

A CRISPR-based technique achieves projection- and function-specific gene editing in the rat brain.
Abstract
A genome editing technique based on the clustered regularly interspaced short palindromic repeats (CRISPR)–associated endonuclease Cas9 enables efficient modification of genes in various cell types, including neurons. However, neuronal ensembles even in the same brain region are not anatomically or functionally uniform but divide into distinct subpopulations. Such heterogeneity requires gene editing in specific neuronal populations. We developed a CRISPR-SaCas9 system–based technique, and its combined application with anterograde/retrograde AAV vectors and activity-dependent cell-labeling techniques achieved projection- and function-specific gene editing in the rat brain. As a proof-of-principle application, we knocked down the cbp (CREB-binding protein), a sample target gene, in specific neuronal subpopulations in the medial prefrontal cortex, and demonstrated the significance of the projection- and function-specific CRISPR-SaCas9 system in revealing neuronal and circuit basis of memory. The high efficiency and specificity of our projection- and function-specific CRISPR-SaCas9 system could be widely applied in neural circuitry studies.
INTRODUCTION
Neuronal ensembles with distinct genetic, morphological, and functional features are organized into complex neuronal networks in the mammalian brain. Precise genetic manipulation in specific neuronal subtypes and/or circuits is crucial for pinpointing causal relationships between neuronal activity and behavior. In vivo methods based on DNA antisense oligonucleotides and RNA interference have been commonly used for gene silencing in the brain (1, 2). However, it remains challenging to achieve stable gene knockout or gene modification in neuronal subpopulations with specific connectional or functional features, especially in rats and nonhuman primates. Conditional recombination systems have been widely used to study brain function with spatiotemporal accuracy, but animal model construction could be labor-intensive and time-consuming, especially for transgenic rats.
A genome editing technique based on the CRISPR-Cas9 system enables rapid, efficient, and convenient modification of endogenous genes in various cell types, resulting in frameshifting insertion/deletion (indel) mutations and allowing for functional analysis of specific genes in the brain (3). More intriguingly, in vivo gene editing would allow for systematic genetic dissection of neuronal circuits while bypassing the need for transgenic strains. However, neuronal ensembles even in the same brain region could divide into distinct subpopulations, either anatomically by their afferent/efferent connection or functionally by their recruitment in various tasks. Such heterogeneity would require techniques for controlled perturbation in specific neuronal populations, e.g., projection- and function-specific CRISPR-Cas9–based approaches, to facilitate rapid gene editing in neural circuitry studies.
Another obstacle in applying the CRISPR-Cas9 system in the nervous system is the limited capacity of the viral vector. Adeno-associated virus (AAV) is one of the most commonly used vectors. The widely used endonuclease Cas9 from Streptococcus pyogenes (SpCas9) is limited by the capacity limitation (usually less than 4.4 to 4.7 kb) of the highly versatile AAV delivery vehicle and the inefficient packaging. The Cas9 ortholog from Staphylococcus aureus (SaCas9), by contrast, is more than 1 kb shorter but edits the genome with an efficiency similar to SpCas9 (4).
In the present study, we aimed to develop a CRISPR-SaCas9 system that could edit target genes in neuronal subpopulations with specific connectional or functional features. As proof of concept, we chose the cbp (CREB-binding protein), a transcriptional coactivator with histone acetyltransferase activity essential for neuronal excitability and memory formation (5–7), as a target gene and sought to achieve projection- and function-specific gene knockdown in neural circuits relevant to fear memory.
RESULTS
SaCas9 inactivates cbp in vitro with high efficiency
We first examined the performance of SaCas9 in rat glioma cells using cbp as our target gene. Considering the inefficient transfection of AAV vehicle in vitro, we designed a lentivirus vector that packaged SaCas9 fused to mCherry via a P2A self-cleaving peptide, driven by the cytomegalovirus (CMV) promoter and a U6–guide RNA (gRNA) expression cassette. SaCas9 was further labeled with a flag epitope tag (Fig. 1A). We designed five gRNAs with the highest proposed score, targeting exons 2, 4, and 14 of the rat cbp gene (Fig. 1B). Three days after lentivirus transfection, we observed robust expression of SaCas9 and gRNAs in F98 and C6 cell lines (Fig. 1, C and D). Using Western blot analysis, we observed a corresponding decrease in CBP expression (Fig. 1E). In addition, CBP was strongly knocked down by multiple gRNA-1+4+5 (hereafter referred to as gRNA-mix) spacing less than 200 base pairs (bp) apart in exon 2 compared to the scrambled gRNA (Fig. 1E), suggesting that shorter distance between adjacent gRNAs led to more efficient gene knockout (8). As a downstream target, the acetylation of histone 3 on Lys14 (H3K14) decreased after cbp knockdown (Fig. 1F). T7 endonuclease assay confirmed indel generation in the cbp locus (Fig. 1G). Using immunocytochemistry, we also verified a corresponding decrease in the CBP and the acetylation of H3K14 in SaCas9+ and gRNA+ cells (Fig. 1, H and I). To further confirm its efficacy, we applied the SaCas9 system on another target gene, the p300 (E1A-binding protein), a homolog of cbp. SaCas9 could efficiently knock down the p300 in glioma cells (fig. S1), indicating the high efficiency and potentially wide application of the SaCas9 system in gene editing in vitro.
Fig. 1. SaCas9 inactivates the cbp in vitro with high efficiency.
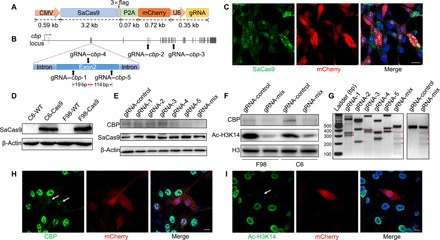
(A) A schematic of CMV-SaCas9-P2A-mCherry-U6-gRNA lentivirus vector. SaCas9 was labeled with a flag epitope tag. mCherry was cleaved from SaCas9 through the P2A self-cleaving peptide, indicating the expression of vector. (B) Graphical representation of the rat cbp gene locus showing gRNA target sites, with three gRNAs spacing less than 200 bp apart in exon 2. (C and D) Expression of SaCas9 was evaluated by immunofluorescence (C) and immunoblot (D) 3 days after lentivirus transfection in rat glioma cells. Scale bar, 20 μm. WT, wild type. (E) Western blot analysis showing decreased CBP expression in F98 cells 1 week after lentivirus transfection. (F) Western blot analysis showing decreased CBP expression and acetylation of H3K14 in cells expressing SaCas9 and multiple gRNA-1+4+5 (gRNA-mix). (G) T7 endonuclease assay for Cas9-mediated cleavage in F98 cells transfected with SaCas9 and indicated gRNA, with red triangles indicating cleavage fragments. (H and I) Immunofluorescence staining of CBP (H) and Ac-H3K14 (I) 1 week after CRISPR-SaCas9 targeting of cbp locus in F98 cells, with white arrows indicating decreased CBP expression (H) and acetylation of H3K14 (I) in SaCas9+ and gRNA+ cells. Scale bars, 10 μm.
