

Abstract
Aims
AJM300 is an oral antagonist of α4‐integrin that reduces inflammation by blocking leucocyte trafficking. This study aimed to investigate safety, tolerability, pharmacokinetics and pharmacodynamics of AJM300 in healthy male subjects.
Methods
A total of 23 subjects were randomised to receive 240 mg (n = 6), 480 mg (n = 5), 960 mg (n = 6) of AJM300 or the corresponding placebo (n = 2 per group). The study drugs were taken orally 3 times daily after each meal on the first day followed by a 4‐day washout period. Thereafter, multiple‐dose administration was conducted for 6 consecutive days. The pharmacokinetic parameters of AJM300 and its active metabolite (HCA2969) were assessed, and total white blood cells and the differential cell count were used to determine the pharmacodynamic effects. Adverse events (AEs) were also monitored.
Results
The plasma AJM300 and HCA2969 concentration–time curves displayed a triphasic pattern on Day 1 (single‐day administration) and Day 10 (last day of multiple dosing), whereas the concentration of HCA2969 was much higher than that of AJM300. A significant but transient increase in lymphocyte count was observed after AJM300 dosing at all dosages tested compared with the placebo. The increase was sustained over a 24‐h period only at the 960‐mg dosage. In particular, a significant increase in the lymphocyte count compared to placebo (mean, 50.58%; 95% confidence intervals, 20.40–80.76) was observed at the first 960‐mg dose on Day 10. Six (26.1%) subjects reported ≥1 AEs, all of which were mild and resolved spontaneously.
Conclusion
The maximal and 24‐h sustained pharmacodynamic effects were demonstrated at the 960‐mg dosage after oral administration of AJM300 3 times daily for 6 days, which was also found to be safe and well tolerated.
Keywords: clinical trial, pharmacodynamics, pharmacokinetics, Phase I, randomised controlled trial
What is already known about this subject
AJM300 is a novel, orally administrable α4‐integrin antagonist that inhibits leucocyte trafficking into inflamed tissues.
The anti‐α4 integrin antibody, natalizumab, has been used to treat patients with multiple sclerosis and Crohn's disease.
AJM300 has shown a preventative effect on worsening neurological deficits in a rat experimental autoimmune encephalomyelitis model.
What this study adds
HCA2969 was found to have a short half‐life of 14.1 h at the 960‐mg dosage.
Multiple oral administration of AJM300 at the 960‐mg dosage increased lymphocyte count by 125–170%. This pharmacodynamic effect was sustained over a 24‐h period and is almost comparable to that of natalizumab.
1. INTRODUCTION
Targeting of immune cell‐trafficking molecules has been a burgeoning area for the development of anti‐inflammatory drugs. Anti‐integrin therapy, which inhibits the extravasation of circulating leucocytes into inflamed tissues, has been used to treat various inflammatory diseases such as multiple sclerosis (MS), inflammatory bowel disease,1, 2 asthma and atopic dermatitis.3 However, approved therapeutics are mostly monoclonal antibodies which are invariably delivered by injection because of their high molecular weight. Immunogenicity, which leads to a decrease in the effectiveness of drugs and an increase in the risk of hypersensitivity reactions due to the development of persistent anti‐drug antibodies, also remains a concern.1, 4
A widely used anti‐integrin medication with well‐documented efficacy is https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6591.5, 6, 7 It is a humanised monoclonal antibody against the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2443 of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2580 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2770 integrins on leucocytes, acting as a selective adhesion molecule inhibitor. Natalizumab significantly reduces rate of relapse and progression of disability in patients with relapsing–remitting MS.8, 9, 10, 11 In patients with Crohn's disease, natalizumab increases rates of clinical remission and response and improves patients' quality of life.12 However, natalizumab treatment has been associated with a risk of progressive multifocal leucoencephalopathy (PML), an opportunistic brain infection caused by reactivated John Cunningham virus.13 Three risk factors of developing natalizumab‐associated PML are known: the presence of anti‐John Cunningham virus antibodies, previous immunosuppression, and treatment duration, especially beyond 2 years.14, 15, 16
https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=10510 (INN; https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=10510) is a small‐molecule α4‐integrin antagonist that can be administered orally and is almost nonimmunogenic. An orally administered active drug would have the additional advantage of potentially increasing drug adherence. AJM300 is an esterified prodrug that has limited pharmacological action by itself but is specifically designed to enhance oral bioavailability. After absorption in the gastrointestinal tract, the majority of AJM300 is hydrolysed by carboxylesterase 1 to its active metabolite, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=105101 in the liver. This HCA2969 should be widely distributed throughout the body, uptaken by hepatic organic anion‐transporting polypeptide 1B1 or 1B3, primarily excreted in bile via transporters such as multidrug resistance‐associated protein 2, minimally reabsorbed into the enterohepatic circulation, and thereby eliminated in faeces (unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan). It shares the same mechanism of action as natalizumab, inhibiting leucocyte transmigration into inflamed tissue by blocking the interaction of α4β1 or α4β7 integrins and their counter‐receptors (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6758 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9339) and ligand (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6754). In a rat experimental autoimmune encephalomyelitis—a rodent model of MS—administration of AJM300 prevented worsening of the neurological deficit. The lymphocyte count at trough level in the blood showed a significant increase in rats administered AJM300 compared with the vehicle‐treated group.17 AJM300 has also entered clinical development for induction therapy of active ulcerative colitis (UC).18
In a previous single‐dose study, we found that a single oral dose of AJM300 is safe and tolerable up to 960 mg.19 In this phase 1 study, we aimed to assess the safety of AJM300 and HCA2969 and to investigate their pharmacokinetic (PK) and pharmacodynamic (PD) properties after oral multiple dosing of AJM300 3 times daily in healthy male volunteers.
