a CpG is used as model adjuvant in numerous preclinical trials, while the limited endosomal localization of TLR9 in DCs and B cells in humans restricted the therapeutic efficacy of CpG molecules in patients (29). Formulations combining different immunostimulants from a large panel of adjuvants can act synergistically, representing a potent strategy to develop cancer vaccines (41). Here, we incorporated TLR7/8a (R848) into CpG molecules’ self-assembled nanoparticle. Costimulatory molecules and cytokine production elaborated the priority of combination use of both TLR7/8a and TLR9a, which is probably due to (i) the engagement of TLR7/8 on DCs as well as macrophages and NK cells and (ii) the main pathway “TLR7-MyD88-IRF7” played by TLR7/8a for the production of type I interferons (42). Our results of markedly higher uptake of vaccines especially the model antigens (SIINFEKL) in macrophages may partially explain the additional mechanistic insights into high-magnitude immune activation induced by banNVs. Although the mode is not completely understood, our study extended the earlier findings that combination of TLR7/8a and TLR9a can enhance the immunogenicity of neoantigen and showed that nanoparticulate vaccines efficiently improve the retention of neoantigen in draining LNs and the uptake of dual adjuvants by APCs, whereas additional studies need to be performed to carefully clarify the underlying mechanisms of banNVs.
Despite the involvement of TLRa, our study indicated that aPD-1 also plays a vital role in enhancing the immunogenicity of neoantigen. It has been shown that the distinct tumor therapeutic efficacy of immune checkpoint inhibitors may have also improved the therapeutic efficacy of neoantigen nanovaccines by boosting immune response (26). PD-1 engagement mediates T cell receptor (TCR) signaling components with CD28 as the primary target. aPD-1 treatment blocks the signaling pathway of TCR/CD28 and increased the secretion of associated cytokines. In line with our study, Khleif’s recent study showed that simultaneous treatment of cancer vaccines and aPD-1 invigorated CD8+ T cells during the immune induction phase. However, aPD-1 treatment before vaccine treatment restrained CD8+ T cell priming and restricted the therapeutic efficacy of aPD-1 due to the induction of PD-1+CD38+CD8+ T cells (43).
Although we are intrigued by the results of neoantigen-specific immune responses induced by banNVs, there were still a fraction of tumor-bearing mice that could not be cured even by combined banNVs and immune checkpoint blockade, for which many factors should be taken into account. One of the prevailing notions underlying this phenomenon is that tumor immunologic heterogeneity influences immune response to cancer vaccines (44). Although the precise mechanisms of immunologic heterogeneity interfering with immune response against neoantigen-specific malignant cancers are not fully understood, we speculate the existence of immunologic heterogeneity that parallels the genetic and environmental changes in immunogenic mice. The current challenge in evaluating the potential contribution of heterogeneity as an incentive of tumor progression is the fact that the small size of preclinical studies cannot fully recapitulate the condition under which tumor heterogeneity is driven and evolved in humans. We surmise that it would be worthwhile to increase the numbers of model mice that will or will not benefit from neoantigen-based cancer vaccines to further interpret tumor heterogeneity against immunotherapy. Moreover, it is conceivable that new technologies including genome editing might be applied in tumor immunologic heterogeneity modeling to study the factors that influence responses to cancer immunotherapy (45). Furthermore, although we make a prediction here that neoantigens derived from indicated tumor cells can potentiate T cell receptor recognition and T cell activation for neoantigen-specific tumor immunotherapy, it should be kept in mind that the efficient delivery of neoantigen does not equal the effective T cell response. Thus, there will be cases in which despite higher immunity elicited by banNVs, neoantigen-specific T cell cytotoxicity is limited or absent due to lack of MHC expression on tumor cells.
Together, this study suggests the great potential of banNVs to potentiate the immunogenicity of cancer neoantigens for personalized tumor combination immunotherapy.
MATERIALS AND METHODS
Experimental design
This study aimed to develop a banNV to enhance immunogenicity against colorectal cancer. For this, dual adjuvants (TLR7/8 agonist: R848 and TLR9 agonist: CpG) and MC38 colorectal cancer cell–specific neoantigen (Adpgk) were programmably assembled and strategically encapsulated into multistructured nanoparticles, enabling efficient codelivery of adjuvants and neoantigens. To validate the immune activation induced by banNVs, in vitro APC stimulation and antigen presentation experiments and additional in vivo tumor prophylaxis and immunotherapy using MC38 tumor mouse models were designed. The number of mice per experimental group is indicated in the respective figure legends.
