

Abstract
H-RasV12 oncogene has been shown to promote autophagic cell death. Here we provide evidence of a contextual role for H-RasV12 in cell death that is varied by its effects on miR-130a. In E1A-immortalized murine embryo fibroblasts, acute expression of H-RasV12 promoted apoptosis but not autophagic cell death. miRNA screens in this system showed that miR-130a was strongly downregulated by H-RasV12 in this model system. Enforced expression of miR-130a increased cell proliferation in part via repression of PTEN. Consistent with this effect, miR-130a overexpression in human breast cancer cells promoted Akt phosphorylation, cell survival and tumor growth. In clinical specimens of multiple human cancers, expression of miR-130 family members correlated inversely with PTEN expression. Overall, our results defined miR-130a as an oncogenic microRNA that targets PTEN to drive malignant cell survival and tumor growth.
Keywords: H-RasV12, Autophagy, miR-130a, PTEN
Introduction
MicroRNAs (miRNAs) are a class of small noncoding RNAs that negatively regulate mRNA stability and/or repress mRNA translation (1). miRNAs mediate their effects via the posttranscriptional repression of targeted gene expression containing complementary sites in the 3’-untranslated region (UTR), resulting in degradation of the targeted mRNA or inhibition of its translation (2). The low stringency for miRNA binding to complimentary sequences of target mRNAs gives each miRNA the capacity to regulate a largely non-overlapping target mRNAs. Additionally, targeted mRNAs often contain several recognition sites for each miRNA, further increasing the complexity of the miRNA regulatory network. Given that miRNAs act as important regulators of a multitude of biological processes, aberrant expression of miRNAs has been implicated in the development of various diseases, including cancer (3). The function of miRNAs can be efficiently and specifically inhibited by chemically modified antisense oligonucleotides, thereby highlighting their potential as targets for the development of novel therapies (4).
Autophagy, an evolutionarily conserved cellular process that controls cytoplasmic homeostasis, is induced by starvation and other stresses where it induces the bulk degradation of unnecessary or damaged proteins and organelles. Dysfunctions in autophagic processes have been associated with a variety of human diseases, including cancer, metabolic and neurodegenerative disorders, autoimmune diseases, cardiovascular and pulmonary diseases (5–12). Interestingly, Elgendy et al found that transient expression of H-RasV12 in a series of normal as well as neoplastic cell populations triggered autophagic cell death (13).
In this study, we find that the inducible expression of H-RasV12 in E1A immortalized MEF cells causes increased cell apoptosis and proliferation, but not autophagic cell death. While examining changes in the miRNAs profile of H-RasV12-induced cells, we identified miR-130a as a dysregulated miRNA. Further studies demonstrate that upregulated miR-130a expression stimulated cell proliferation, increased Akt phosphorylation and promoted in vivo tumor growth of breast cancer cells by targeting PTEN (phosphatase and tensin homolog deleted on chromosome 10), one of the most common tumor suppressor genes involved in a wide spectrum of human cancers (14,15). Together, these studies identify a miR-130a/PTEN axis as a potential pathophysiologic regulator of tumor cell behavior.
Materials and Methods
Mice, and cell culture
All studies involving mice were performed according to the National Institute of Health Animal Care and Use Guidelines. HEK293T cells, MCF7 and MDA-MB-231 cells, all from ATCC, as well as MEFs isolated for the current study, were maintained in DMEM with 10% FBS and penicillin-streptomycin. 4T1 cells (ATCC) were kept in RPMI 1640 while human ovarian surface epithelial (HOSE) cells (obtained from J. Downward at The Francis Crick Institute) maintained in DMEM/F12 with 10% FBS and penicillin-streptomycin. All cell lines were determined to be mycoplasma-free.
Constructs, retroviral or lentiviral transduction, transfection
H-RasV12 in pLERFP-C1, constructed as described previously (16,17), was cloned into TRMPVIR-puro vector [derived from TRMPVIR, a gift from S. Lowe (Addgene plasmid 27994), with a replacement of venus gene by puromycin resistant gene via overlapping PCR], wherein H-RasV12 expression is under the control of a Dox-inducible promoter. The precursors of mouse miR-130a amplified by PCR from genomic DNA using primers (5’-CGCGGGATCCACCACCCCTCAAGAAAAAGGTGA-3’ and 5’-GGCCGAATTCAGGAGCTCTACAGCCTTAGCTTC-3’), or human miR-130a using primers (5’-GCGCGGATCCAGGCGGCAAAAGGAAGAGTGGTG-3’ and 5’-CGGCGAATTCCACAAGCACTGCATACAGAAGTAG-3’) were cloned into BamHI/EcoRI sites of TRMPVIR-puro or miR-130a precursors fused with CMV promoter (5’-GGCGCAGATCTGAAACAGCTATGACCATGATTACG-3’ / 5’-TCTAGAGTCGACCTGCAGAAGC-3’) from pKH3 plasmid as described previously (18) inserted into the BglII/EcoRI sites of MSCVpuro (Clontech) vector. Two 0.7 kb fragments of human PTEN 3’-UTRs using the primers (5’-GCGCCTCGAGGATCAGCATACACAAATTACAAAAGTCTG-3’ / 5’-GGCCGCGGCCGCAGCCCATTCTTTGTTGATAGCCTCCAC-3’ or 5’-GCGCCTCGAGGTTGACACGTTTTCCATACCTTGTCAG-3’/ 5’-GGCCGCGGCCGCCAAGTAACTTCAGACATGTAAGCTGCTGC-3’) were amplified by PCR from the genomic DNA and cloned into XhoI/NotI sites immediately downstream of the renilla luciferase cassette of psiCHECK2 vector. Mutations of the predicted miR-130a binding sites in the 3’-UTR of human PTEN were introduced by site-directed mutagenesis. Four different shRNAs containing the 22 nt target sequence (PTEN.995, 5’-CGAAGGTGTATACAGGAACAAT-3 ‘; PTEN. 1071, 5’-CACAATCTATGTGCTGAGAGAC-3’; PTEN.1778, 5’-CTGCAGTATAGAGCGTGCAGAT-3 ‘; and PTEN.2996, 5’-CCTGCTCCATCTCCTATGTAAT-3’) for mouse PTEN were amplified by PCR using miR-E primers as described (19), and inserted into TRMPVIR at the XhoI/EcoRI sites to generate constructs capable for knocking-down mouse PTEN in a Dox-inducible manner, as described previously (20). sgRNAs against mouse PTEN were cloned into pLX-sgRNA vector [a gift from E. Lander and D. Sabatini (Addgene plasmid # 50662)]. The Cas9-expressed plasmid, pLCRISPR.EFS.GFP, was a gift from B. Ebert (Addgene plasmid # 57818). The pLEGFP-LC3 construct has been described previously (17). Recombinant retroviral packaging and transduction were carried out as previously described (18,21). Lentiviral particles were produced by transient transfection of 293T cells as described (22,23). For PTEN deletion using the CRISPR-Cas9 system, infected cells were clonally selected, amplified and verified for PTEN deletion by Western blotting. Anti-miR-130a (#4464084) and control (#AM17010) (Thermo Fisher Scientific) were transfected with Lipofectamine 2000 at a final concentration of 50 nM.
Preparation of MEFs
MEFs were generated from 13.5-day-old embryos of wild-type FVB/n mice according to standard protocol (16,17). MEFs were then immortalized by E1A, and subsequently infected with H-RasV12 in TRMPVIR-puro, pLERFP-C1 as well as control vectors as described previously (16,17). Generated MEFs either expressed Dox-inducible or consistent H-RasV12, or served as control MEFs.