2. METHODS
2.1. Study participants
The study population consisted of 23 healthy, non‐smoking, Japanese male subjects aged 20–39 years, with a body mass index ≥18.5 and <25.0 kg m−2. Eligible subjects had normal vital signs, laboratory tests and electrocardiogram (ECG), had no clinically significant medical history and were taking no medication or supplements that would interfere with the procedures or compromise subject safety. Subjects were ineligible if their white blood cell (WBC) count was <4.0 × 103/μL and/or they were seropositive for anti‐HTLV‐1 antibody. Other exclusion criteria included neurological symptoms and serious infectious diseases, including opportunistic infection within 1 year before administration of the drug. All subjects provided written informed consent prior to study enrolment. The study was approved by the local institutional review board (CPC2007–23, at CPC Clinic ethics committee, Kagoshima, Japan) and was conducted in accordance with Good Clinical Practice and the Declaration of Helsinki.
2.2. Study design
This was a single‐centre, randomised, placebo‐controlled, double‐blind study conducted at the CPC clinic between June and August 2008. Eligible subjects were admitted to the study centre according to the dose groups. One subject who was allocated to the 480‐mg dose (or placebo) group was excluded due to not meeting the criteria (WBC count increased) on the first day of drug administration (Day 1). Subjects were randomly assigned to receive either the active drug (240 mg, n = 6; 480 mg, n = 5; 960 mg, n = 6) or a corresponding placebo (n = 2 per group) as described in Figure 1 and Figure S1. Subjects received the study drug orally 3 times daily after each meal on Day 1 followed by a 4‐day washout (observation) period. Thereafter, they took multiple doses of the study drug for 6 consecutive days according to the investigator's safety evaluation. The washout period was not only for safety purposes, but also to observe the PK properties of AJM300 after each meal as a previous single dose and food effect study suggested that absorption of AJM300 could be affected by food consumption (Fukase et al. 2019. manuscript in preparation). The 240‐mg, 480‐mg and corresponding placebo administration were performed in parallel, and the subsequent 960‐mg administration was determined based on the investigator's safety evaluation.
Figure 1.
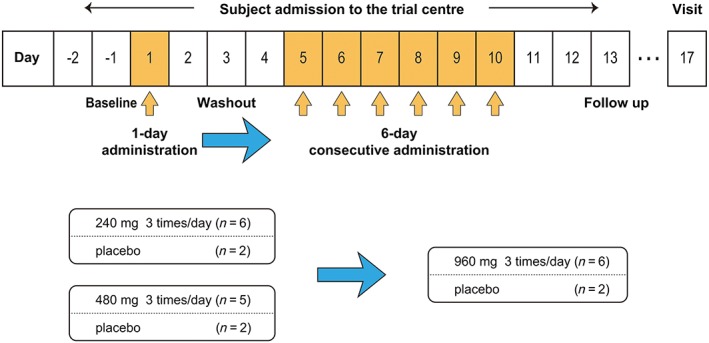
Study design. All subjects were admitted to the trial centre from day −2 (evening) until day 13 (morning). The orange arrow indicates 3 times daily administration of the study drug. Administration of 240 and 480 mg of the study drug was conducted in parallel. The blue arrow indicates a safety examination performed to determine the further progress of the study
The subjects were randomised by using a permuted block method. Blinding was achieved by providing a placebo tablet identical to the AJM300 tablet in both appearance and weight. Subjects, investigators and data analysts were masked to treatment allocation. All subjects were housed at the study centre on Day −2 for baseline assessments, discharged on Day 13 and visited the study centre for follow‐up examinations on Day 17. All food and drink provided at the study centre were standard meals (approx. energy, 2000 kcal; protein, 80 g; fat, 54 g; carbohydrate, 290 g; sodium, 8 g per day), no special dietary meal was provided and any other food or drink (alcohol, caffeine and grapefruit‐containing products in particular) were not permitted during admission. Subjects were required to finish their meals 30 min before dosing and were not allowed to take any additional food or drink other than water until 4 h postdose. They were also not allowed to lie down for at least 2 h after dosing.
2.3. Sample collection and analytical methods
For the quantification of plasma AJM300 and HCA2969 concentrations, 2‐mL venous blood samples were collected from an intravenous cannula at the following timing points after the first dose: −0.5, 0.5, 1, 2, 2.5, 3, 3.5 and 4 h and every hour thereafter until 15 and 21 h (on Days 1, 8 and 10); −0.5 h (on Days 2, 3, 5, 6, 7, 9 and 13); −0.5 and 11 h (on Days 11 and 12). For the WBC count and differential, 2‐mL venous blood samples were collected at the following timing points after the first dose: −0.5 h (from Day −1 to Day 13 except for Day 4); 0.5, 1, 2, 2.5, 3, 3.5 and 4 h and every hour thereafter until 15 and 21 h (on Days −1, 1, 8 and 10); 11 h (on Days 11 and 12). Urine samples were collected at predose and 0–5, 5–11, 11–16 and 16–24 (until the next day's administration) h after the first dose on Days 1, 7 and 9, and the samples were stored at ≤−20°C under acidic conditions (pH 3.0) in 5% 1 M sodium dihydrogen phosphate buffer until analysis. Plasma and urinary concentrations of AJM300 and HCA2969 were measured using a liquid chromatography–tandem mass spectrometry (LC–MS/MS) assay at Toray Research Center, Inc. (Tokyo, Japan). The method was developed and validated according to the recommendations of the US Food and Drug Administration (FDA) guideline on bioanalytical method validation guidance for industry.20 The analytical range was linear with a 1/x2 weighting between 0.5 and 500 ng mL–1 for plasma analyte (AJM300 and HCA2969) concentrations, and between 2.11 and 526 ng mL–1 for urinary analyte concentrations based on an r value ≥0.998. Inter‐day and intra‐assay accuracy for plasma concentrations with low (1 ng mL–1), medium (10 ng mL–1) and high (400 ng mL–1) quality control samples were 107.0–114.0% for AJM300 and 103.7–113.9% for HCA2969, and the precision (% coefficient of variation) was ≤2.5% for AJM300 and ≤ 5.4% for HCA2969.
