

Abstract
Aims
Population pharmacokinetic models of Δ9‐tetrahydrocannabinol (THC) have been developed for THC plasma and blood concentration data. Often, only the metabolites of THC are measurable when blood samples are obtained. Therefore, we performed a population pharmacokinetic analysis of THC, 11‐OH‐THC and THCCOOH plasma concentration data from a Phase I clinical trial of THC smoking.
Methods
Frequently obtained plasma THC, 11‐OH‐THC and THCCOOH concentration data were obtained over 168 h from 6 subjects who smoked low (15.8 mg) and high dose (33.8 mg) THC cigarettes on 2 occasions. Bayesian estimates of the THC pharmacokinetic model from each individual for each dose were fixed prior to the sequential pharmacokinetic analysis of the metabolites.
Results
A 3‐compartment model of THC was developed that has a steady‐state volume of distribution (VdSS) of 3401 ± 788 L and a clearance of 0.72 ± 0.10 L/min. 11‐OH‐THC was characterized by 50 ± 6% of the THC being directly cleared to a 3‐compartment model with a VdSS of 415.2 ± 4.3 L and clearance of 0.78 ± 0.05 L/min. The THCCOOH model shared the central compartment of the 11‐OH‐THC model with a VdSS of 29.1 ± 0.05 L and a clearance of 0.12 ± 0.02 L/min. First order kinetics were observed for THC and THCCOOH between the low and high doses, but a nonlinear pattern was observed for 11‐OH‐THC.
Conclusion
We describe the pharmacokinetics of THC, 11‐OH‐THC and THCCOOH including inter‐ and intraindividual variability of the parameter estimates of the model.
Keywords: Δ9‐tetrahydrocannabinol, THC, CB1 agonist, population pharmacokinetics, metabolite pharmacokinetics
What is already known about this subject
Population pharmacokinetic studies of Δ9‐tetrahydrocannabinol (THC) have demonstrated its rapid removal from the blood via extensive distribution to tissues and metabolic clearance.
Studies have also well documented that THC metabolites persist in the blood and urine for days even after a single administration.
What this study adds
A population pharmacokinetic model of both THC and its 11‐OH‐THC and THCCOOH metabolites from a Phase I clinical trial, based on frequent blood sampling for a week after a single dose of THC
The metabolism of THC to THCCOOH can be well represented by their plasma pharmacokinetics, while the metabolism of the intermediate 11‐OH‐THC appears to be less reliably described from its plasma concentration data alone
1. INTRODUCTION
Recreational and medical use of cannabis is on the rise in North America with its medical legalization in 34 states, full legalization in 11 states, and full legalization in Canada,1, 2 yet scientific and public uncertainty exists regarding the effects of widespread cannabis use on a range of behavioral and medical outcomes. It is, therefore, likely that some subjects and patients in clinical trials of other therapeutics may also be users of cannabis. Understanding these effects of cannabis is hampered by difficulties in assessing the systemic exposure to cannabis' active ingredients, specifically Δ9‐tetrahydrocannabinol (THC). Questionnaire methods (e.g. timeline followback) showed a correlation with biometric measures of THC exposure based on empiric ratios of THC and its metabolites (11‐OH‐THC, THCCOOH and THCCOOH‐glucuronide) blood concentrations.3, 4 Biometric techniques were proposed to estimate the time of last consumption of cannabis for use in forensic toxicology.5, 6, 7 These techniques typically employ sparse, often single blood THC and metabolite determinations with exposure and time course since consumption imputed by empiric formulae.5, 6
Detailed pharmacokinetic models of THC were developed to allow for simulation of single or chronic dosing as well as providing the theoretical basis for interpreting forensic data.8, 9 Such models were extended to population (nonlinear mixed effects) pharmacokinetic approaches to better estimate variability of THC disposition among individuals,10 bioavailability by smoking and vaping,10 as well as to develop pharmacokinetic–pharmacodynamic models of the effect of THC on heart rate and the central nervous system (e.g. body sway, feeling high).11
Because THC is often not present in measurable concentrations in blood after only a few hours since last use5 and because its metabolites, which have longer half‐lives, are measureable,5, 6, 7, 12 it is desirable to extend the pharmacokinetic model of THC to include its metabolites. Also, the first metabolite, 11‐hydroxy‐THC, is equipotent to THC and should be included in any analysis. As with proprietary drug development, population pharmacokinetic modeling of dense Phase I data is the first step.13 Such a structural population pharmacokinetic model of THC and its metabolites would not only describe the relationships among their variable concentration histories, it would establish the basis for exploring interindividual variability in THC pharmacokinetics and pharmacodynamics, testing for pharmacokinetic variability between low‐ and high‐potency cannabis products, and for interpreting sparse data from phase 3‐like observational cannabis studies.13 To these ends we have taken existing plasma concentration vs time data of THC, 11‐OH‐THC and THCCOOH from a carefully controlled Phase 1 clinical trial of 2 National Institute on Drug Abuse (NIDA) cannabis smoking products,14 which, unlike previous THC models, includes key blood sampling during the smoking event, in a randomized crossover study design in order to develop a population pharmacokinetic model of THC and its metabolites.
