

Technical errors have resulted in the false discovery of the DNA modification N6-methyladenine in mammals.
Abstract
N6-methyladenine (6mdA) is a widespread DNA modification in bacteria. More recently, 6mdA has also been characterized in mammalian DNA. However, measurements of 6mdA abundance and profiles are often very dissimilar between studies, even when performed on DNA from identical mammalian cell types. Using comprehensive bioinformatics analyses of published data and novel experimental approaches, we reveal that efforts to assay 6mdA in mammals have been severely compromised by bacterial contamination, RNA contamination, technological limitations, and antibody nonspecificity. These complications render 6mdA an exceptionally problematic DNA modification to study and have resulted in erroneous detection of 6mdA in several mammalian systems. Together, our results strongly imply that the evidence published to date is not sufficient to support the presence of 6mdA in mammals.
INTRODUCTION
The covalent attachment of methyl groups to nucleotides, DNA methylation, is a key epigenetic mechanism in mammals. The most common DNA modification in mammals is 5-methylcytosine (5mC), comprising ~3 to 6% of the total cytosines in human DNA (1). 5mC is essential for early mammalian development, playing a central role in lineage-specific gene silencing, X inactivation, genomic imprinting, and suppression of repetitive elements (2, 3). In contrast, 5mC is rare in prokaryotes, in which N6-methyladenine (6mdA) is the most prevalent form of DNA methylation (4). 6mdA is a fundamental component of bacterial restriction-modification systems that allow prokaryotes to distinguish between benign host DNA and potentially pathogenic nonhost DNA (5). Despite the lack of a known restriction-modification system in mammals, the presence of 6mdA has recently been reported in the DNA of mouse and human cells (4, 6–10). Although most studies have reported vanishingly low 6mdA levels in mammals (0.0001 to 0.01% 6mdA/adenine), the range of reported 6mdA abundance between mammalian species and tissues is considerable (0.0001 to 1% 6mdA/dA; range, ~10,000-fold) (4, 6–10). In contrast, several other studies have been unable to detect 6mdA in mammals using highly sensitive methods (11–13). While the function of 6mdA in mammalian DNA is currently unknown, reports have suggested a potential role for 6mdA in lineage specification (8, 9), similar to the role of its RNA counterpart, N6-methyladenosine (m6A) (14). To further dissect the role of 6mdA in lineage specification in mammals, we leveraged the ability to differentiate human naïve T helper (NTH) cells into TH cell subsets in vitro (8, 10, 15). This system has several advantages for studying DNA modifications: (i) TH differentiation is associated with profound epigenetic remodeling; (ii) NTH cells can be isolated in high purity from blood (~98%) and are synchronized in the G1 phase of the cell cycle, reducing the effects of cell heterogeneity; and (iii) the short-term culture of primary cells reduces the risk of cell culture contamination with bacterial species rich in 6mdA.
RESULTS
Artifactual detection of 6mdA by anti-6mdA antibodies
In contrast to previous studies of 5mC and 5-hydroxymethylcytosine (5hmC) (15), no global changes in 6mdA abundance were observed during TH differentiation (Fig. 1A and fig. S1A). Moreover, 6mdA levels in differentiating T cells were identical to that of unmethylated whole-genome-amplified (WGA) DNA (Fig. 1A), suggesting that 6mdA was not present at detectable levels in human T cells. We failed to detect 6mdA in any mouse or human tissue assayed with all samples having 6mdA signals similar to that observed in corresponding WGA DNA lacking 6mdA (Fig. 1B). We next analyzed 6mdA in DNA isolated from six human cell lines and identified two cell lines with 6mdA levels above background (Fig. 1C). Unexpectedly, two separate cultures of the 293T cell line had different 6mdA levels, suggesting that the 6mdA signal observed was not intrinsic to the cell type itself. The two cell lines with elevated 6mdA levels were found to be contaminated with Mycoplasma spp. (Fig. 1C), a common bacterial cell culture contaminant rich in 6mdA (16). Treatment with the mycoplasma-specific antibiotic Plasmocin (17) reduced the 6mdA signal to that of the negative WGA control (fig. S1B), confirming that Mycoplasma spp. were the source of the positive 6mdA signal.
Fig. 1. Artifactual detection of 6mdA in mammalian DNA.
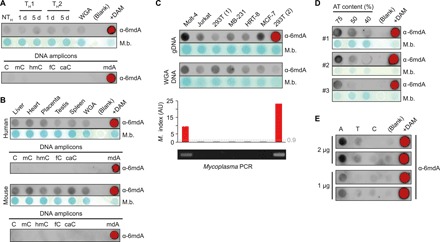
(A to C) Immunodot blot showing relative 6mdA abundance in human CD4+ T cells during in vitro differentiation (A), primary human and mouse DNA (B), and cultured cell lines (top); their WGA controls (middle) and mycoplasma contamination status (bottom). The dotted line indicates threshold for mycoplasma positivity when using the MycoAlert kit (Lonza) (C). gDNA, genomic DNA; AU, arbitrary units. (D) Immunodot blots showing relative signal strength of three unmodified double-stranded DNA amplicons (2 μg) with decreasing adenine content. (E) Immunodot blot showing relative signaling strength of 100-bp single-stranded DNA oligonucleotides of poly-adenine (A), poly-thymine (T), or poly-cytosine (C). (A to E) Anti-6mA antibody: Synaptic Systems, no. 202003. (A to D) M.b., methylene blue. (A and B) As internal controls, 10 ng of modified DNA amplicons (data S2) equivalent to 16 fmol of the indicated modification is shown. (A and B) WGA, WGA CD4+ T cell DNA. (A, B, D, and E) deoxyadenosine methylase–positive (DAM+), human CD4+ T cell DNA treated with bacterial DAM.