Microarray analyses of miRNAs
Total RNA including miRNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s protocol. Mircoarray was performed at The Ohio State University Comprehensive Cancer Center Microarray Core Facility. Each array was normalized using the relative median over all genes.
Cell assays
Cell growth assays were carried out by trypsinizing cells and determine cell number by manual counting with trypan blue exclusion according to the manufacturer’s protocol (ThermoFisher Scientific). Apoptotic cell death was measured by Annexin V-FITC staining (eBioscience). 3’-UTR-reporter assays were performed in HEK293T cells following transient transfection. Firefly and renilla luciferase activities were measured 48 h after transfection using the Dual-Luciferase Reporter Assay Kit (Promega), according to the manufacturer’s instruction.
BrdU incorporation assays
For BrdU incorporation assays, cultured cells were serum-starved overnight and then incubated with growth medium supplemented with 100 μM BrdU (Sigma). BrdU uptake was determined using anti-BrdU antibody (#5292, Cell Signaling Technology) according to the manufacturer’s protocol. DAPI was used for counterstaining, and coverslips mounted on glass slides with Prolong® Gold Antifade Mountant (Thermo Fisher Scientific).
Quantitative RT-PCR
Total RNA was isolated by TRIzol reagent (Invitrogen) and miR-130a determined by qRT-PCR with TaqMan microRNA Reverse Transcription kit (Applied Biosystems). RNU6B was used as an endogenous control for miRNA normalization. miR-130a levels were quantified with the 2(−ΔΔct) relative quantification method that was normalized to RNU6B. For quantitative RT-PCR of PTEN, RNA samples were reverse transcribed using the SuperScript III First-Strand Synthesis System (Invitrogen) with oligo-dT as primer. The resulting templates were subjected to PCR. mRNA expression levels were normalized to HPRT.
Western blotting
Whole-cell lysates were prepared as previously described and analyzed by SDS/PAGE and immunoblotting (18,21). The following primary antibodies were used: Ras (#3339), PTEN (#9559), TSC1 (#6935), Phospho-Erk (Thr202/Tyr204)(#9101), Erk (#4695), Phospho-Akt (Ser473)(#4060), Akt (#4691), cleaved caspase-3 (#9661), all from Cell Signaling Technology; LC3B (#2775) from Cell Signaling Technology or LC3B (NB100–2220) from Novus Biologicals; p62 (#BML-PW9860) from Enzo Life Sciences; and vinculin (#V4505) from Sigma.
Xenograft
The mouse breast cancer cell line, 4T1, or the human breast cancer cell line, MDA-MB-231, was transduced with mouse or human miR-130a, respectively, under the control of CMV promoter from pKH3 (18) in pMSCVpuro plasmid or control vector, and then selected with puromycin. Tumor cells were harvested, washed twice in DMEM, and then subcutaneously injected (5×105 cells in 1:1 PBS:Matrigel [BD Bioscience]) into the flanks of 7- to 8-week-old female BALB/c mice (Charles River) for 4T1 cells, or athymic nude mice (Jackson Laboratory) for MDA-MB-231 cells. Tumor size was measured as described (17) and tumor volume calculated using the formula: length × width2/2.
Correlation analysis
The TCGA miRNA-seq, RNA-seq data for breast invasive carcinoma (BRCA), colon adenocarcinoma (colon ADC) and lung adenocarcinoma (lung ADC) along with clinical information were downloaded from the TCGA website. Only log-2 transformed level 3 data were used for analyses. For the correlation analyses between miR-130a/miR-130b and PTEN gene expression, Spearman or Pearson correlation coefficients were calculated.
Statistical analysis
Statistical significance was evaluated by unpaired Student’s T-test, using p < 0.05 as indicative of statistical significance.
Results
Acute expression of H-RasV12 in E1A-immortalized MEF cells causes increased cell apoptosis and proliferation, rather than autophagic cell death
Acute expression of H-RasV12 has been reported to increase autophagic cell death, but not apoptosis (13). To assess the potentially broad applicability of these observations to other cell systems, E1A-immortalized wild-type MEFs were transduced with a tetracycline-induced H-RasV12 expression vector to generate MEF-iRas cells. In the absence of a tetracycline derivative, doxycycline (Dox), these cells grew normally (Fig. 1A and B). However, after induction of H-RasV12 expression, cell growth rapidly declined, followed by their detachment from the monolayer (Fig. 1A and B). By contrast, Dox-treatment alone had no effect on E1A-immortalized MEF cells (Fig. 1A and B). As these findings confirm that H-RasV12 transient expression inhibits cell survival, we next evaluated the ability of H-RasV12 to induce autophagic cell death (13). Although H-RasV12 expression was increased upon Dox treatment and its expression associated with the increased phospho-Erk (P-Erk) levels (Fig. 1C), we detected the increased protein levels of p62 and LC3-I (i.e., likely caused by reduced LC3-I-to-LC3-II conversion), suggesting that autophagy was inhibited. Additionally, starvation-induced GFP-LC3 autophagosome puncta were not increased in MEF-iRas cells with Dox-induced H-RasV12 expression (Supplementary Fig. S1), reinforcing the fact that H-RasV12 expression has no significant effect on autophagic processes. Rather, we detected increased levels of cleaved caspase-3 (Casp3) in the presence of Dox, reflecting an apparent increase in apoptotic cell death (Fig. 1C). We further confirmed these findings by flow cytometric analysis of Annexin V, an additional marker of apoptosis. Under these conditions, both apoptotic (Ann-V+/DAPI+) and necrotic (Ann-V−/DAPI+) cell populations were significantly increased in MEF-iRas cells following 3 days of Dox treatment (Fig. 1D). Consistent with reports that E1A can overcome the induction of a senescent state following H-RasV12-induced oncogenic stress (24,25), senescence was not increased in E1A-immortalized MEFs following H-RasV12 induction (Supplementary Fig. S2). Surprisingly, while we anticipate that H-RasV12 overexpression would trigger proliferation arrest, cell proliferation increased as assessed by BrdU incorporation (Fig. 1E) despite the fact that H-RasV12 induction reduced cell number (Fig. 1B). Apparently, H-RasV12 expression in E1A-immortalized MEFs increased cell apoptosis and proliferation in tandem with an overall effect of reduced cell survival rather than autophagic cell death. These findings were not confined to immortalized MEFs as transient induction of H-RasV12 expression in HOSE cells or MCF7 cells, cell lines used previously by Elgendy et al (13), did not trigger reduction in either p62 or LC3-I protein levels, suggesting a lack of autophagic cell death, despite an evident change in cell morphology and a robust increase in phosphor-Erk levels (Supplementary Fig. S3A and S3B). However, unlike the effects observed in E1A-immortalized MEFs, H-RasV12 expression had no evident effect on cleaved caspase-3 levels in either HOSE cells or MCF7 cells (Supplementary Fig. S3B).
Figure 1. Acute expression of H-RasV12 in ElA-immortalized MEFs promotes apoptotic cell death and cell proliferation, rather than autophagic cell death.