2. METHODS
As described previously,14 6 healthy male cannabis users provided written informed consent for a clinical research study approved by the Institutional Review Board of the NIDA and were admitted to the secure residential facility for 4–6 weeks at the Intramural Research Program of NIDA. Urine drug testing for THC metabolites and other drugs of abuse were assayed throughout residence to assure lack of exposure to other drugs and to reach a baseline below the limits of quantification for THC and its metabolites prior to controlled cannabis administration. At weekly intervals, cigarettes containing 0, 15.8 or 33.8 mg were smoked according to a computer‐paced protocol (i.e. 2‐s inhale, 10‐s hold, 72‐s exhale/rest) for a total of 8 puffs. Plasma THC, 11‐OH‐THC and THCCOOH concentrations were measured by gas chromatography–mass spectrometry with a lower limit of quantification of 0.5 ng/mL for each compound. The data are published in their entirety in this report.14 Metabolite concentrations were normalized for their respective molecular weight ratios with THC (e.g. [THCCOOH] x MWTHC/MWTHCCOOH (314.45/344.4) = [THCCOOH] × 0.913). Correcting for molecular weight differences among parent drug and metabolites permits their mutual incorporation of pharmacokinetic model parameters.
2.1. Pharmacokinetic data analysis
The pharmacokinetic analysis was performed using Phoenix NMLE 8.1 with the FOCE ELS algorithm (Certara, Princeton, NJ). Model parameters (θj, theta, for model parameter j) were assumed to be log‐normally distributed across the population with a central, typical value (θTV) allowing for assessment of between subject variability (ηj,I, eta, for individual i and model parameter j) such that:
| (1) |
Residual within‐subject error was calculated with relative error. THC concentration data from both doses in the 6 subjects of the study were combined and tested for 1, 2 and 3 compartment models based on visual inspection of the data fits, the minimum value of the objective function with a decrease in the objective function (−2 log likelihood) of 6.63 (considered significant at the p < 0.01 level for χ2) and a decrease in the Akaike information criterion (AIC)15 for additional model parameters. As the inhaled bioavailability is not known, we assumed it was 0.25 for simplicity, in accordance with previous studies,10, 16 and to assess pharmacokinetic parameter estimates in terms of physiological volumes and processes.17 Potential intrasubject differences of inhaled bioavailability between the 2 doses was tested with the introduction of a parameter estimating the bioavailability ratio between the 2 smoking sessions in the same manner as applied to adding additional compartments and was compared to interoccasion variability for ability to reduce the objective function. Upon selection of the best model, based on the −2 log likelihood model objective function and AIC selection criteria, interindividual variability parameters (ηj) with shrinkage values above 0.9 were sequentially removed, beginning with the highest value until all shrinkage values were below 0.9.18 Subject covariates, such as body weight, were not considered given the small number of subjects and their demographic similarity.
In a sequential manner, the individual Bayes parameter estimates, of the plasma THC pharmacokinetic model were fixed prior to fitting the 11‐OH‐THC and THCCOOH concentration data.19 The parent drug and metabolite pharmacokinetic model (Figure 1) was constructed based on the following assumptions: (i) the elimination clearance of THC equaled the conversion clearance to 11‐OH‐THC plus direct conversion to THCCOOH via a single, fixed delay element; (ii) 11‐OH‐THC and THCCOOH shared equal volumes of distribution and tested for 1‐, 2‐ and 3‐compartment models as described above for the THC model; and (iii) the elimination clearance of 11‐OH‐THC equaled its conversion clearance to THCCOOH. Residual within‐subject error was calculated with both additive and relative error. Metabolite models were tested for parsimony in the manner described above for THC.
Figure 1.
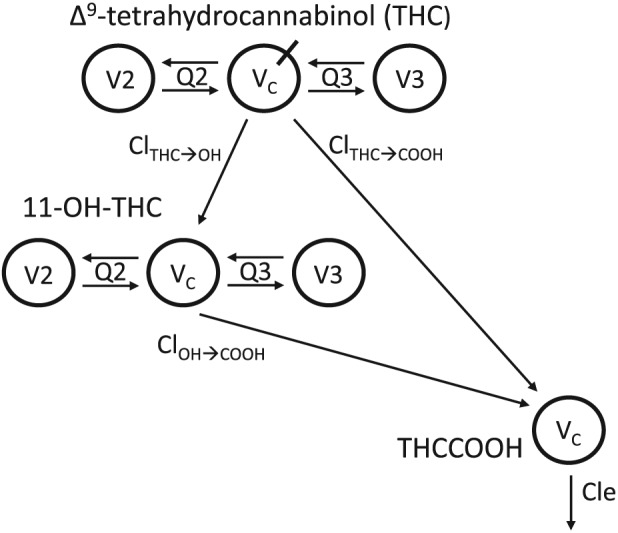
Final compartmental model used to fit plasma Δ9‐tetrahydrocannabinol (THC), 11‐OH‐THC and THCCOOH concentrations. Drug is inhaled into VC of a 3‐compartment THC model with rapidly and slowly equilibrating peripheral compartments V2 and V3, intercompartmental clearances Q2 and Q3, and elimination clearances to the metabolites 11‐hydroxyTHC and carboxyTHC, ClTHC➔OH and ClTHC➔COOH for the respective metabolites. Note that the sum of ClTHC➔OH and ClTHC➔COOH was assumed to be equal to the total elimination clearance (Cle) for THC. The respective metabolites are introduced into 3‐ and 1‐compartment models of 11‐OH‐THC and THCCOOH. The elimination clearance for 11‐OH‐THC, ClOH➔COOH also introduces THCCOOH into its 1‐compartment model, from which it is eliminated by its Cle
THC and metabolite concentrations below the lower limit of quantitation (i.e. less than 0.5 ng/mL) were analyzed according to the M3 method of Beal20 for including data outside the limits of quantitation.