Materials
MAL-PEG3000-b-PLA2000 was purchased from JenKem Technology Co. Ltd. All DNA sequences, fluorophore-modified CpG sequences, and thiol-modified primer were synthesized in Integrated DNA Technologies. T4 DNA ligase (4000 U/μl), phi29 DNA polymerase (10 U/μl), deoxynucleotide triphosphate (dNTP) solution mix (10 mM each nt), and 10× bovine serum albumin (BSA) in buffer solution were all purchased from New England Biolabs, and Cy5-modified dUTP was purchased from Enzo Life Sciences. R848 was purchased from Sigma-Aldrich. aPD-1 antibody was purchased from Bio X Cell Inc. All the enzyme-linked immunosorbent assay (ELISA) kits used in this study were purchased from Thermo Fisher Scientific. Staining antibodies including anti–CD8-APC-Cy7, PD1-BV421, F4/80-PE (phycoerythrin), and CD11c-APC were purchased from BioLegend. Anti–CD80-PerCP-Cy5.5 and anti–CD86-PE were purchased from PeproTech. Anti-mouse SIINFEKL/H-2Kb-PE antibody was purchased from eBioscience. Dextramer-PE was purchased from Immudex. MC38 cell line was purchased from ATCC and cultured in Dulbecco’s modified Eagle’s medium, and DC2.4 cells were cultured in RPMI 1640 medium with 2 mM l-glutamine.
Preparation and characterization of primer-PEG-PLA NPs
In a typical experiment, primer-PEG-PLA nanoparticles were synthesized using a two-step approach: (i) self-assembly of amphiphilic PEG-PLA micelles and (ii) surface modification of primer DNA inspired by chemical conjugation. First, MAL-PEG3000-b-PLA2000 can spontaneously self-assemble into amphiphilic micelles in water. After assembly, primer-PEG-PLA conjugates were synthesized using MAL-PEG3000-b-PLA2000 and terminal thiol-modified DNA primer. Thiol-modified DNA primer (0.06 mM) was pretreated with dithiothreitol (DTT; 0.1 M) in 1 × PBS for 1 hour at 37°C to cleave the dithiol bond, and then NAP5 columns were used to purify the DNA primer by removing the DTT and cleaved thiol-appending fragments in 1% sodium ascorbate buffer. The cleaved DNA products (15 μM) were mixed and reacted with PEG-PLA micelles dissolved in 1 × PBS for 30 min at room temperature. The unreacted MAL-PEG3000-b-PLA2000 was subsequently removed using a C18 column in reversed-phase HPLC (Thermo Fisher Scientific). The final products named primer-PEG-PLA conjugates were then quantified by UV absorbance at 260 nm using a Genesys 10S UV-Vis spectrometer. Primer-PEG-PLA conjugation was determined by running 2% agarose gel electrophoresis containing 20% GelRedTM in tris-acetate-EDTA buffer at 100 V. The resulting agarose gel was visualized using a UV transilluminator. The morphology of primer-PEG-PLA nanoparticles was observed using TEM images collected on a Jeol JEM 2010 electron microscope at 300 kV. DLS of primer-PEG-PLA NPs was conducted using a Delsa Nano C instrument.
