

Abstract
Nucleic acid vaccines (NAVs) have recently been tested as a cancer therapy. DNA and mRNA vaccines deliver genetic information encoding tumor antigens (TAs) to the host, which then produces immune responses against cancer cells that express the TAs. Although NAVs are easy, safe, and simple to manufacture, they have not so far been considered viable alternatives to peptide vaccines. Choosing the right TAs, insufficient immunogenicity, and the immunosuppressive nature of cancer are some challenges to this approach. In this review, we discuss approaches that been used to improve the efficiency of anticancer NAVs.
Keywords: cancer vaccines, DNA vaccines, mRNA vaccines, tumor antigens, safety, combination therapy
Introduction
Cancer remains a challenging medical problem affecting millions of people around the world. Treatment strategies, such as surgery, chemotherapy, radiotherapy, and hormonal therapies, are applied alone or in combinations. More recently, targeted drugs and immunotherapy have gained research attention. Nevertheless, innovative cancer treatments are still being investigated in ever-increasing numbers [1]. The immune system has the potential to fight cancer and, thus, immunotherapy is designed to educate the immune system to identify and eliminate tumors; as a result, it has fewer adverse effects compared with chemotherapy [2]. Cancer immunotherapy strategies, such as cancer vaccines, bispecific antibodies, chimeric antigen receptors (CAR) T cells, checkpoint inhibitors, and other cell-based therapies, are important platforms [2,3]. Conventional vaccines based on live attenuated and inactivated pathogens, synthetic peptides, and recombinant subunit vaccines are widely used to prevent many infectious diseases. However, the manufacturing procedures of vaccines are not completely safe, and have a high risk of contamination with living pathogens. Therefore, the development of alternative vaccines is necessary for both infectious diseases and for non-infectious diseases, such as cancer [4,5]. Cancer vaccines have been studied for decades with some sporadic success, but have yet to penetrate the oncological mainstream. They include peptide vaccines, cell-based vaccines, viral vector vaccines, and NAVs. All these vaccines are designed to trigger or augment an immune response toward antigens expressed more or less specifically on tumor cells [6]. Among the different types of cancer vaccine tested, NAVs, such as DNA or mRNA vaccines, have been considered attractive because of their safe, simple, and rapid manufacturing process [7]. Cancer vaccines are more often used as a therapeutic approach, compared with infectious diseases, where prophylactic vaccines are more common. Cancer vaccines are designed to induce an immune response against tumor-derived antigens or TAs [8]. TAs can have a central role in tumor initiation, progression, and metastasis [6,8]. They can include tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs). Oncofetal antigens, cancer-testis antigens (CTAs), and overexpressed self-antigens have been considered to be TAAs. CTAs and oncofetal antigens are considered to be good candidates for cancer immunotherapy because they display zero or low expression in normal adult somatic cells; moreover, they are shared by many cancerous tumors in different patients to various extents, and are also expressed in different pathological types of epithelial tumor [9]. By contrast, mutated self-antigens or TSAs/neoantigens require expensive and laborious identification in the tumors of individual patients, but have shown improved efficacy in clinical trials compared with TAAs. Generally speaking, TAAs have been more often studied than TSAs in NAVs used for cancer [10].
In this review, we discuss current approaches using NAVs for cancer, compare DNA and mRNA cancer vaccines (Box 1 provides an introduction to DNA and mRNA cancer vaccines), summarize the latest challenges and recent successes, and offer perspectives for the future application of NAVs in cancer therapy.
Box 1. DNA and mRNA cancer vaccines.
Cancer DNA vaccines are engineered DNA molecules that encode one or several predetermined TAs, with or without other immunomodulatory molecules [12]. DNA vaccines must pass through the cell membrane of APCs to the cytoplasm and migrate to the nucleus to initiate transcription. The resulting mRNAs translocate to the cytoplasm, where they are translated to TAs. These proteins can be degraded by proteasomes and processed through the endoplasmic reticulum as intracellular antigens, which are presented as peptides bound to MHC I. Alternatively, the proteins can be degraded in endosomes as extracellular antigens, producing peptides that are bound to MHC II. APCs can present the epitopes to CD4+ (helper) or CD8+ (cytotoxic) T cells and also to B cells [24]. The final destinations for encoded antigens are lymphatic organs, such as spleen and lymph nodes, where resident B and T cells are activated by APCs [53]. Given the general low immunogenicity of DNA vaccines, and poor translation of preclinical results to clinical trials, many optimization strategies concerning vaccine structure and delivery have been tested and are summarized in this review.
mRNA vaccines also deliver genetic information encoding TAs. They are produced by in vitro transcription of template DNA using RNA polymerase [15,25]. mRNA collected from tumor samples can be amplified by PCR, yielding a large amount of complementary DNA encoding patient-specific TAs [54]. Recent progress using mRNA vaccines in preclinical and clinical trials has resulted from improvements in mRNA stability, structure, transfection methods, and purification techniques, which remove impurities and dsRNAs. mRNA vaccines need to only cross the cell membrane and the overall immunogenicity is slightly better than that achieved with DNA vaccines [7,33].