Projection-specific gene knockdown with CRISPR-SaCas9: Perturbing cbp in postsynaptic PL neurons receiving dCA1 inputs
We next evaluated in vivo efficacy of SaCas9-mediated gene editing, first by projection-specific knockdown of the cbp in postsynaptic prelimbic cortical (PL) neurons receiving dorsal hippocampal CA1 (dCA1) inputs (9–11). To confirm the presence of direct dCA1-PL innervation, anterograde trans-synaptic AAV1–CaMKII (calcium/calmodulin-dependent protein kinase II)–Cre-P2A-GFP (12) was injected bilaterally into dCA1, and AAV9-hSyn-DIO-mCherry was injected in bilateral PL (Fig. 2A). Expression of Cre-dependent mCherry was detected in bilateral PL 4 weeks after AAV delivery. Sparse green fluorescent protein (GFP) expression indicating axon terminals of dCA1 projection neurons was also observed in the PL (Fig. 2B). Direct projection from the dCA1 to the PL was further verified by retrograde rAAV2-retro labeling (13): Injection of rAAV2-retro-hSyn-Cre in the PL resulted in labeling of dCA1 neurons with Cre-dependent mCherry (Fig. 2, C and D).
Fig. 2. Perturbing cbp in postsynaptic PL neurons receiving dCA1 inputs.
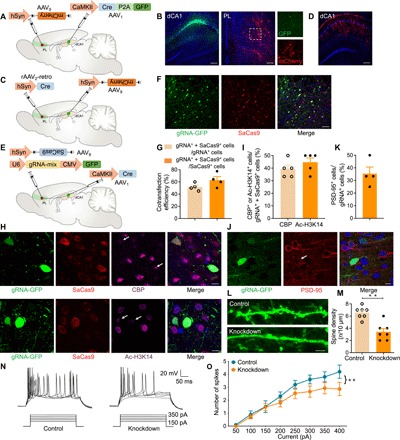
(A) A schematic of the experiment: AAV1-CaMKII-Cre-P2A-GFP injection in bilateral dCA1 and AAV9-hSyn-DIO-mCherry injection in bilateral PL. (B) Coronal sections showing dCA1 labeled with GFP and PL labeled with Cre-dependent mCherry and dCA1 axons expressing GFP. Scale bars, 200 μm. (C) A schematic of the experiment: rAAV2-retro-hSyn-Cre injection in bilateral PL and AAV9-hSyn-DIO-mCherry injection in bilateral dCA1. (D) Coronal sections showing dCA1 labeled with Cre-dependent mCherry. Scale bar, 200 μm. (E) A schematic of the experiment: AAV1-CaMKII-Cre was injected bilaterally into the dCA1, and a mixture (1:1 ratio) of AAV9-hSyn-DIO-SaCas9 and AAV9-U6-gRNA-mix-CMV-GFP was injected into bilateral PL. (F) Expression of SaCas9 and gRNA-GFP in the PL 4 weeks after AAV delivery. Scale bar, 50 μm. (G) Cotransfection efficiency of gRNA and SaCas9 (n = 4 slices from three animals). (H) Immunofluorescence staining of CBP (top) and Ac-H3K14 (bottom) in the PL 4 weeks after AAV injection. The white arrows indicate decreased CBP expression and H3K14 acetylation in SaCas9- and gRNA-GFP+ neurons. Scale bars, 10 μm. (I) Quantification of CBP- or Ac-H3K14+ cells in the targeted neuronal population (gRNA+ + SaCas9+) (n = 5 slices from three animals). (J) Immunofluorescence staining of PSD-95 in the PL 4 weeks after AAV injection, with the white arrow indicating decreased PSD-95 expression in gRNA-GFP+ neurons. Scale bar, 10 μm. (K) Quantification of PSD-95+ cells in the targeted neuronal population (gRNA+) (n = 4 slices from three animals). (L) Decreased dendritic spines in PL neurons after cbp knockdown by SaCas9 and gRNA-mix. Control neurons were transfected with the same AAV with gRNA-mix replaced with gRNA-control. Scale bar, 5 μm. (M) Spine density quantification (t test, t12 = 4.88, **P < 0.01; n = 3 animals per group, two to three slices per animal). (N) An example of AP responses to positive current steps recorded from PL neurons of control and cbp knockdown groups. (O) Number of APs induced by injected currents in PL neurons from control and cbp knockdown rats [n = 10 and 15 neurons; **P < 0.01, two-way analysis of variance (ANOVA)]. Group effect: F1,72 = 11.38, P < 0.01; current intensity effect: F7,112 = 18.95, P < 0.01; interaction: F7,72 = 0.84, P > 0.05.
To knock down cbp in PL neurons innervated by dCA1, AAV1-CaMKII-Cre was injected bilaterally into the dCA1, with a mixture (1:1 ratio) of AAV9-hSyn-DIO-SaCas9 and AAV9-U6-gRNA-mix-CMV-GFP injected into bilateral PL (Fig. 2E). Expression of SaCas9 and gRNA in the PL was detected 4 weeks after injection (Fig. 2, F and G). Consistent with efficient cbp knockdown by CRISPR-SaCas9 in vitro, we observed a substantial decrease in both CBP and acetylation of H3K14 in infected neurons in the PL (Fig. 2, H and I).
We further examined the molecular, morphological, and electrophysiological consequences of cbp knockdown. We observed decreased expression of postsynaptic density protein-95 (PSD-95), a molecular marker for neuroplasticity, and decreased density of dendritic spines in gRNA+ neurons (Fig. 2, J to M, and fig. S2). We also assessed the intrinsic excitability of neurons via whole-cell patch-clamp recording to examine biophysical characteristics of action potentials (APs) after cbp knockdown. APs were evoked by superimposed positive current steps from a baseline potential of −70 mV (Fig. 2N). A rightward shift in the input-output curve appeared in cbp knockdown neurons compared to control cells, showing significant differences with current injection ranging from 300 to 400 pA (Fig. 2O). These results indicate that a proportion of PL neurons are directly innervated by dCA1 and that perturbing the cbp gene in this projection-specific neuronal subpopulation with CRISPR-SaCas9 efficiently reduces neuronal excitability.