Polymerization and condensation of CpG microflowers (MFs)
To synthesize the CpG MFs, 5′-phosphorylated linear CpG templates (12 μM) were mixed with 20 μl of primer-PEG-PLA nanoparticles (10 μM). Ligation buffer (1× DNA) was added to the sample to reach a total volume of 40 μl. The mixed solution was heated at 95°C for 5 min and cooled to 25°C gradually in 1.5 hours. One microliter of T4 DNA ligase (4000 U/μl) was added to the above mixed solution and incubated at room temperature for 3 hours. The nick in the hybridized CpG templates was then permanently closed. Subsequently, an in situ RCA assembly was conducted on the surface of circularized primer-PEG-PLA NPs. Rolling circle replication (RCR) reaction system contained ligated circular CpG templates (0.3 μM), phi29 DNA polymerase (1000 U/μl), 2 mM dNTP, and 1 × BSA in DNA polymerization buffer solution with a total volume of 80 μl. RCA reaction was carried out at 30°C for 48 hours. All the enzymes were heat-inactivated at 90°C for 15 min to terminate the reaction. For further application, Cy5 was integrated into CpG MFs by adding Cy5-modified dUTP (0.075 mM) into the RCA reaction system. CpG amplification folds were quantified by measuring the UV absorbance at 260 nm. Then, CpG MFs were used to encapsulate hydrophobic adjuvants. CpG MFs (50 μM CpG equivalent) were prepared in PBS (1 ml). To the solution, R848 (1 mM) in 500 μl of DMF solution was added. The reaction mixture was transferred into an Eppendorf vial and stirred for 1 hour at room temperature. Subsequently, DMF and free small molecular adjuvants were removed by centrifugation (13,500 rpm for 10 min). The resulting products were washed with PBS twice and resuspended in PBS before use. Afterward, MPs were condensed into NPs by incubating MPs with a specific designed cationic polypeptide (PPT-g-PEG). The loading efficiency of antigen peptides in banNVs was confirmed by FITC-modified model antigen and analyzing the fluorescence of banNV, while the loading efficiency of R848 was measured by using HPLC. In a typical procedure, MPs were then dissolved in 1 ml of PBS, and PPT-g-PEG (2.5 mg/ml) in PBS (0.5 ml) was added into MP solution on a vortex for 24 hours. Condensation was terminated by centrifugation to remove the unreacted supernatant, followed by washing with PBS twice. Furthermore, Adpgk peptides (1 mM) were added to obtained NPs to get the final banNVs by washing them with PBS twice. The sizes and morphologies of MPs and NVs were characterized using TEM and SEM (Hitachi SU-70). DLS of MFs and NVs was measured as indicated above. Fluorescence imaging of Cy5-labeled NVs was monitored using a Zeiss LSM 780 confocal microscope.
DC activation and antigen presentation
DC2.4 cells were plated at 96-well plates. After 24 hours, DCs were incubated with 200 nM CpG and/or 500 nM R808 in various formulations in complete media for 48 hours. Some costimulatory factors on DCs including CD80 and CD86 were analyzed by flow cytometry. DCs incubated with adjuvants were harvested after 48 hours and resuspended and stained with anti–CD80-PerCP-Cy5.5 and anti–CD86-PE in 100 μl of PBS for 1 hour. DCs were then washed three times with PBS for flow cytometry using a BD Beckman Coulter flow cytometer. Proinflammatory factors such as IL-6, IL-12p40, and TNF-α secreted from DCs were evaluated by ELISA kits. Immature DCs were plated at 96-well plates. After 24 hours, DCs were incubated with 200 nM CpG and/or 500 nM R848 in various formulations in complete media for 48 hours at 37°C with 5% CO2. DCs were harvested, washed with FACS buffer, and then stained on ice with fluorophore-labeled antibodies against CD80 and CD86. Cell culture medium of DCs incubated with adjuvants for 48 hours were collected and diluted according to the ELISA instruments. Cytokine secretion quantification was also determined using the ELISA manufacturer’s instruments. In vitro antigen presentation was first studied using the Zeiss LSM 780 confocal microscope. CpG NP/CSIINFEK(FITC)L or soluble CSIINFEK(FITC)L was incubated in DC2.4 cells for a series of time points. DC2.4 cells were then stained with LysoTracker Red and Hoechst 33342 for regular fluorescence imaging. Furthermore, DC2.4 cells were incubated with CpG NP/SIINFEKL as mentioned above. After incubation, DC2.4 cells were stained with PE-labeled anti-SIINFEKL/H-2Kb antibody for 1 hour on ice and then analyzed using a BD Beckman Coulter flow cytometer.