Having failed to detect 6mdA in any noncontaminated mammalian DNA, we sought to verify the sensitivity of our approach. First, we confirmed that both 6mdA antibodies tested were highly specific to 6mdA (fig. S1C). Second, we generated a 6mdA positive control by methylating the adenines in all 5′-GATC-3′ sequences in a human genomic DNA sample (fig. S1D), resulting in a global 6mdA abundance of ~0.8% 6mdA/dA. We determined the detection limit of immunodot blot as ~0.003% 6mdA/dA, far below the levels previously reported for several of the tested tissues (fig. S1E). However, 6mdA antibodies consistently gave a clear signal for unmodified WGA DNA, which was not evident for non-6mdA antibodies (fig. S1F), suggesting that 6mdA antibodies have an uncharacterized affinity for unmodified bases. Dot blots of unmodified polymerase chain reaction (PCR) products with increasing AT content suggested an affinity for unmodified adenine (Fig. 1D), consistent with the cross-reactivity of several commercial dA antibodies with 6mdA (e.g., BioVision, no. 6652; Synaptic Systems, no. 202103). Binding of 6mdA antibodies to dA was further confirmed by dot blot of poly-adenine, poly-thymine, and poly-cytosine oligonucleotides, revealing a pronounced preference of 6mdA antibodies for binding to unmodified adenine (Fig. 1E). Specificity to guanine could not be tested as even short [4 base pairs (bp)] poly-guanine sequences form strong secondary structures (guanine tetraplexes), precluding synthesis of poly-guanine oligonucleotides. Given the vanishingly low levels of 6mdA reported in mammalian DNA (4, 18), even a very low affinity for the far more abundant dA would result in detectable signal when using 6mdA antibodies. Given the affinity of 6mdA antibodies for unmethylated DNA, the frequent contamination of cultured mammalian cells with 6mdA-rich bacteria and lack of appropriate controls, the results of immunodot blot determination of 6mdA abundance in mammals is rendered invalid.
Artifactual mapping of 6mdA by DNA immunoprecipitation sequencing
6mdA antibodies are also used in DNA immunoprecipitation sequencing (DIP-seq) to generate genome-wide profiles of 6mdA (8–10, 19). We recently reported major technical errors related to off-target binding of antibodies to DNA repeats in DIP-seq resulting in almost exclusively false-positive signals for 6mdA in vertebrate species Xenopus laevis, Danio rerio, and Mus musculus (20). Comparing published human 6mdA DIP-seq data (7) for paired genomic and WGA DNA revealed highly similar profiles with over 70% of natively enriched 6mdA peaks also found in the WGA sample (Fig. 2A), clearly indicating misidentification of 6mdA. Next, we extended this analysis to 16 distinct 6mdA DIP-seq samples across four independent studies (6, 7, 9, 19), revealing that most of the enriched peaks in all studies were located in repetitive elements overrepresented in both low complexity regions and simple repeats compared to other genomic fractions (52.7- and 27.7-fold, respectively) (fig. S2, A and B), consistent with off-target binding (20). Although studies have suggested localization of 6mdA to long interspersed nuclear elements (LINEs) in mammals (7, 8, 21), our analysis showed highly inconsistent enrichment of 6mdA DIP-seq over LINEs between samples and studies (−8.9- to 3.6-fold over genomic) (fig. S2B), suggesting that localization to LINEs is not a general feature of 6mdA, if present. Furthermore, unlike 5mC, genome-wide enrichment of 6mdA was not associated with LINE1 elements but typically found near assembly gaps, such as centromeres and telomeres (fig. S2C), suggesting that the 6mdA enrichment signal was driven by poor mappability. The 6mdA peaks displayed considerably lower average mappability than 5mC DIP-seq peaks or random genomic regions (Fig. 2B). Collectively, our data show that off-target binding and sequence mismapping are the major determinants of 6mdA profiles in mammals when using DIP-seq.
Fig. 2. Artifactual detection of 6mdA related to mismapping in DIP-seq.
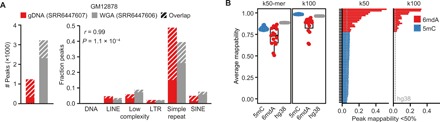
(A) Number of enriched peaks (left) and fraction of peaks located in RepeatMasker repetitive element classes (right) for 6mA DIP-seq samples in genomic (n = 1) or WGA (n = 1) DNA for GM12878 cells. Correlation was calculated using Pearson correlation. LTR, long terminal repeat; SINE, short interspersed nuclear element. (B) Average mappability of peaks using k50- or k100-mers for 5mC (n = 45), 6mA (n = 15), or randomly sampled regions from hg38 (n = 100) (left) and percentage of peaks with mappability <50% for each sample (right). Box plots represent median and first and third quartiles with whiskers extending 1.5 × interquartile range.
Several studies have reported an association between transcription and 6mdA enrichment, which could not be explained by the off-target binding and/or sequence mismapping outlined above (8, 9, 19). Upon visual inspection, we noticed that the reads in some of these samples were located primarily in exons and did not span exon-intron boundaries (Fig. 3A). Surprised by this, we aligned the data using a splice-aware aligner, revealing that up to 20% of all reads at splice junctions in these samples were spliced (Fig. 3B), confirming that the sequenced reads were, in fact, of RNA in origin, not DNA. As antibodies raised against nucleoside modifications cannot distinguish between RNA and DNA (22), cross-contamination of highly abundant nucleic acid modifications, such as RNA m6A, would be a major confounder in 6mdA DIP-seq. The samples containing the highest degree of spliced reads showed preferential enrichment over 3′ untranslated regions (3′UTRs) (fig. S3A) and were enriched for DRACH consensus motifs (fig. S3B), both hallmarks of mRNA m6A profiles (23, 24). RNA contamination is particularly troublesome in the study of 6mdA, as most of the proposed 6mdA methylases and demethylases are known writers and erasers of m6A in RNA (e.g., ALKB and METTL family proteins) (4). Consequently, observations (dot blots and DIP-seq) in functional studies of 6mdA can be a direct result of the methylation/demethylation of RNA, not DNA, rendering it impossible to link observations to mechanisms of adenine methylation on DNA.
Fig. 3. Artifactual detection of 6mdA related to RNA contamination in DIP-seq.
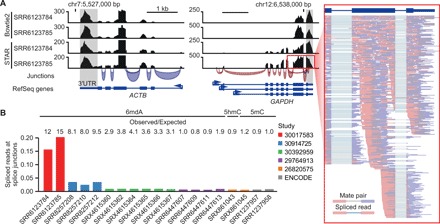
(A) DIP-seq signal tracks for reads aligned with Bowtie2 or splice-aware STAR. Splice junctions correspond to STAR-aligned SRR6123785, colored by strand. The inset shows paired-end read pileups for SRR6123785. (B) Fraction of spliced reads at splice (exon-intron) junctions for 6mdA (n = 15), 5hmC (n = 1), and 5mC (n = 3) DIP-seq samples. Expected fraction calculated per sample based on all reads using bootstrap resampling (n = 10,000).
