

Activated platelets cause microvascular inflammation via direct activation of perivascular mast cells.
Abstract
Platelet responses have been associated with end-organ injury and mortality following complex insults such as cardiac surgery, but how platelets contribute to these pathologies remains unclear. Our studies originated from the observation of microvascular platelet retention in a rat cardiac surgery model. Ensuing work supported the proximity of platelet aggregates with perivascular mast cells (MCs) and demonstrated that platelet activation triggered systemic MC activation. We then identified platelet activating factor (PAF) as the platelet-derived mediator stimulating MCs and, using chimeric animals with platelets defective in PAF generation or MCs lacking PAF receptor, defined the role of this platelet-MC interaction for vascular leakage, shock, and tissue inflammation. In application of these findings, we demonstrated that inhibition of platelet activation in modeled cardiac surgery blunted MC-dependent inflammation and tissue injury. Together, our work identifies a previously undefined mechanism of inflammatory augmentation, in which platelets trigger local and systemic responses through activation of perivascular MCs.
INTRODUCTION
More than 225,000 cardiac surgeries are performed annually in the United States (1). While these procedures provide life-saving corrections of coronary blood flow or valvular abnormalities, the inherent combination of surgical trauma, extracorporeal perfusion, and ischemia/reperfusion (I/R) injury often evoke harmful systemic inflammatory responses (2). These inflammatory responses prominently manifest as acute loss of vascular tone (approximately 25% of patients) (3) but are also linked to an ongoing, high incidence of end-organ damage such as acute kidney injury [up to 54% for all stages (4)]. As a consequence, anti-inflammatory interventions have been identified as a key to improve disease outcomes. Since the underlying basis for inflammatory activation remains poorly defined, appropriate ways to modify inflammation during cardiac surgery have remained elusive.
Platelets are increasingly recognized as circulating immune cells, which intimately associate with activated microvascular endothelia (5) and which have the capacity to markedly influence inflammation through direct cell-cell communications and the secretion of inflammatory mediators [reviewed in (6)]. The occurrence of platelet activation during cardiac surgery is a well-established phenomenon (7), and there is mounting evidence that platelet-dependent inflammatory responses are relevant to patient outcomes. Hence, we have recently observed that platelet responses, measured as a drop in platelet count, are an independent risk factor to acute kidney injury and mortality following coronary bypass grafting surgery (4). However, how platelets contribute to the harmful responses elicited by cardiac surgery remains undefined. Here, we sought to investigate whether platelets have a specific role in activating mast cells (MCs) and to depict the implications of such an interaction in a preclinical model of extracorporeal circulation.
In our previous work, which focused on the early events following cardiopulmonary bypass (CPB), we established that MCs are critical effector cells for injurious and inflammatory responses in a rat model (8) as well as in patients undergoing cardiac surgery (9). These observations linked perioperative inflammatory responses to a cell type that is increasingly recognized as a master regulator of early inflammation (10). Strategically located at endothelial and epithelial interfaces, MCs assume a critical role in organizing responses to pathogens and tissue stress such as I/R through the release of powerful preformed and de-novo synthesized effector molecules, which promote recruitment of inflammatory cells and facilitate their tissue infiltration [reviewed in (10)]. Dysregulated, widespread activation of MCs is a critical determinant of mortality, e.g., in anaphylaxis (11) and hemorrhagic fever (12), by causing shock and vascular leakage. What defines the role of MCs in these pathologies is their close association with blood vessels, which ensures that MC products—beyond their local tissue effects—act directly on endothelial cells and can enter the circulation to rapidly propagate systemic and distant-site inflammation. The significant MC activation observed during cardiac surgery therefore constitutes an important event, which may lead toward an augmented systemic inflammatory response and singles out MC activation as a novel therapeutic target in the ongoing attempt to blunt harmful inflammation in these patients. However, what causes initial activation of MCs within the complex sequence of events elicited by cardiac surgery remains unknown, limiting our understanding of cardiac surgery–associated inflammation and, specifically, our ability to develop new therapeutic interventions to improve outcomes.
In the present study, we sought to identify the factor(s) contributing to MC activation during cardiac surgery using CPB circulation. Since MCs have a perivascular location, we investigated the possibility that the MC-activating factor acts from within the circulation.
RESULTS
Platelet aggregation occurs at sites of early tissue injury following rat CPB
The rat deep hypothermic circulatory arrest (DHCA) model recapitulates several pathophysiologic stimuli present during cardiac surgery such as (nonpulsatile) CPB, blood contact to artificial surfaces of the extracorporeal circulation, cooling, and whole-body I/R. Using this DHCA model, we have previously demonstrated that one of the earliest signs of tissue injury is evidenced in the intestines and that MCs at this site are crucial effectors of pathology through release of preformed mediators (8). Therefore, we examined this site for signs of increased platelet aggregation after DHCA and observed in hematoxylin and eosin staining several platelet-rich thrombi in small and large intestinal sections (Fig. 1A). This observation was confirmed by immunostaining for the platelet-specific marker CD41 in samples obtained 2 hours after completion of CPB (Fig. 1, B to D). Aggregation of platelets was specifically associated with tissue experiencing injury because no platelet aggregation was observed in the lung and brain, where no appreciable tissue injury was observed at this early time point [(8) and fig. S1]. As a consequence, these findings motivated a more detailed investigation into a possible link between platelet deposition and early MC-mediated injury.
Fig. 1. Platelet microvascular deposition and MC activation.
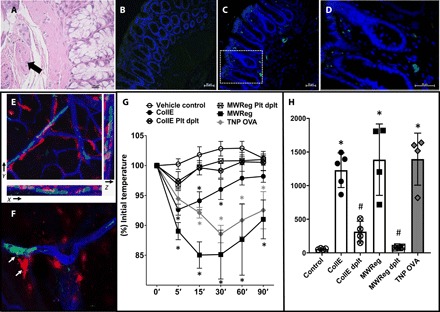
(A) Hematoxylin and eosin staining of rat colons 2 hours after completion of the DHCA model. Arrow indicates platelet-rich thrombus in a small submucosal vessel. Immunofluorescence staining for CD41 (green) in sham (B) or experimental (C and D) animals 2 hours after completion of DHCA model (4′,6 diamidino-2-phenylindole nuclear counterstain). Representative images of n = 4 per condition; magnification, ×200. (D) Insert in (C) in 600×. (E) Z-stack confocal laser scanning micrographs of whole-mount sections (ear) taken from Mcpt5-Cre tdTomatofl/fl mice 60 min after intravenous injection of a collagen and epinephrine mixture. Endogenously expressed tdTomato (red) outlines MCs with additional staining performed against CD41 (green) and CD31 (blue). An animated three-dimensional reconstruction can be viewed in the Supplementary Materials. (F) MC granule staining [tetramethyl rhodamine isothiocyanate (TRITC)–avidin, red; arrows indicate released MC granules] reveals activated MCs in close vicinity to intravascular (anti-CD31, blue) platelet aggregates (anti-CD41, green). Representative images of n = 4 per condition. (G) Rectal temperature following collagen and epinephrine injection (CollE) in comparison to anaphylaxis after sensitization with trinitrophenol (TNP)–specific IgE and exposure to TNP-conjugated ovalbumin (TNP OVA) or vehicle control treatment. In addition, platelets were activated with a monoclonal antibody against mouse integrin αIIb, clone MWReg30 (MWReg). A subset of animals was platelet-depleted before receiving collagen and epinephrine (CollE Plt dplt) or MWReg (MWReg Plt dplt). n = 6 per condition. *P < 0.05 versus vehicle control, two-way ANOVA. (H) Plasma chymase levels following collagen and epinephrine, or MWReg injection or TNP OVA anaphylaxis. Data are represented as the means ± SD. n = 4 to 6 per condition. *P < 0.05 versus vehicle control and #P < 0.05 versus respective treatment group, one-way ANOVA and Tukey’s multiple comparisons test.