LNs draining of banNVs and functional assessment of antigen-specific T cells
For LN delivery studies, C57BL/6 mice were injected with soluble Cy5-CPG + CSIINFEK(FITC)L and Cy5-CPG NP/CSIINFEK(FITC)L. After 12 hours, inguinal LNs were harvested, and a regular LNs fluorescence imaging was conducted using Maestro optical imaging system. Subsequently, LNs were dissociated in collagenase D and DNase I according to the manufacturer’s instructions. Single cells were collected after dissociation and then stained with CD11c for DCs and F4/80 for macrophages. In vivo delivery of banNVs into APCs was analyzed by a BD Beckman Coulter flow cytometer. For assessment of the elicited antigen-specific T cell responses, C57BL/6 mice were injected with different formulations of vaccines on days 0, 7, and 14. Peripheral blood cells were collected on day 21. First, red blood cells were lysed using ACK lysis buffer for 5 min and then blood clot was removed by gentle centrifugation. Next, the left cells were stained with 4′,6-diamidino-2-phenylindole in fetal calf serum buffer for 10 min to block the Fc segment. Then, cells were stained with mixed antibodies including anti–CD8-APC-Cy7, dextramer-PE (Immudex), and PD1-BV421 for 1 hour on ice. Cells were subsequently washed three times and resuspended in Cytofix for 20 min at 4°C. Last, cells were washed with Perm/Wash buffer twice for flow cytometry quantification using a BD LSRFortessa X-50 flow cytometer.
In vivo cancer immunotherapy and tumor challenge studies
All work conducted on animals were cared for following the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals under a protocol approved by the NIH Clinical Center Animal Care and Use Committee. Female C57BL/6 mice (6 to 8 weeks) were purchased from the Jackson Laboratory. For cancer immunotherapy studies, C57BL/6 mice were subcutaneously injected with 0.1 × 106 MC38 cells on the right shoulder. On day 9, C57BL/6 mice were immunized with different formulations containing antigen peptides (20 μg per mouse), CpG (2 nmol per mouse), and R848 (8 μg per mouse) in 100-μl volume by subcutaneous injection at the tail base on days 9, 15, and 21. For the combinatorial immunotherapy, anti-mouse PD-1 (200 μg per mouse) antibodies were administered intraperitoneally on days 9, 12, 15, 18, and 21. Tumor growth was monitored every 3 days, and the tumor volume throughout this study was calculated by caliper measurement using the following equation: tumor volume = length × width2/2. Animals were euthanized when the maximum tumor diameter reached 1.8 cm or more. For prophylaxis studies, C57BL/6 mice (6 to 8 weeks) were immunized with tumor vaccines on days 0, 7, and 14. Vaccinated mice were challenged 6 days later after last immunization by inoculation with 0.3 × 106 MC38 cells on the right shoulder per mouse. Tumor growth was monitored as indicated above. Mice were euthanized if the tumor diameter exceeded 1.8 cm.
Systemic toxicity evaluation was assessed by measuring the splenomegaly
C57BL/6 mice were administered with different formulations of vaccines (antigen peptides: 20 μg, CpG: 2 nmol, and R848: 8 μg) on days 0, 2, and 4. One day after last treatment, spleens and LNs were harvested from mice and then weighed and photographed. Fixed spleen and LN tissues were used for paraffin embedding and slide scanning. Fixed LN tissues were frozen for slide preparation and immunofluorescence staining.
Statistical analysis
On the basis of the pilot study results and literature, four to five mice per group were used for T cell response evaluation and six to seven mice per group were used for cancer immunotherapy study. All values shown in the manuscript were means ± SD. Comparisons between two groups or several groups were analyzed using unpaired Student’s t test or one-way analysis of variance (ANOVA), followed by Bonferroni post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001 were considered statistically significant. All statistical analyses were carried out using Prism 7.0 (GraphPad Software, La Jolla).