A visual predictive check was performed by using the final model parameter estimates to simulate data for 1,000 virtual subjects and calculating their 5th, 50th and 95th percentiles at all sampling times. The distributions of the simulated THC, 11‐OH‐THC and THCCOOH concentrations were visually compared with the measured concentrations at each sampling time.
3. RESULTS
As previously reported, the 6 male cannabis‐using subjects had a mean age of 31.3 years (range, 29–36 years) and a mean weight of 77.6 kg (range, 64.8–93.4 kg). The drug and metabolite concentration vs time data used in this analysis are also previously reported.14
Model building and testing are summarized in Table 1. Each increase in model complexity of THC from 1 to 2 and to 3 compartments significantly decreased the −2 log likelihood: 2200, 1989 and 1863, respectively. Introducing interoccasion variability among the model parameters, except intercompartmental clearances, and proportional weighting significantly decreased the −2 log likelihood further to 1785 with an AIC of 1823. Substituting the interoccasion variability with a parameter for the estimate of the ratio of inhaled dose between the smaller and the larger cigarette THC‐content reduced the −2 log likelihood to 1781 with an AIC of 1813 (Table 1). Thus, each individual's 3‐compartment parameter estimates for THC, adjusted for their varying fraction of dose inhaled were carried as fixed parameters into the metabolite modeling.
Table 1.
Results of the model fitting and selection process. For Δ9‐tetrahydrocannabinol (THC), 1‐, 2‐ and 3‐compartment models followed by addition of the interoccasion variability and bioavailable dose ratio between the high THC dose and the low THC dose to the 3‐compartment model. For the metabolites, 1, 2, and 3‐compartment models with all parameters shared, except elimination or metabolic clearances, between the models for 11‐OH‐THC and THCCOOH. This is followed by a model with 3 compartments for 11‐OH‐THC, THCCOOH model sharing only the central and fast peripheral compartments of 11‐OH‐THC, the THCCOOH model sharing only the central compartment of 11‐OH‐THC, and the last model in which the ratio of the THC elimination clearance is split between production of 11‐OH‐THC and THCCOOH varied between the high and low THC dosages
| –2LL | AIC | |
|---|---|---|
| THC | ||
| 1‐compartment | 2200 | 2212 |
| 2‐compartment | 1989 | 2009 |
| 3‐compartment | 1863 | 1891 |
| + occasion | 1785 | 1823 |
| + dose ratio | 1781 | 1811 |
| Metabolites | ||
| 1‐compartment | 2593 | 2625 |
| 2‐compartment | 2515 | 2555 |
| 3‐compartment | 2379 | 2427 |
| 3/2‐compartment | 2306 | 2354 |
| 3/1‐compartment | 2292 | 2340 |
| 3/1‐compartment + split ratio | 2284 | 2336 |
–2LL = the −2 log likelihood model objective function; AIC = Akaike information criterion.
Similarly, each increase in metabolite model complexity from 1 to 2 to 3 compartments significantly decreased the −2 log likelihood objective function, respectively. Additionally, incorporation of a parameter to estimate the plasma‐apparent split between clearances from THC to 11‐OH‐THC and THCCOOH further reduced the −2 log likelihood significantly. The peripheral second and third compartments for 11‐OH‐THC were not carried forward to THCCOOH as doing so increased the −2 log likelihood. From the final metabolite model, η‐values (Equation 1) with very small distributions were sequentially removed (i.e. the η‐values for V2, Q2, Q3 and Cl11‐OH‐THC‐ > THCCOOH of the metabolite models) until all the η shrinkages were below 0.9.
The pharmacokinetic parameter values are given in Table 2 along with ω standard deviations and shrinkage values. For the 3‐compartment THC model, all typical value parameter estimates had a CV% <25%. The typical value for the low:high inhaled dose was estimated to be 0.89 (range: 0.62–1.46). For the metabolite models all estimated typical values also had CV% <20%. Of note, many of the standard errors of the estimates are quite low and are probably due to the small number of subjects, which also precludes a bootstrap analysis to better estimate this descriptive statistic.