Platelets aggregate in microvasculature in close proximity to activated MCs
Platelets, like MCs, can release large amounts of preformed inflammatory mediators; thus, they have the potential to rapidly initiate responses upon activation (6). We hypothesized that platelets may trigger early MC-mediated tissue injury in the DHCA model through their capacity to directly activate MCs. Since platelets are typically intravascular while MCs are extravascular, we sought to examine how this purported interaction could occur. Using microscopy of whole-mount vascular beds, we examined the spatial relationship between activated platelets and perivascular MCs following platelet activation in Mcpt5-Cre tdTomatofl/fl mice. In these animals, MCs harbor a red fluorescent dye, which allows for direct microscopic visualization. We induced specific activation of platelets in these mice by intravenous administration of a cocktail of collagen and epinephrine, carefully titered to cause systemic platelet activation but not mortality. We then examined the vasculature of the mouse ear for microvascular platelet aggregation using microscopy. We found numerous platelet aggregates at the inner walls of blood vessels in close proximity to perivascular MCs (Fig. 1E). Because the genetic elements of Mcpt5 drive tdTomato expression in MCs, red fluorescence is mostly cytoplasmic and does not readily denote degranulation. Therefore, to evaluate the actual degranulation of MCs, we stained MC granules by using avidin, which is routinely used for MC granule staining (8, 13), and found that several of these MCs showed signs of degranulation (Fig. 1F). To confirm that significant MC activation occurred following platelet activation, we examined collagen and epinephrine–treated mice for signs of shock, a classical manifestation of systemic MC activation. It is known that when mice are subjected to immunoglobulin E (IgE)–mediated MC activation, they experience anaphylaxis, which is indicated by a sharp drop in core body temperature (13). We observed that collagen and epinephrine–treated mice experienced a sharp drop in core body temperature, which was attributable to MC activation based on the high levels of chymase—a major prestored MC mediator (10)—detected in the plasma of these mice (Fig. 1, G and H). To confirm the contribution of platelets to this MC-mediated anaphylaxis response, we depleted platelets before administration of the collagen and epinephrine cocktail and found that it abrogated the hypothermic response (Fig. 1G). Thus, specific activation of platelets results in degranulation of perivascular MCs.
Products of activated platelets can induce MC degranulation
We next investigated the mechanism of platelet-mediated MC activation. The close spatial relationship between platelet aggregates and perivascular MCs on apposing sides of the vasculature raised the possibility that activated platelets release bioactive agents that traverse the endothelial barrier to stimulate MC degranulation. Therefore, we examined whether any secreted products of platelets had MC-activating properties by applying conditioned medium from activated human platelets on to two different human MC lines [ROSA (14) and LAD2 (15)]. Cell-free conditioned medium from platelets activated by either thrombin, collagen, or convulxin, but not from resting platelets, evoked a comparable MC degranulation response from both MC lines (Fig. 2A). The magnitude of the MC responses was dependent on the concentration of platelets in the conditioned medium and the duration of time that the platelets were activated (Fig. 2A). To further define the nature of the platelet factor(s), we ultracentrifugated the conditioned medium after platelet activation and observed that MC-stimulating activity was contained in the supernatant and not in the microparticle pellet. Furthermore, the soluble portion obtained from freeze-thawed resting platelets (to release their cellular content without activation) only marginally activated MCs, suggesting that the MC-activating factor(s) was not stored in significant quantities as a preformed mediator. Together, these in vitro observations suggest that MC activation is not dependent on direct contact and that soluble factors formed after platelet activation can directly trigger MC degranulation.
Fig. 2. Activated platelets secrete PAF to stimulate MC degranulation in vitro.
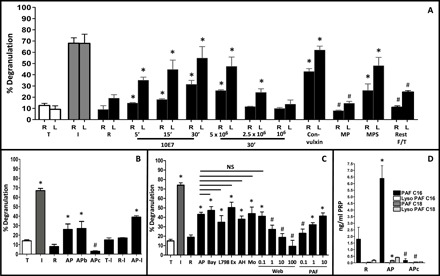
(A) Isolated platelets were activated for indicated periods (5, 15, and 30 min) and at indicated concentrations (1 × 107, 5 × 106, 2.5 × 106, and 1× 106 platelets/ml) with thrombin (or convulxin where indicated). Two MC lines [ROSA (R) and LAD2 (L)] were then exposed to cell-free supernatant from this reaction and MC degranulation measured by tryptase activity assay. In addition, supernatant from activated platelets was ultracentrifuged, and the pellet—resuspended in Tyrode’s buffer (MP, microparticle pellet)—or the supernatant (MPS, microparticle supernatant) was added to MCs. Last, resting platelets (1 × 107) were freeze-thawed and centrifuged, and debris-free supernatant was tested on MCs (Rest F/T). T, Tyrode’s buffer; I, ionomycin positive control; R, resting platelet supernatant. (B) For biochemical characterization of MC-activating effect, LAD2 cells were exposed to supernatant from activated platelets without further treatment (AP), after boiling for 30 min (APb), incubation on activated charcoal (APc), or following isolation of lipid fraction (AP-l). T-l, lipid fraction from Tyrode’s buffer; R-I, lipid fraction from resting platelet supernatant. (C) LAD2 cells were pretreated with antagonists against various lipid mediators [BAY-u 3405 (10 μM): Bay; L798,106 (100 nM): L798; Ex26 (10 μM): Ex; AH 6809 (10 μM): AH; montelukast (100 μM): Mo; WEB2086 (0.1 to 100 μM): Web] before exposure to heat-treated activated-platelet supernatant. Purified PAF was added at 0.1 to 10 μM. Degranulation was measured using β-hexosaminidase assay. NS, not significant. (D) Quantitative determination of PAF in supernatants from resting and activated platelets and activated platelet supernatant absorbed with activated charcoal. Data are represented as the means ± SD. *P < 0.05 versus resting platelet supernatant and #P < 0.05 versus respective activated platelet supernatant, one-way ANOVA and Tukey’s multiple comparisons test. All data derived from four independent experiments were performed in triplicate wells. PRP, platelet-rich plasma.
Platelet activating factor is the platelet product causing MC degranulation
We next sought to determine the identity of the MC-activating factor(s) in the platelet-conditioned medium. Notably, boiling of the platelet supernatant did not reduce MC-activating activity, whereas absorption with activated charcoal abrogated it, suggesting that the active component was a lipid compound (Fig. 2B). We confirmed this by preparing a lipid extract of the conditioned medium and observed that MC-activating activity was largely contained in this extract. Since platelets are already known to produce several prominent bioactive lipid mediators, we undertook a screening experiment of possible MC-activating candidates. Using antagonists to leukotriene receptors (montelukast: 1 to 100 nM), the prostaglandin EP1/EP2 receptor (AH 6809: 1 to 100 μM), the EP3 receptor (L-798,1016: 1 to 100 nM), the dual thromboxane TP/prostaglandin DP2 receptor (BAY-u 3405: 0.1 to 10 μM), or the shingosine-1-phosphate receptor 1 (Ex-26: 1 to 100 μM) before exposure to the activated platelet-conditioned medium, we observed no appreciable decline in MC activation (Fig. 2C). However, when we pretreated MCs with WEB2086, an inhibitor of the platelet activating factor (PAF) receptor, we observed a dose-dependent inhibition of MC activation. To verify that PAF is the active factor in the platelet-conditioned medium, we conducted liquid chromatography–mass spectrometry (LC-MS) lipid quantification of the medium and found that platelet activation caused the release of significant amounts of PAF C16 and C18 (Fig. 2D). Consistent with the functional properties of PAF, absorption of platelet medium with activated charcoal significantly reduced PAF levels. Last, we exposed MCs to increasing doses of purified PAF and observed dose-dependent MC degranulation (Fig. 2C and fig. S2). Together, our data indicate that PAF is the predominant platelet product responsible for MC degranulation.
These findings led us to question whether platelet-derived PAF can directly act on MCs, which are found on the apposing side of the endothelium. We therefore applied PAF at concentrations shown previously to cause MC activation to the apical side of human umbilical vein endothelium cells (HUVECs) grown on semipermeable supports. Starting from a transendothelial resistance (TEER) of 146.9 ± 22.7 ohm·cm2, we observed that PAF itself significantly but transiently disrupted endothelial integrity. However, when PAF was added to HUVECs in the presence of human MCs (ROSA) in the basal compartment of the Transwell system, the drop in TEER did not reverse during the experiment (Fig. 3A). At the same time, we observed that these basal MCs degranulated after addition of apical PAF and that the extent of degranulation was comparable to that when PAF was added to Transwell inserts without a HUVEC cell layer (Fig. 3B). Together, these results suggest that intravascular PAF can act on perivascular MCs and that this contact is made possible likely by a collaborative effort of PAF and MC products on endothelial barrier tightness. Moreover, endothelial cells do not appear to directly participate in the signaling events.
Fig. 3. PAF disrupts endothelial integrity and activates MCs across endothelial barriers.
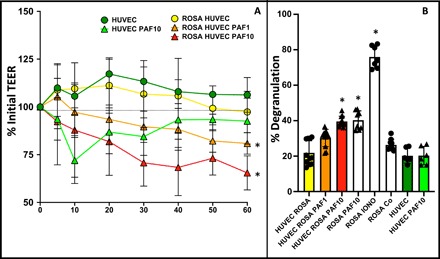
(A) HUVEC cells were grown to confluency on permeable supports, and then 1 × 106 ROSA cells were added to some of the basal compartments (ROSA HUVEC) followed by addition of PAF at 1 or 10 μM or vehicle control to the apical compartment. TEER was measured for 1 hour. *P < 0.05 versus untreated ROSA/HUVEC cocultures by two-way ANOVA. (B) β-Hexosaminidase from supernatants of HUVEC endothelial cells, ROSA MC cells, and cocultured HUVEC/ROSA cells with or without addition of PAF (at 1 or 10 μM) to the apical side of the endothelia. n = 8 per condition. Results shown as average ± SD, *P < 0.05 versus untreated ROSA/HUVEC cocultures.