Supplementary Material
Acknowledgments
We thank the Flow Cytometry Core Facility at the Vaccine Research Center (VRC) of the National Institute of Allergy and Infectious Diseases (NIAID) for the use of flow cytometer and the provision of MC38 cells from the Dr. Robert Seder Laboratory at VRC. We also thank V. Schram and L. Holtzclaw of Eunice Kennedy Shriver National Institute of Child Health (NICHD) for the use of the confocal microscope. Funding: This research was supported partially by the Intramural Research Program (IRP) of the NIBIB, NIH (1ZIAEB000073-09); the National Key Basic Research Program of the PRC (2014CB744501 and 2014CB744504), Key projects of the National Natural Science Foundation of China (81230032), and the Major International (Regional) Joint Research Program of China (81120108013); start-up funds from the VCU School of Pharmacy and NCATS Clinical and Translational Science Award (CTSA) (no. KL2TR002648 under UL1TR002649); and a pilot grant from VCU Massey Cancer Center (P30 CA106059). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. Author contributions: Q.N. and G.Z. designed the project. Q.N., F.Z., Y.L., Z.W., G.Y., B.L., and G.Z. performed the experiments. Q.N., F.Z., and G.Z. interpreted and analyzed data. Q.N., F.Z., G.N., T.S., G.Z., G.L., L.Z., and X.C. cowrote the paper. G.Z., G.L., L.Z., and X.C. supervised all studies. Competing interests: X.C. has a patent currently pending (issuing agency: China National Intellectual Property Administration; filing date: 4 Jan 2020; application no: 202010007226.X; patent authors: Q.N. and X.C.). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaaw6071/DC1
Supplementary Materials and Methods
Fig. S1. Characteristics of primer-PEG-PLA conjugates.
Fig. S2. TEM images of CpG MPs.
Fig. S3. Fluorescence confocal microscopy images of CpG MPs labeled with Cy5 via Cy5-dUTP.
Fig. S4. Co-stimulatory factors expression on DCs.
Fig. S5. Photographs and H&E staining slice images of inguinal lymph nodes.
Fig. S6. Immunofluorescence images of LNs (10×) collected on day 6 from C57BL/6 mice treated with PBS, soluble forms of CpG + R848 + Ag (Adpgk), CpG NP encapsulated with Ag (Adpgk), GpC NP encapsulated with R848 and Ag (Adpgk), and banNVs (CpG: 2 nmol, R848: 8 μg per mouse, Adpgk: 20 μg) on days 0, 2, and 4 (n = 3 mice per group).
Fig. S7. Scheme of dextramer staining and gating tree flow cytometric analysis.
Fig. S8. PD-1 median intensity of fluorescence gated from PD-1+ CD8+ peripheral T cells.
Fig. S9. Tumor challenge of C57BL6 mice after immunization of banNVs.
Fig. S10. Hematoxylin and eosin (H&E) staining of the spleen sections (10×) on day 6 from C57BL/6 mice (with photographs of spleens inserted) treated with PBS, soluble forms of CpG + R848 + Ag (Adpgk), CpG NP encapsulated with Ag (Adpgk), GpC NP encapsulated with R848 and Ag (Adpgk), and banNVs (CpG: 2 nmol, R848: 8 μg per mouse, Adpgk: 20 μg) on days 0, 2, and 4.
Fig. S11. Experimental design of immune depletion study.
Table S1. DNA sequences.
Table S2. Definition of abbreviations used in the manuscript.
Table S3. Tumor progression rate and regression rate.
REFERENCES AND NOTES
- 1.Rosenberg S. A., Restifo N. P., Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mellman I., Coukos G., Dranoff G., Cancer immunotherapy comes of age. Nature 480, 480–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu Y., Liu Q., Wei J., Liu B., Personalized cancer neoantigen vaccines come of age. Theranostics 8, 4238–4246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irvine D. J., Hanson M. C., Rakhra K., Tokatlian T., Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 115, 11109–11146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swartz M. A., Hirosue S., Hubbell J. A., Engineering approaches to immunotherapy. Sci. Transl. Med. 4, 148rv149 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Neelapu S. S., Locke F. L., Bartlett N. L., Lekakis L. J., Miklos D. B., Jacobson C. A., Braunschweig I., Oluwole O. O., Siddiqi T., Lin Y., Timmerman J. M., Stiff P. J., Friedberg J. W., Flinn I. W., Goy A., Hill B. T., Smith M. R., Deol A., Farooq U., McSweeney P., Munoz J., Avivi I., Castro J. E., Westin J. R., Chavez J. C., Ghobadi A., Komanduri K. V., Levy R., Jacobsen E. D., Witzig T. E., Reagan P., Bot A., Rossi J., Navale L., Jiang Y., Aycock J., Elias M., Chang D., Wiezorek J., Go W. Y., Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stephan S. B., Taber A. M., Jileaeva I., Pegues E. P., Sentman C. L., Stephan M. T., Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol. 33, 97–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gubin M. M., Zhang X., Schuster H., Caron E., Ward J. P., Noguchi T., Ivanova Y., Hundal J., Arthur C. D., Krebber W.