Comparison of DNA and mRNA vaccines
Cellular processing and delivery methods
The first crucial step for the success of NAVs is their internalization into the cytoplasm and nucleus (if necessary) of the host cells, especially dendritic cells (DCs), which are the most important type of antigen-presenting cell (APC). Improved delivery systems for nucleic acids have been designed to enhance the efficiency of gene therapy and also NAVs [11]. There are two general delivery approaches for NAVs: in vivo delivery and ex vivo delivery. The first approach involves administering NAVs directly to tissues of the body, whereas the second involves transfection of isolated autologous APCs outside of the body, which are then loaded and returned back to the body [12]. DC-based vaccines using isolated DCs that have been either transfected with NAVs encoding TAs or loaded with tumor antigenic peptides, are often used in cancer vaccine approachess [13]. Ex vivo loading of DCs with mRNAs that are either synthetic or have been isolated from whole tumor cells is the main approach used in clinical trials. To achieve high transfection efficiency, numerous gene delivery methods and transfection reagents have been used for both DNA and mRNA vaccines. Electroporation (EP) is the most common physical method used to transfect DCs by creating temporary pores in the cell membranes through which NAVs can enter the cell [14]. Although successful personalized DC-based vaccines have been reported using either DNA or mRNA, this delivery approach is expensive and complex. Moreover, the natural maturation process of DCs in the laboratory does not occur to the same extent in the natural proinflammatory environment encountered in the body; hence, direct, in vivo administration using physical or chemical methods are more preferable. Compared with DNA vaccines, mRNA (which is a large negative hydrophilic molecule with possible secondary and tertiary structures) showing thermodynamically unfavorable diffusion across membranes, causes more problems with chemical delivery approaches, such as cationic nanoparticles, cationic peptide protamine, cationic lipids, and other polymers or biomaterials [15,16]. In addition to physical and chemical methods, biological strategies (such as recombinant viruses) have been widely used to deliver genes encoding TAs for cancer vaccines in preclinical models and clinical trials. Even though there have been many promising results for the above-mentioned delivery methods and virus-like particles, each type has limitations and risks, including a possible unfavorable immunological response against the components of the vectors (especially viruses) and the complexity of carrier production, which requires extra precautions to be taken [16–18]. There are also a variety of delivery routes, including subcutaneous (SC), intradermal (ID), intramuscular (IM), intravenous (IV), intralymphatic (IL), or intranodal injection (IN) [19]. Special delivery methods or carriers can be used for each administration route to overcome extracellular barriers and achieve a desirable outcome. For instance, mucoadhesive carriers with hydrophilic surfaces have been used to target nasal-associated lymphoid tissue to overcome impediments such as poor tissue permeability and mucociliary clearance in the nose [5,20]. Naked NAVs have been injected into various tissues (ID and IN injection for mRNA; IM injection for DNA vaccines) where they primarily transfect different cells, such as muscle cells for IM injection rather than APCs, where the antigen must be cross-presented by an APC. However, when naked NAVs are injected, they mostly remain extracellular, where they are rapidly degraded by nucleases and, therefore, require frequent repeated injections. The formulation of NAVs can significantly improve their stability and result in higher vaccine efficacy. An example are PEGylated lipid nanoparticles that carried a melanoma mRNA vaccine encoding tyrosinase-related protein 2 (TRP2), and were functionalized with mannose as a targeting ligand to facilitate preferential uptake by DCs in lymph nodes after SC administration in mice [21]. Table 1 summarizes the barriers and cellular uptake mechanisms of DNA and mRNA vaccines by their target cells.
Table 1.
Similarities and differences between barriers to DNA and mRNA cancer vaccines
| Barrier | DNA vaccine | mRNA vaccine | Refs |
|---|---|---|---|
| Extracellular barriers | Serum nucleases ECM Poor lymphatic drainage at injection site Poor tissue permeability and mucociliary clearance Tissue nucleus Tissue pH |
[55] | |
| Intracellular barriers | Cell membrane Nuclear membrane Nucleases (DNase) |
Cell membrane Nucleases (RNase) |
[55] |
| Cellular uptake of naked NAVs mechanisms | Temperature and dose dependent Endocytosis Endosomal pathway to reach cytosol Entrance to nucleus by nuclear localization signal sequences |
Temperature and dose dependent Through caveolae and/or lipid rafts Macropinocytosis Endosomal pathway to reach cytosol |
[31,56] |
Structures and manufacturing processes
DNA and mRNA have different structures. DNA has a double-stranded structure, presence of deoxythymidine, a C2’-endo conformation, and 2’-H in deoxyribose. mRNA has a single-stranded structure, presence of uridine, a C3’-endo conformation, and 2’-OH in ribose. Plasmid DNA structures are used for DNA vaccines, whereas DNA molecules or libraries of cDNA are used as templates for the preparation of mRNA vaccines. DNA vaccines contain both prokaryotic sequences (for replication and selection in bacteria) and eukaryotic sequences (for the encoded TAs), whereas mRNA vaccines mostly comprise eukaryotic sequences. DNA vaccines include a nuclear localization signal (NLS) recognized by nuclear import receptors to transport the DNA into the nucleus [22]. Both DNA and mRNA vaccines need to incorporate a strong promoter sequence to allow efficient transcription, an open reading frame (ORF) encoding the TA, a ‘Kozak consensus sequence’, or binding site for ribosomes [23].
Steps in the preparation and function of DNA vaccines include bacterial fermentation for amplification of the recombinant plasmid, plasmid isolation and purification, transfection into the target cells, and finally transcription and translation to produce TAs within the target cells. By contrast, the manufacture of mRNA vaccines is cell free and simpler than that of DNA vaccines, because it uses linear DNA molecules or libraries of cDNA for in vitro transcription, followed by purification of the chemically synthesized mRNAs [23,24]. Figure 1 illustrates these processes.
Figure 1.