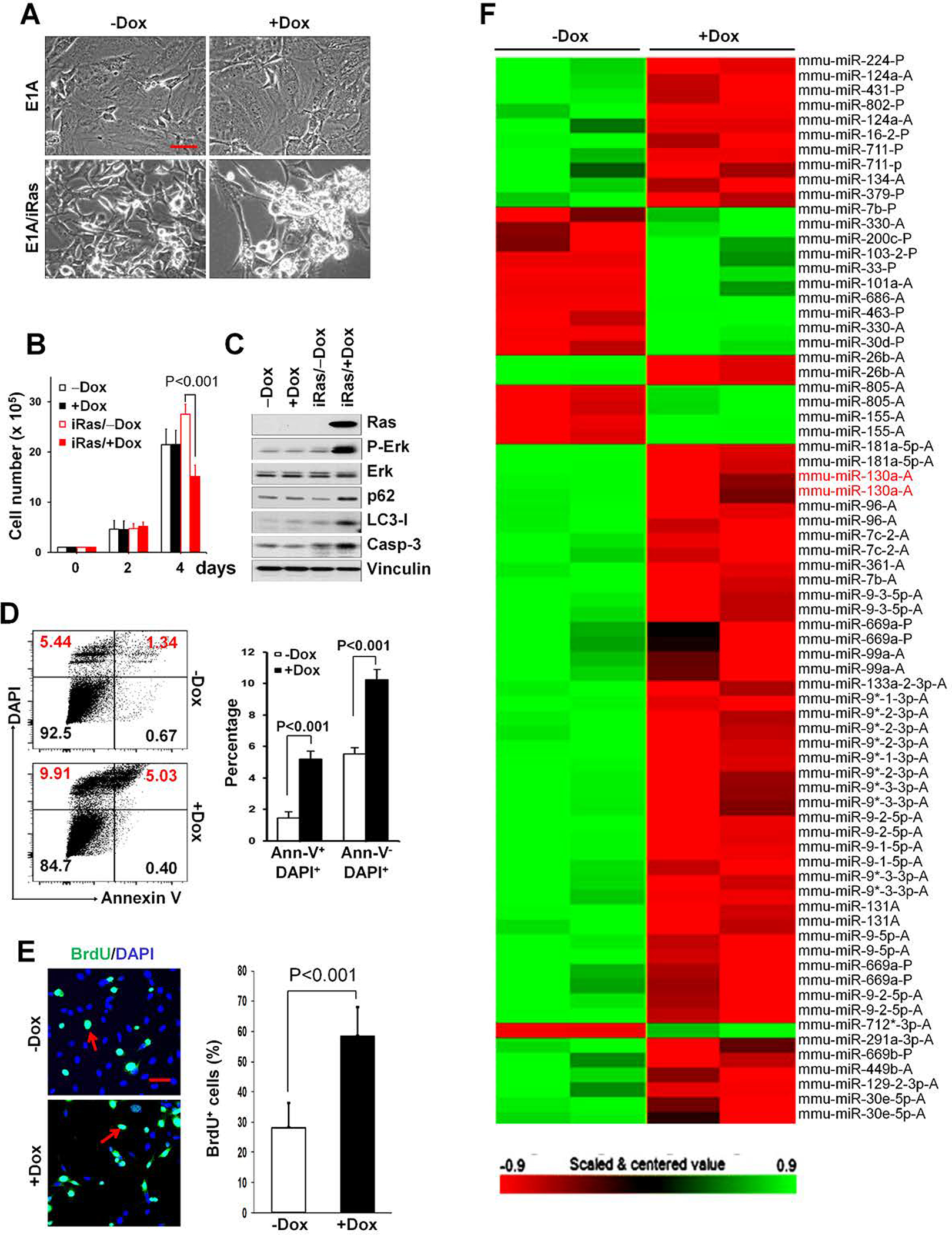
(A, B) ElA-immortalized MEF cells transduced with control vector or Dox-induced H-RasV12 expression vector (MEF-iRas cells) were either untreated or treated with 1 μg/mL doxycycline (Dox) to induce H-RasV12 expression. Representative phase-contrast images are shown for E1A- or E1A and inducible H-RasV12 (iRas)-transduced MEFs after treatment with ± Dox for 3 days. Bar, 50 μm (A). Cell growth of ElA-immortalized MEFs transduced with inducible H-RasV12 expression vector or empty vector. Cells were cultured with ± Dox, and cell number determined at the indicated time points (B). (C) Cell lysates from serum-starved MEF cells were analyzed by Western blotting as indicated after 3 days with ± Dox treatments. Anti-vinculin is used as a loading control. (D) Flow cytometric analysis for Annexin V (FITC) and DAPI after 3 days treatment with ± Dox for MEF-iRas cells. Representative dot plots (left-hand panel) from one of three independent experiments are shown. Data presented in the right-hand panel represent mean ± SD of the percentage cells undergoing apoptotic cell death (Ann-V+/DAPI+) or necrotic cell death (Ann-V−/DAPI+) for indicated treatments. (E) MEF-iRas cells were either untreated or treated with 1 μg/mL Dox to induce H-RasV12 expression. Cells were cultured under serum-starved conditions overnight, and replaced by growth medium that included 100 μM BrdU and incubated 5 hrs. Representative images (left-hand panel) and the mean ± SD (right-hand panel) of BrdU-positive staining (green) indicated by red arrows are shown. DAPI (blue) was used for counterstaining. Bar, 50 μm. (F) miRNA heatmap showing dysregulated miRNAs in MEF-iRas cells treated with ± Dox.
In an effort to identify miRNAs associated with acute H-RasV12 expression in MEF-iRas cells, we next performed genome-wide miRNA expression profiling on vehicle- and Dox-treated MEF-iRas cells at 48 hrs upon induction. Two independent samples were analyzed with Dox-treated MEF-iRas cells against two control samples without Dox treatment. In this analysis, 44 miRNAs were aberrantly expressed following H-RasV12 induction with 31 miRNAs up-regulated and 13 down-regulated using a minimum two-fold cutoff in MEF-iRas cells without Dox-treatment compared to those with Dox-treatment (Fig. 1F and Supplementary Table 1).
miR-130a stimulates cell proliferation
Considering abrupt H-RasV12 expression in E1A-immortalized MEF cells led to a decreased cell survival (Fig. 1A and B), we wondered whether these dysregulated miRNAs, either positively or negatively, involved in the process of decreased cell survival. In our preliminary screening, we selected miRNAs demonstrating major fold changes and transduced a series of miRNA precursors under the control of Dox-responsive promoter, into E1A/H-RasV12 transformed MEF cells by retroviral transduction as described previously (18,21) in order to assess their effects on cell survival. In this screen, only miR-130a significantly promoted cell growth. Having confirmed that miR-130a with H-RasV12 induction in MEF-iRas cells was down-regulated by approximately 65% relative to controls by real-time reverse transcriptase-PCR (RTPCR) (Fig. 2A), we next purposely enforced miR-130a expression in these cells by Dox treatment. While, Dox treatment itself had no effect on expression levels of endogenous miR-130a in cells transduced with the control vector, miR-130a levels increased almost 2-fold with Dox treatment in cells transduced with inducible miR-130a expression vector (Fig. 2B). Consistent with the ability of miR-130a to promote cell response, miR-130a expression significantly increased cell number over 6 days relative to vehicle-treated cells. As expected, Dox-treatment itself did not display effect on cell survival in the transformed MEFs infected with control vector (Fig. 2C). Likewise, miR-130a overexpression significantly increased cell proliferation as assessed by BrdU incorporation assay (Fig. 2D) with no evident effect on cell apoptosis as determined by Annexin V FACS assay (Fig. 2E).
Figure 2. miR-130a promotes cell proliferation.