Function-specific gene knockdown with CRISPR-SaCas9: Perturbing cbp in PL engram cells impairs remote memory
In the PL, only a small portion of neurons, referred to as engram cells (14, 15), are necessary to encode remote memory. We next extended the application of the CRISPR-SaCas9 system to manipulating the cbp gene in functionally specific PL engram cells. We injected AAV9–c-fos–rtTA-U6-gRNA-mix with AAV9-TRE3G-SaCas9 in the PL (Fig. 3A). This system directly coupled the promoter of c-fos, an immediate early gene frequently used as a biomarker of neuronal excitation, to the doxycycline (Dox) system for inducible gene expression (16, 17). With this approach, the absence of Dox inhibited c-fos promoter–driven reverse tetracycline transactivator (rtTA) from binding to a tetracycline-responsive element (TRE) site, which subsequently prevented SaCas9 expression. In the presence of Dox, by contrast, training-induced neuronal excitation selectively labeled c-Fos–expressing PL neurons with SaCas9 and consequently induced indel generation in the cbp locus.
Fig. 3. Perturbing cbp in PL engram cells impairs remote memory.
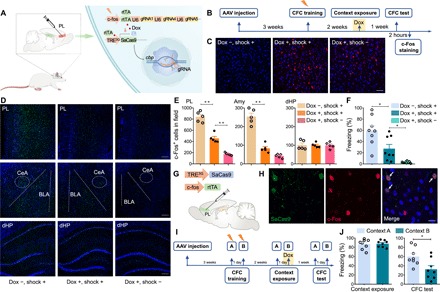
(A) A schematic of the experiment: A mixture (1:1 ratio) of AAV9–c-fos–rtTA-U6-gRNA-mix and AAV9-TRE3G-SaCas9 was injected into bilateral PL. With Dox, training induced the expression of rtTA, which bound to TRE, drove the expression of SaCas9, and induced indel generation in the cbp locus of activated PL cells. (B) Scheme of behavioral test. Rats were trained in the context for fear conditioning 3 weeks after AAV injection. Two weeks later, the rats were given Dox for 1 day and then exposed to the context for engram cell labeling. Recall test was performed 1 week later. (C) Immunofluorescence staining of SaCas9 in the PL 2 days after context exposure. Left (Dox −, shock +): with shock during CFC training and without Dox during context exposure; middle (Dox +, shock +): with shock during CFC training and with Dox during context exposure; right (Dox +, shock −): without shock during CFC training and with Dox during context exposure. Scale bar, 50 μm. (D) Immunofluorescence staining of c-Fos in PL, amygdala, and dHP 2 hours after the CFC test. Scale bars, 200 μm. (E) c-Fos+ cell counts in PL, amygdala, and dHP following the CFC test (n = 5 per group, one-way ANOVA. PL: F2,12 = 107.90, P < 0.01. “Dox −, shock +” versus “Dox +, shock +”, **P < 0.01. “Dox +, shock +” versus “Dox +, shock −”, **P < 0.01; amygdala: F2,12 = 81.45, P < 0.01. “Dox −, shock +” versus “Dox +, shock +”, **P < 0.01. “Dox +, shock +” versus “Dox +, shock −”, P > 0.05; dHP: F2,12 = 0.01, P > 0.05). (F) Rats showed less freezing during the recall test after cbp knockdown (n = 8 per group, one-way ANOVA, F2,21 = 16.64, P < 0.01. “Dox −, shock +” versus “Dox +, shock +”, *P < 0.05. “Dox +, shock +” versus “Dox +, shock −”, *P < 0.05). (G) A schematic of the experiment: AAV9–c-fos–rtTA and AAV9-TRE3G-SaCas9 were injected in the PL. With Dox, training induced the expression of rtTA, which bound to TRE and labeled the activated cells with SaCas9. There was no indel generation in the cbp locus due to the absence of gRNA, ensuring the normal function of neurons. (H) Representative coronal section of the PL labeled with SaCas9 and c-Fos 2 hours after the CFC test. SaCas9 and c-Fos double-positive neurons were indicated by arrows. Scale bar, 20 μm. (I) Scheme of behavioral test. Three weeks after AAV injection as shown in (A), the rats were fear-conditioned in contexts A and B. Two weeks later, the rats were exposed to context A, returned to home cage, and taken on Dox for 1 day before exposure to context B. The recall test for contexts A and B was performed 1 week later. (J) Rats showed less freezing during the CFC test in context B than in context A (t test, n = 8 per group. Context exposure: t14 = 0.20, P > 0.05; CFC test: t14 = 2.63, *P < 0.05).
To verify the inducible and activity-dependent expression of SaCas9, we injected AAV9–c-fos–rtTA-U6-gRNA-mix and AAV9-TRE3G-SaCas9 in the PL and performed contextual fear conditioning (CFC) training 3 weeks after injection (Fig. 3B). Two weeks after training, the rats received Dox from food pellet for 1 day and then were exposed to the context for activity-dependent labeling of PL engram cells with SaCas9. We confirmed the Dox-dependent expression of SaCas9 by immunofluorescence 2 days after context exposure (Fig. 3C). During the CFC probe test for remote memory, the labeled rats exhibited fewer c-Fos+ cells in the PL, but not in the dorsal hippocampus (dHP), storage sites for remote and recent memory, respectively (Fig. 3, D and E) (18–20). cbp knockdown rats also showed significantly reduced level of freezing compared to those without exposure to Dox (Fig. 3F). In addition, after injection of AAV9–c-fos–rtTA and AAV9-TRE3G-SaCas9 in the PL (Fig. 3G), most of the c-Fos+ neurons also colabeled with SaCas9 2 hours after CFC test, indicating that activity-dependent labeling with SaCas9 recapitulated the induction of endogenous c-Fos in our system (Fig. 3H). Last, we examined whether the activity-dependent cell labeling was context specific. Three weeks after AAV delivery (Fig. 3A), the rats were fear-conditioned in contexts A and B. Two weeks later, the rats were taken on Dox for 1 day before exposure to context B (Fig. 3I). During the CFC probe test, the rats showed significantly reduced levels of freezing to context B, but not to context A (Fig. 3J). Thus, we knock down the cbp in functionally specific PL engram cells and observe corresponding behavioral impairment.