Table 2.
| Pharmacokinetic Model Parameters | Typical value | ± | SEE | ω2 | ± | SEE | Shrinkage | |
|---|---|---|---|---|---|---|---|---|
| THC | ||||||||
| VC (L) | 28.3 | ± | 3.61 | 0.09 | ± | 0.04 | 0.03 | |
| V2 (L) | 45.5 | ± | 5.37 | − | ||||
| V3 (L) | 3327 | ± | 736 | − | ||||
| Cl2 (L/min) | 1.35 | ± | 0.27 | 0.11 | ± | 0.06 | 0.11 | |
| Cl3 (L/min) | 1.30 | ± | 0.30 | 0.21 | ± | 0.09 | 0.21 | |
| Cle (L/min) | 0.72 | ± | 0.10 | − | ||||
| f | 0.89 | ± | 0.13 | 0.11 | ± | 0.04 | 0.14 | |
| δLowDose | 0.23 | ± | 0.03 | |||||
| δHighDose | 0.29 | ± | 0.01 | |||||
| 11‐OH‐THC | ||||||||
| Clthc‐ > oh (L/min) | 0.36 | ± | 0.02 | 0.05 | ± | 0.001 | 0.01 | |
| High dose split (f) | 0.88 | ± | 0.03 | 0.02 | ± | 0.002 | 0.19 | |
| VC (L) | 29.1 | ± | 0.16 | 0.14 | ± | 0.01 | 0.07 | |
| V2 (L) | 113.8 | ± | 0.66 | 3.02 | ± | 0.12 | 0.09 | |
| V3 (L) | 272.3 | ± | 3.50 | − | ||||
| Cl2 (L/min) | 4.77 | ± | 0.05 | − | ||||
| Cl3 (L/min) | 1.90 | ± | 0.02 | − | ||||
| Cloh‐ > cooh (L/min) | 0.78 | ± | 0.05 | − | ||||
| δLowDose | 0.24 | |||||||
| δHighDose | 0.19 | |||||||
| δA‐LowDose (ng/mL) | 1.91 | |||||||
| δA‐HighDose (ng/mL) | 0.41 | |||||||
| THCCOOH | ||||||||
| Cle (L/min) | 0.12 | 0.02 | 0.12 | ± | 0.001 | 0.01 | ||
| δLowDose | 0.18 | |||||||
| δHighDose | 0.24 | |||||||
| δA‐LowDose (ng/mL) | 0.68 | |||||||
| δA‐HighDose (ng/mL) | 0.51 | |||||||
THC = Δ9‐tetrahydrocannabinol; Cle = elimination clearance; Clthc‐ > oh = metabolic clearance from THC to 11‐OH‐THC; Cloh‐ > cooh = metabolic clearance from 11‐OH‐THC to THCCOOH; Q = intercompartmental clearance between the central compartment, Vc, and the peripheral compartments, V2 and V3; f = factor or ratio between the inhalation bioavailability of the high THC dose and the low dose; High dose split (f) = fraction of the low dose Clthc‐ > oh that is estimated for the high dose (the remaining clearance is assigned to Clthc‐ > thccooh; δA = additive intrasubject variability; δ = proportional (relative) intrasubject variability; ω2 = intersubject variability; SEE = standard error of the estimate.
The observed vs final posthoc individual model predictions for THC, 11‐OH‐THC, and THCCOOH for each dose are presented in (Figure 2). The conditional weighted residuals vs time and predicted concentration relationships are presented in Figure 3. The visual predictive checks were performed as described and are presented in Figure 4.
Figure 2.
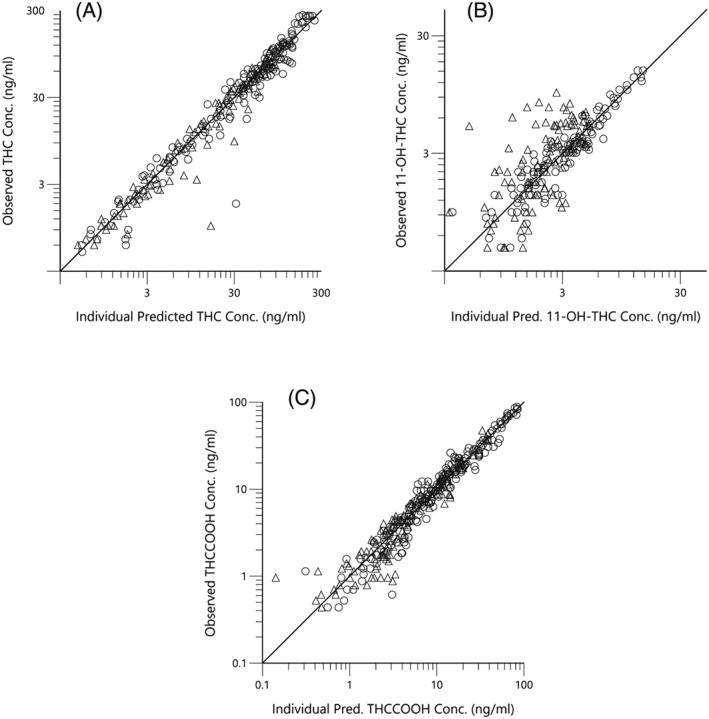
Observed (open triangles, low dose and open circles, high dose) vs predicted Δ9‐tetrahydrocannabinol (THC; a), 11‐OH‐THC (b) and THCCOOH (c) concentrations for the posthoc individual compartment models. The lines of identity are the black lines
Figure 3.