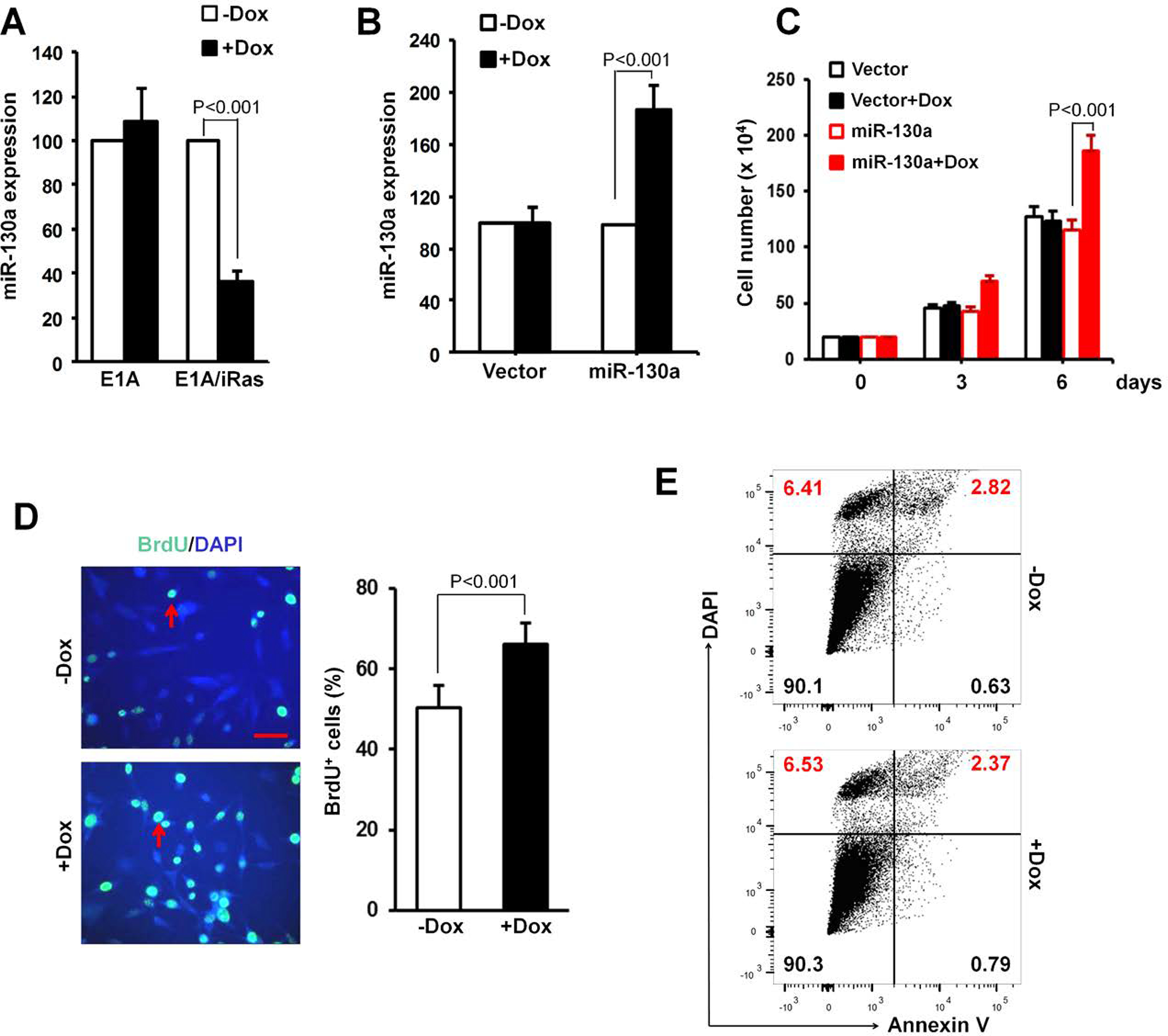
(A) Expression levels of miR-130a in E1A- or E1A/iRas-transduced MEFs cultured with ± Dox for 2 days were determined using TaqMan miRNA assays. (B-E) E1A/H-RasV12 transformed MEFs were infected with miR-130a expression vector or control vector. miR-130a expression levels were detected by TaqMan miRNA assay after the MEFs were treated with ± Dox for 2 days (B). Cell number (C), BrdU incorporation (green) indicated by red arrows after 5 hrs uptake of BrdU (D), and apoptosis by Annexin V assay (E), were determined. DAPI was used for counterstaining. Bar, 50 μm.
miR-130a attenuates the expression of the tumor suppressor, PTEN
To identify potential direct target genes affected by miR-130a overexpression, we first performed miRNA target-prediction analyses using several miRNA target-prediction software, including TargetScan (http://www.targetscan.org/) and MIRDB (http://mirdb.org/miRDB/). Among hundreds of miR-130a targets, we first focused on two tumor suppressors, PTEN and TSC1, as both play important roles in suppressing cell and tumor growth (14,26). Indeed, miR-130a expression led to a significant decrease in endogenous levels of PTEN protein, while having no effect on TSC1 protein levels (Fig. 3A). Likewise, miR-130a expression reduced PTEN mRNA levels as determined by qRT-PCR, suggesting miR-130a targets PTEN mRNA for degradation (Fig. 3B). Consistent with this interpretation, anti-mir-130a transfection increased PTEN protein levels (Fig. 3C).
Figure 3. PTEN is a direct target of miR-130a.
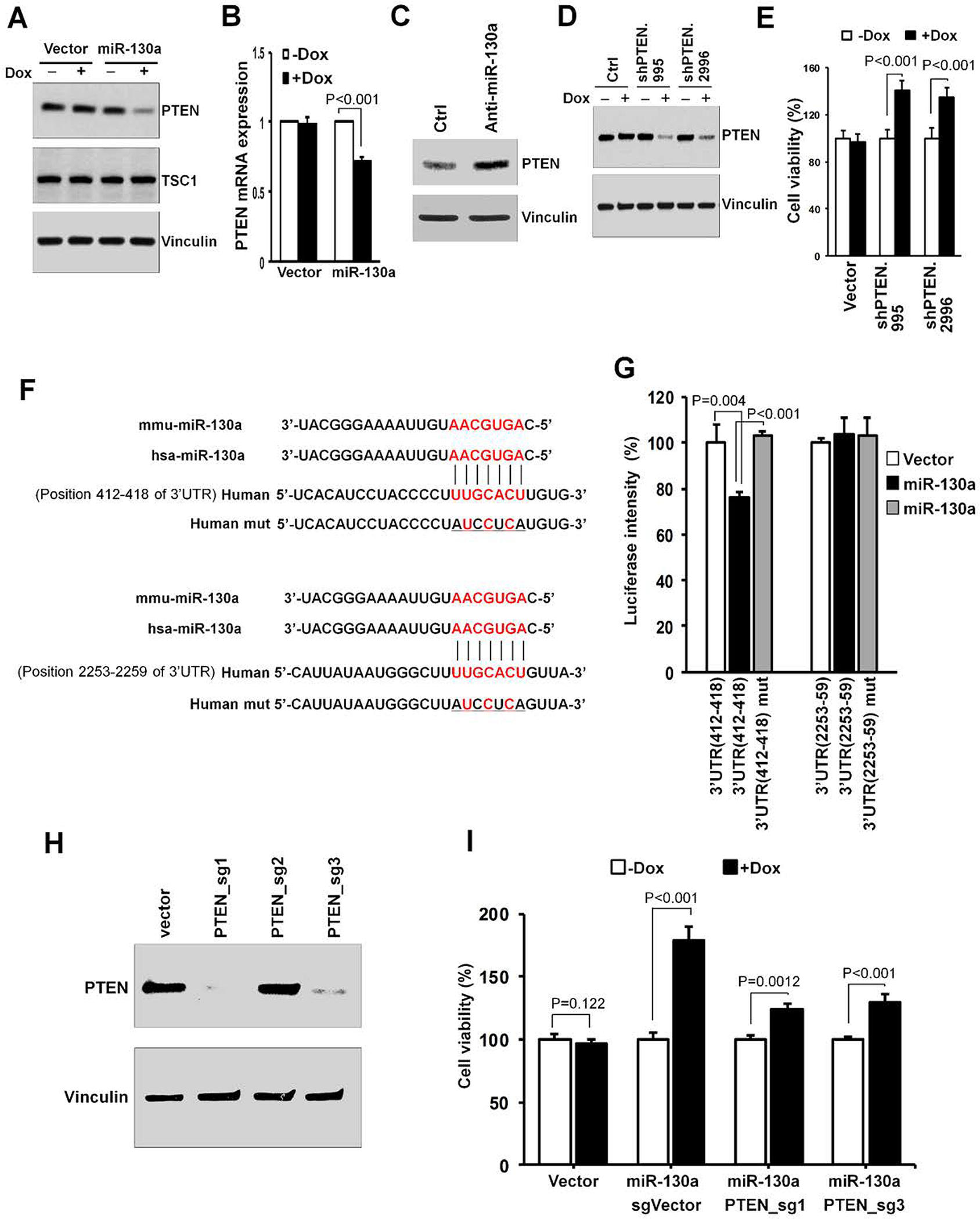
(A, B) E1A/H-RasV12 transformed MEF cells transduced with vector control or Dox-inducible mouse miR-130a expression vector were treated with ± Dox (1 μg/mL) for 2 days. Cells were lysed for immunoblotting using the antibodies as indicated (A). Mouse PTEN mRNA levels were determined by qRT-PCR (B). (C) Transformed MEF cells were transfected with miR-130a inhibitor and the control, and PTEN levels determined by Western blotting. (D) Transformed MEFs were cultured for 2 days with ± Dox, as indicated. Cell lysates were analyzed by Western blotting for indicated proteins. (E) Transformed MEFs (5 × 104) were seeded into six-well plates, and the percent (normalized to cell number under -Dox condition) of the control cell number determined after 5 days in DMEM + 10% FBS culture medium under the indicated conditions. (F) Schematic of predicted miR-130a binding sites in the PTEN 3’-UTR. (G) Luciferase reporter constructs containing wild-type or mutated PTEN 3’-UTR were co-transfected with miR-130a or control plasmids, as indicated, into 293T cells. 48 hrs later cells were lysed for luciferase assay. (H) sgRNA efficiency was assessed by immunoblotting using the indicated antibodies. (I) Transformed MEFs with inducible miR-130a expression were transduced with PTEN sgRNAs or control together with Cas9 construct to produce PTEN-null or -intact cells, respectively. The cells were incubated in growth medium with ± Dox. After 5 days, the cells were counted, and the mean ± SD of relative growth (normalized to cell number under - Dox condition) was determined.