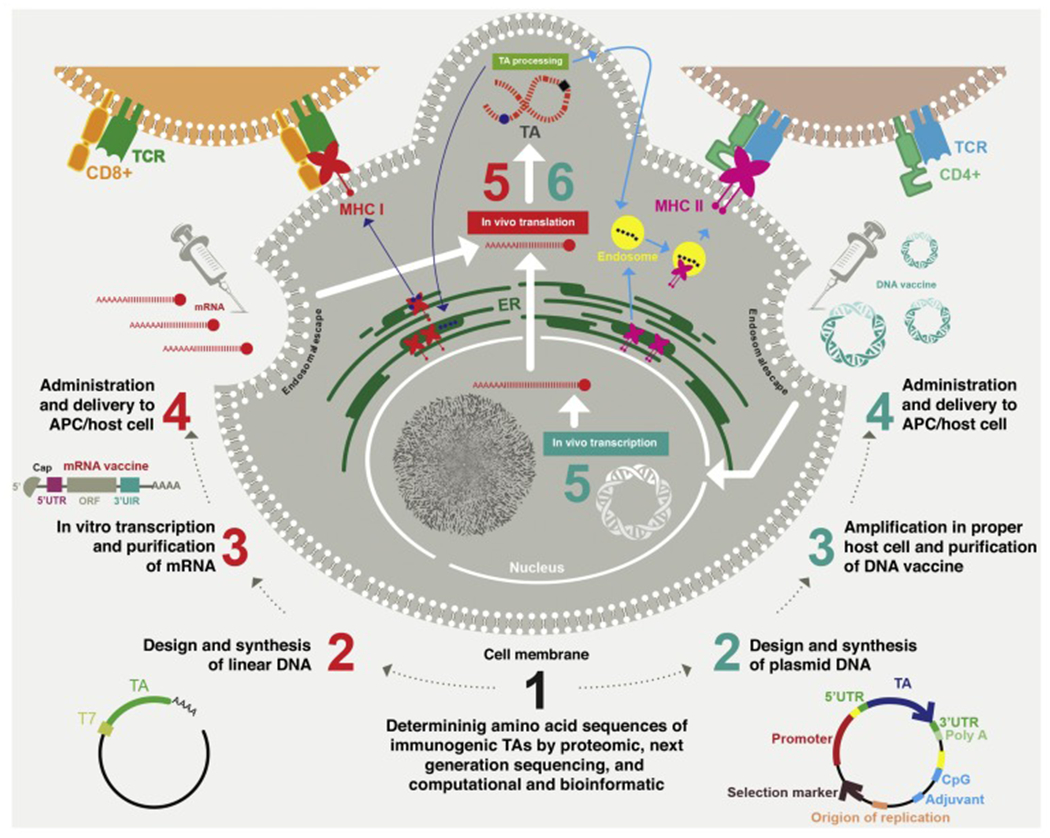
The production and function of naked DNA and mRNA vaccines. After determining the sequences of suitable tumor antigens (TAs), the required DNA or mRNA backbone is used in both types of vaccine. For DNA vaccines, amplification is performed in a suitable host cell, such as bacteria, and, after transfection of the purified DNA plasmid, transcription and translation are performed in the nucleus and cytoplasm of the host target cell, respectively (right side). For mRNA vaccines, T7 RNA polymerase is commonly used to amplify the mRNA vaccine in vitro, leaving only translation of TAs to be carried out after transfection (left side). The intracellular TA protein can be proteasomally degraded and routed to the endoplasmic reticulum, where antigenic peptide epitopes (square and sphere shapes) are loaded onto major histocompatibility complex (MHC) I and then presented to CD8+ T cells. Extracellular TAs can be taken up by endocytosis and loaded onto MHC II for presentation to helper T cells. Antigen-presenting cells (APCs), such as dendritic cells (DCs), can present antigens (endogenous and exogenous) on both MHC I and MHC II. Most other cells present antigens only on MHC I. Abbreviations: ORF, open reading frame; UTR, untranslated region.
Stability and degradation
Similar to other vaccines and unlike gene therapy, the nucleic acids in NAVs only need to function for a limited time within the body. However, NAVs can be degraded and excreted so rapidly that they cannot provide a long-lasting immune response. Given the ubiquitous presence of RNase enzymes, and structural differences between DNA and mRNA, the half-life of DNA vaccines is longer than that of mRNA vaccines. Likewise, the better heat stability of DNA vaccines allows for better subcellular sorting and transportation. The improved stability of plasmid DNA compared with mRNA resulted in higher numbers of DNA vaccine studies during the first years of their discovery (post-1990s). Despite the better stability of DNA vaccines, improvements in formulation and carrier loading are still required [16].
Structural elements of in vitro transcribed mRNA vaccines, including the 5’ and 3’ UTRs and the length of the 3’ poly (A) tail, can influence both the stability and translation efficiency of mRNAs [15,25]. However, there are ongoing studies to improve the stability of mRNA vaccines to ovecome their easy degradation by RNAses, such as using carrier molecules for mRNA vaccines, enrichment of the guanine-cytosine (GC) content, nucleotide-modified mRNAs, and using a circular mRNA instead of a linear structure [15,24,26]. Interestingly, some transfection reagents or carriers used for mRNA can also function in an adjuvant role (as an immunostimulant). Protamine is a polycationic peptide with simultaneous carrier and adjuvant (via Toll-like receptor 7 (TLR7) activation) roles, and has shown favorable activation of the innate immune system, leading to improved CD4+ T cells (humoral immunity) or CD8+ T cells (cell-mediated immunity) in multiple preclinical animal models [15].
Self-amplifying mRNA (SAM) vaccines are derived from the alpha-virus genome, and can provide both adjuvancity (by dsRNA replication intermediates) and a high amount of expressed antigen (by self-replication) simultaneously [27]. SAM vaccines have been termed ‘replicon mRNA vaccines’, because the engineered RNA virus genome contains nonstructural proteins (for mRNA replication) and a target antigen cassette substituting the genes encoding the structural proteins. Intracellular replication of SAM vaccines results in the need for only low doses (as little as a few hundred nanograms) of replicon mRNA vaccine to produce high amounts of antigen. Interestingly, immunization with alpha-virus DNA vectors only required up to 1000-fold lower concentrations of DNA, compared with conventional plasmid DNA to elicit equivalent tumor-specific immune responses [28]. However, compared with conventional nonreplicative NAVs, self-amplifying vaccines have some size constraints for the insert (especially in the case of multi-epitope vaccines). There might also be potential immunogenicity of the vaccine vectors after repeated use, as well as possible host immune reactions against dsRNA [15,29].