Artifactual detection of 6mdA in mammalian DNA by mass spectrometry
While immunodot blots are used to measure relative abundances of DNA modifications, mass spectrometry (MS) has been used to quantify global DNA levels of 6mdA. Despite many studies using ultrahigh-performance liquid chromatography (UHPLC)–tandem MS, meta-analysis of published MS data revealed profound disparity (10- to 1000-fold) in 6mdA abundances in mammals, even within the same species and cell type (Fig. 4A), suggesting large study-specific effects. Whereas study explained >70% of the variance in mammalian 6mdA abundance (P = 0.001), neither species (P = 0.7) nor tissue type (P = 0.6) was associated with 6mdA abundance. These observations are consistent with the findings of several recent studies detailing the potential of MS to generate false-positive 6mdA signals in eukaryotic DNA (13, 18, 25). As sample contamination with bacterial material can easily arise from both cell culture infection and reagents produced in bacterial systems (13, 18, 26), study-specific 6mdA identification could perhaps be explained by different reagent batches, as several highly sensitive studies have failed to detect 6mdA in mammalian cells (11–13, 18).
Fig. 4. Artifactual detection of 6mdA in MS and single-molecule real-time sequencing.
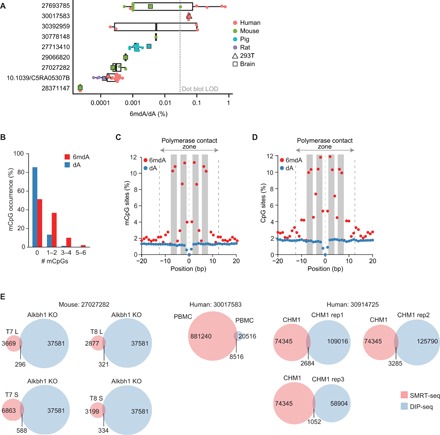
(A) Meta-analysis of published 6mdA abundance (% 6mdA/dA) in mammalian DNA as determined by high performance liquid chromatography–tandem MS for species human (n = 34), mouse (n = 18), pig (n = 11), and rat (n = 7) from nine independent studies. LOD, limit of detection. (B) Number of methylated CpGs (mCpGs) flanking (±12 bp) single-molecule real-time sequencing (SMRT-seq) 6mdA sites in human blood or matched random adenines (N = 881,240; equal chromosomal distribution). (C) mCpG distribution around SMRT-seq 6mdA sites in human blood or matched random adenines (N = 881,240; equal chromosomal distribution). (D) CpG distribution around SMRT-seq 6mdA sites in a human lymphoblastoid cell line (AK1) or matched random adenines (N = 80,560; equal chromosomal distribution). (E) Venn diagrams of SMRT-seq and DIP-seq peak overlaps for 6mdA in mouse (n = 4) and human (n = 4) samples. KO, knockout; PBMC, peripheral blood mononuclear cell.
Artifactual detection of 6mdA in mammalian DNA by single-molecule real-time sequencing
Single-molecule real-time sequencing (SMRT-seq) measures both the fluorescence pulse generated by incorporation of individual nucleotides in a sequence and the time between nucleotide incorporation events, the interpulse duration (IPD) (27). Changes in IPD value from that expected for a given nucleotide have been associated with specific DNA modifications, allowing direct, single-base resolution sequencing of DNA methylation. However, given the vanishingly low levels of 6mdA in mammals and a lack of any consistent sequence context for 6mdA, calling of 6mdA events from IPDs in mammals is associated with exceptionally high false discovery rates (FDRs) (7). A per-strand coverage of >100 reads per adenine would, at best, achieve a 6mdA FDR between 30 and 65% in mammalian DNA assuming an abundance of 0.01% 6mdA/dA (7). On the basis of the predicted 6mdA abundance in mammalian tissues and reported sequencing coverage, 6mdA SMRT-seq studies published to date have an FDR rate between 88 and 99.9% (table S1) (7–9, 19). Furthermore, other local sequence modifications (e.g., 5mC) can also give rise to false-positive signals (28), making the interpretation of SMRT-seq data at heavily methylated regions difficult, for instance, gene promoters, imprinted regions, the inactive X chromosome, and repetitive elements, such as LINE1 retrotransposons (29). A recent SMRT-seq study found all bases (A, C, T, and G) to be modified at the typically heavily 5mC-marked 5′ region of LINE1 elements (7), suggesting a high degree of false positives due to preexisting 5mC. In a comprehensive comparison of SMRT-seq, DIP-seq, and MS in fungi with varying 6mdA content, Mondo and colleagues (25) reported that SMRT-seq consistently overestimated 6mdA levels in comparison to MS, even in species with high abundance (2.8% 6mdA/dA), and for species with 0.05% 6mdA/dA, SMRT-seq could no longer reliably detect 6mdA. Critically, 95% of mammalian samples assayed to date have reported 6mdA levels far less (2- to 1000-fold less) than 0.05% 6mdA/dA (Fig. 4A).