2.4. PK assessments
PK parameters were analysed by non‐compartmental methods using WinNonlin Professional Version 5.0.1 (Pharsight Corporation, St. Louis, MO, USA), and the following parameters were included: peak plasma concentration from zero to 24 h (Cmax 24h); the time to reach Cmax 24h (Tmax 24h); trough plasma concentration (Ctrough) which was obtained as a minimum plasma concentration just before the first dose on the next day (24 h after the initial dose); the area under the concentration–time curve from zero to 24 h (AUC24h) which was estimated via the linear trapezoidal rule; the apparent terminal elimination half‐life (t1/2); the cumulative fraction of the dose excreted in the urine over each collection interval (fe).
2.5. Statistical analyses
Descriptive statistics were provided for all PK, PD, demographic and safety parameters. All statistical analyses were performed using SAS 8.2 (SAS Institute Inc., Tokyo, Japan) at BELLSYSTEM24, Inc. (Tokyo, Japan). Statistical tests for significance were 2‐sided, and the significance level was set at α = 0.05.
A natural logarithmic transformation of PK parameters, except for Tmax and fe, was applied for all statistical inference. The PK dose‐proportionality with regard to Cmax, AUC24h was assessed using a power model, and it was considered to have been demonstrated if the corresponding 95% confidence intervals (CIs) were within the 0.7–1.3 window.21 The variability of median Tmax was assessed using a Kruskal–Wallis test.
For the PD analyses, considering the daily fluctuation of biomarkers, the total WBC and differential counts at baseline (Day −1) were measured at the same timing points for plasma concentrations measured on Day 1. We first analysed the changes in the PD markers (i.e. lymphocyte count) from baseline value as well as the percentage change from baseline. The percentage change was calculated using the following equation:
where X = the day after dosing of AJM300. Secondly, we estimated 3 area under the effect curves (AUECs) where the percentage change in the lymphocyte count were ≥ 0%, ≥30% and ≥ 50% at each dosage.
The statistical significance of the change in the PD markers between AJM300 and placebo was assessed using a t‐test. In addition, a planned orthogonal comparison was performed using a maximum contrast method22 with 5 sets of polynomial contrast coefficients as described Table S3B. The strength of the relationship described as η 2 and the P value were calculated to evaluate the polynomial trends (upper bound, half up, linear, half down, lower bound) for the dose–response to the AUEC of the percentage change in the lymphocyte count.
Safety was assessed by monitoring the type, severity and incidence of adverse events (AEs) and by evaluation of standard clinical laboratory parameters, physical and neurological symptoms, and ECG. Clinical safety was addressed by evaluating the number/proportion of subjects experiencing AEs and by investigating any clinically significant changes from baseline in laboratory tests, vital signs (pulse, blood pressure, body temperature and weight) and ECG. The AEs were classified as mild, moderate or severe, and the relationship to the study drug was judged by the investigator.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal fordata from the IUPHAR/BPS Guide to PHARMACOLOGY.
3. RESULTS
3.1. Subjects
Of the 78 subjects enrolled, 23 eligible subjects were randomised, and all of them completed the study. No subject was withdrawn; hence, all 23 subjects were included in the PK, PD and safety analyses. Baseline demographics were generally similar across all treatment groups (mean ± standard deviation); age (24.3 ± 3.7), height (171.5 ± 7.3), weight (64.3 ± 6.8) and body mass index (21.8 ± 1.8). The complete CONSORT flow chart and baseline demographics are shown in Figure S1 and Table S1 of the online supporting documents.
3.2. PK
The main PK parameters and the concentration–time profiles of AJM300 and its active metabolite, HCA2969 on Day 1 and Day 10 (the last of 6 consecutive days of administration) are summarised in Table 1 and Figure 2. While the concentration–time profiles of both AJM300 and HCA2969 displayed a similar triphasic pattern after 3 times daily administration on Day 1 and Day 10, the systemic exposure based on AUC24h was much higher to HCA2969 than to AJM300 (Figure 2). For example, HCA2969 AUC24h was 6.34‐fold higher than AJM300 AUC24h at the 960‐mg dosage on Day 10. The mean t1/2 of AJM300 and HCA2969 at the 960‐mg dosage on Day 10 was 5.60 and 14.07 h, respectively. Both AJM300 and HCA2969 concentrations declined soon after the last dose. Particularly, the plasma AJM300 concentration reached the lower limit of quantification within 37 h on Day 10 after the last 960‐mg dose of AJM300. The fe of AJM300 and HCA2969 after 960 mg of AJM300 administration on Day 9 were 0.001 and 0.537%, respectively; both the parent compound and its metabolite were excreted at very low levels in the urine. Plasma AJM300 and HCA2969 trough concentrations were stable from Day 6 at all dosages tested and reached steady state on the second day of consecutive administration (Figure 3).
Table 1.