In vivo platelet activation and release of PAF results in shock
In view of our in vitro results, we now sought to determine whether the platelet-mediated MC activation in vivo was dependent on PAF. We therefore first examined the systemic response to collagen and epinephrine injection after pretreatment with the PAF inhibitor WEB2086. Although we noticed a marked blunting of the shock response, it was not totally abolished (Fig. 4A), and thus, we could not readily infer that this response was fully PAF-mediated. A possible explanation for this finding is that collagen and epinephrine injections cause systemic thrombosis and thus evoke temperature changes independent of inflammatory reactions. Therefore, we next examined a second, distinct model of systemic platelet activation (16). This model achieves platelet activation with a monoclonal antibody against mouse integrin αIIb (clone MWReg30), which causes thrombocytopenia within a few minutes (Fig. 4, B and C). We found that platelet activation in this manner also resulted in a sharp drop in core body temperature, which was abrogated in mice pretreated with WEB2086, indicating that it was PAF dependent (Fig. 4D). We further confirmed that this severe inflammatory response was platelet dependent by showing that depletion of platelets in these mice before MWReg30 antibody administration protected from shock. Further support for the notion that the shock response following platelet activation was PAF dependent was provided by the finding that phospholipase A2 knockout (Pla2-KO) mice, which display a significantly reduced PAF production (17), failed to succumb to shock. Similarly, deficiency of PAF sensing, as observed in PAF receptor knockout (Pafr-KO) mice (18), also protected from shock following MWReg30 antibody administration (Fig. 4D). In agreement, injection of purified PAF was sufficient to cause shock in wild-type (WT) and Pla2-KO mice but not in Pafr-KO mice (Fig. 4E). Together, in vivo platelet activation and systemic release of platelet-derived PAF results in shock.
Fig. 4. Platelet-secreted PAF stimulates MC degranulation in vivo.
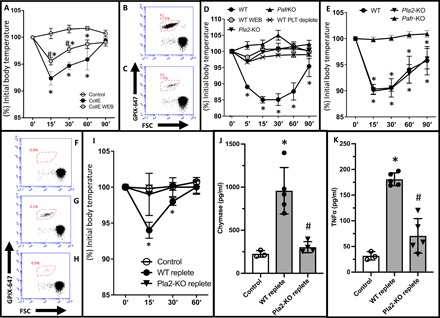
(A) Temperature measurement following intravenous administration of collagen and epinephrine with (CollE WEB) or without (CollE) pretreatment with the PAF inhibitor WEB2086; control animals received intravenous phosphate-buffered saline (PBS). Administration of anti–integrin aIIb (MWReg30) antibody caused significant thrombocytopenia [representative anti-glycoprotein IX (GPIX) stain versus forward scatter (FSC) before (B) and after (C) treatment]. (D) Temperature measurement following intravenous administration of MWReg30 in untreated mice (WT), in WEB2086-pretreated (WT-WEB) and platelet-depleted (WT-PLT deplete) mice, and in phospholipase A2–(Pla2-KO) and PAF receptor–(Pafr-KO) knockout animals. (E) Temperature measurement after administration of purified PAF (2 mg/g bodyweight) in WT, Pla2, or Pafr-knockout (Pafr-KO) mice. Adoptive platelet transfer was performed in hIL-4Rα/GPIbα–transgenic mice. These were platelet-depleted through administration of anti–IL-4R antibody (F) and repleted with platelets from either Pla2-KO or WT mice (G) before administration of MWReg30 (H). Fluorescence-activated cell sorting (FACS) data are shown as anti-GPIX stain versus forward scatter and is representative of experimental findings. Systemic response to MWReg30 was measured as change in body temperature (I) and by quantification of plasma levels of MC-specific chymase (J) and TNFα (K). Data are represented as the means ± SD. n = 4 to 6 per condition. *P < 0.05 versus control-treated animals and #P <0.05 versus respective stimulated WT or WT-repleted animals. (A, B, and G) Two-way ANOVA; (H and I) one-way ANOVA and Tukey’s multiple comparisons test.
Platelet-released PAF is sufficient to induce shock
Because PAF is released by various immune cells into the circulation (19), we sought to verify that it is indeed platelet-secreted PAF that causes MC activation and shock in vivo. For this, we used an adoptive transfer model where platelets were infused into human interleukin-4 receptor alpha/platelet glycoprotein Ibα (hIL-4Rα/GPIbα) transgenic mice whose endogenous platelets had been depleted by administration of anti–IL-4R antibody as described previously (20). This allowed repletion with platelets from either WT or Pla2-KO mice that lack the ability to generate PAF (17). We then administered MWReg30 antibody to activate platelets in both of these repleted groups and examined the mice for MC activation and shock. Here, the exposure of the WT-platelet reconstituted mice to MWReg30 elicited an overall blunted systemic response compared to our previous experiments, likely due to the reduced platelet numbers achieved after reconstitution (Fig. 4, F to H). However, this response was altogether absent in mice reconstituted with Pla2-KO platelets (Fig. 4I). Furthermore, only mice reconstituted with WT platelets displayed increases in plasma-chymase and plasma–tumor necrosis factor–α (TNFα) levels, which are indicators of MC activation (Fig. 4, J and K). These results support that the PAF responsible for MC activation and shock following administration of MWReg30 antibody is specifically platelet derived.
MCs are critical effectors of platelet-derived PAF effects
Since shock as indicted previously is a characteristic inflammatory response linked to MCs, we aimed to further define the specific role of MCs in the propagation of PAF-mediated platelet responses. We compared MWReg30 antibody–induced shock responses in WT and MC-deficient Sash mice and in Mcpt5-cre+iDTR+ mice depleted of MCs (21). We found that whereas MC-competent mice evoked a significant shock response to MWReg30 administration, both groups of MC-deficient mice experienced blunted shock response (Fig. 5A). Since shock is preceded by vascular leakage, another well-known MC-mediated inflammatory response, we compared vascular leakage in WT and MC-deficient mice following MWReg30 administration. We found that WT mice experienced severe vascular leakage but not MC-deficient or MC-depleted mice (Fig. 5, B to E). Last, products of activated perivascular MCs can also significantly affect the organ they are proximal to. To demonstrate the potential of PAF-activated MCs to affect surrounding tissue responses, we examined expression of inflammatory genes in the intestines of MC-deficient Sash mice reconstituted with bone marrow–derived MCs (BMMCs) from WT or Pafr-KO mice following administration of MWReg30. BMMC injection into Sash mice does not reconstitute tissue MC uniformly but achieves good reconstitution in the intestines (22). For comparison, we also included MC-deficient Sash mice that were not reconstituted in this assay. As shown in Fig. 5F, animals reconstituted with WT BMMC, but not unreconstituted mice or mice reconstituted with BMMCs from Pafr-KO animals, demonstrated a significant increase in tissue inflammatory gene expression in intestinal samples, suggesting that PAFR (PAF receptor)–competent MCs are necessary for platelets to promote tissue inflammation. Thus, platelet-induced MC degranulation serves to amplify platelet-initiated inflammatory signals and translate these into tissue responses.
Fig. 5. MCs propagate tissue responses after platelet activation.
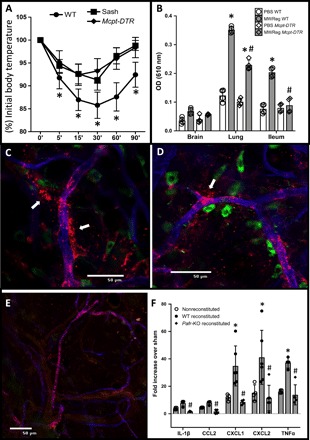
(A) Temperature measurement following intravenous administration of MWReg30 in WT mice and in MC-deficient KitW-sh/W-sh (Sash) or MC-depleted Mcpt5-cre + iDTR + (Mcpt-DTR) mice. n = 6 per condition. *P < 0.05 versus MWReg30-treated WT, two-way ANOVA. (B) Extravasation of Evans blue after intravenous administration of MWReg30 in control or MC-depleted Mcpt-DTR mice. OD, optical density. n = 5 per condition. *P < 0.05 versus unchallenged WT; #P < 0.05 versus MWReg30-treated WT, one-way ANOVA and Tukey’s multiple comparisons test. Immunofluorescence in whole-mount tissue (ear) in animals administered 150 kDa of TRITC-dextran intravenously before MWReg30 administration [blue, endothelium (anti-CD31); green, MCs (fluorescein isothiocyanate–avidin)] shows tracer extravasation (arrows) in WT mice (C and D) but not in Sash mice (E). (F) Transcriptional analysis of small intestinal tissue expression of inflammatory markers in Sash mice or in Sash mice reconstituted with Pafr-KO (Pafr-KO reconstituted) or WT (WT reconstituted) bone marrow–derived MCs and treated with MWReg30. Data are represented as the means ± SD. n = 4 to 6 per condition. *P < 0.05 versus nonreconstituted and #P < 0.05 versus WT-reconstituted animals. One-way ANOVA and Tukey’s multiple comparisons test.