Perturbing cbp in postsynaptic PL engram cells receiving dCA1 afferents via the CRISPR-SaCas9 system impairs remote memory
We next sought to examine the contribution of the dCA1-PL pathway to memory with the CRISPR-SaCas9 system. Over time, declarative memory gradually transfers from the dHP (recent memory) to the PL (remote memory) (18–20). Most of the dCA1 efferents reach the PL through the entorhinal cortex (EC), but it is not clear whether the direct dCA1-PL projection contributes to remote memory. We combined the function- and projection-specific features of our CRISPR-SaCas9 system to target postsynaptic PL engram cells that received direct dCA1 inputs. To identify whether PL engram cells received direct dCA1 inputs, AAV1-hSyn-Cre-P2A-GFP was injected bilaterally into the dCA1, with a mixture of AAV9–c-fos–rtTA-U6-gRNA-mix and AAV9-TRE3G-DIO-SaCas9 injected into bilateral PL (Fig. 4A). Similar to Fig. 3B, CFC training was performed 3 weeks after injection (Fig. 4B). Expression of Cre- and activity-dependent SaCas9 was detected in the PL 2 days after context exposure in the presence of Dox (Fig. 4, C and D), indicating that a proportion of PL engram cells were directly innervated by dCA1. During the CFC probe test, cbp knockdown rats exhibited significantly reduced levels of freezing compared to the control group (Fig. 4E). Together, our CRISPR-SaCas9 system could efficiently knock down cbp in postsynaptic engram cells and demonstrates that PL neurons receiving monosynaptic inputs from dCA1 contribute to remote memory.
Fig. 4. PL engram neurons receiving monosynaptic inputs from dCA1 contribute to remote memory.
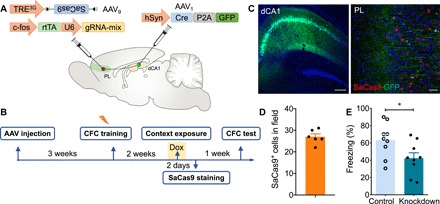
(A) Schematic of the experiment: AAV1-hSyn-Cre-P2A-GFP was injected bilaterally into the dCA1, and a mixture (1:1 ratio) of AAV9-c-fos-rtTA-U6-gRNA-mix and AAV9-TRE3G-DIO-SaCas9 was injected into bilateral PL. (B) Scheme of behavioral test. Rats were trained in the context for fear conditioning 3 weeks after AAV injection. Two weeks later, the rats were given Dox for 1 day and exposed to the context for cell labeling. Recall test was performed 1 week later. (C) Coronal sections showing dCA1 labeled with GFP and PL labeled with Cre- and activity-dependent SaCas9 and dCA1 axons expressing GFP. Scale bars, 200 μm (left) and 50 μm (right). (D) SaCas9+ cell counts in the PL 2 days after context exposure (n = 6 slices from three animals). (E) cbp knockdown in PL engram cells with direct dCA1 inputs decreased the duration of freezing in the probe test (t test, t16 = 2.31, *P < 0.05; n = 9 per group). Rats that were never exposed to Dox served as controls.
Perturbing cbp in presynaptic extinction-ensemble cells via CRISPR-SaCas9 system impairs extinction learning
We next sought to knock down cbp in functionally specific presynaptic neuronal ensembles. Within the medial prefrontal cortex (mPFC), the PL mediates the expression of conditioned fear, whereas the infralimbic cortex (IL) plays a critical role in fear extinction learning via its projection to the amygdala (21–23). To achieve genome perturbation in extinction-ensemble cells, we first injected AAV9–c-fos–rtTA-U6-gRNA-mix and AAV9-TRE3G-SaCas9 into the IL (Fig. 5A). We detected increased number of IL c-Fos+ neurons 2 hours after context exposure, confirming the involvement of the IL in extinction learning–related plasticity (Fig. 5, B to D). In the extinction test performed 1 week later, rats with cbp knockdown in extinction-ensemble IL cells showed impaired extinction learning (Fig. 5E).
Fig. 5. Perturbing cbp in presynaptic IL extinction-ensemble cells via the CRISPR-SaCas9 system impairs extinction learning.
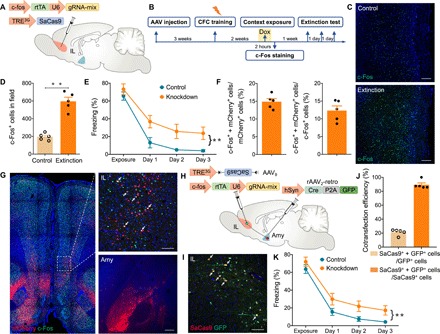
(A) A schematic of the experiment: A mixture (1:1 ratio) of AAV9–c-fos–rtTA-U6-gRNA-mix and AAV9-TRE3G-SaCas9 was injected into bilateral IL. (B) Scheme of behavioral testing. Rats were trained in the context for fear conditioning 3 weeks after AAV injection. Two weeks later, the rats were given Dox for 1 day, followed by exposure to the context for labeling IL extinction-ensemble cells. Rats were reexposed to the context for three consecutive days for extinction test. (C) Immunofluorescence staining of c-Fos in the IL after context exposure. Scale bars, 200 μm. (D) c-Fos+ cell counts in the IL following context exposure (t test, t8 = 8.18, **P < 0.01; n = 5 slices from three animals in either group). (E) Averaged freezing time during the recall test for cbp knockdown and control groups (two-way ANOVA, **P < 0.01; n = 8 per group. Group effect: F1,28 = 19.45, P < 0.01; time effect: F3,28 = 42.97, P < 0.01; interaction: F3,28 = 0.76, P > 0.05). Rats not exposed to Dox served as control. (F) Graph depicting the percentage of c-Fos+ + mCherry+/mCherry+ (left) and c-Fos+ + mCherry+/c-Fos+ (right) neurons after context exposure (n = 5 slices from three animals). mCherry+: amygdala-projecting IL neurons; c-Fos+: activated neurons. Amygdala-projecting IL neurons (14%) were activated (c-Fos+ + mCherry+/mCherry+), and 12% of the activated neurons projected to amygdala (c-Fos+ + mCherry+/c-Fos+). (G) Illustration of amygdala-projecting IL neurons (mCherry+) that showed immunolabeling of c-Fos protein 2 hours after context exposure. Scale bars, 100 μm (IL) and 200 μm (Amy). (H) Schematic of the experiment: rAAV2-retro-hSyn-Cre-P2A-GFP was injected bilaterally into the amygdala, and a mixture (1:1 ratio) of AAV9–c-fos–rtTA-U6-gRNA-mix and AAV9-TRE3G-DIO-SaCas9 was injected into bilateral IL. (I) Coronal sections showing some amygdala-projecting IL neurons (GFP+) colabeled with Cre- and activity-dependent SaCas9. Scale bar, 100 μm. (J) Cotransfection efficiency of GFP and SaCas9 (n = 5 slices from three animals). Dependent expression of SaCas9 on Cre-GFP resulted in 90% SaCas9+ + GFP+/SaCas9+ neurons. (K) Averaged freezing time during extinction test for cbp knockdown and control groups (two-way ANOVA, **P < 0.01; n = 8 per group. Group effect: F1,28 = 10.64, P < 0.01; time effect: F3,28 = 88.17, P < 0.01; interaction: F3,28 = 0.14, P > 0.05). Rats that were never exposed to Dox served as control.
