

Abstract
Acetaminophen (APAP) is the most common antipyretic analgesic worldwide. However, APAP overdose causes severe liver injury, especially centrilobular necrosis, in humans and experimental animals. At therapeutic dosage, APAP is mainly metabolized by sulfation and glucuronidation, and partly by cytochrome P450–mediated oxidation. However, APAP overdose results in production of excess reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI), by cytochromes P450; NAPQI overwhelms the level of glutathione (GSH), which could otherwise detoxify it. NAPQI binds covalently to proteins, leading to cell death. A number of studies aimed at the prevention and treatment of APAP-induced toxicity are underway. Rats are more resistant than mice to APAP hepatotoxicity, and thus mouse models are mainly used. In the present study, we compared the toxic responses induced by APAP overdose in the liver of ICR mice obtained from three different sources and evaluated the usability of the Korl:ICR stock established by the National Institute of Food and Drug Safety Evaluation in Korea. Administration of APAP (300 mg/kg) by intraperitoneal injection into male ICR mice enhanced CYP2E1 protein expression and depleted hepatic GSH level 2 h after treatment accompanied with significantly increased level of hepatic malondialdehyde, a product of lipid peroxidation. Regardless of the source of the mice, hepatotoxicity, as evidenced by activity of serum alanine aminotransferase, increased from 8 h and peaked at 24 h after APAP treatment. In summary, hepatotoxicity was induced after the onset of oxidative stress by overdose of APAP, and the response was the same over time among mice of different origins.
Keywords: Acetaminophen, Hepatotoxicity, GSH, ICR mouse
Introduction
Acetaminophen (APAP) is the most popular analgesic and antipyretic. Generally, the recommended dose is 4 g/day for adults and 60–75 mg/kg/day for children. However, overuse of APAP can cause acute liver failure followed by hepatocellular necrosis [1–4]. Clinically, the most common cause of APAP addiction are suicide attempts (47.8%) in adolescents and accidental addiction (42.2%) in infants [5]. The only medication in cases of APAP overdose is N-acetyl cysteine, a well-known antioxidant. Thus, the development of new drugs superior to NAC is urgently required. Our PubMed search using the MeSH terms ‘liver injury’, ‘hepatotoxicity’, and ‘acetaminophen’, suggested that intensive research has been undertaken to understand the mechanism of APAP-induced acute hepatic injury and develop novel drug candidates.
The cause of hepatotoxicity induced by over-dosage of APAP is the excessive generation of toxic substances mediated by xenobiotic metabolism, also known as drug metabolism, in the liver [6, 7]. At the suggested dosage, APAP is mainly metabolized by sulfation and glucuronidation, and partly by cytochrome P450–mediated oxidation. However, APAP overdose results in production of excess reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI), by cytochromes P450. NAPQI overwhelms the level of the endogenous tripeptide glutathione (GSH), which could otherwise detoxify it. NAPQI binds covalently to proteins, leading to cell death [8–11]. Interestingly, the toxic effects of acetaminophen differ in different mammalian species [12–16]. For example, mice are highly vulnerable to liver injury induced by APAP in comparison with rats [16]. Although the overall metabolism of APAP is similar in both species, mice exhibit severe mitochondrial dysfunction accompanied by high oxidative stress, similar to what occurs in humans [17]. Therefore, it is reasonable to use mice in preclinical studies of APAP-induced hepatotoxicity.
Outbred stocks of experimental animals are commonly used to study genetics, oncology, toxicology, and pharmacology in academy and industry. ICR mice, originated from albino non-inbred mice in Switzerland, are a vigorously outbred stock [18]. The National Institute of Food and Drug Safety Evaluation (NIFDS) in Korea has established the “Korl:ICR” stock and it was used for the last 50 years in the NIFDS. Recently, it has been proved that Korl:ICR mice are genetically independent from commercially available ICR stocks from Japan and USA; however, no biological differences have been observed so far [19]. The purpose of the present study was to characterize the Korl:ICR stock in terms of hepatotoxicity and determine its utility in evaluating the safety of preclinical drug candidates. Because APAP-induced hepatotoxicity is involves excessive oxidative stress following GSH depletion [20], we examined the level of GSH and lipid peroxidation in the liver as well as the parameters of hepatotoxicity including the activity of alanine aminotransferase (ALT) in serum and histopathological analysis.