Next, we examined whether the down-regulation of PTEN by miR-130a in transformed MEF cells was responsible for increased cell growth and/or survival. To this end, we expressed PTEN shRNAs from an inducible tetracycline-responsive element promoter (20) to mimic miR-130a-mediated PTEN down-regulation in our transformed MEFs. Under these conditions, two of four PTEN shRNAs reduced PTEN protein levels in the transformed MEF cells (Fig. 3D) while simultaneously enhancing cell survival (Fig. 3E) and proliferation as assessed by BrdU incorporation (Supplementary Fig. S4A) without affecting apoptosis (Supplementary Fig. S4B). Taken together, the results support a model wherein reduction of PTEN levels by miR-130a exerts positive effects on cell growth.
To further validate PTEN regulation by miR-130a, we next sought to determine whether the inhibition of PTEN expression by miR-130a is due to bona fide miRNA targeting or alternatively, a result of indirect protein-mediated crosstalk. There are 3 putative miR-130a binding sites for both the human and mouse PTEN 3’-untranslated region (3’-UTR) wherein two binding sites on the PTEN 3’-UTR are also highly conserved in other species (Fig. 3F). As such, we generated luciferase reporter constructs harboring 2 various fragments, including respectively conserved 3’-UTR binding sites of human PTEN transcripts. Using luciferase reporter assays, overexpression of miR-130a resulted in a modest, but significant, decrease in luciferase expression with the PTEN 412–418 3’-UTR site, but not 2253–2259 binding site. Furthermore, mutations affecting the 412–418 site fully abrogated miR-130a-mediated repression (Fig. 3G). These findings suggest that direct binding between miR-130a and PTEN 3’-UTR, likely through binding site 412–418, is required for PTEN repression by miR-130a.
To further determine whether the miR-130a effect on cell proliferation is dependent on PTEN status, we disrupted PTEN by CRISPR-Cas9 technology in miR-130a-expressing cells. Two of three sgRNAs targeting mouse PTEN significantly reduced PTEN protein expression after 2 weeks of selection (Fig. 3H, and Supplementary Fig. S5). We then selected each cell line from a single cell colony having confirmed PTEN deletion by Western blotting. As predicted, under PTEN-deleted conditions, the ability of miR-130a to enhance cell growth was repressed, though not completely (Fig. 3I). As such, these results suggest that cell growth promotion by miR-130a expression is largely dependent on PTEN status.
miR-130a overexpression leads to increased tumor growth in vivo
Given the effects exerted by miR-130a on transformed MEFs, we sought to determine its functional effects in additional tumor cell lines. In this regards, we elected to use breast cancer cells as a tumor growth model to examine miR1–30a function in vivo as miR-130a and miR-130b, a close family member of miR-130a with a seed sequence identical to that of miR-130a, display a higher frequency of amplification in breast cancer xenografts (http://www.cbioportal.org) (Supplementary Fig. S6A and S6B). Consistent with our model, mouse or human miR-130a overexpression significantly down-regulated endogenous PTEN levels in the 4T1 mouse breast cancer cells as well as the MDA-MB-231 human breast cancer cells, respectively (Fig. 4A). In turn, miR-130a expression increased Akt phosphorylation without affecting total Akt levels (Fig. 4A). Finally, overexpression of either mouse miR-130a in 4T1 cells or human miR-130a in MDA-MB-231 cells increased cell growth during in vitro culture (Fig. 4B).
Figure 4. miR-130a expression causes increased tumor growth in vivo.