Selected plasma AJM300 and HCA2969 pharmacokinetic parameters
| Day 1 | Day 10 | |||||
|---|---|---|---|---|---|---|
| 240 mg (n=6) | 480 mg (n=5) | 960 mg (n=6) | 240 mg (n=6) | 480 mg (n=5) | 960 mg (n=6) | |
| AJM300 | ||||||
| C trough (ng mL −1 ) | 0.49 (0.43) | 2.19 (1.10) | 2.77 (2.11) | 0.60 (0.52) | 1.49 (0.36) | 2.00 (1.57) |
| C max 24h (ng mL −1 ) | 121.90 (72.71) | 292.20 (257.31) | 527.33 (551.13) | 195.00 (40.03) | 278.20 (87.51) | 501.17 (269.59) |
| AUC 24h (ng·h mL −1 ) | 755.33 (310.80) | 1667.26 (559.66) | 3134.36 (1307.14) | 1186.95 (176.07) | 2408.00 (617.91) | 3120.62 (788.25) |
| T max 24h (h) | 10.00 (9.00, 13.00) | 10.00 (10.00, 13.00) | 6.00 (4.00, 10.00) | 10.00 (9.00, 10.00) | 9.00 (8.00, 9.00) | 10.00 (9.00, 10.00) |
| t 1/2 (h) | 1.43 (0.14)‡ | 6.01 (6.64) | 3.98 (5.87) | 1.45 (0.14)† | 24.83 (46.14) | 5.60 (8.24) |
| fe (%) | 0.0003 (0.0004) | 0.0007 (0.0002) | 0.0007 (0.0005) | 0.0005 (0.0003) | 0.0006 (0.0006) | 0.0005 (0.0005) |
| HCA2969 | ||||||
| C trough (ng mL −1 ) | 53.98 (5.98) | 103.38 (45.51) | 191.97 (115.43) | 48.52 (4.89) | 103.72 (29.33) | 168.67 (64.95) |
| C max 24h (ng mL −1 ) | 465.50 (200.11) | 1251.00 (612.71) | 1989.17 (1104.53) | 824.17 (129.86) | 1732.00 (354.43) | 2246.67 (1018.56) |
| AUC 24h (ng mL −1 ) | 4709.99 (1591.89) | 11 312.82 (3603.00) | 18 978.42 (7684.07) | 7660.10 (1916.43) | 17 457.26 (3476.98) | 19 790.21 (5600.69) |
| T max 24h (h) | 10.00 (10.00, 10.00) | 10.00 (10.00, 11.00) | 10.50 (10.00, 11.00) | 9.50 (7.00, 10.00) | 9.00 (9.00, 10.00) | 10.00 (10.00, 10.00) |
| t 1/2 (h) | 6.54 (3.08) | 8.86 (4.07) | 9.56 (4.11) | 18.32 (12.44) | 26.87 (23.96) | 14.07 (7.90) |
| fe (%) | 0.7488 (0.1978) | 0.8223 (0.3075) | 0.6315 (0.2196) | 0.9655 (0.1853) | 0.8232 (0.2319) | 0.5374 (0.1231) |
Selected pharmacokinetic parameters are presented as means (standard deviation, SD) unless otherwise stated.
Tmax is presented as median (interquartile range, IQR)
fe (%) was obtained from the data on Days 1 and 9. †n = 4, ‡n = 3.
AUC24h, area under the plasma concentration–time curve from zero to 24 h; Cmax 24h, maximum plasma concentration from zero to 24 h; Ctrough, plasma trough concentration; fe, cumulative fraction of the dose excreted unchanged into urine; t1/2, terminal half‐life; Tmax 24h; time to reach Cmax 24h from zero to 24 h.
Figure 2.
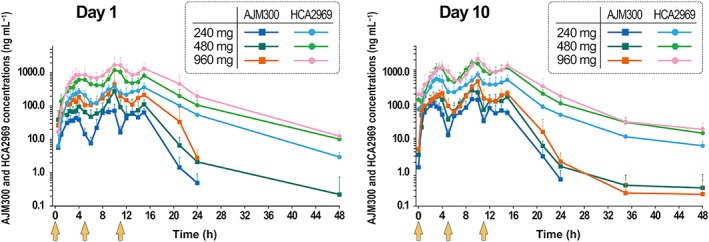
Plasma AJM300 and HCA2969 concentration–time profiles on day 1 (single day 3 times daily administration of AJM300) and day 10 (after multiple doses). Note that the concentration of HCA2969 (the active metabolite) was much higher than that of AJM300 (prodrug) at each dosage. Data are presented as the logarithmic mean (+ standard deviation) of the concentration. AJM300 (blue, green and dark orange) and HCA2969 (light blue, light green and pink) concentrations are indicated after administration of 240, 480 and 960 mg of AJM300, respectively. Light orange arrows indicate the time of drug administration (0, 5 and 11 h)
Figure 3.
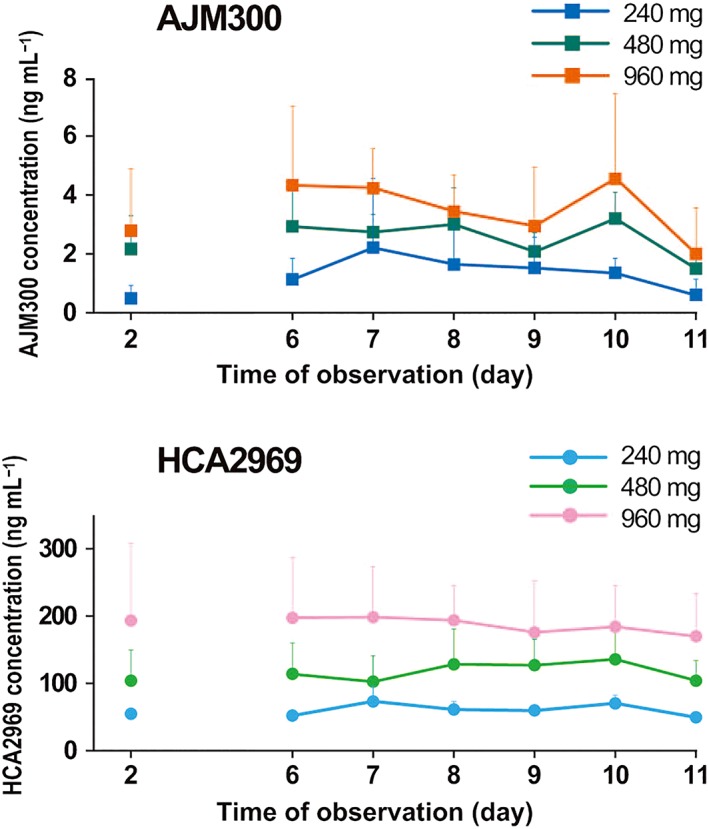
AJM300 (top) and HCA2969 (bottom) trough concentration. Both AJM300 and HCA2969 trough concentrations were stable from day 6 and reached steady state on day 7. Note that HCA2969 trough concentration at the 960‐mg dosage was over IC50 (a half maximal inhibitory concentration; 170 ng mL−1) at all observation days except for day 11. Data are presented as mean + standard deviation
No dose proportionality was concluded with regard to Cmax and AUC24h of HCA2969 on Day 10 in the 960‐mg dosage group because the mean slopes of the linear regression were both 0.678 using a power model analysis, and the 95% CIs were outside the predefined window (Table S2). However, the ratio of the 480‐mg to 240‐mg dosage for HCA2969 Cmax was 2.1; the ratio of 960‐mg to 480‐mg dosage was 1.3. Similarly, the ratio of 480‐mg to 240‐mg dosage for HCA2969 AUC24h was 2.3; the ratio of 960‐mg to 480‐mg dosage was 1.1. Hence, dose proportionality for Cmax and AUC24h of HCA2969 up to 480 mg was indicated. No apparent difference with regards to Tmax among dosage groups was confirmed (data not shown).