Materials and method
Animals and treatments
Male ICR mice were obtained from three different sources. Korl:ICR was supplied by the Department of Laboratory and Animal Resources at the NIFDS (Cheongju, Korea). The other two groups of ICR stocks were purchased from several suppliers located in the United States (A: ICR) and Japan (B: ICR). The use of animals complied with established guidelines and was approved by the Institutional Animal Care and Use Committee of Pusan National University (PNU-2018-1992). The basic conditions such as facility environment and diet were as described previously [21]. Mice were acclimated to room temperature (22 ± 2 °C) and humidity (55 ± 5%) with a 12-h light–dark cycle for 1 week prior to use in the animal facility of the university. They were provided free access to water and standard irradiated chow diet (Samtako Inc., Osan, Korea) consisting of moisture (12.5%), protein (25.43%), fat (6.06%), fiber (3.9%), ash (5.31%), calcium (1.14%), and phosphorus (0.99%). Mice received a single intraperitoneal injection of APAP (300 mg/kg) or vehicle, and samples were collected at 2, 8, 24, and 48 h after APAP administration.
Examination of APAP-induced hepatotoxicity
Blood samples were collected at 2, 8, and 24 h after injection and immediately transferred to BD Microtainer Blood Collection Tubes (BD Life Sciences, Franklin Lakes, NJ, USA). They were centrifuged at 3000×g for 15 min to separate the serum, and the activity of serum ALT was examined by the method of Reitman and Franke [22]. For histological evaluation, liver samples were fixed in 10% phosphate buffered formalin overnight and embedded in paraffin. The tissue was cut into 4–mm-thick sections and stained with hematoxylin and eosin: the nucleus was stained blue with Mayer’s hematoxylin, while the cytoplasm was stained red with eosin (Sigma-Aldrich). Histopathological analysis was performed using an inverted microscope (Olympus, Tokyo, Japan).
Measurement of GSH in liver tissue
Total GSH was determined as reported previously [21]. The liver was homogenized with 4 volumes of ice-cold 1 M perchloric acid. The denatured protein was removed by centrifugation at 10,000×g for 10 min and the supernatant was analyzed for total GSH concentration using HPLC separation followed by fluorescence detection. After addition of 10 μL of 10% (w/v) tris (2-carboxyethyl) phosphine hydrochloride solution, the samples were incubated for 30 min. Next, 90 μL of trichloroacetic acid solution containing 1 mM EDTA was added, and the sample was centrifuged (13,000×g, 10 min). Fifty microliter of supernatant was then added to 10 μL of NaOH (1.55 M), 125 μL of 0.125 M borate buffer (pH 9.5) and 50 μL of 0.1% (w/v) 7-fluorobenzofurazan-4-sulfonic acid ammonium salt (0.125 M with 4 mM EDTA). Samples were incubated at 60 °C for 1 h and then injected into HPLC equipped with a fluorescence detector (excitation, 385 nm; emission, 515 nm) (FLD-3100; Thermo Scientific, Sunnyvale, CA, USA). Chromatographic separation was carried out using a Hector M-C18 column (3 μm × 4.6 mm × 150 mm) (Chiral Technology Korea, Daejeon, Korea).