As both high-resolution SMRT-seq and whole-genome bisulfite sequencing data has recently become available for the same human blood sample, we directly tested the association between SMRT-seq–predicted 6mdA signals and flanking cytosine modifications. As the DNA sequence context ± 12 bp of a given nucleotide can affect the IPD signals observed in SMRT-seq (28, 30), we focused on the 24 bp flanking each 6mdA site. 6mdA sites were highly enriched for methylated CpGs (mCpGs) (~3.5-fold) in their flanking sequences compared to background (Fig. 4B). Moreover, the distribution of mCpG sites surrounding reported 6mdA sites revealed a distinct pattern of mCpG enrichment at exactly 2 to 3 bp or 6 to 7 bp from the 6mdA site (Fig. 4C). Unlike 6mdA, which generates an IPD signal at the modified adenine, methylated cytosines generate IPD signals exactly 3 and 6 bp from methylated cytosine, suggesting that the observed 6mdA IPD signal originates from the flanking mCpG and not the adenine itself (28, 30). To assess the generalizability of this observation, we analyzed recently published SMRT-seq 6mdA profiles of a male human-immortalized lymphoblastoid cell line (AK1) (19). Although DNA methylation data are not available for this sample, we hypothesized that the distribution of CpGs flanking the reported 6mdA sites would be an accurate proxy of cytosine methylation. Again, we observed a pronounced enrichment of CpG sites at 2 to 3 bp and 6 to 7 bp from the reported 6mdA sites in both SMRT-seq datasets (Fig. 4D). The consistent and highly unusual pattern of mCpGs/CpGs flanking reported 6mdA sites strongly suggests that much of the reported 6mdA signal in mammals is an artifact. The tendency of SMRT-seq to overestimate 6mdA content was further demonstrated in a recent study comparing MS-based and SMRT-seq–based estimates of 6mdA in the DNA of 15 prokaryotic and eukaryotic genomes (18). Although SMRT-seq and DIP-seq are routinely used to cross-validate one another, closer inspection reveals very little overlap (1 to 8%) between these techniques when applied to mammalian DNA (Fig. 4E) (8, 9, 19), whereas species with high 6mdA abundance such as algae, fungi, and worms show high overlap around 85 to 95% (4, 7, 25). Thus, although SMRT-seq has the theoretical potential to generate base-resolution maps of 6mdA in mammals, the rarity of 6mdA combined with the limitations of IPD-based modification calling renders the results of SMRT-seq profiling of 6mdA in mammals currently uninterpretable (7, 18).
DISCUSSION
The reported discovery of 6mdA in mammals has generated an intense research effort with the aim of dissecting the role of this enigmatic DNA modification in mammalian biology. Here, we show that a combination of RNA and bacterial contaminations, antibody cross-reactivity, and other technical limitations have resulted in the repeated misidentification of 6mdA in mammalian DNA. The ability to readily generate antibodies against DNA modifications makes antibody-based assays particularly attractive when investigating DNA modifications for which modification-specific methods may be lacking. However, antibodies raised against unmodified adenosine (A) often exhibit a stronger affinity for m6A than for adenosine itself (i.e., BioVision, no. 6652; Synaptic Systems, no. 202103). Consequently, we hypothesized that antibodies raised against m6A were also likely to have a minor affinity for unmethylated adenosine, as supported by our dot blots of unmodified DNA with increasing AT content (Fig. 1D) and dot blots of poly-adenine, poly-thymine, and poly-cytosine showing affinity for unmodified adenine (Fig. 1E). These findings highlight the importance of using appropriate controls in all assays of rare DNA modifications. Whereas modified oligonucleotides are often used to test antibody specificity in immunodot blots (fig. S1C), a single oligonucleotide presents the modification of interest in a unique sequence context, thus failing to present potential off-target binding sequences to the antibody, resulting in exaggerated specificity. Consequently, the generation of appropriate positive (6mdA methylated genomic DNA) and negative (WGA human DNA) genomic DNA controls is crucial to characterizing 6mdA in mammals but has been lacking from most of the studies to date.
Unexpectedly, we found that several DIP-seq libraries were clearly contaminated with mammalian mRNA (Fig. 3). How RNA ended up in these sequencing libraries remains an open question; however, the two studies with increased amounts of spliced reads have seemingly adapted protocols based on m6A RNA immunoprecipitation sequencing (RIP-seq) where adapters are attached after immunoprecipitation (9, 19) unlike standard DIP-seq protocols (31), allowing incorporation of enriched RNA into the subsequent library preparation steps.
Bacterial contamination poses a particularly complex problem when assessing global 6mdA levels in mammals. Whereas Mycoplasma spp. are a well-known contaminant of cultured cells, it is less appreciated that they are also common commensals of mucosal surfaces in mammals and can also be internalized in the mammalian host cell (32). Mycoplasma spp. are readily detected in over 80% of healthy individuals but appear to be particularly prevalent in the context of solid tumors including glioma, prostate cancer, lung cancer, renal cell carcinoma, and ovarian cancer (32). The presence of Mycoplasma spp. in normal human tissues and their elevated numbers in cancerous tissues highlight the extreme caution required for global measurements of 6mdA abundance in healthy and diseased primary human samples (6, 9). Even in the absence of bacterial contamination in the original biological sample, the downstream preparation of samples may lead to introduction of bacterial contaminants. As most of the recombinant enzymes used in preparation of DNA samples for downstream 6mdA analysis are produced in bacterial expression systems, it is possible that these reagents introduced sufficient bacterial DNA to produce a false 6mdA signal in highly sensitive MS assays. Schiffers et al. (13, 18) reported that MS measurements containing only commercial enzyme preparations, but no DNA, gave a weak but clear signal for 6mdA when using highly sensitive triple quadrupole mass spectrometry (UHPLC-QQQ-MS) (13, 18). However, it remains unclear whether this important control is included in most MS studies of 6mdA.
SMRT-seq has the potential to directly detect DNA modification status by measuring changes in the rate of polymerase processivity caused by modified bases. Whereas several recent studies have revealed that SMRT-seq consistently overestimates 6mdA levels in eukaryotic DNA, the cause of this overestimation was unresolved. Here, we show that almost 50% of reported 6mdA sites in human blood DNA contain a flanking mCpG site exactly 2 to 3 bp or 6 to 7 bp from the predicted 6mdA. As 5mC generates IPD signals 2 to 3 bp and 6 to 7 bp downstream of its occurrence, it is likely that many of the reported 6mdA sites are artifacts caused by modification of flanking cytosines and not the adenine itself (28, 30). Whereas this observation calls into question the veracity of 6mdA calls in mammalian DNA, knowledge of this confounder will allow for improved bioinformatics protocols to identify and exclude these false positives, substantially improving the accuracy of 6mdA prediction from SMRT-seq data.
As the family of DNA and RNA modifications continues to grow and the abundance of previously unknown modifications becomes increasingly rare, we highlight the potential for false discovery even with the use of multiple complementary approaches. We suggest the use of modified and unmodified genomic DNA standards as a minimum requirement in future studies of rare DNA modifications, as well as more thorough validation of antibody specificity before their use. Unfortunately, in the absence of a 6mdA detection technique robust to the multiple sources of error that we have outlined, we conclude that the evidence published to date is not sufficient to support the presence of 6mdA in mammals.