To test whether IL neurons projecting to the amygdala were activated during extinction learning, rAAV2-retro-hSyn-mCherry was injected into the amygdala to label projection neurons in the IL. We found that extinction training increased the number of c-Fos+ neurons. Fourteen percent of amygdala-projecting IL neurons colabeled with c-Fos, indicating enhanced IL-amygdala connectivity during extinction (Fig. 5, F and G, and fig. S3). To target the extinction-ensemble cells in amygdala-projecting IL neurons, rAAV2-retro-hSyn-Cre-P2A-GFP was injected in the amygdala, whereas AAV9–c-fos–rtTA-U6-gRNA-mix and AAV9-TRE3G-DIO-SaCas9 were injected in the IL (Fig. 5H). Expression of Cre- and activity-dependent SaCas9 was confirmed by immunofluorescence 2 days after context exposure (Fig. 5, I and J). Rats with cbp knockdown in presynaptic extinction-ensemble IL cells exhibited impaired extinction learning (Fig. 5K). These results demonstrate the high efficiency in modulation of fear extinction learning via genome editing in presynaptic projection neurons in the IL.
CRISPR-SaCas9 has superior targeting specificity in adult neurons
Using sequence similarity–based off-target search, we sought to investigate the targeting specificity of SaCas9 at false gene loci with slight differences from the correct target. For the three cbp targets described above, we computationally selected 33 candidate off-target sites in the rat reference genome that were followed by an NNGRRT protospacer-adjacent motif (PAM) with less than five mismatched bases. We stereotactically injected AAV9-hSyn-SaCas9 and AAV9-U6-gRNA-mix-CMV-GFP to the PL and purified GFP+ neurons with fluorescence-activated cell sorting (FACS) (Fig. 6A). We observed highly specific enrichment of SaCas9 in the sorted neurons (Fig. 6B). Deep sequencing analysis revealed that most of the off-target sites exhibited indel generation at least two magnitudes lower than that of corresponding on-targets (Fig. 6C). Consistent with previous in vitro studies (4), SaCas9 was highly resistant to DNA mismatching, thus ensuring a high targeting specificity in vivo. Using deep sequencing, we also confirmed indel formation in the targeted cbp locus at the single-cell level (Fig. 6D).
Fig. 6. CRISPR-SaCas9 has superior targeting specificity in vivo.
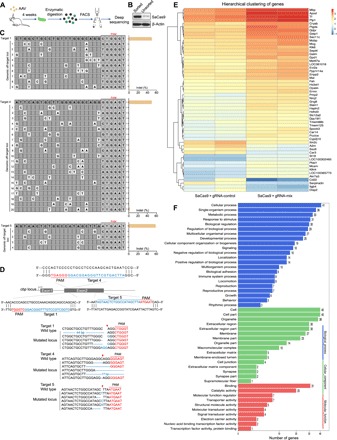
(A) A flowchart showing purification of GFP+ neurons from the rat brain 4 weeks after AAV9-hSyn-SaCas9 and AAV9-U6-gRNA-mix-CMV-GFP injection in the PL. (B) Western blot analysis showing highly enriched SaCas9 within sorted GFP+ neurons. (C) Cleavage levels at putative genomic off-target loci containing fewer than five mismatches (white cells) for cbp targets 1, 4, and 5 were analyzed by deep sequencing. (D) Graphical representation of the rat cbp locus showing gRNA target location. Targeted genomic locus is marked as blue. PAM sequence is marked as red. The red arrowheads indicate CRISPR-SaCas9 cutting site. Representative mutation patterns detected by sequencing of cbp locus are shown below. Top: wild-type sequence; blue dashes: deleted bases; blue bases: insertion or mutations (indel). (E) Hierarchical clustering of differentially expressed genes (log2 fold change >2 and false discovery rate <0.01 as significant difference) detected by RNA sequencing (RNA-seq) 4 weeks after AAV delivery. Relative log [FPKM (fragments per kilobase of transcript per million fragments mapped)] expression levels of genes are displayed in red-blue color scale. (F) The differentially expressed genes from control and cbp knockdown groups were classified by Gene Ontology analysis.
In vivo genome editing could be used to investigate transcription dynamics within intact biological contexts. As a transcriptional coactivator with histone acetyltransferase activity, abundant downstream genes are regulated by cbp. To test whether depletion of CBP would lead to genome-wide transcriptional dysregulation, we sequenced mRNA from GFP+ neurons. We found 54 differentially expressed genes between neurons receiving gRNA-mix and scrambled gRNA (Fig. 6E). Gene Ontology analysis revealed that some genes were related to signaling and synaptic function (Cryab and Sez6), which might contribute to memory deficits upon cbp knockdown (Fig. 6F).
DISCUSSION
There are numerous neuronal subtypes in the mammalian brain, each with distinct morphological, biochemical, biophysical, and functional identities. Malfunction of specific cell types in different neural circuits contributes to diverse behavioral phenotypes, highlighting the need for conditional genome editing in the heterogeneous neuronal population. Here, we develop a CRISPR-SaCas9 system for projection- and function-specific gene perturbation, a rapid and powerful method to decipher complex neuronal networks. We manipulate neuronal activity by targeting cbp in mPFC neurons involved in learning and memory. Decreased spine density and neuronal activity indicate efficient knockdown of cbp by the CRISPR-SaCas9 system. Unlike previous studies that transiently activate or inhibit neural circuits or engram cells with optogenetics (24), we develop an efficient system to achieve controlled permanent gene editing in specific neurons bearing remote memory storage or extinction. This function-specific gene editing provides a powerful method to explore the kinetics of transcription state of engram cells during memory encoding and subsequent consolidation.
Declarative memories are initially stored within the dHP (recent memory) and, over time, slowly transferred to the mPFC for permanent storage (remote memory) via the EC (18–20). It has been reported that a small population of dCA1 neurons directly project to the PL (9–11). However, the role of these monosynaptically targeted PL neurons in remote memory is not clear, given the technical difficulty in manipulating functionally specific PL neurons (PL engram cells) receiving direct dCA1 projection. Combining the projection- and function-specific features of our CRISPR-SaCas9 system, we reduced the excitability of postsynaptic engram cells in the PL after the consolidation of remote memory and showed that beyond the dCA1-EC-mPFC pathway, PL engram cells directly targeted by dCA1 contribute to the storage of remote memory as well. Combining with retrograde rAAV2-retro, we further restricted the SaCas9-mediated genome editing in presynaptic extinction-ensemble cells. In addition, deep sequencing analyses reveal that most of the off-target sites exhibit indel generation at least two magnitudes lower than corresponding on-targets, indicating that SaCas9 has superior targeting specificity in postmitotic neurons.