Unwanted immune responses
The aim of cancer NAVs is to induce an immune response (adaptive and innate) only against the encoded TAs, and not against the delivery vehicle. DNA and mRNA vaccines can also induce unwanted additional immune responses directed toward the NAVs themselves that could decrease their potency and reduce the production of target protein. However, the additional immune response can sometimes be somewhat beneficial for vaccines, by improving the adjuvanticity and boosting the activation and maturation of APCs, thus reducing the immune tolerance to the TAs [15].
Hypomethylated CpG motifs in DNA and the transient double-stranded structure of RNA can bind to TLR9 acting as an innate immune receptor, and induce production of type I interferons (type I IFNs, IFN-α and β). IFNs and IL-12 produced by innate immune cells stimulate T cells. Type I IFNs and IL-12 have a positive effect on DC maturation, and on the activation of B and T cells. However, excessive activation of the innate immune system has been associated with inhibition of antigen expression and of cellular translation in some studies [30]. Type I IFNs (despite their positive effects) can also cause inhibition of T cells through overactivation of type I IFN receptors on T cells. Given the above-mentioned effects of type I IFNs, alphaviruses, in common with other RNA viruses, use various antagonistic effects against IFNs and their downstream signaling pathways to overcome the innate antiviral responses [31]. The double-stranded structure of DNA is also reported to be a trigger of the innate immune response through non-TLR signaling pathways, resulting in the production of inflammatory cytokines and chemokines, such as type I IFNs [32]. The B form right-handed helical structure of DNA can also activate TLR-9, resulting in DC maturation and activation, and stimulation of B cells and T cells. Therefore, TLR-9 activation can have either positive or negative effects depending on the vaccine type, host cells targeted, level of type I IFN production, and other unknown factors.
In addition to dsRNA, endocytosis of NAVs can lead to the production of type I IFN (through activation of endosomal TLR7 and TLR8 by single-stranded oligoribonucleotides) [33]. Moreover, because mRNA vaccines are constructed using in vitro transcription, they can contain different types of immunogenic contaminant. Thus, strategies have been reported that avoid unwanted additional immune responses, while increasing TA production from mRNA vaccines, as discussed in the following section.
Optimization and improvement
NAVs should lead to a sufficient production of the target proteins, and optimized antigen expression to stimulate immune system. To achieve this goal, several manipulations have been tested to increase the potency of NAVs. In the case of DNA vaccines, transcription is performed inside the host cells and, therefore, an appropriate promoter for RNA polymerase II is needed upstream of the target antigen. Virus-derived promoters, such as SV40 or CMV promoters, are more effective than endogenous promoters. One point to be considered regarding viral promoters is their downregulation by cytokines such as tumor necrosis facotr (TNF)-α and IFN-γ. Thus, some studies have used nonviral promoters, such as the major histocompatibility (MHO) class II promoter, as an alternative to viral promoters in DNA vaccines. The hybrid structure of DNA vaccines needs improvement, because DNA vaccines with fewer prokaryotic sequences are more effective, and it was also suggested that reduction of the prokaryoticity of DNA vaccines would result in smaller vectors and easier transfection into host cells. Moreover, DNA vaccines with shorter bacterial sequences (such as minicircle vectors compared with plasmid vectors) could have no or fewer inhibitory effects on TA expression in eukaryotic host cells [30]. Codon optimization is another optimization strategy that is frequently performed for improving both DNA and mRNA vaccines [6]. For instance, the presence of G or C at the third codon position increased expression efficiency in mammalian cells. Often by replacing a rare codon with frequently used synonymous codons, protein translation can be increased. Using the codon preference of mammalian cells for the selected antigen could result in enhanced antigen expression and stronger immune response [15].
Delivery methods and routes of vaccines are also among the factors that affect the amount of target antigens produced; for example, the IM route yields higher amounts of protein for both DNA and mRNA vaccines [24]. Most NAVs also contain an immunostimulant either as protein or peptides encoded by DNA or mRNA, or as adjuvant proteins. Adjuvants such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin 2 (IL2) cause strong activation of the innate immune system, which leads to a potent adaptive immune response [24]. Van Lint et al. showed that mRNA cannot induce the full activation of DCs and, therefore, the authors used a combination of mRNAs encoding TA and adjuvants, including CD70, CD40 ligand, and the constitutively active form of TLR4. This approach was termed the ‘TriMix adjuvant’ strategy to increase tumor-specific T cell responses [34]. These reports show that not only the amount of target antigen, but also the content and structure of vaccines are important factors in optimization of NAVs and immune response. Chudley et al. used a DNA vaccine encoding fusion proteins including TA (prostate-specific membrane antigen) and a T helper cell stimulator (a domain of fragment C of tetanus toxin) in patients with prostate cancer that stimulated high-frequency CD8+ and CD4+ T cells [35].
Chimeric DNA vaccines have also been reported for enhancement of DNA vaccine immunogenicity. These vaccines encode xenogeneic antigens or nonself-antigens compared with autologous antigens to overcome immune tolerance. For instance, murine xenogeneic melanosomal antigens encoded by the DNA vaccine induced immune responses to the syngeneic protein in patients with malignant melanoma in a Phase I clinical trial [36].
A heterologous prime-boost strategy has been used as an approach to magnify the immune response against target antigens, mostly in the case of DNA vaccines (because DNA vaccines do not often induce antivector immunity). In this approach, a DNA vaccine including an immune stimulator and a TA is administered as a primer, whereas a recombinant viral vector or subunit vaccine is used as the booster. Kim et al. used this approach in a Phase I clinical trial in patients with HER2-expressing breast cancer, and showed that the DNA vaccine induced HER2-specific immunity [37].