MATERIALS AND METHODS
Cell culture
Jurkat and Molt-4 cells were acquired from the Leibniz Institute DSMZ. We thank B. Ingelsson, O. Stål, and J. Ernerudh for providing human embryonic kidney (HEK) 293T, MCF-7, MDA-MB-231, and HTR-8/SVneo cells. All cell lines were kept in tissue culture–treated flasks in a humidified incubator at 37°C with 5% CO2. HEK293T cells were cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM) (Gibco Thermo Fisher Scientific, no. 41965), MCF-7, and MDA-MB-231 in standard DMEM (Gibco Thermo Fisher Scientific, no. 31053), and HTR-8/SVneo, Jurkat, and Molt-4 cells were kept in the American Type Culture Collection–modified RPMI 1640 medium (Gibco Thermo Fisher Scientific, no. A10491). All culture media were supplemented with 10% fetal bovine serum (FBS) (Gibco Thermo Fisher Scientific, no. 10500) and 1% penicillin-streptomycin [penicillin (100 U/ml) and streptomycin (100 μg/ml)] (Gibco Thermo Fisher Scientific, no. 15140). Cells were passaged every 2 to 3 days during which adherent cells were detached with trypsin (Gibco Thermo Fisher Scientific, no. 15400). For decontamination, cells infected with Mycoplasma were treated with indicated concentrations of Plasmocin (InvivoGen, ant-mpt) for up to 7 days. Every 2 to 3 days, when cells were passaged, fresh antibiotic was added.
T cell isolation and stimulation
T cell isolation and stimulation were performed as previously described by us (15). Briefly, peripheral blood mononuclear cells were isolated from buffy coats from healthy human donors by density gradient centrifugation using Lymphoprep (Axis-Shield). NTH cells were isolated using a cocktail of biotinylated antibodies (Miltenyi Biotec, no. 130-094-131) and negative magnetic separation over columns (Miltenyi Biotec, no. 130-042-401). Primary NTH were activated by incubation with anti-CD3 (500 ng/ml) and anti-CD28 (500 ng/ml) in addition to interleukin-12 (IL-12) (5 ng/ml), anti–IL-4 (5 μg/ml), and IL-2 (10 ng/μl) for TH1 or IL-4 (10 ng/ml), anti–IL-12 (5 μg/ml), anti–interferon-γ (5 μg/ml), and IL-2 (10 ng/ml) for TH2. Cells were cultured for up to 5 days in RPMI medium (Gibco Thermo Fisher Scientific, no. 22400) with 10% FBS and 1% penicillin-streptomycin. Cytokines and neutralizing antibodies were replenished after 3 days.
DNA extraction
DNA was extracted using Mini Spin Columns (EconoSpin, no. 1910) with reagents from Quick-DNA/RNA Kit (Zymo Research, no. D7001). Instructions from Zymo Research’s kit were followed. Whenever high yields of DNA were required, i.e., Jurkat DNA, phenol/chloroform was used for DNA extraction.
Detection of mycoplasma
All cell lines were periodically tested for mycoplasma with MycoAlert kit (Lonza, LT07), which uses luminescence to detect mycoplasma-specific enzymes. The manufacturer’s instructions were followed with slight modifications, namely, only 50 μl of cell supernatant, substrate, and reagent were used. Mycoplasma was also detected by PCR using multiple primers specific for a range of the most common mycoplasma species (table S2) (17). PCR reactions were performed for 24 cycles using 50 ng of DNA, primers (0.5 μM), and Phusion polymerase (New England Biolabs, no. M0530).
Immunodot blot
Immunodot blot was performed as previously described by us (15, 20) with slight modification of antibody concentrations. Briefly, DNA was denatured at 95°C for 15 min in 400 mM NaOH and 10 mM EDTA at a volume of 200 μl, followed by immediate cooling on wet ice and brief centrifugation. In the case of DNA amplicons, denaturation was performed at 99°C, followed by snap-freezing in a dry ice/ethanol bath. Samples were blotted onto a positively charged membrane under vacuum using a Dot Blot Hybridization Manifold (Harvard Apparatus). Membranes were washed shortly in 2× SSC buffer (300 mM NaCl and 30 mM sodium citrate), ultraviolet (UV) cross-linked (UV Stratalinker 1800, Stratagene) for 1 min, and baked at 80°C for 2 hours. After blocking with 0.5% casein buffer (Thermo Fisher Scientific, no. 37582) diluted in tris-buffered saline (TBS) [20 mM tris base and 150 mM NaCl (pH 7.6)], membranes were stained with primary antibodies against 6mdA (1:1000; Synaptic Systems, no. 202-003 or EMD Millipore Merck, no. ABE572), 5mC (1:3000; Zymo Research, no. A3001), 5hmC (1:3000; Active Motif, no. 39791), 5caC (1:3000; Active Motif, no. 61229), or immunoglobulin G (IgG) (1:1000; Abcam, no. 171870) for 1 hour on ice. Membranes were washed in TBS-Tween (0.1%) three times for 5 min each and stained with either goat anti-rabbit secondary antibody (1:3000; Bio-Rad, no. 1706515; anti-6mdA, anti-5hmC, anti-5caC, and anti-IgG) or goat anti-mouse antibody (1:5000; Bio-Rad, no. 170651; anti-5mC) for 1 hour at room temperature. Membranes were washed twice in TBS-Tween and once in TBS for 5 min, then incubated with enhanced chemiluminescence substrate (Bio-Rad, no. 1705060) for 4 min, and imaged using a ChemiDoc MP System (Bio-Rad). Methylene blue was used to control for DNA loading. After washing in 99.9% ethanol for 15 min and incubation with methylene blue (Molecular Research Center, MB119) for 10 min while lightly shaking, membranes were rinsed in Milli-Q water three times, washed for an additional 2 to 5 min, and then scanned.
Human 6mdA positive control DNA
Using dA methylase (DAM) (New England Biolabs, M0222), a methyl group was added to the adenine (N6) in every 5′-GATC-3′ sequence. One microgram of DNA, 1× methyltransferase reaction buffer, 80 μM S-adenosylmethionine, 1 μl of DAM, and distilled water in 10 μl were incubated at 37°C for 1 hour, followed by 65°C for 15 min. The reaction was scaled up as needed. To control for successful methylation, 500 ng of DNA were digested with either Dpn I (New England Biolabs, R0176) or Mbo I (New England Biolabs, R0147) at 37°C for 15 min. Dpn I cuts at 5′-GATC-3′ if 6mdA is present, while Mbo I cuts the same sequence in the absence of adenine methylation. The resulting products were run on a 1% agarose gel.