Measurement of malondialdehyde (MDA) in liver tissue
Hepatic level of MDA was determined as reported previously [23]. The liver was homogenized with a triple volume of 1.15% KCl. The dispensed homogenate was mixed with 0.2% thiobarbituric acid in 2 M sodium acetate buffer containing 5% butylated hydroxytoluene. The mixture was incubated at 95 °C for 45 min. The supernatant after centrifugation of mixture was injected into the HPLC equipped with a fluorescence detector and a 5 μm Symmetry C18 reverse phase column (4.6 mm × 150 mm, Eka Chemicals, Bohus, Sweden). The mobile phase consisted of 35% methanol and 65% sodium phosphate buffer. The MDA–thiobarbituric acid complex was monitored by fluorescence detection with excitation at 515 nm and emission at 553 nm.
Western blotting
Western blotting was accomplished as reported previously [23]. Liver was lysed with ice-cold protein extract solution (iNtRON; Sungnam, Gyunggi, Korea), and the protein concentration was examined by the BCA reagent (Thermo Scientific, Sunnyvale, CA, USA). Equal amounts of protein were separated by SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). The membranes were incubated with TBS-T containing 5% milk and the primary antibodies against CYP2E1 (Detroit R&D, Detroit, MI, USA), GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, the membrane was incubated with the horseradish peroxidase-conjugated secondary antibodies. The antigen was detected using a Western Bright ECL HRP substrate kit (Advansta, Menlo Park, CA, USA).
Results
APAP-induced hepatic injury in mice of different sources increased ALT
ALT, a biochemical parameter of hepatotoxicity, increased from 8 h after APAP injection and peaked at 24 h in all mice regardless of their source (Fig. 1). However, it returned to the normal level at 48 h after treatment.
Fig. 1.

Effect of APAP on serum activity of ALT. Each value represents the mean ± standard deviation for six mice. *** Significantly different from the corresponding control (Student’s t-test, p < 0.001)
Histopathological changes in the liver of APAP-treated ICR mice
Based on the result of serum ALT activity, we accomplished histopathological analysis using liver tissues obtained from APAP-treated mice for 24 h and compared with vehicle-treated control mice. At 24 h after injection, the necrotic area of the central vein in the liver of APAP-treated mice was considerably larger than that in the liver of vehicle-injected mice (Fig. 2). There were no notable differences among the three sources of mice.
Fig. 2.
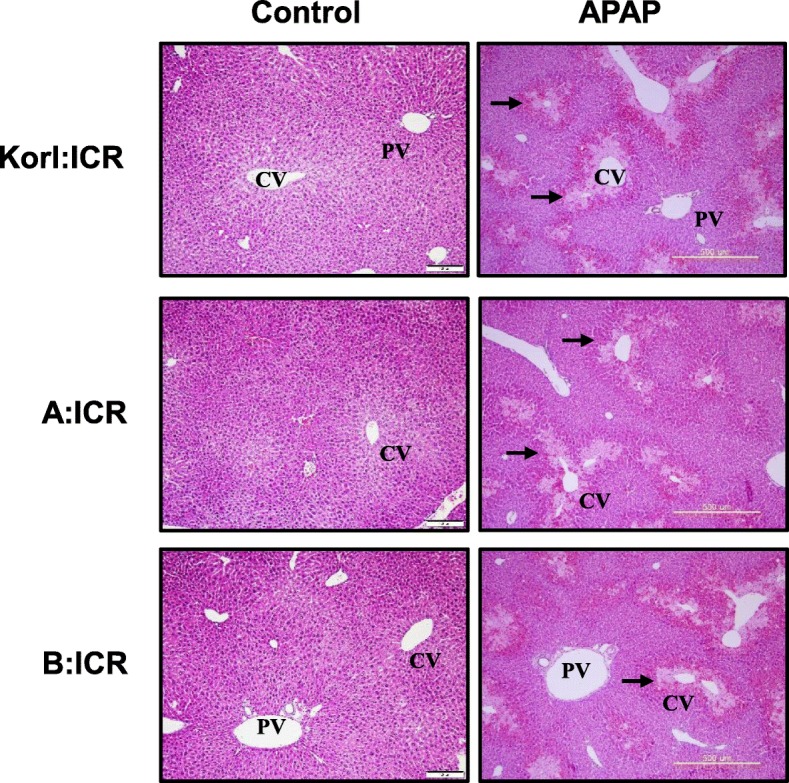
Histopathological changes in liver tissues at 24 h after APAP treatment. Liver sections from the control and APAP-treated Korl:ICR, A:ICR and B:ICR mice were stained with hematoxylin and eosin. CV, Central vein; PV, portal vein. Arrows indicate necrotic area. Original magnification, × 100
Dynamics of hepatic CYP2E1 protein expression and GSH levels after APAP treatment