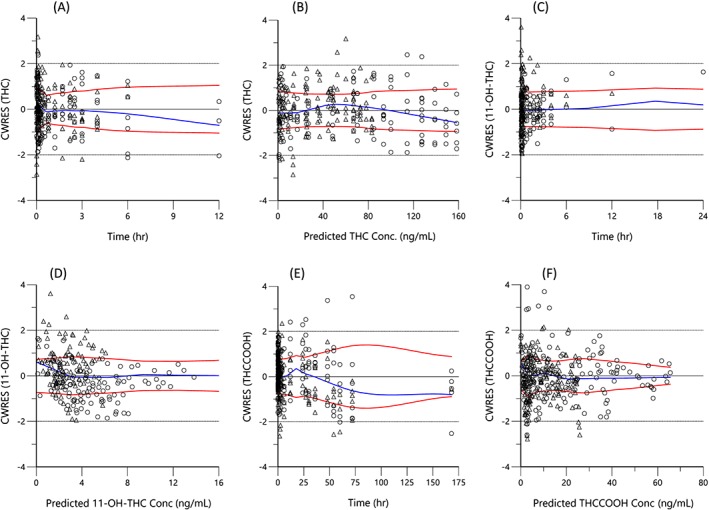
Conditional weighted residual (CWRES) vs time for THC (a), 11‐OH‐THC (c), and THCCOOH (e) and vs predicted concentration for THC (b), 11‐OH‐THC (d), and THCCOOH (f), for the low (triangles) and high doses (circles). For each plot the blue line is the locally weighted scatterplot smoothing (lowess) line and the red lines are the lowess lines for the absolute residuals and its mirror
Figure 4.
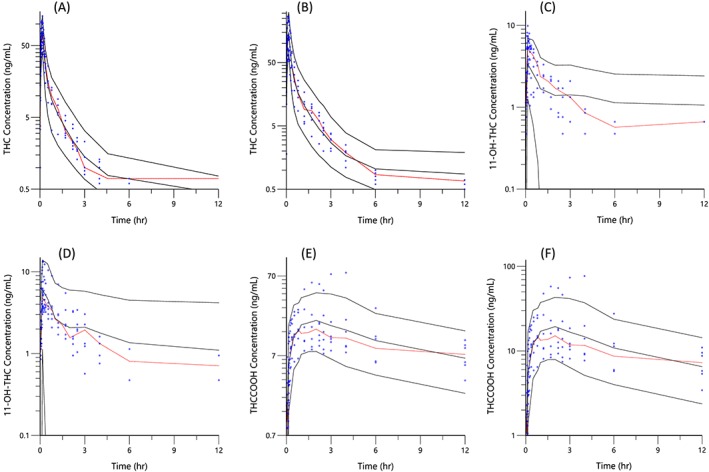
Visual predictive checks. The blue circles represent the observed data for THC (a and b), 11‐OH‐THC (c and d) and THCCOOH (e and f) concentrations for the low and high doses for the first 12 h and THCCOOH (g and h) for the full 168 h of blood sampling. The black lines represent the 95th, 50th and 5th percentiles of the simulated concentrations at each time point
4. DISCUSSION
The aim of the current study was to develop a population pharmacokinetic model for THC and its 11‐hydroxy and carboxy metabolites, which could fit the rising and falling phase I plasma concentration vs time data from healthy volunteers who undertook paced smoking of 2 doses of THC at least 1 week apart. We assumed a fraction absorbed of 0.25 of the smoked dose on all occasions so that we could compare our results to a similar phase I population pharmacokinetics of THC reported by Heuberger et al., which included intravenously administered THC10 and, thus, pharmacokinetic parameters for all modes of administration were expressed in terms of absolute bioavailability. Our estimate of the typical value of THC elimination clearance of 0.72 L/min is similar to their estimate of 0.65 L/min. This concordance indicates that the subjects in this study had a fraction of the dose absorbed from smoking 1 cannabis cigarette very near our assumed value of 0.25. To account for possible bioavailability differences between the 2 cannabis smoking events, the model estimated the fractional difference in relative absorption between the low and high doses to be 0.89 (±0.13, standard error of the estimate). Even though the subjects' smoking was carefully paced, the trend towards lower absorption at higher doses could be indicative of increased side‐stream loss due to decreased smoking efficiency with greater central nervous system effects or unintended dose titration by the subjects. Similar findings of decreased inhalation bioavailability at higher concentrations was observed with aerosolized ketamine.21
In contrast to similar clearance estimates between this study and that of Heuberger et al., 10 our estimate of VSS (sum of V1, V2 and V3) was 3401 L and was considerably larger than their estimate of 236 L. Using the THC typical value parameters in Table 2, the estimated terminal phase half‐life for THC is calculated to be approximately 3.5 days, indicating its distribution to deep, presumably lipid tissues. Despite the reliance of only a few data points near the low limit of quantification near the terminal slope (Figure 4), this estimate is consistent with the persistence of THC and observed similar half‐lives after cessation of chronic cannabis use.12, 16, 22
From the perspective of plasma sampling we estimated that 74% of inhaled THC is metabolized to 11‐OH‐THC. This percentage should be viewed as the minimum amount of THC converted first to 11‐OH‐THC as models of hepatic metabolism estimated from plasma samples model only the 11‐OH‐THC that circulated in the blood before returning to the liver for subsequent metabolism to THCCOOH or other products. The remaining 26% of 11‐OH‐THC produced from THC was modeled as converted to THCCOOH without reaching the circulation. In the current analysis, the assumption was made that all THC was either metabolized to 11‐OH‐THC or appeared to be metabolized to THCCOOH without intermediate conversion to 11‐OH‐THC as the detailed metabolic kinetics involved in the liver are not identifiable from merely sampling the blood (Figure 1).23 Other metabolic pathways and metabolic products of THC have been described (e.g. THC‐glucuronide), but in toto make up no more than 2–5% of THC metabolism and excretion.16
To fully describe metabolite pharmacokinetics, it would be required to administer the metabolite as a separate dose or, better, as a stable isotope at the same time as the parent compound so that the dose, or amount produced by metabolism, is known to the model.24 Alternatively, assumptions regarding metabolite production or distribution volumes must be made. Accordingly, for the current analysis we assumed that all THC is converted to 11‐OH‐THC or directly to THCCOOH, all 11‐OH‐THC is converted to THCCOOH and that V1 is identical for the pharmacokinetic models of 11‐OH‐THC and THCCOOH so that parameter estimates for the entire model could be obtained. The respective estimates of steady‐state volume of distribution of 3401, 581.1 and 22.8 L for THC, 11‐OH‐THC and THCCOOH, respectively, are also consistent with the lipophilic characteristics of THC, the neutrality of 11‐OH‐THC and the acidic nature of THCCOOH. It is known that a small percentage (i.e. 2%) of THC is metabolized to THC‐glucuronide and other molecular products have been identified with in vitro microsomal studies16, 25; thus, their exclusion from our model does not introduce a clinically relevant error. Additionally, most THCCOOH is conjugated, becoming THCCOOH‐glucuronide, but was not included as these data were not reported.14