3.3. PD
The daily fluctuation of WBC and differential counts showed great inter‐subject variability in which no specific trends were observed (data not shown). The total WBC counts were increased after administration of AJM300 across all dosage groups; however, no change in the neutrophil count was observed. This was because resting neutrophil lacks α4‐integrin subunits.23, 24 Under normal conditions, α4β1 integrin is expressed at substantial levels on most mononuclear leucocytes.25, 26 Both α4β1 and α4β7 integrins are expressed at moderate levels on most of naïve CD4+ and CD8+ T cells27 as well as on most resting B cells in peripheral blood28; thus, an increase in the lymphocyte count that was remarkable but clinically within the normal range was observed after administration of AJM300 on both Day 1 and Day 10 (Figure 4). This pharmacological effect was consistent with results obtained for another α4‐integrin inhibitor, natalizumab.12 The noticeable finding was a 50.58% increase in lymphocyte count at the first 960 mg dose on Day 10. A sustained increase of 25.18–72.30% from the baseline was elicited following 3 times daily doses of 960 mg on Day 10. The significant increases observed on Day 10 returned to baseline levels 13 h after the last 240‐mg and 480‐mg doses, and 24 h after the last 960‐mg dose. The AUECs where the percentage change in the lymphocyte count from baseline was ≥0%, ≥30% and ≥ 50% at each dosage on Day 1 and Day 10 is shown in Figure S2. The strongest relationship between the linear trend (contrast coefficient: −3, −1, 1, 3) and the dose–response curve was concluded when the mean AUECs in which the lymphocyte count increased ≥30% and ≥ 50% on Day 10 was compared to each dose. The η 2 describing the strength of the relationship was 97.8% for a 30% increase and 96.4% for a 50% increase (Table S3B).
Figure 4.
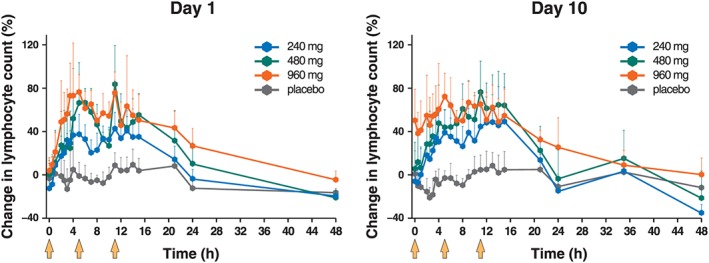
Significant increase in the lymphocyte counts in the peripheral blood following administration of AJM300 on day 1 and day 10. Note that the significant increase was sustained at the 960‐mg dosage until 13 h after the last dose (24 h on graph; P = .014 compared to placebo) but no significance was confirmed 24 h after the last dose (35 h on the graph; P = .399 compared with placebo) on day 10. Data are presented as the proportion of the mean (+standard deviation) change from baseline in the lymphocyte counts. Light orange arrows indicate the time of drug administration (0, 5 and 11 h)
The mean percentage change in lymphocyte count at Ctrough on Day 10 was 1.03, −6.53, 6.27 and 50.58% in the placebo, 240‐, 480‐ and 960‐mg dosage groups, respectively.
3.4. Safety and tolerability
No subject experienced PML, serious AE or an AE that led to treatment discontinuation. A total of 6 (26.1%) subjects reported ≥1 AE after administration of AJM300. The number (percentage) of subjects who reported ≥1 AE was 1 (16.7%), 2 (33.3%), 1 (20.0%) and 2 (33.3%) in the placebo, 240‐, 480‐ and 960‐mg dosage groups, respectively, and at least 1 treatment‐related AE (adverse drug reaction) was reported by 1 (16.7%), 1 (16.7%), 1 (20.0%) and 0 (0.0%) subject, respectively. Observed AEs were all mild in severity, transient and resolved spontaneously. No relevant differences in the incidence or severity of AEs were identified among treatment groups (Table 2).
Table 2.
Adverse events (AEs) and adverse drug reactions (ADRs) reported in the study
| System organ class | Placebo (n = 6) | AJM300 dose | ||
|---|---|---|---|---|
| 240 mg (n = 6) | 480 mg (n = 5) | 960 mg (n = 6) | ||
| Preferred term | n (%) | n (%) | n (%) | n (%) |
| Any AEs | 1 (16.7) | 2 (33.3) | 1 (20.0) | 2 (33.3) |
| Nervous system disorders | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) |
| Headache | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) |
| Gastrointestinal disorders | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) |
| Nausea | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) |
| Investigations | 1 (16.7) | 2 (33.3) | 1 (20.0) | 0 (0.0) |
| White blood cell count increased | 0 (0.0) | 1 (16.7) | 1 (20.0) | 0 (0.0) |
| Alanine aminotransferase increased | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Aspartate aminotransferase increased | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| C‐reactive protein increased | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) |
| Blood glucose increased | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) |
| Blood triglycerides increased | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) |
| Any ADR | 1 (16.7) | 1 (16.7) | 1 (20.0) | 0 (0.0) |
| Investigations | 1 (16.7) | 1 (16.7) | 1 (20.0) | 0 (0.0) |
| White blood cell count increased | 0 (0.0) | 1 (16.7) | 1 (20.0) | 0 (0.0) |
| Alanine aminotransferase increased | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Aspartate aminotransferase increased | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| C‐reactive protein increased | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) |
ADR refers to an AE related or possibly related to the study drug.