Prevention of platelet activation during rat CPB abrogates MC activation and associated tissue injury
In view of the potential for platelets to trigger MC-mediated inflammatory responses, we investigated the consequences of blocking platelet activation in the rat DHCA model, and specifically the impact of such platelet inhibition on subsequent MC activation. Because of the close correlation between the site of tissue damage and deposition of aggregated platelets during DHCA, we first sought to map the major sites of platelet deposition following this procedure. For these studies, we isolated platelets from donor rats and labeled them with NIR78 (23), a near-infrared label, and then intravenously administered these cells immediately after completion of the CPB. After 2 hours of recovery, the major organs of the rats were harvested and imaged for platelet deposition. We found that a major deposition site of platelets following DHCA was the intestines (Fig. 6A). This observation supports our earlier finding indicating preferential deposition in the intestines as a primary site of tissue damage following DHCA (8). As confirmed by immunofluorescence using a platelet-specific marker, imaging revealed platelet deposition also in the kidneys (fig. S3). However, as reported previously, we found no histological (8) or biochemical (urine neutrophil gelatinase-associated lipocalin) (fig. S3) evidence of renal injury at this time point. Kidneys feature a similar susceptibility to hypoxia as the gut (24) but contain very few MC in their parenchyme (25).
Fig. 6. Platelet inhibition reduces MC activation and local and systemic inflammatory responses in a rat DHCA model.
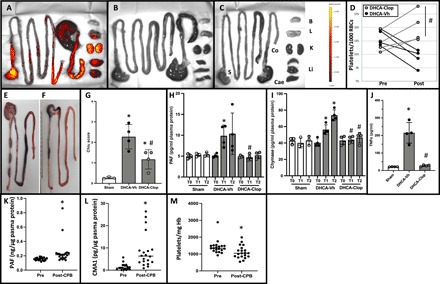
In vivo platelet labeling documents significant tissue platelet retention in vehicle-pretreated animals (A) but not in clopidogrel-pretreated animals (B) or in sham animals (C). Organs are identified in (C): stomach (S), colon (Co), caecum (Cae), brain (B), lungs (L), kidney (K), and liver (Li). (D) Platelet count before and after CPB in clopidogrel-pretreated (gray lines) or vehicle-pretreated (black lines) animals. To account for dilutional effects during extracorporeal circulation, data are presented as the ratio of platelet over red blood cell (RBC) count. #P < 0.05 versus DHCA-Vh, by unpaired Student’s t test of delta baseline values. Each line represents one animal. Representative images of the macroscopic intestinal phenotype in vehicle-pretreated (E) and in clopidogrel-pretreated animals (F). (G) Microscopic injury score in sham and DHCA-treated vehicle control (DHCA-Vh) or clopidogrel-pretreated (DHCA-Clop) animals. (H) Plasma PAF and (I) plasma chymase levels (normalized to plasma protein to adjust for on-bypass dilution effects) at baseline (T0), after CPB (T1), and after a 2-hour recovery period (T2). (J) Plasma TNFα levels after a 2-hour recovery period. Data are represented as the means ± SD. n = 4 per condition and n = 3 for sham. *P < 0.05 versus sham and #P < 0.05 versus DHCA-Vh, one-way ANOVA and Tukey’s multiple comparisons test. Plasma PAF (K) and chymase (L) levels were determined by ELISA in patients undergoing cardiac surgery with DHCA. Samples were collected after induction of anesthesia (baseline: pre) or after completion of CPB perfusion (post-CPB). To account for dilution effects, values are normalized for plasma protein content. (M) Platelet counts were obtained from medical records at time of baseline or post-CPB blood draw and normalized to hemoglobin (Hb) concentration to account for perioperative blood loss and dilution. n = 20, values shown with median, *P < 0.01 by Wilcoxon signed-rank test. Photo credit: Jörn Karhausen, Duke University.
We found that when we pretreated animals with clopidogrel, a potent P2Y12 inhibitor that has been extensively studied in systemic conditions involving platelet activation (26), we could completely block platelet deposition in the intestines and other sites after DHCA (Fig. 6B and fig. S3). Consistent with this finding, rats pretreated with clopidogrel maintained their platelet count throughout the experiment, whereas in control animals, we observed a drop in platelet numbers after completing CPB (Fig. 6D). Furthermore, clopidogrel pretreatment significantly reduced intestinal pathology compared to vehicle-treated DHCA mice as evidenced by both macroscopic (Fig. 6, E and F) and microscopic (Fig. 6G) examination. Plasma PAF levels, which displayed a sharp rise after DHCA in control animals, did not change significantly in the clopidogrel-treated group (Fig. 6H). Supporting the notion that abrogation of platelet activation also blocks MC activation, we observed limited levels of circulating chymase and TNFα levels in the clopidogrel-treated rats compared to controls (Fig. 6, I and J). Thus, during modeled DHCA, platelet activation and aggregation are critical preceding events to MC activation and the severe inflammation and tissue injury in the intestines. To provide clinical support to our findings, we measured PAF- and MC-specific chymase in 20 consecutive patients undergoing DHCA for repair of proximal aortic pathologies. Consistent with our findings in the rat model, we observed a significant increase of plasma PAF {median baseline (plasma protein), 0.154 ng/μg [interquartile range (IQR), 0.138 to 0.169 ng/μg] versus post-CPB, 0.213 ng/μg (IQR, 0.174 to 0.236 ng/μg); P < 0.01; Fig. 6K} and chymase levels [baseline (plasma protein), 1.038 pg/μg (IQR, 0.502 to 2.085 pg/μg) versus post-CPB, 6.322 pg/μg (IQR, 3.630 to 8.929 pg/μg); P < 0.01; Fig. 6L]. During the same period, platelet numbers significantly decreased relative to the hemoglobin (Hb) concentration [baseline, 1344.5 platelets/mg Hb (IQR, 1226.2 to 1482.4 platelets/mg Hb) versus post-CPB, 1045.6 platelets/mg Hb (IQR, 855.3 to 1251.0 platelets/mg Hb); P < 0.01], which suggests that the drop in platelet numbers was not due to bleeding or dilution but potentially due to concurrent platelet activation (Fig. 6M). These observations document that our experimental data align with events observed in patients undergoing comparable procedures.
DISCUSSION
Rapid activation of multiple, powerful inflammatory pathways during and after cardiac surgery has been identified as a key to the ongoing, high incidence of end-organ injury associated with these procedures. To elucidate the critical factors that shape decisions of tissue inflammation and injury in this setting, we focused on early events, reasoning that by limiting our studies to this window, we would identify mediators that are initiators of the inflammatory cascade and prime candidates for therapeutic intervention. This approach revealed a critical role of MCs and linked MC activation to early tissue injury and inflammation in a rat model (8) and to intraoperative hypotension in cardiac surgical patients (9). However, a major obstacle to further develop the therapeutic potential of MC modulation is that it currently remains unclear how MCs are activated in this setting. Therefore, we further examined the injury phenotype of the intestine as a primary site of tissue injury and MC activation in the setting of extracorporeal circulation (8, 27) and found significant platelet deposition on the luminal sides of small blood vessels. This microvascular placement positions platelets in close proximity to perivascular MCs, and indeed, our ensuing work demonstrated that platelets, through release of the lipid mediator PAF, actively triggered MC activation and thus caused shock, vascular leakage, and tissue inflammation. Hence, our results outline a powerful, previously undefined mechanism of inflammatory augmentation by establishing the collaboration of two cell types that have, relatively recently, come into focus as important immune sentinel cells—platelets within the intravascular compartment (6) and MCs at the tissue/microvascular interface (10).
What determines the preferential platelet deposition in the gut in our model is unclear, but evidence exists demonstrating that changes of splanchnic blood flow during nonpulsatile CPB result in substantial intestinal hypoperfusion [reviewed in (24)]. In agreement, we and others had shown intestinal I/R injury and intestinal MC activation to be the earliest signs of end-organ injury in rat and porcine models of extracorporeal circulation (8, 27). Following I/R, endothelial cell surfaces undergo significant changes, resulting in the rapid and sustained adherence of platelets to postcapillary venules upon reperfusion (5). Consequently, it is likely that the particularly hypoperfusion-prone intestinal vasculature provides a unique locale that facilitates the inflammatory collaboration between platelets and perivascular MCs thus making the gut a “hotspot” for the generation of inflammatory mediators in conditions such as cardiac surgery.