Given the compatibility with the Cre-loxP tools, our system allows researchers to adopt cell type–specific promoter and transgenic mice to perform genome editing in various neuronal subtypes and even glial cells. Different gRNAs could be used simultaneously to introduce mutations in multiple genes for modeling multigenic disorders and investigating genetic interactions. Furthermore, promoting memory extinction bearing a traumatic event via genome editing in extinction-ensemble cells might be a potential therapy for anxiety disorders, phobias, and posttraumatic stress disorder.
In summary, the present study is the first application of CRISPR-based technique, in combination with the anterograde/retrograde AAV and activity-dependent cell-labeling technique, to achieve gene editing in projection- and function-specific patterns. Integrated with electrophysiology, behavioral analyses, FACS, and deep sequencing approaches, this CRISPR-SaCas9 system provides a powerful strategy for precise genomic perturbations of brain functions under physiological and pathological conditions.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley rats (230 to 250 g at the start of experiments) were provided by the Department of Laboratory Animal Sciences, Peking University Health Science Center. Rats were maintained in a 12-hour light/12-hour dark cycle with a maximum of four animals per cage and with ad libitum access to food and water. All experimental procedures complied with the guidelines of the Animal Care and Use Committee of the Peking University Health Science Center. Rats were handled for at least 3 days before any experiments were conducted. By the end of the experiment, animals were dissected to perform immunofluorescence staining analysis to verify virus infection, and animals with inappropriate virus infection were excluded from the analysis.
gRNA design and AAV production
For SaCas9 target selection and generation of single gRNA, the 21-nucleotide target sequences were selected to precede a 5′-NNGRRT PAM sequence. The gRNAs were designed using the CRISPR RGEN Tools (www.rgenome.net/cas-designer/) to minimize the off-target effect. gRNAs used in this study are listed in table S1. AAV used in vivo were packaged and purchased from Vigene Biosciences (Jinan, China) or OBiO Technology (Shanghai, China). AAV vectors used in this study are listed in table S2.
Cell culture and lentivirus transfection
F98 and C6 rat glioblastoma cell lines and human embryonic kidney cell line 293T were purchased from the American Type Culture Collection (Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (HyClone) in a humidified atmosphere with 5% CO2 and 95% air at 37°C.
Lentiviral particles were generated by cotransfecting 293T cells with virus packaging vectors (pMD2.G and psPAX2). Transfection was performed using VigoFect (Vigorous Biotechnology Beijing Co. Ltd.) according to the manufacturer’s protocols. Six hours after transfection, the medium was changed. Virus supernatant was harvested 36 and 60 hours after transfection, filtered with a 0.45-μm polyvinylidene difluoride (PVDF) filter (Millipore), ultracentrifuged at 25,000 rpm using a P28S rotor (Hitachi Ltd., Japan), and stocked in a final volume of 100 μl. The titer of the lentivirus used in all cell culture experiments was at least 5.0 × 108 infectious units/ml. F98 and C6 cells were infected with lentivirus at 50 to 60% confluency, followed by selection in DMEM containing puromycin (1 or 6 μg/ml) for 2 to 5 days.
Immunoblot analysis
Cells were collected and lysed in radioimmunoprecipitation assay buffer (Beyotime Biotechnology, Shanghai, China) containing protease and phosphatase inhibitor cocktail (Roche Life Science) on ice for 30 min. Cell lysates were clarified by centrifugation at 4°C for 20 min. Total protein concentration was measured by Coomassie Protein Assay Kit (Pierce Chemical, Dallas, TX). Equal amounts of protein in each sample were separated on 8 or 10% SDS–polyacrylamide gel electrophoresis gels and then electrotransferred to PVDF membranes (Millipore). After blocking in 5% nonfat milk for 1 hour at room temperature, the membranes were incubated overnight at 4°C with specified primary antibody against CBP (7389S, Cell Signaling Technology), histone H3 (ab1791, Abcam), Ac-H3K14 (ab52946, Abcam), flag (F1804, Sigma-Aldrich), p300 (sc-48343, Santa Cruz Biotechnology), and β-actin (M177-3, MBL), respectively. After washing three times with tris-buffered saline containing 0.1% Tween 20, the membranes were incubated with the horseradish peroxidase–conjugated goat anti-rabbit/mouse immunoglobulin G antibody (111-035-003/115-035-003, Jackson ImmunoResearch) at room temperature for 1 hour. Protein bands were detected using Western blotting luminol reagent (sc-2048, Santa Cruz Biotechnology). Data were analyzed with the ImageJ software.
T7 endonuclease assays for genome modification
Genomic DNA was extracted 3 days after lentivirus transfection using the Universal Genomic DNA Kit (CW2298S, CWBIO) following the manufacturer’s protocol. The genomic region flanking the target sites for each guide sequence was amplified with polymerase chain reaction (PCR) by Q5 Hot Start High-Fidelity 2× Master Mix (E3321S, New England Biolabs) with the following program: preheat at 98°C for 30 s, 35 cycles of three-step amplification (98°C for 5 s, 62°C for 10 s, and 72°C for 20 s), and final extension at 72°C for 2 min. The primers used are listed in table S3. A total of 200 ng of the purified PCR products was mixed with buffer 2 and ultrapure water to a final volume of 19 μl. Hybridization reactions were performed with the following program: 95°C for 5 min, 95° to 85°C at −2°C/s, and 85° to 25°C at −0.1°C/s. Then, 1 μl of EnGen T7 Endonuclease I was added, and the mixture was incubated at 37°C for 15 min. One microliter of Proteinase K was added to stop the reaction followed by gel electrophoresis on a 2% agarose gel.
Stereotactic injection of AAV
The rat was anesthetized by intraperitoneal injection of 1% pentobarbital sodium (0.1 g/kg, intraperitoneally) and positioned in a stereotactic instrument equipped with a heating pad (RWD Life Science, Shenzhen, China). The scalp was shaved, a small incision was made along the midline to expose the skull, and a small hole was drilled in the skull above the requisite injection site. A total of 0.5 μl of AAV was injected into the dCA1 [anteroposterior (AP): −3.6 mm; mediolateral (ML): ±2.2 mm; dorsoventral (DV): −2.8 mm], PL (AP: 2.9 mm; ML: ±0.5 mm; DV: −3.0 mm), IL (AP: 2.8 mm; ML: ±0.5 mm; DV: −4.5 mm), or amygdala (AP: −2.8 mm; ML: ±4.8 mm; DV: −8.4 mm) at a flow rate of 0.1 μl/min via a microinjection pump. After each injection, the needle was held in place for 5 min to allow for virus diffusion and gradually withdrawn to prevent possible leakage from the needle track. The incision was sutured, and rat was returned to its home cage for 1-week recovery before subsequent experiments.