The Medical Dictionary for Regulatory Affairs (MedDRA) was used to code the adverse events of the trial.
4. DISCUSSION
The primary objective of this study was to evaluate the PK, PD properties, and safety of AJM300 and its active metabolite, HCA2969—a novel α4‐integrin antagonist—after oral administration of 240, 480 and 960 mg of AJM300 in healthy male subjects. The results of PD assessments provided supporting evidence that 960 mg administered orally 3 times daily is essential to sustain an increase in circulating lymphocytes over a 24‐h period. This study also revealed that multiple administration of AJM300 3 times daily is safe and tolerable up to 960 mg.
Following 3 times daily administration, the plasma HCA2969 concentration was much higher than the AJM300 concentration at each dosage, whilst the triphasic pattern was maintained, which suggested that most of the parent drug (AJM300) was hydrolysed to the active metabolite (HCA2969) after absorption. In addition, the finding that AJM300 was scarcely excreted in urine as unchanged (0.001% on Day 9 after doses of 960 mg) indicated that metabolism is a primary elimination route of AJM300. This was also verified by a previous animal experiment in which 14C‐labelled AJM300 was hardly excreted in urine, bile or faeces. Following intravenous administration of a single dose of 14C‐AJM300 (3 mg/kg) in rats, the cumulative excretion of radioactivity as unchanged from time zero to 72 h was detectable in neither faeces nor urine of the animals administered the 14C‐dose, whereas cumulative recovery of 14C‐HCA2969 was 87.3% in faeces but not detectable in urine (unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan). Comparably, a limited amount (0.537%) of HCA2969 was excreted in urine in this human study. This minimal excretion in urine corroborates the observation that HCA2969 is primarily eliminated via the biliary route.
The AJM300 Tmax was generally not rapid, 3–4.5 h after each drug administration, and appeared to be prolonged after a meal (e.g. the median Tmax 5h, Tmax 5–11h and Tmax 11–24h were 3.75, 9.50 and 15.00 h, respectively at the 960‐mg dosage on Day 10). Given that subjects took meals about 1 h before each administration of AJM300, it is very likely that food consumption interfered with the absorption of AJM300.
The AJM300 and HCA2969 Ctrough reached a steady state on the second consecutive day of administration (Day 6), which indicated the absence of drug accumulation, and greater exposure was observed with a higher dosage. The power model concluded that there was no dose‐proportionality with regards to Cmax and AUC24h of HCA2969 across dosages tested. The absence of dose‐proportionality among the 3 dosages can be explained by the comparison between 2 dosages with the ratio of these PK parameters, which indicated that Cmax and AUC24h would reach a plateau at the 480‐mg dosage. Nevertheless, it should be emphasised that Ctrough on Day 10 at the 960‐mg dosage, which far exceeded those of the 240‐ or 480‐mg dosages, is of the utmost importance for the PD effects that lead to clinical efficacy.
AJM300, an oral form of the anti‐integrin drug, was developed as a safe alternative to currently available anti‐α4 integrin antibodies such as natalizumab whilst providing comparative PD effects. Natalizumab has been shown to be effective at reducing brain inflammation in MS patients by preventing activated leucocytes from accessing the brain parenchyma. In a clinical study on Crohn's disease, natalizumab elicited a 1.3–1.9‐fold increase in the lymphocyte count from baseline.12 In our multiple dose study, administration of 960 mg of AJM300 3 times daily was able to prompt a 24‐h sustained increase of circulating lymphocytes in the range of 1.25–1.7‐fold. Hence, AJM300 at the 960‐mg dosage appears to have a PD effect almost comparable to that of natalizumab. In a CytoTox 96 Non‐Radioactive Cytotoxicity Assay using Jurkat cells expressing human α4β1 integrins, we found that HCA2969 inhibited the binding of VCAM‐1 to α4β1 integrin subunit under the human serum condition with a half maximal inhibitory concentration (IC50) of 170 ng mL−1 (unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan). In this study, only a 960 mg dosage regimen was able to achieve this IC50 level at steady state (refer to Table 1 HCA2969 Ctrough and Figure 3).
A comparison using the maximum contrast method concluded the linear trend of the dose response to the mean AUECs where the increase in lymphocyte count was ≥30% and ≥ 50% on Day 10, indicating that 960‐mg dosage should ensure a maximal PD effect. Furthermore, at the 960‐mg dosage, the mean percentage change in the lymphocyte count at Ctrough on Day 10 was much higher compared with other dosages (51% vs −7–6%), implicating a sustained PD effect induced by multiple dosing of 960 mg. These results suggested that a sustainable IC50 or greater obtained at the 960‐mg dosage would be crucial to acquire clinical efficacy dependent on the PD effect. We also believe that the sustained PD effect shown at the Ctrough at the 960‐mg dosage in this study indicates clinical efficacy in phase 2 clinical trials in patients with active UC at the 960‐mg dosage18 but not at 480 mg (unpublished data on file, EA Pharma Co., Ltd., Tokyo, Japan)
Multiple doses of AJM300 up to 960 mg 3 times daily for 6 days in healthy male subjects were safe and well tolerated. All AEs reported were mild, and no serious AE or PML was identified. To date, there has been no reported case of PML after treatment with AJM300 in clinical trials.18 One of the reasons could be the short‐term treatment used for UC in order to avoid the occurrence of PML. It is unknown whether long‐term treatment with AJM300 is associated with a risk of PML. Natalizumab has a long elimination half‐life (249 ± 105 h [mean ± standard deviation] following a single 300‐mg fixed‐dose infusion29), which may require plasma exchange or immunoadsorption for its removal.30, 31 However, AJM300, which has a much shorter elimination half‐life (AJM300, 5.6 h; HCA2969, 14.1 h at 960‐mg dosage) and a shorter PD effect up to 24 h after the last dose, may make it a more favourable treatment option when considering serious side effects, including PML.