Platelets are increasingly appreciated as central immune regulatory cells that directly interact with both endothelium and intravascular immune cells and perform multifaceted inflammatory functions such as regulating neutrophil recruitment, extracellular trap formation, or cytokine release [reviewed in (6)]. In our work, the close spatial relationship between platelet aggregates and perivascular MCs on apposing sides of the vasculature raised the possibility that activated platelets release bioactive agents that traverse the endothelium to stimulate MC degranulation. Consequently, through a series of fractionation studies and biochemical assays, we established that platelets can activate MCs through secretion of PAF, a potent proinflammatory phospholipid implicated in various pathological reactions including anaphylaxis (28). PAF is released by various cells of the host defense system with neutrophils, basophils, endothelial cells, and MCs previously identified as major producers (19). Consistent with our results, platelet-dependent release of PAF has been described (29), but the relative contribution of platelets to overall PAF levels and whether PAF from other sources (e.g., endothelia) contributes to platelet-triggered responses remain to be further defined.
Consistent with our findings, PAF is not stored in the preformed state but rather is rapidly synthesized in response to cell-specific stimuli by remodeling of cellular phosphatidylcholine (30). Receptor-induced activation of the key enzyme, cytosolic phospholipase A2 (PLA2), is crucial for the acute lipid membrane remodeling during platelet activation and not only constitutes the first step in generating lipid mediators such as PAF but also provides important substrates required to support the energetic demands during platelet activation (31). PLA2 functions are not exclusive to PAF metabolism and Pla2-knockout (Pla2-KO) mice appear to have abnormalities, e.g., in thromboxane A2 synthesis (32). However, our studies involving the specific PAF antagonist WEB2086 and chimeric mice, in which we reconstituted platelet-depleted mice with platelets lacking PLA2 and therefore PAF production, strongly suggest that platelet-derived PAF causes MC activation. Conversely, we also provide evidence on the specific role of MC sensing of such platelet-derived PAF by use of MC-deficient and MC-depleted animals as well as of MC-deficient animals repleted with Pla2-knockout (Pla2-KO) BMMC. Together, these experiments support our notion that platelet-derived PAF triggers MC activation. Although the bioavailability of PAF in the circulation is very limited (33), it is conceivable that platelets create a protective microenvironment where PAF, because of its lipid nature, is able to traverse the endothelial walls and reach MCs. Consistent with reported evidence (34), we showed that platelet-derived PAF significantly alters endothelial barrier integrity and can thus act on MCs on the apposing side of the endothelium. In vivo, such contact may further be facilitated by the fact that MCs appear to form protrusions across the endothelial cell layer to directly survey intravascular events (35).
MC differentiation is highly tissue specific, and PAF receptors have been found in lung MCs and peripheral blood–derived but not skin MCs (36). Therefore, while our work demonstrates that a platelet-specific stimulus can cause release of PAF and resultant perivascular MC activation, these responses may vary in different tissues depending on the receptor equipment of local MC populations. In addition, MCs are not only sensors of PAF but also an important source of this mediator. During anaphylaxis, high levels of PAF are detected (28), and it is believed that hematogenous dissemination of this agent may be pivotal for rapid systemic MC activation after localized allergen exposure (36). This highlights that similarly following platelet-triggered MC activation, MC-autocrine production of PAF (37), as well as of further powerful mediators, may be instrumental in spreading and magnifying an initially limited response.
While our work identifies MC stimulation through platelet-derived PAF as an important, previously unknown proinflammatory mechanism, a limitation of our study is that it remains difficult to ascertain its relative contribution in complex conditions such as cardiac surgery. As highlighted by the work of Cloutier et al. (38) and, more recently, of Mauler et al. (39), PAF-independent mechanisms exist by which platelets trigger systemic responses. These mechanisms appear to have overlapping and distinct effects, e.g., platelet serotonin release after FcγRIIA receptor activation (38) caused vasodilatation and shock but not vascular leakage as observed in our anti-gpIIb/IIIa model. Furthermore, systemic responses in our model were independent of serotonin effects, as previously shown (16) and as documented by the fact that shock was fully prevented by pretreatment with a PAF receptor antagonist, or in Pafr- and Pla2-KO mice. However, we did not test the role of serotonin in systemic responses in the rat model and cannot exclude that, in this more complex preclinical model, multiple platelet-dependent mechanisms contributed.
Last, our finding that platelet activation following rat DHCA is responsible for much of the subsequent early pathology suggests that targeting platelet-dependent inflammatory responses may be an effective strategy to reduce morbidity and mortality. Platelet activation is not routinely determined but has been inferred from the drop in platelet count often observed after cardiac surgery. The possibility that such thrombocytopenia occurs in the context of increased platelet reactivity has been suggested by its association with blood clot formation leading to stroke (40). A drop in platelet count is associated with inflammatory derangements in various conditions including cardiac surgery (4). Evidence from our rat DHCA model that the platelet antagonist clopidogrel stopped microvascular platelet deposition, prevented the associated drop in platelet count, and reduced MC-mediated inflammatory and tissue injurious responses thus is of significant translational interest. Hence, our data add important mechanistic insights to clinical observations, suggesting beneficial effects from controlling the platelet contribution to tissue injury and systemic inflammatory derangements in cardiac surgery (41). However, an inherent problem with these approaches is both the variable pharmacodynamic efficacy of commonly used antiplatelet agents and the fact that, especially in the perioperative setting, more potent inhibitors pose a substantial bleeding risk. As suggested by our data using a MC inhibitor in modeled DHCA (8), targeting downstream events, such as MC activation, may be a safer approach to improve outcomes.
METHODS
Animal models
All procedures performed for this study were approved by the Animal Care and Use Committee of Duke University and the University of North Carolina, Chapel Hill, respectively, and conformed to National Institutes of Health guidelines for animal care.
Rat model
Adult male Sprague-Dawley rats (436.5 ± 34 g, 10 to 12 weeks old) underwent deep hypothermic arrest in association with CPB (referred to in this paper as DHCA for simplicity) as described previously (8). For imaging purposes, animals were transitioned to an alfalfa-free diet (LabDiet, St. Louis, MO) 7 days before the start of the experiment and were randomized to receive two oral doses of either clopidogrel (3 mg/kg bodyweight) (MilliporeSigma, Burlington, MA) or normal saline 12 hours before and immediately after induction of anesthesia.
Anesthesia was induced with isoflurane (2 to 2.5 volume %), and animals were intubated and mechanically ventilated (45% O2/balance N2 and 35 to 45 mmHg of PaCO2). The tail artery and right external jugular vein were then cannulated, and 150 IU of heparin and 5 μg of fentanyl were administered. Physiologic measurements, including mean arterial pressure, pericranial and rectal temperature, and blood gases [adjusted to the measured temperature (pH strategy) and maintaining 31 to 40 mmHg of PaCO2], were recorded (table S1). After initiation of CPB, animals were cooled for 30 min, and at a pericranial temperature of 16° to 18°C, the bypass machine was stopped for a circulatory arrest period of 45 min. CPB was then reinitiated for rewarming and stopped at a pericranial temperature of ≥35.5°C. Animals recovered under anesthesia for 2 hours until euthanasia.
For platelet labeling, we modified the technique of Flaumenhaft et al. (23) using two donor rats per experimental animal, which were pretreated as the experimental animal, i.e., with clopidogrel or vehicle. Donors were anesthetized with isoflurane, and whole blood in a volume of approximately 10% of the total donor blood volume was removed. Platelets were then isolated by centrifugation in the presence of apyrase (0.2 U/ml) and prostaglandin E1 (1 μM) (MilliporeSigma) throughout. The targeted platelet count was approximately 1.8 × 108. Platelets were washed in Tyrode’s buffer and labeled with 2 μM IR-786 (H.W. Sands Corp., Jupiter, FL) for 30 min at 37°C. After additional washing, these platelets were brought up in phosphate-buffered saline (PBS), and an aliquot was tested by fluorescence-activated cell sorting (FACS) to verify absence of surface expression of the platelet activation marker CD62P. Labeled platelets were transfused to recipient rats at the time point of reperfusion after circulatory arrest.
For imaging and tissue harvest, animals were euthanized by isoflurane overdose and perfused with 200 ml of PBS to wash out circulating platelets. Organs were removed, and tissue retention of labeled platelets was visualized using the IVIS Kinetic in vivo imaging system (Caliper Life Sciences, Hopkinton, MA) by setting the excitation to 795 to 815 nm and absorption to 760 to 780 nm.