Generation of 6mdA-containing double-stranded DNA controls
A 500-bp (50% AT content) PCR product was incubated with the Eco GII adenine methyltransferase (New England Biolabs, M0222). The reaction was performed according to the manufacturer’s recommended protocol, at a ratio of 5 U of Eco GII per 1 μg of DNA. The sample was incubated at 37°C for 4 hours, followed by inactivation at 65°C for 15 min. The modified product was then purified with a spin column kit (Zymo Research, D4013) and subsequently incubated with Eco GII for a second time, in the same reaction conditions for 2 hours, followed by another purification step. Modification efficiency was estimated by Dpn I and Mbo I digestion via agarose gel (1.5%) electrophoresis.
WGA 6mdA negative control
Since no methyl groups are added during WGA of genomic DNA, amplified DNA was used as a negative control in dot blots. DNA was extracted, and 50 to 100 ng were amplified using the REPLI-g Mini Kit (QIAGEN, 150023) according to the manufacturer’s instructions.
DNA fragment amplification
DNA fragments of varying AT content were custom-ordered from Integrated DNA Technologies and were amplified by high-fidelity PCR. Briefly, the 20-μl reaction mixture contained 1 ng of sample DNA, forward and reverse primers (10 μM), 10 mM deoxynucleoside triphosphate (dNTPs) and 20 U of Phusion High-Fidelity Polymerase (New England Biolabs, no. M0530S) with the provided 5× Phusion High-Fidelity or GC buffer. PCR conditions were as follows: initial denaturation 98°C for 30 s, denaturation at 98°C for 10 s, annealing at 65°C for 20 s, extension at 72°C for 30 s, and final extension at 72°C for 5 min, for a total of 20 cycles. The products were purified using Agencourt AMPure XP Beads (Beckman Coulter, no. 63881), at 0.6× beads-to-product ratio.
Public data
Published raw human DIP-seq data was obtained from studies of 6mdA for accessions SRR6123784, SRR6123785 (9), SRR6447605, SRR6447606, SRR6447607, SRR6447608, SRR6447609, SRR6447610, SRR6447611, SRR6447612, SRR6447613, SRX4615359, SRX4615360, SRX4615361, SRX4615362, SRX4615363, SRX4615364, SRX4615365, SRX4615366, SRX4615367 (6), SRR8257208, SRR8257209, SRR8257210, SRR8257211, SRR8257212, and SRR8257213 (19); 5hmC and 5mC for accessions SRX861043 and SRX861045 (33); ENCODE 5mC for accessions SRR1237957 and SRR1237958 (34); and RNA m6A for accessions SRR494613, SRR494614, SRR494615, SRR494616, SRR494617, and SRR494618 (24). The 5hmC and 5mC data were selected to be comparable to 6mdA data in the same or similar lymphoblastoid cell lines. Aligned 5mC DIP-seq reads were obtained from the Roadmap Epigenomics Project for accessions GSM493615, GSM543017, GSM543019, GSM543021, GSM543023, GSM543025, GSM543027, GSM613843, GSM613846, GSM613847, GSM613853, GSM613856, GSM613857, GSM613859, GSM613862, GSM613864, GSM613911, GSM613913, GSM613914, GSM613917, GSM669606, GSM669607, GSM669608, GSM669609, GSM669610, GSM669611, GSM669612, GSM669613, GSM669614, GSM669615, GSM669619, GSM707020, GSM707021, GSM707022, GSM707023, GSM817248, GSM817249, GSM941725, GSM941726, GSM941727, GSM958180, GSM958181, GSM958182, GSM1517153, and GSM1517154 (35). Raw whole-genome bisulfite sequencing data for human sample HX1 were downloaded from the short-read archive: SRX5716730 and SRX5716740. The 5hmC and 5mC data were selected to be comparable to 6mdA data in the same or similar lymphoblastoid cell lines. Data S1 presents an outline of all analyzed datasets and their relationship to the figures.
Processing of DIP-seq data
Raw sequencing data was aligned to the human hg38 reference (GCA_000001405.15_GRCh38_no_alt_analysis_set) using Bowtie2 (36) (-N 1 --local) or STAR (37) (--outSAMstrandField intronMotif --outSAMattributes NH HI AS nM XS). Read pileups were visualized using Integrative Genomics Viewer (38) by grouping read pairs. Enriched peaks were called using MACS2 (-g hs) (39), using input controls. For preprocessed Roadmap data, reads in BED format were formatted to BED6 format if needed, followed by peak calling using MACS2 as outlined above but without control samples. Peak locations were then transferred from hg19 to hg38 assemblies using the University of California Santa Cruz (UCSC) LiftOver tool.
Processing of whole-genome bisulfite sequencing data
Bisulfite sequencing data (416 million 150-bp paired-end reads) were obtained from the National Center for Biotechnology Information short read archive (accession: SRX5716730 and SRX5716740) and aligned to a bisulfite-converted hg38 index with Bismark (bismark --N 1). Subsequently, methylation levels of cytosines in both CpG and non-CpG contexts were extracted (bismark_methylation_extractor --p --comprehensive --bedgraph). Summary methylation files were then postprocessed to remove all sites with less than 5× coverage. CpG sites with greater than 20% methylation were considered as “methylated.” The distribution of mCpGs was overlapped with the published genomic positions of 6mdA sites from the indicated studies using the R programming language. As we lacked information on the strandedness of the SMRT-seq reads, the overlapping analysis was not done in a strand-specific manner, resulting in the symmetrical nature of the plots.
Repeat annotations
Annotations of repetitive elements were obtained from the UCSC table browser RepeatMasker (rmsk) track.
Mappability in peaks
Umap (40) per-base-multiread 50-mer and 100-mer mappability scores were overlapped and averaged per DIP-seq peak. Genomic background mappability was estimated from randomly sampled genomic regions of same size and chromosome.
Peak density across chromosomes
Chromosomes were tiled into 1-Mbp bins, and counts per bin were calculated as a fraction of total. A local polynomial regression (LOESS) model was fit to the data to plot multiple replicates. Location of assembly gaps and LINE1 elements were obtained from the UCSC “gap” and “rmsk” tables for hg38, respectively.