The 3‐compartment THC model diagnostics are shown in Figures 2–4. In Figure 2a, the observed vs predicted THC plasma concentrations for the individual subjects for both THC dosages indicate good agreement and demonstrate the good performance of the model. This conclusion is extended to the conditional weighted residual plots (Figure 3), which demonstrate even scatter across time and concentration except for a slight tendency to over predict concentrations at the 6‐ and 12‐h time points for the larger dose. The good general agreement as well as the slight tendency to over predict at late time points is evident in the visual predictive check for the larger dose (Figure 4a vs 4b). The THC plasma concentrations at late time points are notably near the lower limit of quantitation for this assay. Indeed for the smaller dose only 1/3 were measurable at 6 h and none were measurable at 12 h. Without a washout period, frequent users have measurable plasma THC, 11‐OH‐THC and THCCOOH concentrations for weeks due to high postconsumption concentrations at steady‐state. Better estimation of metabolite pharmacokinetic events may be better delineated in later studies with improved assay sensitivity, larger THC doses with higher THC cannabis content, and in studies of chronic users in which THC and its metabolites are already detectable in plasma at the time of the studied THC consumption.16
The metabolite (11‐OH‐THC and THCCOOH) model diagnostics also demonstrate good performance. In Figure 2b–c, higher and midrange concentrations of THCCOOH are well characterized by the model, but are over predicted at low concentrations for both doses. The high‐dose THC study showed excellent agreement of observed vs individual predicted 11‐OH‐THC plasma concentrations, however the lower dose study demonstrated consistent under prediction. This is not surprising as the observed 11‐OH‐THC concentration range for both doses was similar instead of being higher, proportionate to the dosage difference (i.e. double), in the high dose arm of the study. Interestingly, unlike 11‐OH‐THC, both the observed THC and THCCOOH concentrations were proportionately higher for the larger dose, displaying linear, first‐order kinetics. Accordingly, the 5th and 95th percentiles of the visual predictive checks for THC and THCCOOH were well‐bounded while those for 11‐OH‐THC were not (Figure 4c,d). We attempted nonlinear (or saturation) kinetics to account for similar 11‐OH‐THC plasma concentrations following both low and high doses, but overall model performance actually diminished. Additional studies with more subjects and, perhaps, a larger range of doses may allow better exploration of structural models, including metabolism pathways and the nonlinear kinetics observed by Strougo et al.11
Complete population pharmacokinetic models of THC and its metabolites are important for better interpretation of sparse observational data commonly obtained from individuals in studies of cannabis users across a variety of areas of interest such as medical, recreational and forensic.22, 26 For instance, a blood sample that only has measurable THCCOOH can yield estimates of daily THC consumption and exposure, assuming chronic use and approximate steady‐state conditions (CSS), using the basic clinical pharmacokinetic principle of:
If the measured [THCCOOH] is 100 ng/mL, then from Table 2 the THCCOOH ‘dosing rate’ equals approximately (100 ng/mL × 43.2 L/hr) 4320 μg/hr or 3834 μg/hr after correcting for the ratio of THC and THCCOOH molecular weights. A further 5% correction is added to account for the minor THC metabolic pathways not included in the current model, yielding 3642 μg/hr and corresponding to 87.4 mg/day of bioavailable THC, or 0.35 g/day THC product smoked. Assuming 25% bioavailability and a THC content of 10%, the estimated grams of cannabis smoked per day for this individual is 3.5 g. Additionally, the mean [THC] needed to result in a steady‐state [THCCOOH] of 88.75 ng/mL (100 ng/mg corrected for the respective molecular weights) is estimated by multiplying this THCCOOH by the ratio of the elimination clearances of THCCOOH and THC, yielding a [THC] of 14.2 ng/mL and an AUC0–24 for THC exposure of 340.8 ng mL−1 hr.