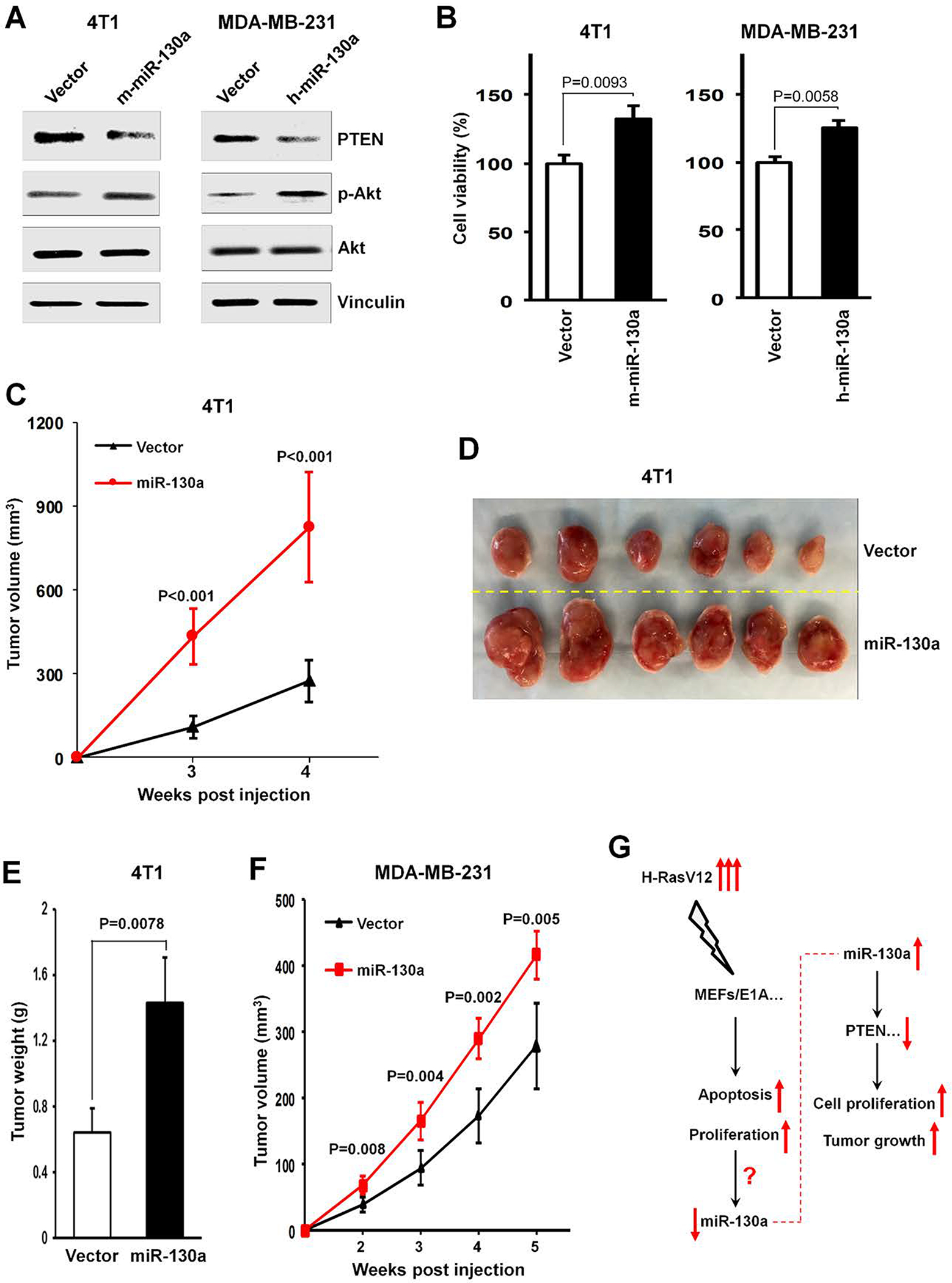
(A, B) 4T1 or MDAMB-231 cells were retrovirally transduced with mouse or human miR-130a expression vectors versus control vector as indicated. Cell lysates were prepared for Western blotting using the indicated antibodies (A). Cell numbers were determined after 5 days of culture. The percent of control cell growth is shown (B). (C) Tumor growth by 5 × 105 4T1 cells transduced with mouse miR-130a or control vectors. (D, E) Tumor images (D) and tumor weight (E) from mice following subcutaneous injection of 5 × 105 4T1 cells transduced with mouse miR-130a or control vectors at 4 weeks after implantation. (F) Tumor growth by 5 × 105 subcutaneously injected MDA-MB-231 cells transduced with human miR-130a or control vectors. (G) A schematic outlines the effect of H-RasV12 transient expression in ElA-immorlatalized MEFs (MEFs/E1A) on mediating increased apoptosis and increased proliferation, accompanying by down-regulated miR-130a expression. While oncogenic stress induces a decrease in miR-130a expression, miR-130a triggers increased cell proliferation in vitro and enhanced tumor growth in vivo, by primarily targeting the tumor suppressor, PTEN.
To next define the impact of miR-130a on tumor behavior in vivo, we subcutaneously implanted miR-130a- or control vector-transduced 4T1 or MDA-MB-231, respectively, into the flanks of BALB/c or athymic nude mice and monitored tumor growth. Under these conditions, mice bearing miR-130a-transduced 4T1 cells displayed significantly increased tumor growth rate relative to vector-transduced control cells as reflected in a 3-fold increase in tumor volume (Fig. 4C, D) and a 2.2-fold increase in tumor weight (Fig. 4E) at 4 weeks after tumor cell injection. Similarly, mice bearing human miR-130a-transduced MDA-MB-231 cells exhibited an increased tumor growth rate relative to the control population (Fig. 4F). Taken together, these data suggest that enforced expression of miR-130a can likewise increase tumor growth in the in vivo setting (Fig. 4G).
miR-130a and miR-130b inversely correlate with PTEN expression
To identify potential correlations between miR-130a and its target gene, PTEN, using TCGA datasets. Invasive breast (n=509), colon ADC (n=224), and lung ADC (n=487) carcinoma samples with both miR-130a/miR-130b and PTEN expression data were selected for analysis. While, miR-130a did not show anti-correlation with PTEN in breast carcinoma (Fig. 5A), miR-130b displayed significant anti-correlation to PTEN in breast cancer (Fig. 5B). Extending these analyses to breast cancer subtypes, ER-positive (Supplementary Fig. S7A), PR-positive (Supplementary Fig. S7B) and HER2-positive (Supplementary Fig. S7C) breast cancers did not display an inverse correlation between miR-130a and PTEN. By contrast, in ER-negative (Supplementary Fig. S7D), PR-negative (Supplementary Fig. S7E) and HER2-negative (Supplementary Fig. S7F) breast cancers, miR-130a exhibited a tendency to correlate negatively with PTEN, particularly, with regard to PR-negative breast cancer. To determine whether miR-130a, miR-130b, and PTEN expression levels are associated with PR expression status, we next compared their expression in PR-positive and -negative breast cancer. Interestingly, miR-130a and miR-130b were significantly upregulated in PR-negative breast cancers (Supplementary Fig. S8A and S8B) while PTEN downregulated significantly in PR-negative breast cancer (Supplementary Fig. S8C). miR-130a and miR-130b also displayed a significant inverse correlation to PTEN levels in either lung ADC (Fig. 5C and 5D) or colon ADC (Fig. 5E and 5F), respectively. Taken together, these results indicate that the miR-130 family inversely correlates with PTEN expression in multiple types of human cancer.
Figure 5. Correlation analyses between miR-130a/miR-130b and PTEN in human cancer samples.