Calculation of read splicing
Spliced reads were identified as STAR-aligned reads containing “N” in compact idiosyncratic gapped alignment report (CIGAR) for the forward or reverse read (if paired-end). Exon-intron junctions were extracted from RefSeq knownGene, and overlap was calculated allowing a maximum gap of 5 bp using R/GenomicRanges (41). Expected distribution was obtained from all reads by bootstrap resampling (n = 10,000).
Identification of RNA m6A consensus motifs in DNA 6mdA DIP-seq data
DRACH ([A/G/T][A/G]AC[A/C/T]) and RRACT ([A/G][A/G]ACT]) motifs were scanned in an assembly gap- and intra-contig ambiguity masked hg38 genome using R/BSgenome (BSgenome.Hsapiens.UCSC.hg38.masked), and hits overlapping peaks were normalized to peak size (counts per base) using R/GenomicRanges (41).
Genic profile
Metagene profiles were produced using deepTools2 (42). First, normalized coverage was calculated (bamCoverage –effectiveGenomeSize 2913022398 –normalizeUsing RPGC); then, count matrices were calculated and plotted to include UTRs using GENCODE (43) version 28 gene transfer format (GTF) annotations (computeMatrix scale-regions -b 1000 -a 1000 --metagene –unscaled5prime 500 --unscaled3prime 500).
Code availability
All associated computer code is freely accessible at https://github.com/ALentini/6mA_Paper.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Swedish Research Council (2015-03495 to C.E.N.), LiU-Cancer Network (2016-007 to C.E.N.), and the Swedish Cancer Society (CAN 2017/625 to C.E.N.). Author contributions: C.E.N. designed and supervised the study and contributed to data analysis. K.D. and M.B. performed all laboratory-based work. A.L. performed all bioinformatics. B.G. contributed to bioinformatics analysis and manuscript writing. C.E.N. and A.L. performed the primary manuscript writing. K.D. and M.B. contributed to manuscript writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaay3335/DC1
Fig. S1. Antibody detection of 6mdA in mammals is flawed.
Fig. S2. Extended analysis of repetitive elements and mappability for 6mdA DIP-seq.
Fig. S3. Extended analysis of 6mdA DIP-seq read splicing and genic location.
Table S1. SMRT-seq read coverage in studies of 6mdA in mammalian DNA.
Table S2. Primers used to detect Mycoplasma species.
Data S1. Summary of analyzed datasets and their relationship to figures.
Data S2. Summary of amplicons used in dot blots.
REFERENCES AND NOTES
- 1.Weisenberger D. J., Campan M., Long T. I., Kim M., Woods C., Fiala E., Ehrlich M., Laird P. W., Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 33, 6823–6836 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li E., Bestor T. H., Jaenisch R., Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 (1992). [DOI] [PubMed] [Google Scholar]
- 3.Smith Z. D., Meissner A., DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 14, 204–220 (2013). [DOI] [PubMed] [Google Scholar]
- 4.O'Brown Z. K., Greer E. L., N6-Methyladenine: A conserved and dynamic DNA Mark. Adv. Exp. Med. Biol. 945, 213–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wion D., Casadesús J., N6-methyl-adenine: An epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 4, 183–192 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie Q., Wu T. P., Gimple R. C., Li Z., Prager B. C., Wu Q., Yu Y., Wang P., Wang Y., Gorkin D. U., Zhang C., Dowiak A. V., Lin K., Zeng C., Sui Y., Kim L. J. Y., Miller T. E., Jiang L., Lee C. H., Huang Z., Fang X., Zhai K., Mack S. C., Sander M., Bao S., Kerstetter-Fogle A. E., Sloan A. E., Xiao A. Z., Rich J. N., N6-methyladenine DNA modification in glioblastoma. Cell 175, 1228–1243.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu S., Beaulaurier J., Deikus G., Wu T. P., Strahl M., Hao Z., Luo G., Gregory J. A., Chess A., He C., Xiao A., Sebra R., Schadt E. E., Fang G., Mapping and characterizing N6-methyladenine in eukaryotic genomes using single-molecule real-time sequencing. Genome Res. 28, 1067–1078 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu T. P., Wang T., Seetin M. G., Lai Y., Zhu S., Lin K., Liu Y., Byrum S. D., Mackintosh S. G., Zhong M., Tackett A., Wang G., Hon L. S., Fang G., Swenberg J. A., Xiao A. Z., DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 532, 329–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao C.-L., Zhu S., He M., Chen D., Zhang Q., Chen Y., Yu G., Liu J., Xie S.-Q., Luo F., Liang Z., Wang D.-P., Bo X.-C., Gu X.-F., Wang K., Yan G.-R., N6-Methyladenine DNA modification in the human genome. Mol. Cell 71, 306–318.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Koziol M. J., Bradshaw C. R., Allen G. E., Costa A. S. H., Frezza C., Gurdon J. B., Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct. Mol. Biol. 23, 24–30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ratel D., Ravanat J.-L., Charles M.-P., Platet N., Breuillaud L., Lunardi J., Berger F., Wion D., Undetectable levels of N6-methyl adenine in mouse DNA: Cloning and analysis of PRED28, a gene coding for a putative mammalian DNA adenine methyltransferase. FEBS Lett. 580, 3179–3184 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Liu B., Liu X., Lai W., Wang H., Metabolically generated stable isotope-labeled deoxynucleoside code for tracing DNA N6-Methyladenine in human cells. Anal. Chem. 89, 6202–6209 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Schiffers S., Ebert C., Rahimoff R., Kosmatchev O., Steinbacher J., Bohne A.-V., Spada F., Michalakis S., Nickelsen J., Müller M., Carell T., Quantitative LC-MS provides no evidence for m6dA or m4dC in the genome of mouse embryonic stem cells and tissues. Angew. Chem. Int. Ed. Engl. 56, 11268–11271 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A. A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y. S., Ben-Haim M. S., Eyal E., Yunger S., Pinto Y., Jaitin D. A., Viukov S., Rais Y., Krupalnik V., Chomsky E., Zerbib M., Maza I., Rechavi Y., Massarwa R., Hanna S., Amit I., Levanon E. Y., Amariglio N., Stern-Ginossar N., Novershtern N., Rechavi G., Hanna J. H., m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347, 1002–1006 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Nestor C. E., Lentini A., Hägg Nilsson C., Gawel D. R., Gustafsson M., Mattson L., Wang H., Rundquist O., Meehan R. R., Klocke B., Seifert M., Hauck S. M., Laumen H., Zhang H., Benson M., 5-Hydroxymethylcytosine remodeling precedes lineage specification during differentiation of human CD4+ T cells. Cell Rep. 16, 559–570 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razin A., Razin S., Methylated bases in mycoplasmal DNA. Nucleic Acids Res. 8, 1383–1390 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uphoff C. C., Denkmann S. A., Drexler H. G., Treatment of mycoplasma contamination in cell cultures with Plasmocin. J. Biomed. Biotechnol. 