Immunofluorescence
Rats were anesthetized with 1% pentobarbital sodium and intracardially perfused with 4% paraformaldehyde. Brains were postfixed with 4% paraformaldehyde for 12 hours and kept in 20 and 30% sucrose solutions in turn for dehydration. Thirty-micrometer sections were sliced coronally using a cryostat microtome (model 1950, Leica). Free-floating sections were washed in phosphate-buffered saline, blocked with a buffer containing 5% bull serum albumin and 0.3% Triton X-100 for 1 hour, incubated with primary antibodies at 4°C overnight, washed three times in phosphate-buffered saline, and then incubated with secondary antibody solution containing 4′,6-diamidino-2-phenylindole for 1 hour at 37°C. The second antibodies used for the staining were Alexa Fluor 488/594/647 goat anti-rabbit/mouse immunoglobulin G (ZSGB-BIO, Beijing). Sections were lastly washed three times and mounted on microscope slides using the mounting medium (ZSGB-BIO, Beijing). Images were captured under a Leica TCS SP8 confocal laser-scanning microscope, and colocalization analysis and merged images were processed according to our previous work (25).
Electrophysiology
Rats were anesthetized with isoflurane and perfused transcardially with chilled sucrose-based cutting solution: 234 mM sucrose, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 0.5 mM CaCl2, 7 mM MgSO4, and 10 mM glucose (pH 7.4; 335 to 340 mosmol). The brain was removed quickly, and coronal bilateral slices that contained the PL were cut at 350 μm with a vibratome (VT1000S, Leica). These slices were incubated in 37°C artificial cerebral spinal fluid solution (aCSF) consisting of 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 2 mM CaCl2, 2 mM MgCl2, and 10 mM glucose (305 to 310 mosmol), aerated with 95% O2 and 5% CO2 to a final pH of 7.4. After 1-hour incubation, slices were transferred carefully to the recording chamber by super-fusing aCSF at room temperature.
A slice was viewed with an upright microscope (Axioskop Fsmot, Zeiss) equipped with infrared differential interference contrast optics. Pyramidal cells of the PL were recognized via a 40× water immersion lens. The recording pipettes (3 to 5 megohms) were filled with solution that contained 140 mM K-gluconate, 8 mM NaCl, 2 mM MgCl2, 1 mM EGTA, 10 mM Hepes, 2 mM Mg–adenosine triphosphate, and 0.3 mM Na–guanosine triphosphate (pH 7.2; 290 to 320 mosmol). Voltage and current signals were recorded from PL neurons using an Axon 200B amplifier. The spike firing pattern of the AP was used to electrophysiologically identify pyramidal neurons, which exhibited significant spike frequency adaptation in response to a depolarization current. The APs were recorded using the current-clamp mode.
Contextual fear conditioning
For CFC, we used a chamber (25 cm wide by 25 cm long by 25 cm high) with distinct visual cues and a grid floor, which consisted of 36 stainless steel rods (fear conditioning and startle system, Panlab, Spain). The chamber was cleaned with 70% ethanol before the introduction of each rat. Rats were kept in the conditioning chamber for a total of 300 s, when foot shocks (2 s, 0.75 mA) were delivered at 120, 180, and 240 s. During the test, rats were placed into the same context and allowed to explore for 300 s to monitor their freezing behavior. Freezing was defined as being motionless and measured by Packwin V2.0 software (Panlab, Spain). The amount of time spent freezing was expressed as a percentage of total session time. For context-specific fear conditioning, chamber A had a black plastic wall and roof, while chamber B was white.
For fear extinction, rats were placed in the conditioning chamber for a total of 300 s, when foot shocks (2 s, 0.50 mA) were delivered at 120, 180, and 240 s. Two weeks later, the rats were taken on Dox (100 mg/kg) for 24 hours to open a window of activity-dependent labeling. Rats were exposed to the context for 300 s for cell labeling, and Dox diets were withdrawn immediately after context exposure. One week later, rats were placed into the conditioned context and allowed to explore for 300 s for three consecutive days to monitor their fear extinction.
Fluorescence-activated cell sorting
Four weeks after delivery of AAV9-hSyn-SaCas9-3×flag and AAV9-U6-gRNA-mix-CMV-GFP into the PL, the rat was anesthetized with 1% pentobarbital sodium, and the brain was quickly removed and sectioned on a vibratome on ice. Individual slices of interest were transferred to a small dish containing cold (4°C) dissection media: 116 mM NaCl, 5.4 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 1.5 mM CaCl2, 1 mM MgSO4, 0.5 mM EDTA, 25 mM glucose, and 1 mM cysteine, bubbled with 95% O2 and 5% CO2. The PL was dissected and treated with dissection media with papain (1 mg/ml; LS003119, Worthington) for 30 min at 37°C. The tissue pieces were dissociated into single cells by gentle trituration and filtered through a 70-μm cell strainer (F613462, BBI). Sorting was performed by a fluorescence-activated cell sorter (BD Biosciences) in the single-cell sorting mode to select neurons with high enhanced green fluorescent protein fluorescence for subsequent deep sequencing or RNA sequencing (RNA-seq) analysis.
Deep sequencing for off-target analysis
Potential off-target sites were identified using CRISPR RGEN Tools (www.rgenome.net/cas-offinder/). For the three CBP targets described above, we computationally selected 33 candidate off-target sites in the rat reference genome (rn5) that were followed by a 5′-NNGRRT PAM with less than five mismatched bases. The genomic region flanking the target sites for each guide sequence was PCR amplified with the following protocol: preheat at 98°C for 30 s, 35 cycles of three-step amplification (98°C for 5 s, 62°C for 10 s, and 72°C for 20 s), and final extension at 72°C for 2 min. The primers for each potential off-target site are listed in table S4. PCR products were purified using DNA Clean-up Kit (CW2301, CWBIO) following the manufacturer’s recommended protocol and sequenced with the MGISEQ-200 (BGI, China). Reads were filtered by an average Phred quality (Q score) > 20 and perfect sequence matches to amplicon forward and reverse primers. Reads from on- and off-target loci were analyzed by first performing alignments against amplicon sequences that included 25 nucleotides upstream and downstream of the target site (a total of 77 bp). Alignments, meanwhile, were analyzed for indels from 5 bp upstream to 5 bp downstream of the target site (37 bp), and indels in this region were discarded with no change among six nucleotides upstream of the PAM sequence (cutting site by SaCas9).
RNA-seq analysis
Total RNA of the neurons by FACS was extracted by TRIzol reagent and quantified with the ND-2000 (NanoDrop Technologies). The libraries were sequenced by BGISEQ-500 (igeneCode Biotech, Beijing), and qualified reads were mapped to the rat reference genome (rn5). The expression level for each transcript was calculated using FPKM (fragments per kilobase of transcript per million fragments mapped). Hierarchical clustering and Gene Ontology were used to analyze differentially expressed genes, with log2 fold change >2 and false discovery rate <0.01 as significantly enriched.