-J., Mulder G. E., Toebes M., Vesely M. D., Lam S. S. K., Korman A. J., Allison J. P., Freeman G. J., Sharpe A. H., Pearce E. L., Schumacher T. N., Aebersold R., Rammensee H.-G., Melief C. J., Mardis E. R., Gillanders W. E., Artyomov M. N., Schreiber R. D., Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strønen E., Toebes M., Kelderman S., van Buuren M. M., Yang W., van Rooij N., Donia M., Böschen M.-L., Lund-Johansen F., Olweus J., Schumacher T. N., Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Sahin U., Derhovanessian E., Miller M., Kloke B.-P., Simon P., Lower M., Bukur V., Tadmor A. D., Luxemburger U., Schrörs B., Omokoko T., Vormehr M., Albrecht C., Paruzynski A., Kuhn A. N., Buck J., Heesch S., Schreeb K. H., Müller F., Ortseifer I., Vogler I., Godehardt E., Attig S., Rae R., Breitkreuz A., Tolliver C., Suchan M., Martic G., Hohberger A., Sorn P., Diekmann J., Ciesla J., Waksmann O., Bruck A. K., Witt M., Zillgen M., Rothermel A., Kasemann B., Langer D., Bolte S., Diken M., Kreiter S., Nemecek R., Gebhardt C., Grabbe S., Holler C., Utikal J., Huber C., Loquai C., Tureci O., Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ott P. A., Hu Z., Keskin D. B., Shukla S. A., Sun J., Bozym D. J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., Chen C., Olive O., Carter T. A., Li S., Lieb D. J., Eisenhaure T., Gjini E., Stevens J., Lane W. J., Javeri I., Nellaiappan K., Salazar A. M., Daley H., Seaman M., Buchbinder E. I., Yoon C. H., Harden M., Lennon N., Gabriel S., Rodig S. J., Barouch D. H., Aster J. C., Getz G., Wucherpfennig K., Neuberg D., Ritz J., Lander E. S., Fritsch E. F., Hacohen N., Wu C. J., An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran E., Robbins P. F., Rosenberg S. A., ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 18, 255–262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuai R., Yuan W., Son S., Nam J., Xu Y., Fan Y., Schwendeman A., Moon J. J., Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv. 4, eaao1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H., Moynihan K. D., Zheng Y., Szeto G. L., Li A. V., Huang B., Van Egeren D. S., Park C., Irvine D. J., Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu G., Lynn G. M., Jacobson O., Chen K., Liu Y., Zhang H., Ma Y., Zhang F., Tian R., Ni Q., Cheng S., Wang Z., Lu N., Yung B. C., Wang Z., Lang L., Fu X., Jin A., Weiss I. D., Vishwasrao H., Niu G., Shroff H., Klinman D. M., Seder R. A., Chen X., Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat. Commun. 8, 1954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ilyinskii P. O., Roy C. J., O’Neil C. P., Browning E. A., Pittet L. A., Altreuter D. H., Alexis F., Tonti E., Shi J., Basto P. A., Iannacone M., Radovic-Moreno A. F., Langer R. S., Farokhzad O. C., von Andrian U. H., Johnston L. P., Kishimoto T. K., Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine 32, 2882–2895 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lynn G. M., Laga R., Darrah P. A., Ishizuka A. S., Balaci A. J., Dulcey A. E., Pechar M., Pola R., Gerner M. Y., Yamamoto A., Buechler C. R., Quinn K. M., Smelkinson M. G., Vanek O., Cawood R., Hills T., Vasalatiy O., Kastenmüller K., Francica J. R., Stutts L., Tom J. K., Ryu K. A., Esser-Kahn A. P., Etrych T., Fisher K. D., Seymour L. W., Seder R. A., In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 33, 1201–1210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo M., Wang H., Wang Z., Cai H., Lu Z., Li Y., Du M., Huang G., Wang C., Chen X., Porembka M. R., Lea J., Frankel A. E., Fu Y.