Large doses of APAP is correlated with increased generation of a reactive metabolite, NAPQI, through enzyme reactions mediated by CYP enzymes [24]. Although various forms of CYP are implicated in the oxidative metabolism of APAP, it is generally accepted that CYP2E1 plays a principal role in production of NAPQI [25]. Subsequently, it is detoxified via conjugation with an endogenous tripeptide, GSH. After APAP injection, protein level of CYP2E1 increased from 2 h and it was highest at 8 h after treatment (Fig. 3). The hepatic GSH level rapidly decreased, reaching minimum at 2 h, but was restored at 8 h to control level (Fig. 4). Changes in hepatic GSH content induced by APAP treatment were similar in ICR mice from the three sources.
Fig. 3.

Effect of APAP on the expression levels of CYP2E1 protein in the liver. (a) Western blot analysis of CYP2E1 in the liver of APAP-treated mice for 48 h and (b) quantification of CYP2E1 levels. Quantitative analysis of blots normalized by GAPDH expression. Each value represents the mean ± standard deviation (SD) for six mice. *** Significantly different from the corresponding control (Student’s t-test, p < 0.001)
Fig. 4.

Effect of APAP on GSH levels in the liver. Each value represents the mean ± standard deviation (SD) for six mice. *** Significantly different from the corresponding control (Student’s t-test, p < 0.001)
Dynamics of hepatic MDA levels after APAP treatment
The depletion of GSH by NAPQI is an important component of APAP-induced oxidative stress followed by liver injury. MDA level, an index of oxidative stress, was significantly increased at 2 h after APAP treatment (the time point of maximum hepatic GSH depletion), but returned to normal at 8 h (Fig. 5), confirming that MDA reflects the changes in antioxidant capacity of the liver. When it was compared among three different sources of ICR mice, No significant difference was observed among ICR mice from the three sources.
Fig. 5.

Effect of APAP on MDA levels in the liver. Each value represents the mean ± standard deviation (SD) for six mice. *** Significantly different from the corresponding control (Student’s t-test, p < 0.001)
Discussion
GSH is a tripeptide composed of cysteine, glutamic acid, and glycine and exists in all mammalian tissues; its levels are particularly high in the liver [26]. It is a major endogenous antioxidant that protects cells from oxidative stress or electrophilic metabolites from xenobiotics. GSH is synthesized by γ-glutamylcysteine ligase (GCL) and subsequently GSH synthetase as a final product in the transsulfuration pathway [27]. APAP is partly metabolized by cytochrome P450 (CYP) enzymes such as CYP1A2, CYP2E1, and isoforms of CYP3A, to the highly reactive metabolite NAPQI [28]. In normal physiological conditions, NAPQI is rapidly conjugated with GSH mediated by glutathione S-transferase. The resulting conjugate, APAP–GSH, can be further metabolized to APAP–cysteine/cysteinylglycine and APAP–mercapturate followed by excretion.