Based on credible evidence, AJM300 has entered further clinical development as an induction therapy for active UC. In a phase 2 randomised controlled trial, oral administration of 960 mg of AJM300 3 times daily for 8 weeks was found to be safe and effective in terms of clinical response, clinical remission, and mucosal healing in 102 patients with moderately active UC.18
The limitation of this study is the small sample size (5–6 subjects per arm) used to evaluate the pharmacological action and safety of AJM300. Nevertheless, this study was conducted in a well‐controlled, in‐house setting; therefore, the results of PK and PD studies should be more accurate than studies conducted in an out‐patient setting. A larger scaled, longer‐term phase 3 study with a 960 mg dosage in patients with active UC is currently ongoing to evaluate the efficacy and safety of AJM300 (http://ClinicalTrials.gov Identifier: NCT03531892).
In conclusion, this study demonstrated that oral administration of AJM300 of 960 mg 3 times daily provided a sustained, maximal PD effect which was represented as an increase in circulating lymphocytes. The percentage change in the lymphocyte count after AJM300 dosing was almost comparable to that observed with natalizumab. AJM300 has the advantage of minimal immunogenicity and hyper‐sensitive reactions as well as better drug adherence. The short elimination half‐life of AJM300 enables control of the duration of pharmacological action followed by cessation. Three times daily administration of AJM300 up to 960 mg for 6 days in healthy male subjects was found to be safe and well tolerated.
CONTRIBUTORS
All authors were involved in study design and carried out the study. H.F. participated in data acquisition and analysis. I.O. carried out statistical analysis and, together with rest of the authors, interpreted the data. All authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this manuscript, critically reviewed and approved the final version of the manuscript for submission.
COMPETING INTERESTS
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). T.K., I.O. and N.I. are employees of EA Pharma Co., Ltd. H.F. received consultancy fees from EA Pharma Co., Ltd. H.F. received funds for conducting clinical investigations in connection with this trial.
Supporting information
Figure S1 CONSORT flow chart.
Table S1 Subject demographics shown as mean (SD).
Table S2 PK dose proportionality assessed using a power model.
Table S3A Mean AUEC where the increase in lymphocyte count was ≥0%, ≥30% and 50%.
Table S3B Strength of the relationship (p‐value) to each contrast coefficient.
Figure S2 The mean AUECs where the increase in lymphocyte count from baseline were ≥0% (top), ≥30% (middle) and ≥50% (bottom) at each dosage tested.
ACKNOWLEDGEMENTS
The authors thank the patients for their participation, the staff at the CPC Clinic and EA Pharma Co., Ltd. for their kind assistance in the conduct of the study as well as data management and processing. The authors acknowledge Toray Research Center, Inc. for the pharmacokinetic analysis, and BELLSYSTEM24 Holdings, Inc. (currently A2 Healthcare Corporation) for the statistical analysis. We would also like to thank Mie Yamamoto, PhD (SunFlare Co., Ltd., Tokyo, Japan), who wrote the first draft of the manuscript based on input from the authors and revised subsequent drafts. This study was funded by EA Pharma Co., Ltd. and the medical writing assistance was funded by EA Pharma Co., Ltd. and Kissei Pharmaceutical Co., Ltd.
Fukase H, Kajioka T, Oikawa I, Ikeda N, Furuie H. AJM300, a novel oral antagonist of α4‐integrin, sustains an increase in circulating lymphocytes: A randomised controlled trial in healthy male subjects. Br J Clin Pharmacol. 2020;86:591–600. 10.1111/bcp.14151
The authors confirm that Dr Hiroyuki Fukase was the principal investigator of this clinical trial and he had direct clinical responsibility for the subjects.
DATA AVAILABILITY STATEMENT
The data underpinning this manuscript cannot be shared openly due to a lack of consent from study participants.
REFERENCES
- 1. Park SC, Jeen YT. Anti‐integrin therapy for inflammatory bowel disease. World J Gastroenterol. 2018;24(17):1868‐1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ghosh N, Chaki R, Mandal SC. Inhibition of selective adhesion molecules in treatment of inflammatory bowel disease. Int Rev Immunol. 2012;31(5):410‐427. [DOI] [PubMed] [Google Scholar]
- 3. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6(12):1182‐1190. [DOI] [PubMed] [Google Scholar]
- 4. Cohen BA, Oger J, Gagnon A, Giovannoni G. The implications of immunogenicity for protein‐based multiple sclerosis therapies. J Neurol Sci. 2008;275(1‐2):7‐17. [DOI] [PubMed] [Google Scholar]
- 5. Pucci E, Giuliani G, Solari A, et al. Natalizumab for relapsing remitting multiple sclerosis. Cochrane Database Syst Rev. 2011; Cd007621(11):1–107. [DOI] [PubMed] [Google Scholar]
- 6. European Medical Agency (EMA) . Tysabri: EPAR ‐ Scientific discussion 2007. https://www.ema.europa.eu/documents/scientific-discussion/tysabri-epar-scientific-discussion_en.pdf. [Accessed in Jan 2019].
- 7. Biogen . Medical information for healthcare professions ‐ patient treated. https://www.tysabrihcp.com/en_us/home/real-world-experience/patients-treated.html. [Accessed in Feb 2019].