Population pharmacokinetic models with estimates of both inter‐ and intraindividual variability can also be used to simulate concentrations of THC, 11‐OH‐THC and THCCOOH that might be obtained from various dosing and usage patterns, both acute and chronic. Additionally, studies of larger populations, whether with sparse data (i.e. only a few blood samples per subject) or dense studies (i.e. many blood samples following drug dose(s)) as with the current analysis, can be used to assess potentially important covariates (e.g. body weight, age, sex, interacting drugs, genetics) and pharmacodynamic outcomes (e.g. ability to safely operate a vehicle). Such modeling tools are in common use by the Food and Drug Administration, European Medicines Agency, the pharmaceutical industry, and by clinicians when studying therapeutic drugs.13
While the current analysis only involved 6 subjects, it provides a potentially useful model of THC and 2 of its major metabolites as well as the expected range of parameter estimates that might occur in a group of healthy male cannabis users. Further studies that would expand on the types of subjects, THC doses, steady‐state (i.e. chronic use) conditions, addition of THCCOOH‐glucuronide measurements and urine data are needed to get a more complete and useful model.
COMPETING INTERESTS
The authors confirm that the Principal Investigator for this paper is M.A.H. and that she had direct clinical responsibility for patients. There was no funding for this research.
CONTRIBUTORS
C.S. was the lead author, responsible for data preparation, pharmacokinetic modeling, statistical analysis, data interpretation and preparation of the draft manuscript. T.K.H. performed the pharmacokinetic analysis, data interpretation and assisted with manuscript preparation. M.A.H. was the principle investigator of the original study, assisted with model development, data interpretation and critically reviewed the manuscript. S.K.M. assisted with the statistical analysis and critically reviewed the manuscript. U.C. and J.K. assisted with data interpretation, model development and critically reviewed the manuscript.
Sempio C, Huestis MA, Mikulich‐Gilbertson SK, Klawitter J, Christians U, Henthorn TK. Population pharmacokinetic modeling of plasma Δ9‐tetrahydrocannabinol and an active and inactive metabolite following controlled smoked cannabis administration. Br J Clin Pharmacol. 2020;86:611–619. 10.1111/bcp.14170
DATA AVAILABILITY STATEMENT
All data are available in reference 14 (DOI: https://doi.org/10.1093/jat/16.5.276).
REFERENCES
- 1. Bowling CM, Glantz SA. Conflict of interest provisions in state Laws governing medical and adult use cannabis. Am J Public Health. 2019;109(3):423‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Windle SB, Wade K, Filion KB, Kimmelman J, Thombs BD, Eisenberg MJ. Potential harms from legalization of recreational cannabis use in Canada. Can J Public Health. 2019;110(2):222‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hjorthoj CR, Fohlmann A, Larsen AM, Arendt M, Nordentoft M. Correlations and agreement between delta‐9‐tetrahydrocannabinol (THC) in blood plasma and timeline follow‐back (TLFB)‐assisted self‐reported use of cannabis of patients with cannabis use disorder and psychotic illness attending the CapOpus randomized clinical trial. Addiction. 2012;107(6):1123‐1131. [DOI] [PubMed] [Google Scholar]
- 4. Smith MJ, Alden EC, Herrold AA, et al. Recent self‐reported cannabis use is associated with the biometrics of Delta‐9‐tetrahydrocannabinol. J Stud Alcohol Drugs. 2018;79(3):441‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huestis MA, Barnes A, Smith ML. Estimating the time of last cannabis use from plasma delta9‐tetrahydrocannabinol and 11‐nor‐9‐carboxy‐delta9‐tetrahydrocannabinol concentrations. Clin Chem. 2005;51(12):2289‐2295. [DOI] [PubMed] [Google Scholar]
- 6. Huestis MA, Elsohly M, Nebro W, Barnes A, Gustafson RA, Smith ML. Estimating time of last oral ingestion of cannabis from plasma THC and THCCOOH concentrations. Ther Drug Monit. 2006;28(4):540‐544. [DOI] [PubMed] [Google Scholar]
- 7. Schwope DM, Karschner EL, Gorelick DA, Huestis MA. Identification of recent cannabis use: whole‐blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration. Clin Chem. 2011;57(10):1406‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet. 2003;42(4):327‐360. [DOI] [PubMed] [Google Scholar]