-X., Chen Z. J., Gao J., A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 12, 648–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan Y., Kuai R., Xu Y., Ochyl L. J., Irvine D. J., Moon J. J., Immunogenic cell death amplified by co-localized adjuvant delivery for cancer immunotherapy. Nano Lett. 17, 7387–7393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melero I., Gaudernack G., Gerritsen W., Huber C., Parmiani G., Scholl S., Thatcher N., Wagstaff J., Zielinski C., Faulkner I., Mellstedt H., Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 11, 509–524 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Moon J. J., Huang B., Irvine D. J., Engineering nano- and microparticles to tune immunity. Adv. Mater. 24, 3724–3746 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu K., He C., Guo N., Chan C., Ni K., Weichselbaum R. R., Lin W., Chlorin-based nanoscale metal-organic framework systemically rejects colorectal cancers via synergistic photodynamic therapy and checkpoint blockade immunotherapy. J. Am. Chem. Soc. 138, 12502–12510 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee I.-H., Kwon H.-K., An S., Kim D., Kim S., Yu M. K., Lee J.-H., Lee T. S., Im S.-H., Jon S., Imageable antigen-presenting gold nanoparticle vaccines for effective cancer immunotherapy in vivo. Angew. Chem. Int. Ed. Engl. 51, 8800–8805 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Gao S., Yang D., Fang Y., Lin X., Jin X., Wang Q., Wang X., Ke L., Shi K., Engineering nanoparticles for targeted remodeling of the tumor microenvironment to improve cancer immunotherapy. Theranostics 9, 126–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuai R., Ochyl L. J., Bahjat K. S., Schwendeman A., Moon J. J., Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seidel J. A., Otsuka A., Kabashima K., Anti-PD-1 and anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 8, 86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Neill L. A., Golenbock D., Bowie A. G., The history of Toll-like receptors-redefining innate immunity. Nat. Rev. Immunol. 13, 453–460 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Vacchelli E., Galluzzi L., Eggermont A., Fridman W. H., Galon J., Sautès-Fridman C., Tartour E., Zitvogel L., Kroemer G., Trial watch: FDA-approved Toll-like receptor agonists for cancer therapy. Oncoimmunology 1, 894–907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guy B., The perfect mix: Recent progress in adjuvant research. Nat. Rev. Microbiol. 5, 505–517 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Pockros P. J., Guyader D., Patton H., Tong M. J., Wright T., McHutchison J. G., Meng T.-C., Oral resiquimod in chronic HCV infection: Safety and efficacy in 2 placebo-controlled, double-blind phase IIa studies. J. Hepatol. 47, 174–182 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Kasturi S. P., Skountzou I., Albrecht R. A., Koutsonanos D., Hua T., Nakaya H. I., Ravindran R., Stewart S., Alam M., Kwissa M., Villinger F., Murthy N., Steel J., Jacob J., Hogan R. J., García-Sastre A., Compans R., Pulendran B., Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470, 543–547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suma T., Cui J., Müllner M., Fu S., Tran J., Noi K. F., Ju Y., Caruso F., Modulated fragmentation of proapoptotic peptide nanoparticles regulates cytotoxicity. J. Am. Chem. Soc. 139, 4009–4018 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Skakuj K., Wang S., Qin L., Lee A., Zhang B., Mirkin C. A., Conjugation chemistry-dependent T-cell activation with spherical nucleic acids. J. Am. Chem. Soc. 140, 1227–1230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shopsowitz K. E., Roh Y. H., Deng Z. J., Morton S. W., Hammond P. T., RNAi-microsponges form through self-assembly of the organic and inorganic products of transcription. Small 10, 1623–1633 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yadav M., Jhunjhunwala S., Phung Q. T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T. K., Fritsche J., Weinschenk T., Modrusan Z., Mellman I., Lill J. R., Delamarre L., Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Decker T., Schneller F., Sparwasser T., Tretter T., Lipford G. B., Wagner H., Peschel C., Immunostimulatory CpG-oligonucleotides cause proliferation, cytokine production, and an immunogenic phenotype in chronic lymphocytic leukemia B cells. Blood 95, 999–1006 (2000). [PubMed] [Google Scholar]