In addition to the aforementioned optimization strategies (promoter selection and prokaryotic sequence reduction), nucleotide modification can be considered as another approach in mRNA vaccines. It was shown that incorporation of modified nucleosides within the mRNA construct could prevent activation of TLR7 and TLR8 and other innate immune receptors [26]. Covalent modification of the mRNA nucleotides (in addition to being one approach to enhance mRNA vaccine stability) also decreases unnecessary immune responses [38]. Likewise, the use of better purification methods, for example high-performance liquid chromatography (HPLC) following in vitro transcription of mRNA, has been shown to decrease nonspecific innate immune response and increase the potency of an mRNA vaccine and TA production in DCs [15]. By using combinations of these modification strategies, the efficiency of mRNA vaccines could be substantially improved. Both DNA and mRNA vaccines need codon optimization, whereas promoter design is important for DNA vaccines and modified nucleotides are needed for mRNA vaccines.
Safety aspects
For NAVs to be accepted for testing in humans, they are required to be shown to be safe. Both DNA and mRNA vaccines are non-infective platforms. However, there are some reported toxicities for both DNA and mRNA vaccines. Although insertional mutagenesis and integration of DNA plasmids could present a drawback of DNA vaccines, so far there have been few concerns expressed regarding possible integration into the host genome [39]. Contaminating microorganisms in mRNA vaccines are less likely than in DNA vaccines because the manufacture of mRNA vaccines does not need bacterial cell culture, and is also quicker compared with DNA vaccines [15]. The proinflammatory nature of mRNA is more pronounced than that of DNA, and could provide a self-adjuvant property for mRNA vaccines. By contrast, the inflammatory activity of mRNA vaccines can result in local and systemic inflammation, and more autoimmune responses in some preclinical studies [24]. As noted earlier, the formulation of mRNA vaccines, its delivery methods and routes, and presence of modified nucleotides are among the factors that affect mRNA vaccine safety, and can decrease the toxicity and inherent inflammatory activity of mRNA [7]. Given that potential toxic effects have been suggested for non-native modified nucleotides in mRNA vaccines, as well as the components of delivery systems used for both types of vaccine, precautions should be taken in preclinical studies and clinical trials [31].
Recent clinical trials of anticancer NAVs
Over the past few years, there have been several clinical trials for cancer (mostly Phase I/II) using both DNA and mRNA vaccines. Cancer DNA vaccines have mostly been applied in cervical, prostate, and breast cancer in clinical studies. By contrast, melanoma, glioblastoma, and prostate cancer have been the most frequent cancers for which mRNA vaccines have been tested. The application of immunotherapy and endocrine therapy in combination with anticancer NAVs, as well as adjuvants and chemotherapy have been tested in clinical studies in recent years. In addition, the use of immune checkpoint inhibitors as a type of immunotherapy has shown an increasing trend in combination with anticancer NAVs to allow more effective T cell killing of cancer cells. Although most clinical studies have been Phase I, I/II, or II, there are now a few Phase III clinical trials, such as VGX-3100 as a DNA vaccine against cervical cancer. This DNA vaccine ( NCT03721978 and NCT03185013) targets human papillomavirus (HPV) E6 and E7 proteins delivered IM followed by EP [6]. The number of clinical trials of DNA vaccines is higher than mRNA vaccines, in accordance with the later introduction of mRNA vaccines into the field (Table 2).
Table 2.
Examples of human clinical trials using anticancer NAVs
| Cancer type | NAV type | Target antigen | Combination therapy | Route of administration | Results or recruitment status | Refs |
|---|---|---|---|---|---|---|
| Breast cancer | DNA | Mammaglobin-A (Mam-A) antigen | IM followed by EP | Expansion of IFN-γ-producing CD4+ T cells with ability to lyse Mam-A-positive breast cancer cells | [57] | |
| Neoantigens | Durvalumab (anti-PD-L1 antibody) | IM | Recruiting | NCT03199040 | ||
| Insulin-like growth factor-binding protein (IGFBP)-2, HER2, and insulin-like growth factor (IGF)-1 receptor (1R) | GM-CSF | ID | NCT02780401 | |||
| mRNA | Alphaviral vector encoding portion of HER2 (VRP-HER2) | Pembrolizumab (anti-PD-1) | IV | Recruiting | NCT03632941 | |
| Shared tumor antigens and patient-specific mutated neoantigens | Surgery and adjuvant chemotherapy | IV as nanoparticulate lipoplex | Recruiting | NCT02316457 | ||
| Melanoma | DNA | Emm55 streptococcal antigen | Intralesion | Recruiting | NCT03655756 | |
| mRNA | Melanoma-associated antigens | GM-CSF | ID | Completed (results not provided) | NCT00204516 | |
| Two TAAs of melanoma (RBL001/RBL002) | IN | Completed (induction of specific CD8+ and CD4+ T cell responses against malignant melanoma TAAs) | NCT01684241 | |||
| Neoantigens [poly-epitopic RNA vaccine (IVAC MUTANOME®)] | RBL001/RBL002 | Active, not recruiting (detection of a strong poly-neoepitopic immune response against vaccine antigens and elicitation of T cell response in 60% of 125 selected neoepitopes) | NCT02035956 | |||
| Four TAAs [RBL001.1, RBL002.2, RBL003.1, and RBL004 (Lipo-MERIT)] | IV | Recruiting | NCT02410733 | |||
| Colorectal cancer | DNA | Oncoprotein MYB (TetMYB) | Tetanus toxoid peptides and anti-PD1 antibody | ID | Not yet recruiting | NCT03287427 |
| Prostate cancer | DNA | PSMA | Fragment C of tetanus toxin | IM or IM followed by EP | Induction of expansion of antigen-specific CD4+ and CD8+ T cells | [35] |
| PAP (pTVG-HP DNA vaccine) | Pembrolizumab (anti-PD-1) | ID and IV | Induction of PAP-specific T cell responses and decrease in PD-1 expression and tumor cell proliferation | [58] | ||
| Androgen receptor ligand-binding domain (AR LBD) | GM-CSF | ID | Active, not recruiting | NCT02411786 | ||
| Neoantigens | Nivolumab (anti-PD-1)/ipilimumab (anti-CTLA-4) and PROSTVAC | IM followed by EP | Recruiting | NCT03532217 | ||