- 8. Miller DH, Khan OA, Sheremata WA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348(1):15‐23. [DOI] [PubMed] [Google Scholar]
- 9. Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta‐1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911‐923. [DOI] [PubMed] [Google Scholar]
- 10. Miller DH, Soon D, Fernando KT, et al. MRI outcomes in a placebo‐controlled trial of natalizumab in relapsing MS. Neurology. 2007;68(17):1390‐1401. [DOI] [PubMed] [Google Scholar]
- 11. Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo‐controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899‐910. [DOI] [PubMed] [Google Scholar]
- 12. Ghosh S, Goldin E, Gordon FH, et al. Natalizumab for active Crohn's disease. N Engl J Med. 2003;348(1):24‐32. [DOI] [PubMed] [Google Scholar]
- 13. Stuve O, Bennett JL. Pharmacological properties, toxicology and scientific rationale for the use of natalizumab (Tysabri) in inflammatory diseases. CNS Drug Rev. 2007;13(1):79‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab‐associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870‐1880. [DOI] [PubMed] [Google Scholar]
- 15. Kornek B. An update on the use of natalizumab in the treatment of multiple sclerosis: appropriate patient selection and special considerations. Patient Prefer Adherence. 2015;9:675‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ho PR, Koendgen H, Campbell N, Haddock B, Richman S, Chang I. Risk of natalizumab‐associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol. 2017;16(11):925‐933. [DOI] [PubMed] [Google Scholar]
- 17. Kageyama S, Ito H, Maruyama S, Kihara H, Suzuki M. An orally active alpha4 integrin antagonist AJM300 prevents the development of experimental autoimmune encephalomyelitis in rats. In: 23rd Congress of the European Committee for Treatment and Research in Multiple Sclerosis, Prague, Czech Republic: Program and abstracts of the ECTRIMS 2007: Denver Poster 838.
- 18. Yoshimura N, Watanabe M, Motoya S, et al. Safety and efficacy of AJM300, an oral antagonist of α4 integrin, in induction therapy for patients with active ulcerative colitis. Gastroenterology. 2015;149(7):1775, e2‐1783. [DOI] [PubMed] [Google Scholar]
- 19. Fukase H, Kajioka T, Oikawa I, Ikeda N, Furuie H. Food effect of a single high dose of carotegrast methyl, an oral antagonist of α 4‐integrin, in healthy male subjects: a randomised, placebo‐controlled, double‐blind study. Clin Drug Invest. 2019; In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. U.S. Department of Health and Human Services Food and Drug Administration . Bioanalytical method validation guidance for industry 2001. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry. Accessed in Mar 2019.
- 21. Igarashi T, Yabe T, Noda K. Study design and statistical analysis of toxicokinetics: a report of JPMA investigation of case studies. J Toxicol Sci. 1996;21(5):497‐504. [DOI] [PubMed] [Google Scholar]
- 22. Stewart WH, Ruberg SJ. Detecting dose response with contrasts. Stat Med. 2000;19(7):913‐921. [DOI] [PubMed] [Google Scholar]
- 23. Kirveskari J, Bono P, Granfors K, Leirisalo‐Repo M, Jalkanen S, Salmi M. Expression of alpha4‐integrins on human neutrophils. J Leukoc Biol. 2000;68(2):243‐250. [PubMed] [Google Scholar]
- 24. Bochner BS, Luscinskas FW, Gimbrone MA Jr, et al. Adhesion of human basophils, eosinophils, and neutrophils to interleukin 1‐activated human vascular endothelial cells: contributions of endothelial cell adhesion molecules. J Exp Med. 1991;173(6):1553‐1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wagner N, Müller W. Functions of alpha 4‐ and beta 7‐integrins in hematopoiesis, lymphocyte trafficking and organ development. Curr Top Microbiol Immunol. 1998;231:23‐32. [DOI] [PubMed] [Google Scholar]
- 26. Lobb RR, Hemler ME. The pathophysiologic role of alpha 4 integrins in vivo. J Clin Invest. 1994;94:1722‐1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Picker LJ, Treer JR, Nguyen M, Terstappen LW, Hogg N, Yednock T. Coordinate expression of beta 1 and beta 2 integrin "activation" epitopes during T cell responses in secondary lymphoid tissue. Eur J Immunol. 1993;23:2751‐2757. [DOI] [PubMed] [Google Scholar]
- 28. Postigo AA, Sanchez‐Mateos P, Lazarovits AI, Sanchez‐Madrid F, de Landázuri MO. Alpha 4 beta 7 integrin mediates B cell binding to fibronectin and vascular cell adhesion molecule‐1. Expression and function of alpha 4 integrins on human B lymphocytes. J Immunol. 1993;151:2471‐2483. [PubMed] [Google Scholar]
- 29. Biogen Canada . TYSABRI® Product Monograph 2017. https://www.biogen.ca/content/dam/corporate/en_CA/pdfs/products/TYSABRI/TYSABRI_PM_E.pdf. Accessed in Feb 2019.
- 30. Pavlovic D, Patera AC, Nyberg F, Gerber M, Liu M. Progressive multifocal leukoencephalopathy: current treatment options and future perspectives. Ther Adv Neurol Disord. 2015;8:255‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan IL, McArthur JC, Clifford DB, Major EO, Nath A. Immune reconstitution inflammatory syndrome in natalizumab‐associated PML. Neurology. 2011;77(11):1061‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 CONSORT flow chart.
Table S1 Subject demographics shown as mean (SD).
Table S2 PK dose proportionality assessed using a power model.
Table S3A Mean AUEC where the increase in lymphocyte count was ≥0%, ≥30% and 50%.
Table S3B Strength of the relationship (p‐value) to each contrast coefficient.
Figure S2 The mean AUECs where the increase in lymphocyte count from baseline were ≥0% (top), ≥30% (middle) and ≥50% (bottom) at each dosage tested.
Data Availability Statement
The data underpinning this manuscript cannot be shared openly due to a lack of consent from study participants.