- 37.Saini S. K., Ostermeir K., Ramnarayan V. R., Schuster H., Zacharias M., Springer S., Dipeptides promote folding and peptide binding of MHC class I molecules. Proc. Natl. Acad. Sci. U.S.A. 110, 15383–15388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho N.-H., Cheong T.-C., Min J. H., Wu J. H., Lee S. J., Kim D., Yang J. S., Kim S., Kim Y. K., Seong S.-Y., A multifunctional core-shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat. Nanotechnol. 6, 675–682 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Kaczanowska S., Joseph A. M., Davila E., TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 93, 847–863 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coffman R. L., Sher A., Seder R. A., Vaccine adjuvants: Putting innate immunity to work. Immunity 33, 492–503 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zitvogel L., Galluzzi L., Kepp O., Smyth M. J., Kroemer G., Type I interferons in anticancer immunity. Nat. Rev. Immunol. 15, 405–414 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Oh N., Park J.-H., Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomedicine 9, 51–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verma V., Shrimali R. K., Ahmad S., Dai W., Wang H., Lu S., Nandre R., Gaur P., Lopez J., Sade-Feldman M., Yizhak K., Bjorgaard S. L., Flaherty K. T., Wargo J. A., Boland G. M., Sullivan R. J., Getz G., Hammond S. A., Tan M., Qi J., Wong P., Merghoub T., Wolchok J., Hacohen N., Janik J. E., Mkrtichyan M., Gupta S., Khleif S. N., PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+ CD38hi cells and anti-PD-1 resistance. Nat. Immunol. 20, 1231–1243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J., Byrne K. T., Yan F., Yamazoe T., Chen Z., Baslan T., Richman L. P., Lin J. H., Sun Y. H., Rech A. J., Balli D., Hay C. A., Sela Y., Merrell A. J., Liudahl S. M., Gordon N., Norgard R. J., Yuan S., Yu S., Chao T., Ye S., Eisinger-Mathason T. S. K., Faryabi R. B., Tobias J. W., Lowe S. W., Coussens L. M., Wherry E. J., Vonderheide R. H., Stanger B. Z., Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49, 178–193.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alizadeh A. A., Aranda V., Bardelli A., Blanpain C., Bock C., Borowski C., Caldas C., Califano A., Doherty M., Elsner M., Esteller M., Fitzgerald R., Korbel J. O., Lichter P., Mason C. E., Navin N., Pe’er D., Polyak K., Roberts C. W., Siu L., Snyder A., Stower H., Swanton C., Verhaak R. G., Zenklusen J. C., Zuber J., Zucman-Rossi J., Toward understanding and exploiting tumor heterogeneity. Nat. Med. 21, 846–853 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaaw6071/DC1
Supplementary Materials and Methods
Fig. S1. Characteristics of primer-PEG-PLA conjugates.
Fig. S2. TEM images of CpG MPs.
Fig. S3. Fluorescence confocal microscopy images of CpG MPs labeled with Cy5 via Cy5-dUTP.
Fig. S4. Co-stimulatory factors expression on DCs.
Fig. S5. Photographs and H&E staining slice images of inguinal lymph nodes.
Fig. S6. Immunofluorescence images of LNs (10×) collected on day 6 from C57BL/6 mice treated with PBS, soluble forms of CpG + R848 + Ag (Adpgk), CpG NP encapsulated with Ag (Adpgk), GpC NP encapsulated with R848 and Ag (Adpgk), and banNVs (CpG: 2 nmol, R848: 8 μg per mouse, Adpgk: 20 μg) on days 0, 2, and 4 (n = 3 mice per group).
Fig. S7. Scheme of dextramer staining and gating tree flow cytometric analysis.
Fig. S8. PD-1 median intensity of fluorescence gated from PD-1+ CD8+ peripheral T cells.
Fig. S9. Tumor challenge of C57BL6 mice after immunization of banNVs.
Fig. S10. Hematoxylin and eosin (H&E) staining of the spleen sections (10×) on day 6 from C57BL/6 mice (with photographs of spleens inserted) treated with PBS, soluble forms of CpG + R848 + Ag (Adpgk), CpG NP encapsulated with Ag (Adpgk), GpC NP encapsulated with R848 and Ag (Adpgk), and banNVs (CpG: 2 nmol, R848: 8 μg per mouse, Adpgk: 20 μg) on days 0, 2, and 4.
Fig. S11. Experimental design of immune depletion study.
Table S1. DNA sequences.
Table S2. Definition of abbreviations used in the manuscript.
Table S3. Tumor progression rate and regression rate.