Mouse lines
Six- to 8-week-old mice were used for our experiments. Mcpt5-cre+iDTR+ mice were from A. Roers, University of Technology, Dresden (21). In these mice, MCs were conditionally depleted through intravenous injections of 200 ng of diphtheria toxin per mouse every other day for 2 weeks (13). Pla2−/− (17) and Pafr−/− (18) mice were provided by Shimizu (University of Tokyo) through the RIKEN BioResource Research Center (RBRC01733 and RBRC05641) and were rederived by the Division of Laboratory Animal Resources, Duke University Medical Center. In addition, the following strains were used: C57BL/6, KitW-sh/W-sh, and Mcpt5-Cre tdTomatofl/fl (42).
Mouse systemic platelet activation models
Systemic platelet activation was induced in two ways. First, we used a systemic mouse thrombosis model with collagen and epinephrine as platelet stimulants. Second, we examined the systemic response elicited by a monoclonal antibody that targets platelet integrin αIIb receptor as previously published (16). Following anesthesia with isoflurane, animals received either 0.275 μg collagen/g bodyweight (MilliporeSigma) together with 1.2 μg of epinephrine (MilliporeSigma) in 200 μl of PBS or 3 μg/g bodyweight of the anti–integrin αIIb receptor antibody clone MWReg30 (BioLegend, San Diego, CA). Animals were then recovered at room temperature, and the rectal temperature was measured in regular intervals.
Platelet depletion and repletion model
Mice (hIL-4Rα/GPIbα–Tg) (43) were rendered thrombocytopenic by retro-orbital injection of anti–hIL-4R (2.5 μg/g body weight, clone 25463; R&D Systems, Minneapolis, MN). Platelet depletion was verified 16 hours after antibody injection by flow cytometry analysis (Accuri C6, BD Biosciences, Franklin Lakes, NJ) of whole blood stained with Alexa Fluor 647–labeled antibodies against glycoprotein IX (GPIX) (2 μg/ml; R&D Systems). Platelet repletion was performed as previously published (20). In short, blood was drawn into heparinized tubes from the retro-orbital plexus of sedated Pla2-KO or WT animals (7.7 μl/g body weight). Platelets were purified by successive centrifugation at 100g for 5 min (to obtain platelet-rich plasma) and at 700g in the presence of PGI2 (2 μg/ml) for 5 min at room temperature. Pelleted platelets were resuspended in modified Tyrode’s buffer [137 mM NaCl, 0.3 mM Na2HPO4, 2 mM KCl, 12 mM NaHCO3, 5 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, and 5 mM glucose (pH 7.3)]. Platelets from several donor mice were pooled, and the platelet count was adjusted to 5 × 109 platelets/ml in 100 μl of Tyrode’s buffer for transfusion. Posttransfusion platelet counts were determined by flow cytometry 30 min after injecting platelets before MWReg30 administration.
MC reconstitution
BMMCs were obtained from bone marrow of WT and Pafr−/− mice and cultured for 8 to 12 weeks as previously described (13). For repletion, 1 × 107 BMMCs were injected intravenously into KitW-sh/W-sh mice and allowed to mature in the tissues for another 8 weeks.
Platelet isolation and ex vivo activation
Whole blood (40 ml) was collected in acid-citrate-dextrose sodium citrate (1:9, v/v) from healthy individuals under a protocol approved by the Institutional Review Board for Human Subject Research at Duke University and centrifuged at 120g for 8 min. To prevent platelet activation, 1 μM prostaglandin E1 and apyrase (0.2 U/ml) (MilliporeSigma) were added to the platelet-rich plasma and at each of the following steps. Platelets were obtained by centrifugation at 650g for 8 min, washed in buffer containing 36 mM citric acid, 5 mM glucose, 5 mM KCl, 1 mM MgCl2, 103 mM NaCl, 2 mM CaCl2, bovine serum albumin (3.5 g/liter), and resuspended in standard Tyrode’s buffer. Platelet activation was performed with either thrombin (0.2 U/ml), collagen (10 μg/ml), type I solution from rat tail (both MilliporeSigma), or convulxin (0.3 ng/ml) (Cayman Chemical, Ann Arbor, MI). To verify platelet activation, a platelet aliquot was fixed in 4% formalin for each condition, washed in PBS, and stained with an anti–CD62-allophycocyanin antibody (BD Biosciences) and analyzed on a FACSCalibur flow cytometer. Isotype control–treated samples were used for comparison (BD Biosciences). The remaining sample was centrifuged to obtain cell-free conditioned media. In a subset of experiments, this supernatant was further processed by incubating on activated charcoal (MilliporeSigma), boiling for 30 min, or ultracentrifugating for 30 min at 20,000g to isolate microparticles. Lipid extraction was performed using the method of Bligh and Dyer (44). In brief, the platelet supernatant was mixed with chloroform-methanol (2:1, v/v), and then successively, chloroform and water were added before centrifugation at 1000 rpm for 5 min. The lower phase was then carefully harvested, dried under airflow, and resolubilized in Tyrode’s buffer.
Determination of PAF by LC-MS
PAFs were extracted and analyzed by LC-MS essentially as described earlier with minor modifications (45). Briefly, the samples were extracted for PAF by methyl tert-butyl ether, and the extracts were fractionated using aminopropyl silica to isolate the PAF fraction. The PAFs coelute with lysophosphatidylcholine in this method. They were resolved by high-performance liquid chromatography using a Luna C18(2) column (2 × 150 mm, 3 μ; Phenomenex) before detecting by LC–tandem MS with unique multiple reaction monitoring combinations as described (45). PAFs were quantified by internal standard quantitation method using PAF C16-d4 as the internal standard added to the sample before processing.
MC lines and assays
The human MC lines ROSA (14) were provided by M. Arock (Laboratoire de Biologie et Pharmacologie Appliquée, CNRS) and LAD2 (15) by A. S. Kirshenbaum (National Institutes of Health). The ROSA cells were maintained in Iscove’s modified Dulbecco’s medium (Invitrogen, Carlsbad, CA) and LAD2 cells in StemPro-34 (Invitrogen) supplemented with recombinant human stem cell factor (100 ng/ml), penicillin and streptomycin (100 U/ml), and 1× GlutaMAX (ThermoFisher, Waltham, MA). MC degranulation was examined by measuring the activity either of tryptase using a commercially available kit (MilliporeSigma) or of β-hexosaminidase as published. In both cases, results were calculated as the percent activity in supernatant versus activity in cell lysate. The following agonists and antagonists were tested at concentrations indicated in Results: WEB2086, montelukast, AH 6809, BAY-u 3405, L798.106, and Ex26 (all Tocris, Bristol, UK); PAF C16 and pertussis toxin (both MilliporeSigma). For IgE-antigen activation, LAD2 cells were passively sensitized by incubating with biotinylated human IgE for 16 hours. Sensitized LAD2 cells were stimulated with different doses of streptavidin as indicated for 1 hour. Supernatant was collected and analyzed for β-hexosaminidase activity. To determine viability, LAD2 cells were treated with indicated concentrations of PAF for 1 hour, and reduction of MTS tetrazolium compound was measured according to the manufacturer’s instructions. Results were calculated as % viable cells.
In vitro endothelial barrier assay
HUVECs were maintained as described (46). For experiments, 1 × 105 cells were seeded on 0.4-μm polyethylene permeable supports (Corning Life Sciences, Tewksbury, MA) and left to adhere until TEER reached approximately 120 ohm·cm2, and then medium was exchanged to Tyrode’s buffer. The basal compartment of the Transwell system was loaded with either buffer or 1 × 106 ROSA cells, and TEER was recorded before and during 1 hour after addition of 0, 1, or 10 μM PAF to the apical surface of the HUVECs. Degranulation of basal ROSA cells was determined at the end of the experiment by β-hexosaminidase assay.
Quantitative polymerase chain reaction
RNA was isolated (Macherey-Nagel, Bethlehem, PA) from snap-frozen tissue. After deoxyribonuclease digestion and reverse transcription (Bio-Rad, Hercules, CA), quantitative polymerase chain reaction (PCR) was performed on a CFX96 Real-Time PCR Detection System (Bio-Rad), with β-actin (NM_007393; F: cccaacttgatgtatgaagg and R: tttgtgtaaggtaaggtgtgc) serving as internal standard. The following primers were used and amplified at 60°C: Cxcl2 (NM_009140.2; F: cagactccagccacacttca and R: ttcagggtcaaggcaaactt), Tnfa (NM_013693; F: ctgaacttcggggtgatcgg and R: ggcttgtcactcgaattttga), Il6 (NM_031168; F: gatggatgctaccaaactgga and R: tgaaggactctggctttgtct), and Il1b (NM_008361.3; F: tgtaatgaaagacggcacacc and R: tcttctttgggtattgcttgg).
Enzyme-linked immunosorbent assay
Plasma samples were analyzed by enzyme-linked immunosorbent assay (ELISA) using rat chymase (LifeSpan Biosciences, Seattle, WA), rat TNFα, mouse MC protease 1, mouse TNFα (all ThermoFisher), and PAF (Lifeome BioLabs, Oceanside, CA) ELISA kits. To account for dilution during bypass, rat samples were normalized to each sample’s protein level (DC Protein Assay, Bio-Rad).