| mRNA | TAAs, including PSA, prostate stem cell antigen, PSMA, and six-transmembrane epithelial antigen of prostate 1 (STEAP1) (CV9103) | ID | Induction of CD4+ and CD8+ T cell responses | [59] | ||
| Glioblastoma | DNA | INO-5401 [three separate DNA plasmids targeting Wilms tumor gene-1 (WT1) antigen, PSMA, and human telomerase reverse transcriptase (hTERT)] | INO-9012 (human IL12), cemiplimab, temozolomide, and radiation | IM followed by EP | Active, not recruiting | NCT03491683 |
| Pancreatic cancer | DNA | Neoantigens | Surgical resection and adjuvant chemotherapy | IM followed by EP | Recruiting | NCT03122106 |
| Cervical cancer | DNA | Modified version of HPV E6 and E7 | IM followed by EP | Generation of potent CD4+ and CD8+ T cell immune responses | [60] | |
| E6/E7 fusion protein of HPV | Unknown | NCT02596243 | ||||
| HPV E6 and E7 (VGX-3100) | IL-12; durvalumab (anti-PD-L1) | IM followed by EP and IV | Recruiting | NCT03439085 | ||
| Nonsmall-cell lung cancer | mRNA | TAAs (five formulated mRNAs) (CV9201) | Not provided | Completed (not provided) | NCT00923312 | |
| TAAs (six formulated mRNAs) (CV9202) | Local radiation | ID | Induction of immune response against six antigens | [61] |
Despite many ongoing efforts to optimize cancer NAVs, researchers still need to deal with many challenges to provide fully effective NAVs for cancer immunotherapy; however with sufficient time, they might be able to solve all of them. Suggested reasons for the lack of convincing evidence of benefit gained by using current NAVs are as follows. First unclear understanding of the biology of cancer cells makes it difficult to identify TAs that can engender a powerful immune response, and deeper investigations remain required in this direction [7]. It appears that NAVs designed against infectious diseases have performed better than anticancer NAVs, because of better defined and more specific TAs [40,41]. It is also possible that, during cancer progression, tumor cells tend to lose expression of a specific neoantigen for which a personalized vaccine had been prepared. Thus, the use of several TAs simultaneously has often been assumed to be beneficial in the design of NAVs. Indeed, there are no fully valid criteria for the choice and identification of suitable immunogenic TAs, because of incomplete understanding of the factors that affect immunogenicity, such as peptide processing by the proteasome complex and stability of epitope binding to MHC I. Moreover, tumor heterogeneity makes this problem even more difficult [42].
Second, the immunosuppressive nature of tumors is regarded as a powerful obstacle to the success of NAVs, especially in patients with advanced stages of cancer. This immunosuppressive environment involves both inhibitory cell surface molecules expressed on cancer cells, such as programmed death-ligand 1 (PD-L1), immunosuppressive cytokines, and various types of immune suppressor cell, such as immature myeloid cells or regulatory T cells inside the tumor microenvironment (TME) [43]. According to previous studies, anticancer drugs, which promote immunogenic cancer cell death (rather than nonimmunogenic cancer cell death) are more favorable for triggering antitumor immunity. Depletion of immunosuppressive cells inside the TME and enhancement of antigen presentation to T cells could be encouraged by therapies that promote immunogenic cell death [44]. Thus, immunological modification of the TME in combination with NAVs that target TAs could yet yield an effective cancer therapy. NAVs against infectious diseases do not need to overcome a particular immunosuppressive environment [40,41].
The third reason for the unsuccessful clinical outcome of NAVs might be that human responses to NAVs can induce unnecessary inflammatory signaling and systemic reactions, such as fever and cytokine release syndrome [40]. The fourth reason is the susceptibility of some individuals to autoimmune reactions triggered by the type I INF response caused by NAVs, and this is among potential safety concerns [45]. The fifth reason is unclear understanding of the immune signaling pathways responding to NAVs, because, in some cases, these signaling mechanisms are regarded as boosting adjuvanticity, whereas they might be considered as unnecessary inflammatory signaling [46,47]. Finally, the existence of several differences between humans and animal models might have encouraged somewhat unrealistic expectations [48]. Some commentators have observed ‘if humans were the same as mice, cancer would have already been cured’.
One suggested solution for decreasing the contrasting results between preclinical and clinical trials is ‘Body-on-a-Chip’ technology. 3D cultures utilizing patient-derived cells for precision medicine applications could provide the best tool for laboratory testing of NAVs and new drugs for a specific patient [49]. It is proposed that organotypic models could mimic the human body better than traditional animal models, and could be used in preclinical and clinical trials to test which NAVs or combined therapies will work in each patient with cancer or group of patients, leading to the maximum success of personalized therapy [50]. Many cancer scientists emphasize the use of combinations (concurrently or sequentially) of NAVs with other types of therapy, including other immunotherapies, targeted drug therapy, chemotherapy, and radiotherapy, to completely eliminate cancer cells [51]. For instance, in one combination study, co-delivery of PD-L1 small interfering (si)RNA (a type of immunotherapy) and a mRNA vaccine caused downregulation of PD-L1 in DCs and tumor cells, and enhanced T cell activation, resulting in a profound inhibitory effect on melanoma growth and metastasis in a preclinical study [21]. PD-L1 is an immune checkpoint that is profoundly expressed on DCs and tumor cells (especially affected by IFN-γ secretion); binding to its receptor, PD-1, on T cells attenuates their cytotoxic function. Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) on T cells has the same function as PD-1 and can inhibit T cell activity. Immune checkpoint blockade using anti-PD-1 monoclonal antibodies (mAbs) and anti-CTLA-4 mAbs can also be used in combination with anticancer NAVs [52].