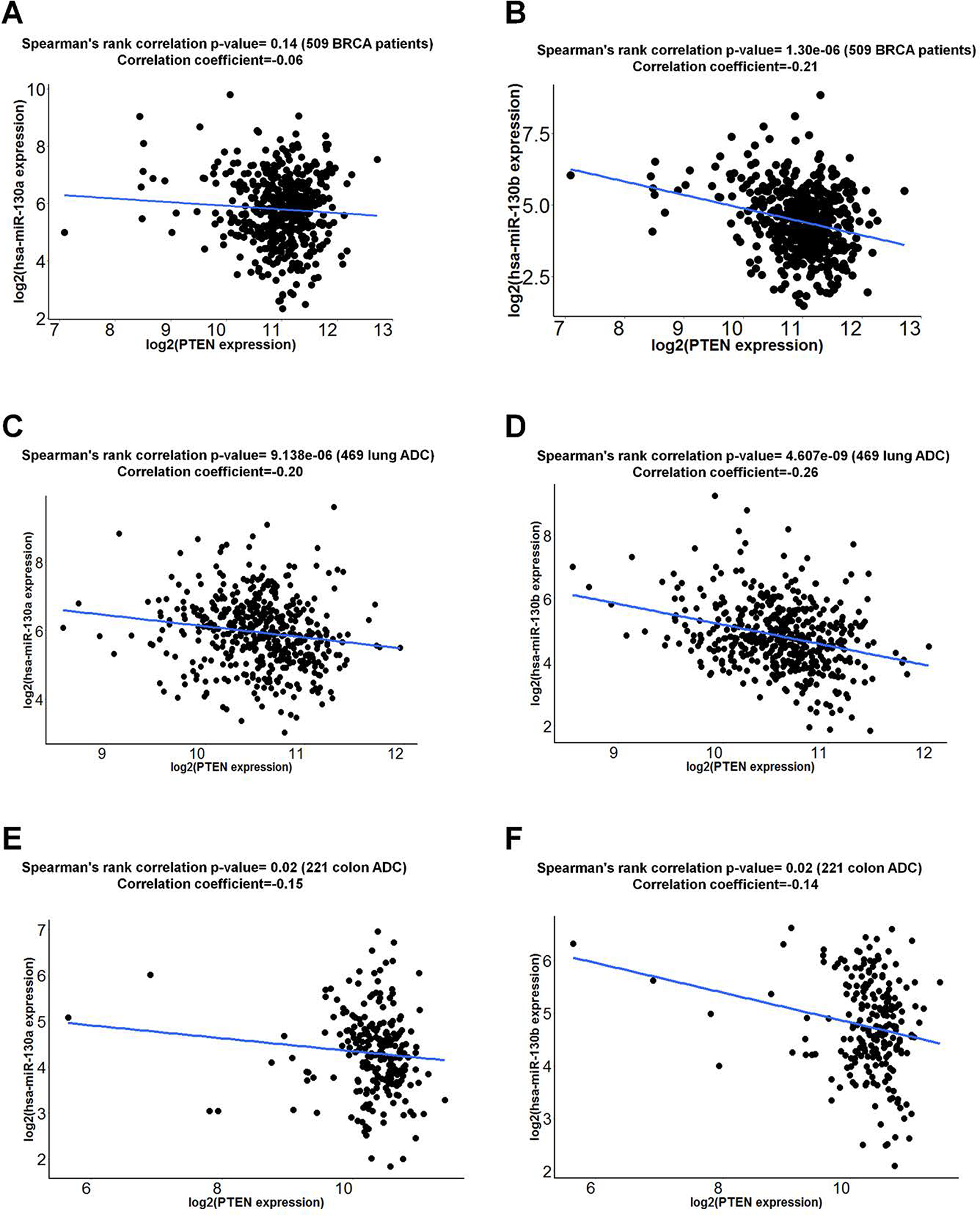
miR-130a or miR-130b expression from TCGA miRNA-seq data and PTEN expression from RNA-seq data were examined for correlation analyses. Correlations between miR-130a/miR-130b and PTEN in BRCA (A and B), lung ADC (C and D) or colon ADC (E and F) were calculated by Spearman rank correlation test.
Discussion
In this report, we find that acute H-RasV12 expression in E1A-immortalized MEF cells leads to an aberrant growth response that coupled increased apoptotic cell death with increased proliferative response. Through integrated analyses of miRNA profile, we identified 44 miRNAs associated in the process of H-RasV12 transient expression in E1A-immortalized MEF cells. After screening these miRNAs for effects on cell growth/survival we find that miR-130a stimulates cell proliferation and in vivo tumor growth in a manner that correlates with its targeting of the tumor suppressor PTEN. Based on this activity, we propose that miR-130 family members can function as pro-oncogenic factors.
Our initial findings contrasted with those reported by Elgendy et al where transient expression of H-RasV12 induced an autophagic cell death program (13). Alternatively, we find that the transient H-RasV12 expression in E1A-immortalized MEFs co-induces cell apoptosis and proliferation. While the previous studies by Elgendy et al reported that transient expression H-RasV12 in HOSE cells and MCF7 cells triggered autophagic cell death [13], we were unable to elicit a similar response in either HOSE or MCF7 cells using Dox-inducible system. It is currently unclear whether the use of different inducible systems or cell culture conditions contributed to these disparate results.
The abrupt expression of H-RasV12 in E1A-immortalized MEF cells was accompanied by down-regulated miR-130a expression. Paradoxically, we found that increased expression of miR-130a stimulated cell proliferation without affecting cell apoptosis. This apparent functional “clash” between the up-regulated cell proliferation and the down-regulated miR-130a during H-RasV12 expression indicates that increased proliferative responses occur independently of the H-RasV12-miR-130a axis in this more complex scenario. We speculate that acute H-RasV12 expression might provoke a negative feedback loop that down-regulates miR-130a expression, perhaps contributing to the less than optimal growth responses that characterized this process.
We next examined miR-130a-mediated posttranscriptional regulation of PTEN and its role in promoting tumorigenesis. A large fraction of human cancer exhibit loss of only one PTEN allele (27), and a reduced expression of this tumor suppressor has been reported to correlate with poor clinical outcomes in breast, ovarian and liver cancers (28). Indeed, mouse models have demonstrated that heterozygous PTEN loss, or even subtler reduction in its expression levels can lead to increased rates of spontaneous tumorigenesis (15). Given the importance of miRNA in tumorigenesis (3) and the fact that PTEN is a direct target of miR-130a, it would be of interest to evaluate whether miR-130a might promote spontaneous tumorigenesis in transgenic mice, especially in concert with the heterozygous inactivation of PTEN.
To date, the function of miR-130 family members in promoting cell proliferation and tumorigenesis has been controversial. While some reports have concluded that miR-130 family miRNAs can promote cell proliferation and are upregulated in several types of cancer (29–39), other studies concluded that miR-130 family members inhibit cell proliferation and are downregulated in cancer (40–45), highlighting the complexity of miR-130 function in tumorigenesis. Here we find that enforced miR-130a expression promotes cell proliferation in either MEFs or breast cancer cell lines while stimulating tumor growth in vivo through a PTEN-regulated process. Although we did not observe a significant inverse correlation between miR-130a and PTEN levels in breast cancer overall, subgroup analyses uncovered a link between miR-130a and PTEN levels in PR-negative breast cancer. Furthermore, miR-130b, a close family member with a seed sequence identical to that of miR-130a, also displays an inverse correlation to PTEN in breast cancer. These findings could also be extended to lung or colon cancer where inverse relationships between miR-130a/miR-130b and PTEN levels could also be established. In this regard, we note that several recent findings likewise confirm that PTEN is targeted by the miR-130 family (36,46–51). Taken together, our results support a model wherein the miR-130 family plays a pro-oncogenic function, in part by targeting PTEN. Targeting miR-130a as well as other family members could provide a novel means for interfering tumor progression.
Supplementary Material
Acknowledgement
We are deeply indebted to Dr. Stephen J Weiss at University of Michigan for him to have supervised the project, designed the study and written the manuscript. We thank Drs. Xiao-Yan Li, Hemant Kumar Bid, Lingxin Zhu, Xinying Jia at University of Michigan and Drs. Yong Peng, Taewan Kim, Hui-Lung Sun, Esmerina Tili at The Ohio State University for discussion and assistance on different methods used in this study. We thank Dr. Julian Downward at The Francis Crick Institute for providing us the HOSE cells. We appreciate Dr. Stefano Volinia at The Ohio State University for the analyses of microRNA profile data.
Financial Support: This research was supported by NIH grant R35 CA197706 to C.M. Croce, and National Natural Science Foundation of China (81672305) to R. Cui.
Footnotes
Conflict of interest. The authors declare no conflict of interest.