Immunohistology from whole-mount and paraffin sections
For whole-mount staining, the inner parts of the ear skin were peeled away from the intervening cartilage and fixed for 1 hour in 1% paraformaldehyde. Ear skin segments were then washed, permeabilized, and blocked in a solution containing 10% donkey serum, 0.3% Triton X, and 1% bovine serum albumin in PBS. Then tissue was incubated with anti-CD31 (BD Biosciences) and anti-CD41 (Novus Biologicals, Centennial, CO) antibody overnight, washed, and stained with fluorescent-labeled secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 2 hours at room temperature. MC granules were visualized with tetramethyl rhodamine isothiocyanate (TRITC)–avidin (MilliporeSigma). Samples were mounted with ProLong antifade with 4′,6-diamidino-2-phenylindole as counterstain (ThermoFisher).
Embedded tissue sections were deparaffinized, and heat-mediated antigen retrieval was performed in sodium citrate buffer. After blocking at room temperature, sections were labeled with antibodies targeting CD41 and E-cadherin (Invitrogen), and binding was visualized with fluorescent-labeled secondary antibody.
Vascular permeability
To examine vascular permeability, mice were injected with 200 μl of dextran-TRITC (10 mg/ml) (150 kDa; MilliporeSigma) at the time of platelet activation (13). After 90 min, animals were euthanized, and whole mounts were prepared and processed as outlined above. In vivo vascular leakage was quantified using the Evans blue dye extravasation technique (13). Briefly, Evans blue (20 mg/kg; MilliporeSigma) was injected intravenously 60 min before euthanasia. Tissue was then harvested, air-dried, weighed, and incubated in tissue formamide (25 μl/mg) at 55°C for 48 hours. The absorption of extracted Evans blue was then measured at 610 nm.
Histology and Chiu score
Hematoxylin and eosin–stained sections from formalin-fixed and paraffin-embedded tissue samples were scored by independent observers blinded to treatment modalities according to the method of Chiu et al. (47).
Patient plasma samples
Plasma samples were obtained in the course of an ongoing, Institutional Review Board–approved clinical study investigating effects of temperature on cognitive function after DHCA procedures for repair of ascending aortic arch pathologies (ClinicalTrials.gov identifier: NCT02834065). Plasma samples from 20 consecutively enrolled patients after induction of anesthesia (baseline) and following completion of the circulatory arrest and CPB period were retrieved and analyzed by ELISA for PAF (Cusabio, Houston, TX) and chymase levels (Cloud Clone Corp., Wuhan, China). Platelet count at corresponding times was obtained from clinical records. Because CPB entails significant blood dilution, measurements were normalized either to plasma protein content (for ELISAs, determined by RC DC Protein Assay, Bio-Rad) or to hemoglobin concentration (for platelet count, from patient electronic records with corresponding time stamp).
Statistical analyses
Statistical analyses were performed using GraphPad Prism v.8 (GraphPad Software). Unpaired Student’s t test, two-way analysis of variance (ANOVA), and one-way ANOVA with Tukey’s multiple comparisons tests were used to calculate statistical significance. P < 0.05 was considered statistically significant. Measured values of PAF, chymase, and platelet count in patients were compared with baseline values using Wilcoxon signed-rank test. Experimental data are presented as median ± SD. Patient data are shown as median (IQR).
Supplementary Material
Acknowledgments
We thank J. Fowler for technical assistance. Funding: This work was funded by U.S. National Institutes of Health grants 1R56HL126891-01 to J.K.; 1R35 HL144976-01 to W.B.; R01-AI096305, R56-DK095198, and U01-AI082107 to S.N.A.; T32HL007149 to R.H.L.; and 1R01HL130443 to J.K. and J.P.M. Further support came from National Center for Research Resources, National Institutes of Health grant S10RR027926 to K.R.M., the American Heart Association grant 15SDG25080046 to J.K., and a Duke Clinical and Translational Science Institute grant (UL1TR002553) to J.K. Author contributions: J.K., W.B., and S.N.A. were involved with conceptualization, development of methodology, and preparation of original draft and reviewing and editing of the manuscript. J.K., H.W.C., Q.M., Y.B., R.H.L., and J.P.M. performed the investigation. K.R.M. performed methodology development and analyses. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaay6314/DC1
Fig. S1. Platelet deposition following rat DHCA model occurs in tissues that are particularly sensitive to I/R injury.
Fig. S2. EC50 and LD50 of PAF on cultured MCs.
Fig. S3. Clopidogrel prevents tissue platelet retention after DHCA.
Table S1. Physiologic variables during rat DHCA.
Movie S1. Platelets aggregate in close proximity to perivascular MCs after activation with collagen and epinephrine.
REFERENCES AND NOTES
- 1.D’Agostino R. S., Jacobs J. P., Badhwar V., Fernandez F. G., Paone G., Wormuth D. W., Shahian D. M., The society of thoracic surgeons adult cardiac surgery database: 2018 update on outcomes and quality. Ann. Thorac. Surg. 105, 15–23 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Warren O. J., Smith A. J., Alexiou C., Rogers P. L. B., Jawad N., Vincent C., Darzi A. W., Athanasiou T., The inflammatory response to cardiopulmonary bypass: Part 1--mechanisms of pathogenesis. J. Cardiothorac. Vasc. Anesth. 23, 223–231 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Levin M. A., Lin H.-M., Castillo J. G., Adams D. H., Reich D. L., Fischer G. W., Early on—Cardiopulmonary bypass hypotension and other factors associated with vasoplegic syndrome. Circulation 120, 1664–1671 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Kertai M. D., Zhou S., Karhausen J. A., Cooter M., Jooste E., Li Y.-J., White W. D., Aronson S., Podgoreanu M. V., Gaca J., Welsby I. J., Levy J. H., Stafford-Smith M., Mathew J. P., Fontes M. L., Platelet counts, acute kidney injury, and mortality after coronary artery bypass grafting surgery. Anesthesiology 124, 339–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massberg S., Enders G., Leiderer R., Eisenmenger S., Vestweber D., Krombach F., Messmer K., Platelet-endothelial cell interactions during ischemia/reperfusion: The role of P-selectin. Blood 92, 507–515 (1998). [PubMed] [Google Scholar]
- 6.Herter J. M., Rossaint J., Zarbock A., Platelets in inflammation and immunity. J. Thromb. Haemost. 12, 1764–1775 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Rinder C. S., Mathew J. P., Rinder H. M., Bonan J., Ault K. A., Smith B. R., Modulation of platelet surface adhesion receptors during cardiopulmonary bypass. Anesthesiology 75, 563–570 (1991). [DOI] [PubMed] [Google Scholar]
- 8.Karhausen J., Qing M., Gibson A., Moeser A. J., Griefingholt H., Hale L. P., Abraham S. N., Mackensen G. B., Intestinal mast cells mediate gut injury and systemic inflammation in a rat model of deep hypothermic circulatory arrest. Crit. Care Med. 41, e200–e210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kertai M. D., Cheruku S., Qi W., Li Y.-J., Hughes G. C., Mathew J. P., Karhausen J. A., Mast cell activation and arterial hypotension during proximal aortic repair requiring hypothermic circulatory arrest. J. Thorac. Cardiovasc. Surg. 153, 68–76.e2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.St John A. L., Abraham S. N., Innate immunity and its regulation by mast cells. J. Immunol. 190, 4458–4463 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metcalfe D. D., Peavy R. D., Gilfillan A. M., Mechanisms of mast cell signaling in anaphylaxis. J. Allergy Clin. Immunol. 124, 639–648 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.St John A. L., Rathore A. P. S., Raghavan B., Ng M.-L., Abraham S. N., Contributions of mast cells and vasoactive products, leukotrienes and chymase, to dengue virus-induced vascular leakage. ELife 2, e00481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi H. W., Suwanpradid J., Kim I. H., Staats H. F., Haniffa M., MacLeod A. S., Abraham S. N., Perivascular dendritic cells elicit anaphylaxis by relaying allergens to mast cells via microvesicles. Science 362, eaao0666 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saleh R., Wedeh G., Herrmann H., Bibi S., Cerny-Reiterer S., Sadovnik I., Blatt K., Hadzijusufovic E., Jeanningros S., Blanc C., Legarff-Tavernier M., Chapiro E., Nguyen-Khac F., Subra F., Bonnemye P., Dubreuil P., Desplat V., Merle-Béral H., Willmann M., Rülicke T., Valent P., Arock M., A new human mast cell line expressing a functional IgE receptor converts to tumorigenic growth by KIT D816V transfection. Blood 124, 111–120 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Kirshenbaum A. S., Akin C., Wu Y., Rottem M., Goff J. P., Beaven M. A., Rao V. K., Metcalfe D. D., Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk. Res. 27, 677–682 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Nieswandt B., Echtenacher B., Wachs F. P., Schröder J., Gessner J. E., Schmidt R. E., Grau G. E., Männel D. N., Acute systemic reaction and lung alterations induced by an antiplatelet integrin gpIIb/IIIa antibody in mice. Blood 94, 684–693 (1999). [PubMed] [Google Scholar]