Concluding remarks
Taken together, more extensive studies need to be carried out that concentrate on molecular and cellular aspects, to translate preclinical research successes of NAVs into clinical trials that will provide efficient therapy. For instance, the use of small-molecule targeting of inflammatory signaling cascades (especially in the case of mRNA vaccines), better selection of immunogenic TAs, improvement of delivery systems, and choice of suitable combination therapies will be needed to ensure the success of anticancer NAVs in the near future.
Highlights.
Cancer vaccines have a long history, but traditional protein/peptide antigens have not had much success
Nucleic acid vaccines based on either plasmid DNA or mRNA encoding tumor antigens are a promising alternative
The present review compares DNA and mRNA vaccines based on preparation, delivery, immunogenicity, and clinical trials
mRNA vaccines allow delivery of individual patient-derived antigens
DNA vaccines are less inflammatory with fewer side effects compared to mRNA vaccines.
Other immunotherapies such as checkpoint inhibitors can be combined with NAVs
Acknowledgments
M.R.H. was supported by US NIH Grants R01AI050875 and R21AI121700. The authors are thankful for the support of the Immunology Research Center, Tabriz University of Medical Science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Teaser: DNA and mRNA cancer vaccines have different characteristics, such as structure, stability, immunogenicity, and safety, and, thus, they require individual optimization to induce effective immune responses against TAs.
Conflicts of interest
M.R.H. declares the following potential conflicts of interest. Scientific Advisory Boards: Transdermal Cap Inc; BeWell Global Inc.; Hologenix Inc.; LumiThera Inc.; Vielight; Bright Photomedicine; Quantum Dynamics LLC; Global Photon Inc.; Medical Coherence; NeuroThera; JOOVV Inc.; AIRx Medical; FIR Industries, Inc.; UVLRx Therapeutics; Ultralux UV Inc.; Illumiheal & Petthera; MB Lasertherapy; ARRC LED; Varuna Biomedical Corp.; and Niraxx Light Therapeutics, Inc. Consulting: Lexington Int.; USHIO Corp.; Merck KGaA; Philips Electronics Nederland; Johnson & Johnson Inc.; and Sanofi-Aventis Deutschland GmbH. Stockholdings: Global Photon Inc. and Mitonix.
References
- 1.Chakraborty C et al. (2018) The novel strategies for next-generation cancer treatment: miRNA combined with chemotherapeutic agents for the treatment of cancer. Oncotarget 9 10164–10174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sathyanarayanan V and Neelapu SS (2015) Cancer immunotherapy: strategies for personalization and combinatorial approaches. Mol. Oncol 9, 2043–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ventola CL (2017) Cancer immunotherapy, Part 1: current strategies and agents. P. T 42, 375–83 [PMC free article] [PubMed] [Google Scholar]
- 4.Guo C et al. (2013) Therapeutic cancer vaccines: past, present, and future. Adv. Cancer Res 119, 421–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rezaei T et al. (2019) Recent advances on HIV DNA vaccines development: stepwise improvements to clinical trials. J. Control. Release 316, 116–137 [DOI] [PubMed] [Google Scholar]
- 6.Lopes A et al. (2019) Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res 38, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNamara MA et al. (2015) RNA-based vaccines in cancer immunotherapy. J. Immunol. Res 2015, 794528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tagliamonte M et al. (2014) Antigen-specific vaccines for cancer treatment. Hum. Vaccin. Immunother 10, 3332–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas R et al. (2018) NY-ESO-1 based immunotherapy of cancer: current perspectives. Front. Immunol 9, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner S et al. (2018) Colorectal cancer vaccines: tumor-associated antigens vs neoantigens. World J. Gastroenterol 24, 5418–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H et al. (2018) Enhanced gene transfection efficiency by low-dose 25 kDa polyethylenimine by the assistance of 1.8 kDa polyethylenimine. Drug Deliv. 25, 1740–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terbuch A and Lopez J (2018) Next generation cancer vaccines-make it personal! Vaccines 6, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santos PM and Butterfield LH (2018) Dendritic cell-based cancer vaccines. J. Immunol 200, 443–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broderick KE and Humeau LM (2015) Electroporation-enhanced delivery of nucleic acid vaccines. Expert Rev. Vaccines 14, 195–204 [DOI] [PubMed] [Google Scholar]
- 15.Pardi N et al. (2018) mRNA vaccines — a new era in vaccinology. Nat. Rev. Drug Discov 17, 261–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jorritsma SHT et al. Wijesundara DK (2016) Delivery methods to increase cellular uptake and immunogenicity of DNA vaccines. Vaccine 34, 5488–5494 [DOI] [PubMed] [Google Scholar]
- 17.Mokhtarzadeh A et al. (2016) P53-Derived peptides conjugation to PEI: an approach to producing versatile and highly efficient targeted gene delivery carriers into cancer cells. Expert Opin. Drug Deliv 13, 477–491 [DOI] [PubMed] [Google Scholar]
- 18.Soltani F et al. (2015) Synthetic and biological vesicular nano-carriers designed for gene delivery. Curr. Pharm. Des 21, 6214–6235 [DOI] [PubMed] [Google Scholar]
- 19.Grunwitz C and Kranz LM (2017) mRNA cancer vaccines—messages that prevail. Curr. Top. Microbiol. Immunol 405, 145–164 [DOI] [PubMed] [Google Scholar]
- 20.Irvine DJ et al. (2015) Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev 115, 11109–11146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y et al. (2018) mRNA vaccine with antigen-specific checkpoint blockade induces an enhanced immune response against established melanoma. Mol. Ther 26, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suschak JJ et al. (2017) Advancements in DNA vaccine vectors, non-mechanical delivery methods, and molecular adjuvants to increase immunogenicity. Hum. Vaccin. Immunother 13, 2837–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rauch S et al. (2018) New vaccine technologies to combat outbreak situations. Front. Immunol 9, 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu MA (2019) A comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines 7, E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahin U et al. (2014) mRNA-based therapeutics--developing a new class of drugs. Nat. Rev. Drug Discov 13 759–780 [DOI] [PubMed] [Google Scholar]