References
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–97 [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 2009;10:704–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov 2014;13:622–38 [DOI] [PubMed] [Google Scholar]
- 5.Mizushima N Autophagy: process and function. Genes Dev 2007;21:2861–73 [DOI] [PubMed] [Google Scholar]
- 6.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009;186:773–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008;9:1004–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008;451:1069–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol 2009;10:461–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabinowitz JD, White E. Autophagy and metabolism. Science 2010;330:1344–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013;368:651–62 [DOI] [PubMed] [Google Scholar]
- 12.Doria A, Gatto M, Punzi L. Autophagy in human health and disease. N Engl J Med 2013;368:1845. [DOI] [PubMed] [Google Scholar]
- 13.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell 2011;42:23–35 [DOI] [PubMed] [Google Scholar]
- 14.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012;13:283–96 [DOI] [PubMed] [Google Scholar]
- 15.Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature 2011;476:163–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011;25:1510–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei H, Wang C, Croce CM, Guan JL. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev 2014;28:1204–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei H, Wang X, Gan B, Urvalek AM, Melkoumian ZK, Guan JL, et al. Sumoylation delimits KLF8 transcriptional activity associated with the cell cycle regulation. J Biol Chem 2006;281:16664–71 [DOI] [PubMed] [Google Scholar]
- 19.Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 2013;5:1704–13 [DOI] [PubMed] [Google Scholar]
- 20.Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol 2011;29:79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei H, Gan B, Wu X, Guan JL. Inactivation of FIP200 leads to inflammatory skin disorder, but not tumorigenesis, in conditional knock-out mouse models. J Biol Chem 2009;284:6004–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014;343:80–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol 2014;32:941–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007;130:223–33 [DOI] [PubMed] [Google Scholar]
- 25.Deng Q, Li Y, Tedesco D, Liao R, Fuhrmann G, Sun P. The ability of E1A to rescue ras-induced premature senescence and confer transformation relies on inactivation of both p300/CBP and Rb family proteins. Cancer Res 2005;65:8298–307 [DOI] [PubMed] [Google Scholar]
- 26.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell 2008;133:403–14 [DOI] [PubMed] [Google Scholar]
- 28.Mester J, Eng C. When Overgrowth Bumps Into Cancer: The PTEN-Opathies. Am J Med Genet C Semin Med Genet 2013 [DOI] [PubMed] [Google Scholar]
- 29.Shao M, Geng Y, Lu P, Xi Y, Wei S, Wang L, et al. miR-4295 promotes cell proliferation and invasion in anaplastic thyroid carcinoma via CDKN1A. Biochem Biophys Res Commun 2015;464:1309–13 [DOI] [PubMed] [Google Scholar]
- 30.Sun L, Wang Q, Gao X, Shi D, Mi S, Han Q. MicroRNA-454 functions as an oncogene by regulating PTEN in uveal melanoma. FEBS Lett 2015;589:2791–6 [DOI] [PubMed] [Google Scholar]
- 31.Ma F, Zhang J, Zhong L, Wang L, Liu Y, Wang Y, et al. Upregulated microRNA-301a in breast cancer promotes tumor metastasis by targeting PTEN and activating Wnt/betacatenin signaling. Gene 2014;535:191–7 [DOI] [PubMed] [Google Scholar]
- 32.Lee SH, Jung YD, Choi YS, Lee YM. Targeting of RUNX3 by miR-130a and miR-495 cooperatively increases cell proliferation and tumor angiogenesis in gastric cancer cells. Oncotarget 2015;6:33269–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang M, Li C, Yu B, Su L, Li J, Ju J, et al. Overexpressed miR-301a promotes cell proliferation and invasion by targeting RUNX3 in gastric cancer. J Gastroenterol 2013;48:1023–33 [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, Chen Z, Zhu C. Endogenous nitric oxide mediates alleviation of cadmium toxicity induced by calcium in rice seedlings. J Environ Sci (China) 2012;24:940–8 [DOI] [PubMed] [Google Scholar]
- 35.Colangelo T, Fucci A, Votino C, Sabatino L, Pancione M, Laudanna C, et al. MicroRNA-130b promotes tumor development and is associated with poor prognosis in colorectal cancer. Neoplasia 2013;15:1218–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu T, Cao R, Li S, Fu M, Ren L, Chen W, et al. MiR-130b plays an oncogenic role by repressing PTEN expression in esophageal squamous cell carcinoma cells. BMC Cancer 2015;15:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma M, Karimiani EG, Byers RJ, Rehman S, Westerhoff HV, Day PJ. Mathematical modelling of miRNA mediated BCR.ABL protein regulation in chronic myeloid leukaemia vis-a-vis therapeutic strategies. Integr Biol (Camb) 2013;5:543–54 [DOI] [PubMed] [Google Scholar]
- 38.Zhu X, Zhao H, Lin Z, Zhang G. Functional studies of miR-130a on the inhibitory pathways of apoptosis in patients with chronic myeloid leukemia. Cancer Gene Ther 2015;22:573–80 [DOI] [PubMed] [Google Scholar]
- 39.Yang F, Miao L, Mei Y, Wu M. Retinoic acid-induced HOXA5 expression is co-regulated by HuR and miR-130a. Cell Signal 2013;25:1476–85 [DOI] [PubMed] [Google Scholar]
- 40.Fang B, Zhu J, Wang Y, Geng F, Li G. MiR-454 inhibited cell proliferation of human glioblastoma cells by suppressing PDK1 expression. Biomed Pharmacother 2015;75:148–52 [DOI] [PubMed] [Google Scholar]
- 41.Pan Y, Wang R, Zhang F, Chen Y, Lv Q, Long G, et al. MicroRNA-130a inhibits cell proliferation, invasion and migration in human breast cancer by targeting the RAB5A. Int J Clin Exp Pathol 2015;8:384–93 [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao G, Zhang JG, Shi Y, Qin Q, Liu Y, Wang B, et al. MiR-130b is a prognostic marker and inhibits cell proliferation and invasion in pancreatic cancer through targeting STAT3. PLoS One 2013;8:e73803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niu G, Li B, Sun J, Sun L. miR-454 is down-regulated in osteosarcomas and suppresses cell proliferation and invasion by directly targeting c-Met. Cell Prolif 2015;48:348–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu C, Fu H, Gao L, Wang L, Wang W, Li J, et al. BCR-ABL/GATA1/miR-138 mini circuitry contributes to the leukemogenesis of chronic myeloid leukemia. Oncogene 2014;33:44–54 [DOI] [PubMed] [Google Scholar]
- 45.Fei J, Li Y, Zhu X, Luo X. miR-181a post-transcriptionally downregulates oncogenic RalA and contributes to growth inhibition and apoptosis in chronic myelogenous leukemia (CML). PLoS One 2012;7:e32834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu C, Wang X, Ha T, Hu Y, Liu L, Zhang X, et al. Attenuation of cardiac dysfunction and remodeling of myocardial infarction by microRNA-130a are mediated by suppression of PTEN and activation of PI3K dependent signaling. J Mol Cell Cardiol 2015;89:87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J, Yan D, Wu W, Zhu J, Ye W, Shu Q. MicroRNA-130a promotes the metastasis and epithelial-mesenchymal transition of osteosarcoma by targeting PTEN. Oncol Rep 2016;35:3285–92 [DOI] [PubMed] [Google Scholar]
- 48.Song CL, Liu B, Shi YF, Liu N, Yan YY, Zhang JC, et al. MicroRNA-130a alleviates human coronary artery endothelial cell injury and inflammatory responses by targeting PTEN via activating PI3K/Akt/eNOS signaling pathway. Oncotarget 2016;7:71922–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egawa H, Jingushi K, Hirono T, Ueda Y, Kitae K, Nakata W, et al. The miR-130 family promotes cell migration and invasion in bladder cancer through FAK and Akt phosphorylation by regulating PTEN. Sci Rep 2016;6:20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang RM, Xu JF, Fang F, Yang H, Yang LY. MicroRNA-130b promotes proliferation and EMT-induced metastasis via PTEN/p-AKT/HIF-1alpha signaling. Tumour Biol 2016;37:10609–19 [DOI] [PubMed] [Google Scholar]
- 51.Gong XC, Xu YQ, Jiang Y, Guan H, Liu HL. Onco-microRNA miR-130b promoting cell growth in children APL by targeting PTEN. Asian Pac J Trop Med 2016;9:265–8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.