- 17.Uozumi N., Kume K., Nagase T., Nakatani N., Ishii S., Tashiro F., Komagata Y., Maki K., Ikuta K., Ouchi Y., Miyazaki J., Shimizu T., Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 390, 618–622 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Ishii S., Kuwaki T., Nagase T., Maki K., Tashiro F., Sunaga S., Cao W. H., Kume K., Fukuchi Y., Ikuta K., Miyazaki J., Kumada M., Shimizu T., Impaired anaphylactic responses with intact sensitivity to endotoxin in mice lacking a platelet-activating factor receptor. J. Exp. Med. 187, 1779–1788 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Triggiani M., Schleimer R. P., Warner J. A., Chilton F. H., Differential synthesis of 1-acyl-2-acetyl-sn-glycero-3-phosphocholine and platelet-activating factor by human inflammatory cells. J. Immunol. 147, 660–666 (1991). [PubMed] [Google Scholar]
- 20.Boulaftali Y., Hess P. R., Getz T. M., Cholka A., Stolla M., Mackman N., Owens III A. P., Ware J., Kahn M. L., Bergmeier W., Platelet ITAM signaling is critical for vascular integrity in inflammation. J. Clin. Invest. 123, 908–916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudeck A., Dudeck J., Scholten J., Petzold A., Surianarayanan S., Köhler A., Peschke K., Vöhringer D., Waskow C., Krieg T., Müller W., Waisman A., Hartmann K., Gunzer M., Roers A., Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Wolters P. J., Clair J. M.-S., Lewis C. C., Villalta S. A., Baluk P., Erle D. J., Caughey G. H., Tissue-selective mast cell reconstitution and differential lung gene expression in mast cell-deficient Kit(W-sh)/Kit(W-sh) sash mice. Clin. Exp. Allergy 35, 82–88 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flaumenhaft R., Tanaka E., Graham G. J., de Grand A. M., Laurence R. G., Hoshino K., Hajjar R. J., Frangioni J. V., Localization and quantification of platelet-rich thrombi in large blood vessels with near-infrared fluorescence imaging. Circulation 115, 84–93 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karhausen J., Stafford-Smith M., The role of nonocclusive sources of acute gut injury in cardiac surgery. J. Cardiothorac. Vasc. Anesth. 28, 379–391 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Ehara T., Shigematsu H., Contribution of mast cells to the tubulointerstitial lesions in IgA nephritis. Kidney Int. 54, 1675–1683 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Liverani E., Rico M. C., Tsygankov A. Y., Kilpatrick L. E., Kunapuli S. P., P2Y12 Receptor Modulates Sepsis-Induced Inflammation. Arterioscler. Thromb. Vasc. Biol. 36, 961–971 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McILwain R. B., Timpa J. G., Kurundkar A. R., Holt D. W., Kelly D. R., Hartman Y. E., Neel M. L., Karnatak R. K., Schelonka R. L., Anantharamaiah G. M., Killingsworth C. R., Maheshwari A., Plasma concentrations of inflammatory cytokines rise rapidly during ECMO-related SIRS due to the release of preformed stores in the intestine. Lab. Invest. 90, 128–139 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vadas P., Gold M., Perelman B., Liss G. M., Lack G., Blyth T., Simons F. E. R., Simons K. J., Cass D., Yeung J., Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N. Engl. J. Med. 358, 28–35 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Sturk A., Schaap M. C. L., Prins A., ten Cate J. W., van den Bosch H., Synthesis of platelet-activating factor by human blood platelets and leucocytes. Evidence against selective utilization of cellular ether-linked phospholipids. Biochim. Biophys. Acta 993, 148–156 (1989). [DOI] [PubMed] [Google Scholar]
- 30.Prescott S. M., Zimmerman G. A., Stafforini D. M., McIntyre T. M., Platelet-activating factor and related lipid mediators. Annu. Rev. Biochem. 69, 419–445 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Slatter D. A., Aldrovandi M., O’Connor A., Allen S. M., Brasher C. J., Murphy R. C., Mecklemann S., Ravi S., Darley-Usmar V., O’Donnell V. B., Mapping the human platelet lipidome reveals cytosolic phospholipase A2 as a regulator of mitochondrial bioenergetics during activation. Cell Metab. 23, 930–944 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong D. A., Kita Y., Uozumi N., Shimizu T., Discrete role for cytosolic phospholipase A(2)alpha in platelets: Studies using single and double mutant mice of cytosolic and group IIA secretory phospholipase A(2). J. Exp. Med. 196, 349–357 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J., Chen R., Marathe G. K., Febbraio M., Zou W., McIntyre T. M., Circulating platelet-activating factor is primarily cleared by transport, not intravascular hydrolysis by lipoprotein-associated phospholipase A2/ PAF acetylhydrolase. Circ. Res. 108, 469–477 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pluskota E., Bledzka K. M., Bialkowska K., Szpak D., Soloviev D. A., Jones S. V., Verbovetskiy D., Plow E. F., Kindlin-2 interacts with endothelial adherens junctions to support vascular barrier integrity. J. Physiol. 595, 6443–6462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng L. E., Hartmann K., Roers A., Krummel M. F., Locksley R. M., Perivascular mast cells dynamically probe cutaneous blood vessels to capture immunoglobulin E. Immunity 38, 166–175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kajiwara N., Sasaki T., Bradding P., Cruse G., Sagara H., Ohmori K., Saito H., Ra C., Okayama Y., Activation of human mast cells through the platelet-activating factor receptor. J. Allergy Clin. Immunol. 125, 1137–1145.e6 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Kulinski J. M., Muñoz-Cano R., Olivera A., Sphingosine-1-phosphate and other lipid mediators generated by mast cells as critical players in allergy and mast cell function. Eur. J. Pharmacol. 778, 56–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cloutier N., Allaeys I., Marcoux G., Machlus K. R., Mailhot B., Zufferey A., Levesque T., Becker Y., Tessandier N., Melki I., Zhi H., Poirier G., Rondina M. T., Italiano J. E., Flamand L., McKenzie S. E., Cote F., Nieswandt B., Khan W. I., Flick M. J., Newman P. J., Lacroix S., Fortin P. R., Boilard E., Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc. Natl. Acad. Sci. U.S.A. 115, E1550–E1559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mauler M., Herr N., Schoenichen C., Witsch T., Marchini T., Härdtner C., Koentges C., Kienle K., Ollivier V., Schell M., Dorner L., Wippel C., Stallmann D., Normann C., Bugger H., Walther P., Wolf D., Ahrens I., Lämmermann T., Ho-Tin-Noé B., Ley K., Bode C., Hilgendorf I., Duerschmied D., Platelet serotonin aggravates myocardial ischemia/reperfusion injury via neutrophil degranulation. Circulation 139, 918–931 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karhausen J. A., Smeltz A. M., Akushevich I., Cooter M., Podgoreanu M. V., Stafford-Smith M., Martinelli S. M., Fontes M. L., Kertai M. D., Platelet counts and postoperative stroke after coronary artery bypass grafting surgery. Anesth. Analg. 125, 1129–1139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mangano D. T.; Multicenter Study of Perioperative Ischemia Research Group , Aspirin and mortality from coronary bypass surgery. N. Engl. J. Med. 347, 1309–1317 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Choi H. W., Abraham S. N., Why serological responses during cystitis are limited. Pathogens 5, 19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanaji T., Russell S., Ware J., Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood 100, 2102–2107 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Bligh E. G., Dyer W. J., A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959). [DOI] [PubMed] [Google Scholar]
- 45.Kim S. J., Back S. H., Koh J. M., Yoo H. J., Quantitative determination of major platelet activating factors from human plasma. Anal. Bioanal. Chem. 406, 3111–3118 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Hoshi H., McKeehan W. L., Brain- and liver cell-derived factors are required for growth of human endothelial cells in serum-free culture. Proc. Natl. Acad. Sci. U.S.A. 81, 6413–6417 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiu C. J., McArdle A. H., Brown R., Scott H. J., Gurd F. N., Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch. Surg. 101, 478–483 (1970). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaay6314/DC1
Fig. S1. Platelet deposition following rat DHCA model occurs in tissues that are particularly sensitive to I/R injury.
Fig. S2. EC50 and LD50 of PAF on cultured MCs.
Fig. S3. Clopidogrel prevents tissue platelet retention after DHCA.
Table S1. Physiologic variables during rat DHCA.
Movie S1. Platelets aggregate in close proximity to perivascular MCs after activation with collagen and epinephrine.