2012, 267678 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Brown Z. K., Boulias K., Wang J., Wang S. Y., O’Brown N. M., Hao Z., Shibuya H., Fady P.-E., Shi Y., He C., Megason S. G., Liu T., Greer E. L., Sources of artifact in measurements of 6mA and 4mC abundance in eukaryotic genomic DNA. BMC Genomics 20, 445 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pacini C. E., Bradshaw C. R., Garrett N. J., Koziol M. J., Characteristics and homogeneity of N6-methylation in human genomes. Sci. Rep. 9, 5185 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lentini A., Lagerwall C., Vikingsson S., Mjoseng H. K., Douvlataniotis K., Vogt H., Green H., Meehan R. R., Benson M., Nestor C. E., A reassessment of DNA-immunoprecipitation-based genomic profiling. Nat. Methods 15, 499–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao B., Cheng Y., Wang Z., Li Y., Chen L., Huang L., Zhang W., Chen D., Wu H., Tang B., Jin P., DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat. Commun. 8, 1122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feederle R., Schepers A., Antibodies specific for nucleic acid modifications. RNA Biol. 14, 1089–1098 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., Sorek R., Rechavi G., Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Meyer K. D., Saletore Y., Zumbo P., Elemento O., Mason C. E., Jaffrey S. R., Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149, 1635–1646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mondo S. J., Dannebaum R. O., Kuo R. C., Louie K. B., Bewick A. J., LaButti K., Haridas S., Kuo A., Salamov A., Ahrendt S. R., Lau R., Bowen B. P., Lipzen A., Sullivan W., Andreopoulos B. B., Clum A., Lindquist E., Daum C., Northen T. R., Kunde-Ramamoorthy G., Schmitz R. J., Gryganskyi A., Culley D., Magnuson J., James T. Y., O'Malley M. A., Stajich J. E., Spatafora J. W., Visel A., Grigoriev I. V., Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 49, 964–968 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Hodge K., Have S. T., Hutton L., Lamond A. I., Cleaning up the masses: Exclusion lists to reduce contamination with HPLC-MS/MS. J. Proteomics 88, 92–103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., Bibillo A., Bjornson K., Chaudhuri B., Christians F., Cicero R., Clark S., Dalal R., DeWinter A., Dixon J., Foquet M., Gaertner A., Hardenbol P., Heiner C., Hester K., Holden D., Kearns G., Kong X., Kuse R., Lacroix Y., Lin S., Lundquist P., Ma C., Marks P., Maxham M., Murphy D., Park I., Pham T., Phillips M., Roy J., Sebra R., Shen G., Sorenson J., Tomaney A., Travers K., Trulson M., Vieceli J., Wegener J., Wu D., Yang A., Zaccarin D., Zhao P., Zhong F., Korlach J., Turner S., Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Schadt E. E., Banerjee O., Fang G., Feng Z., Wong W. H., Zhang X., Kislyuk A., Clark T. A., Luong K., Keren-Paz A., Chess A., Kumar V., Chen-Plotkin A., Sondheimer N., Korlach J., Kasarskis A., Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res. 23, 129–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deniz Ö., Frost J. M., Branco M. R., Author correction: Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 20, 432 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Flusberg B. A., Webster D. R., Lee J. H., Travers K. J., Olivares E. C., Clark T. A., Korlach J., Turner S. W., Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taiwo O., Wilson G. A., Morris T., Seisenberger S., Reik W., Pearce D., Beck S., Butcher L. M., Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 7, 617–636 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Vande Voorde J., Balzarini J., Liekens S., Mycoplasmas and cancer: Focus on nucleoside metabolism. EXCLI J. 13, 300–322 (2014). [PMC free article] [PubMed] [Google Scholar]
- 33.Chowdhury B., Seetharam A., Wang Z., Liu Y., Lossie A. C., Thimmapuram J., Irudayaraj J., A study of alterations in DNA epigenetic modifications (5mC and 5hmC) and gene expression influenced by simulated microgravity in human lymphoblastoid cells. PLOS ONE 11, e0147514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roadmap Epigenomics Consortium, Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y.-C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J. M., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L.-H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karimzadeh M., Ernst C., Kundaje A., Hoffman M. M., Umap and Bismap: Quantifying genome and methylome mappability. Nucleic Acids Res. 46, e120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M. T., Carey V. J., Software for computing and annotating genomic ranges. PLOS Comput. Biol. 9, e1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramírez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J. M., Sisu C., Wright J., Armstrong J., Barnes I., Berry A., Bignell A., Sala S. C., Chrast J., Cunningham F., Domenico T. D., Donaldson S., Fiddes I. T., Girón C. G., Gonzalez J. M., Grego T., Hardy M., Hourlier T., Hunt T., Izuogu O. G., Lagarde J., Martin F. J., Martínez L., Mohanan S., Muir P., Navarro F. C. P., Parker A., Pei B., Pozo F., Ruffier M., Schmitt B. M., Stapleton E., Suner M.-M., Sycheva I., Uszczynska-Ratajczak B., Xu J., Yates A., Zerbino D., Zhang Y., Aken B., Choudhary J. S., Gerstein M., Guigó R., Hubbard T. J. P., Kellis M., Paten B., Reymond A., Tress M. L., Flicek P., GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaay3335/DC1
Fig. S1. Antibody detection of 6mdA in mammals is flawed.
Fig. S2. Extended analysis of repetitive elements and mappability for 6mdA DIP-seq.
Fig. S3. Extended analysis of 6mdA DIP-seq read splicing and genic location.
Table S1. SMRT-seq read coverage in studies of 6mdA in mammalian DNA.
Table S2. Primers used to detect Mycoplasma species.
Data S1. Summary of analyzed datasets and their relationship to figures.
Data S2. Summary of amplicons used in dot blots.