- 26.Pardi N et al. (2018) Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med 215, 1571–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundstrom K (2018) Latest development on RNA-based drugs and vaccines. Future Sci. OA 4, FSO300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundstrom K (2019) Plasmid DNA-based alphavirus vaccines. Vaccines 7, E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maruggi G et al. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther. (2019) 27, 757–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams JA (2013) Vector design for improved DNA vaccine efficacy, safety and production. Vaccines 1, 225–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iavarone C et al. (2017) Mechanism of action of mRNA-based vaccines. Expert Rev. Vaccines 16, 871–881 [DOI] [PubMed] [Google Scholar]
- 32.Herrada AA et al. (2012) Harnessing DNA-induced immune responses for improving cancer vaccines. Hum Vaccin Immunother. 8 1682–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlake T et al. (2019) mRNA as novel technology for passive immunotherapy. Cell Mol. Life Sci 76, 301–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Lint S et al. (2015) The ReNAissanCe of mRNAbased cancer therapy. Expert Rev. Vaccines 14, 235–251 [DOI] [PubMed] [Google Scholar]
- 35.Chudley L et al. (2012) DNA fusion-gene vaccination in patients with prostate cancer induces high-frequency CD8(+) T-cell responses and increases PSA doubling time. Cancer Immunol, Immunother 61, 2161–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan J et al. (2013) Immunologic responses to xenogeneic tyrosinase DNA vaccine administered by electroporation in patients with malignant melanoma. J. Immunother. Cancer 1, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SB et al. (2015) A phase 1 study of a heterologous prime-boost vaccination involving a truncated HER2 sequence in patients with HER2-expressing breast cancer. Mol. Ther. Methods Clin. Dev 2, 15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Czech MP et al. (2011) RNAi-based therapeutic strategies for metabolic disease. Nat. Rev. Endocrinol 7, 473–484 [DOI] [PubMed] [Google Scholar]
- 39.Stenler S et al. (2014) Safety and efficacy of DNA vaccines: plasmids vs. minicircles. Hum. Vaccin. Immunother 10, 1306–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hobernik D and Bros M (2018) DNA vaccines–how far from clinical use? Int. J. Mol. Sci 19, 3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang C et al. (2019) Advances in mRNA vaccines for infectious diseases. Front. Immunol 10, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo Y et al. (2018) Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front. Immunol 9 1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buqué A et al. Trial Watch-Small molecules targeting the immunological tumor microenvironment for cancer therapy. Oncoimmunology. (2016) 5, e1149674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimizu K et al. (2018) Immune suppression and reversal of the suppressive tumor microenvironment. Int Immunol. 30 445–54 [DOI] [PubMed] [Google Scholar]
- 45.Theofilopoulos AN et al. (2005) Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol 23 307–336 [DOI] [PubMed] [Google Scholar]
- 46.Campbell JD (2017) Development of the CpG adjuvant 1018: a case study. Methods Mol. Biol 1494, 15–27 [DOI] [PubMed] [Google Scholar]
- 47.Doener F et al. (2019) RNA-based adjuvant CV8102 enhances the immunogenicity of a licensed rabies vaccine in a first-in-human trial. Vaccine 37, 1819–1826 [DOI] [PubMed] [Google Scholar]
- 48.Shanks N et al. (2009) Are animal models predictive for humans? Philos. Ethics Humanit. Med 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun W et al. (2019) Organ-on-a-chip for cancer and immune organ modeling. Adv. Healthc. Mater 8, e1801363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y et al. (2015) Drug cytotoxicity and signaling pathway analysis with three-dimensional tumor spheroids in a microwell-based microfluidic chip for drug screening. Anal. Chim. Acta 898, 85–92 [DOI] [PubMed] [Google Scholar]
- 51.Liu L et al. (2018) Combination Immunotherapy of MUC1 mRNA nano-vaccine and CTLA-4 blockade effectively inhibits growth of triple negative breast cancer. Mol. Ther 26 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mougel A et al. (2019) Therapeutic cancer vaccine and combinations with antiangiogenic therapies and immune checkpoint blockade. Front. Immunol 10, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas S and Prendergast GC (2016) Cancer vaccines: a brief overview. Methods Mol. Biol 1403, 755–761 [DOI] [PubMed] [Google Scholar]
- 54.Ulmer JB et al. (2012) RNA-based vaccines. Vaccine 30, 4414–4418 [DOI] [PubMed] [Google Scholar]
- 55.Mellott AJ et al. (2013) Physical non-viral gene delivery methods for tissue engineering. Ann. Biomed. Eng 41, 446–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu M and Yuan F (2011) Membrane binding of plasmid DNA and endocytic pathways are involved in electrotransfection of mammalian cells. PLoS ONE 16, e20923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tiriveedhi V et al. (2013) Mammaglobin-A cDNA vaccination of breast cancer patients induces antigen-specific cytotoxic CD4CICOShi T cells. Breast Cancer Res. Treat 138, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scarpelli M et al. (2019) FLT PET/CT imaging of metastatic prostate cancer patients treated with pTVG–HP DNA vaccine and pembrolizumab. J. Immunother. Cancer 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kübler H et al. (2015) Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. J. Immunother. Cancer 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bagarazzi ML. et al. (2012) Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci. Transl. Med 4, 155ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Papachristofilou A et al. (2019) Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. Immunother. Cancer 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]