- 9. Hunault CC, van Eijkeren JC, Mensinga TT, de Vries I, Leenders ME, Meulenbelt J. Disposition of smoked cannabis with high Delta(9)‐tetrahydrocannabinol content: a kinetic model. Toxicol Appl Pharmacol. 2010;246(3):148‐153. [DOI] [PubMed] [Google Scholar]
- 10. Heuberger JA, Guan Z, Oyetayo OO, et al. Population pharmacokinetic model of THC integrates oral, intravenous, and pulmonary dosing and characterizes short‐ and long‐term pharmacokinetics. Clin Pharmacokinet. 2015;54(2):209‐219. [DOI] [PubMed] [Google Scholar]
- 11. Strougo A, Zuurman L, Roy C, et al. Modelling of the concentration‐‐effect relationship of THC on central nervous system parameters and heart rate ‐‐ insight into its mechanisms of action and a tool for clinical research and development of cannabinoids. J Psychopharmacol. 2008;22(7):717‐726. [DOI] [PubMed] [Google Scholar]
- 12. Newmeyer MN, Swortwood MJ, Barnes AJ, Abulseoud OA, Scheidweiler KB, Huestis MA. Free and glucuronide whole blood Cannabinoids' pharmacokinetics after controlled smoked, vaporized, and Oral cannabis Administration in Frequent and Occasional Cannabis Users: identification of recent cannabis intake. Clin Chem. 2016;62(12):1579‐1592. [DOI] [PubMed] [Google Scholar]
- 13. Desai A, Kovanda L, Kowalski D, Lu Q, Townsend R, Bonate PL. Population pharmacokinetics of Isavuconazole from phase 1 and phase 3 (SECURE) trials in adults and target attainment in patients with invasive infections due to aspergillus and other filamentous fungi. Antimicrob Agents Chemother. 2016;60(9):5483‐5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11‐OH‐THC and THCCOOH during and after smoking marijuana. J Anal Toxicol. 1992;16(5):276‐282. [DOI] [PubMed] [Google Scholar]
- 15. Olofsen E, Dahan A. Using Akaike's information theoretic criterion in mixed‐effects modeling of pharmacokinetic data: a simulation study. F1000Res. 2013;2:71‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desrosiers NA, Himes SK, Scheidweiler KB, Concheiro‐Guisan M, Gorelick DA, Huestis MA. Phase I and II cannabinoid disposition in blood and plasma of occasional and frequent smokers following controlled smoked cannabis. Clin Chem. 2014;60(4):631‐643. [DOI] [PubMed] [Google Scholar]
- 17. Avram MJ, Spyker DA, Henthorn TK, Cassella JV. The pharmacokinetics and bioavailability of prochlorperazine delivered as a thermally generated aerosol in a single breath to volunteers. Clin Pharmacol Ther. 2009;85(1):71‐77. [DOI] [PubMed] [Google Scholar]
- 18. Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82(1):17‐20. [DOI] [PubMed] [Google Scholar]
- 19. Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best‐case performance. J Pharmacokinet Pharmacodyn. 2003;30(6):387‐404. [DOI] [PubMed] [Google Scholar]
- 20. Bergstrand M, Karlsson MO. Handling data below the limit of quantification in mixed effect models. AAPS j. 2009;11(2):371‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jonkman K, Duma A, Olofsen E, et al. Pharmacokinetics and bioavailability of inhaled Esketamine in healthy volunteers. Anesthesiology. 2017;127(4):675‐683. [DOI] [PubMed] [Google Scholar]
- 22. Karschner EL, Darwin WD, Goodwin RS, Wright S, Huestis MA. Plasma cannabinoid pharmacokinetics following controlled oral delta9‐tetrahydrocannabinol and oromucosal cannabis extract administration. Clin Chem. 2011;57(1):66‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacquez JA. Parameter identifiability is required in pooled data methods. J Pharmacokinet Biopharm. 1996;24(3):301‐305. [DOI] [PubMed] [Google Scholar]
- 24. Dutcher JS, Strong JM, Lucas SV, Lee WK, Atkinson AJ Jr. Procainamide and N‐acetylprocainamide kinetics investigated simultaneously with stable isotope methodology. Clin Pharmacol Ther. 1977;22(4):447‐457. [DOI] [PubMed] [Google Scholar]
- 25. Mazur A, Lichti CF, Prather PL, et al. Characterization of human hepatic and extrahepatic UDP‐glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos. 2009;37(7):1496‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hartman RL, Brown TL, Milavetz G, et al. Cannabis effects on driving longitudinal control with and without alcohol. J Appl Toxicol. 2016;36(11):1418‐1429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available in reference 14 (DOI: https://doi.org/10.1093/jat/16.5.276).