Statistical analysis
The data were expressed as means ± SEM and analyzed using GraphPad Prism. Statistical analysis of two experimental groups was performed using two-tailed Student’s t tests, and comparisons of two groups with different time points were performed using two-way analysis of variance (ANOVA), with P < 0.05 as statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants from the National Natural Science Foundation of China (81974166, 31872774, 31371119, 91732107, 81571067, and 81821092), the National Basic Research (973) Program of the Ministry of Science and Technology of China (2014CB548200 and 2015CB554503), the “111” Project of Ministry of Education of China (B07001), the Beijing Natural Science Foundation (5182013), and the Interdisciplinary Medicine Seed Fund of Peking University (BMU2018MX011). Author contributions: H.S., M.Y., and Y.W. designed the experiment; H.S., S.F., S.C., X.Y., X.S., K.C., and X.-H.W. performed the experiments; H.S., L.M., J.W., X.Q., F.-Y.L., F.-F.L., and M.Y. analyzed the data; H.S., M.Y., and Y.W. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaay6687/DC1
Fig. S1. SaCas9 inactivates the p300 gene in vitro with high efficiency.
Fig. S2. Neuronal specificity for spine density analysis.
Fig. S3. Amygdala-projecting IL neurons are activated during extinction.
Table S1. gRNA sequences targeting cbp gene or p300 gene.
Table S2. AAV vectors used.
Table S3. Primer sequences for T7 endonuclease assays.
Table S4. Primer sequences for off-target analysis.
REFERENCES AND NOTES
- 1.Xia H., Mao Q., Paulson H. L., Davidson B. L., siRNA-mediated gene silencing in vitro and in vivo. Nat. Biotechnol. 20, 1006–1010 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Smith R. A., Miller T. M., Yamanaka K., Monia B. P., Condon T. P., Hung G., Lobsiger C. S., Ward C. M., McAlonis-Downes M., Wei H., Wancewicz E. V., Bennett C. F., Cleveland D. W., Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 116, 2290–2296 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., Zhang F., In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 33, 102–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ran F. A., Cong L., Yan W. X., Scott D. A., Gootenberg J. S., Kriz A. J., Zetsche B., Shalem O., Wu X., Makarova K. S., Koonin E. V., Sharp P. A., Zhang F., In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korzus E., Rosenfeld M. G., Mayford M., CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42, 961–972 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wood M. A., Kaplan M. P., Park A., Blanchard E. J., Oliveira A. M. M., Lombardi T. L., Abel T., Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn. Mem. 12, 111–119 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen G., Zou X., Watanabe H., van Deursen J. M., Shen J., CREB binding protein is required for both short-term and long-term memory formation. J. Neurosci. 30, 13066–13077 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuo E., Cai Y.-J., Li K., Wei Y., Wang B.-A., Sun Y., Liu Z., Liu J., Hu X., Wei W., Huo X., Shi L., Tang C., Liang D., Wang Y., Nie Y.-H., Zhang C.-C., Yao X., Wang X., Zhou C., Ying W., Wang Q., Chen R.-C., Shen Q., Xu G.-L., Li J., Sun Q., Xiong Z.-Q., Yang H., One-step generation of complete gene knockout mice and monkeys by CRISPR/Cas9-mediated gene editing with multiple sgRNAs. Cell Res. 27, 933–945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye X., Kapeller-Libermann D., Travaglia A., Inda M. C., Alberini C. M., Direct dorsal hippocampal-prelimbic cortex connections strengthen fear memories. Nat. Neurosci. 20, 52–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoover W. B., Vertes R. P., Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct. 212, 149–179 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Xu W., Südhof T. C., A neural circuit for memory specificity and generalization. Science 339, 1290–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zingg B., Chou X.-L., Zhang Z.-g., Mesik L., Liang F., Tao H. W., Zhang L. I., AAV-mediated anterograde transsynaptic tagging: Mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 93, 33–47 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tervo D. G. R., Hwang B.-Y., Viswanathan S., Gaj T., Lavzin M., Ritola K. D., Lindo S., Michael S., Kuleshova E., Ojala D., Huang C.-C., Gerfen C. R., Schiller J., Dudman J. T., Hantman A. W., Looger L. L., Schaffer D. V., Karpova A. Y., A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitamura T., Ogawa S. K., Roy D. S., Okuyama T., Morrissey M. D., Smith L. M., Redondo R. L., Tonegawa S., Engrams and circuits crucial for systems consolidation of a memory. Science 356, 73–78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Josselyn S. A., Köhler S., Frankland P. W., Finding the engram. Nat. Rev. Neurosci. 16, 521–534 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Liu X., Ramirez S., Pang P. T., Puryear C. B., Govindarajan A., Deisseroth K., Tonegawa S., Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature 484, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reijmers L. G., Perkins B. L., Matsuo N., Mayford M., Localization of a stable neural correlate of associative memory. Science 317, 1230–1233 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Squire L. R., Mechanisms of memory. Science 232, 1612–1619 (1986). [DOI] [PubMed] [Google Scholar]
- 19.McClelland J. L., Incorporating rapid neocortical learning of new schema-consistent information into complementary learning systems theory. J. Exp. Psychol. Gen. 142, 1190–1210 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Frankland P. W., Bontempi B., The organization of recent and remote memories. Nat. Rev. Neurosci. 6, 119–130 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Bukalo O., Pinard C. R., Silverstein S., Brehm C., Hartley N. D., Whittle N., Colacicco G., Busch E., Patel S., Singewald N., Holmes A., Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci. Adv. 1, e1500251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Do-Monte F. H., Manzano-Nieves G., Quinones-Laracuente K., Ramos-Medina L., Quirk G. J., Revisiting the role of infralimbic cortex in fear extinction with optogenetics. J. Neurosci. 35, 3607–3615 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sierra-Mercado D., Padilla-Coreano N., Quirk G. J., Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacol. 36, 529–538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fenno L., Yizhar O., Deisseroth K., The development and application of optogenetics. Annu. Rev. Neurosci. 34, 389–412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Y.-Y., Shao S., Zhang Y., Zheng J., Chen X., Cui S., Liu F.-Y., Wan Y., Yi M., Neural pathways in medial septal cholinergic modulation of chronic pain: Distinct contribution of the anterior cingulate cortex and ventral hippocampus. Pain 159, 1550–1561 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaay6687/DC1
Fig. S1. SaCas9 inactivates the p300 gene in vitro with high efficiency.
Fig. S2. Neuronal specificity for spine density analysis.
Fig. S3. Amygdala-projecting IL neurons are activated during extinction.
Table S1. gRNA sequences targeting cbp gene or p300 gene.
Table S2. AAV vectors used.
Table S3. Primer sequences for T7 endonuclease assays.
Table S4. Primer sequences for off-target analysis.