Overdosed APAP leads to increased bioactivation by CYP enzymes and formation of NAPQI, and also causes depletion of cellular GSH. Excess NAPQI may result in hepatotoxicity due to covalent binding to essential cellular macromolecules and mitochondrial dysfunction, and/or other mechanisms such as oxidative stress [27, 29]. In particular, recent studies have focused on the effect of NAPQI on mitochondria, which produce ATP through respiration [16, 17, 30, 31]. NAPQI caused by APAP overdose depletes mitochondrial GSH, and remaining NAPQI binds to mitochondrial proteins including the membrane ones. Subsequently, increased reactive oxygen species generated by GSH-depleted mitochondria activate JNK and induce translocation to mitochondria, where JNK induces permeabilization of the mitochondrial membranes and inhibits mitochondrial bioenergetics, eventually leading to cell death due to depletion of ATP [16, 17, 30, 31].
Drug-induced liver injury (DILI) is a major clinical and regulatory issue worldwide [32]. Control of DILI is still not easy because of the poor understanding of the mechanisms of toxicity caused by different drugs. Without knowledge of these mechanisms, it is difficult to develop ways to predict and prevent problems. To understand the causes of DILI, a research model that can reproduce the clinical response is indispensable. APAP is the most commonly used model drug of dose-dependent and predictable DILI. Indeed, the mechanism of APAP toxicity has been well studied, but the clinical problem has not been solved completely and APAP overdose is the primary cause of acute liver failure in several countries [33]. Early studies that investigated APAP-induced hepatotoxicity used multiple species, including rats, guinea pigs, cats, and hamsters, but currently it is clear that mice are the best available model [17] .
In the present study, we compared the time course of hepatic injury following oxidative stress induction by APAP in ICR mice from three different sources. Serum activity of ALT and histopathological analysis were used to determine liver injury, and hepatic levels of GSH and MDA were analyzed to examine the involvement of oxidative stress. Increased oxidative stress, suggested by increased CYP2E1 expression, hepatic GSH depletion and MDA elevation at 2 h after APAP administration, preceded liver injury, which was observed from 8 h, and hepatotoxicity, which peaked at 24 h. This study confirms that Korl:ICR mice are comparable to ICR mice from the two other sources in terms of APAP-induced hepatotoxicity.
Conclusions
The results of this study clearly show that APAP overdose induces hepatic GSH depletion accompanied by oxidative stress and followed by liver injury. We also found that the responses of Korl:ICR mice established by the NIFDS in Korea are similar to those of ICR mice from other sources, which suggests that it is a useful resource to study the mechanism of DILI and chemical-induced hepatotoxicity.
Acknowledgements
We appreciate NIFDS for providing Korl:ICR mice and its information.
Abbreviations
- ALT
Alanine aminotransferase
- APAP
Acetaminophen;
- CYP
Cytochrome P450
- DILI
Drug-induced liver injury
- GCL
γ-glutamylcysteine ligase
- GSH
Glutathione
- MDA
Malondialdehyde
- NAPQI
N-acetyl-p-benzoquinone imine
- NIFDS
National Institute of Food and Drug Safety Evaluation
Authors’ contributions
JYC, DYH, KSK, JHK, and YSJ were responsible for the study concept and design. TBJ, JHK, SHK, SL, SWS, and YL contributed to data acquisition. TBJ, JHK, SHK, and YSJ assisted with data analysis and interpretation of findings. TBJ, JHK, and YSJ drafted the manuscript. All authors read and approved the final manuscript.
Funding
This project was supported by a grant of NLAR (National Laboratory Animal Resources) from Ministry of Food and Drug Safety in 2018.
Availability of data and materials
The datasets used and/or analyzed in this study are available from the corresponding author on reasonable request.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Tae Bin Jeong, Joung-Hee Kim and Yong Lim contributed equally to this work.
References
- 1.Lee WM. Acute liver failure in the United States. Semin Liver Dis. 2003;23(3):217–226. doi: 10.1055/s-2003-42641. [DOI] [PubMed] [Google Scholar]
- 2.Bunchorntavakul C, Reddy KR. Acetaminophen-related hepatotoxicity. Clin Liver Dis. 2013;17(4):587–607. doi: 10.1016/j.cld.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Michaut A, Moreau C, Robin MA, Fromenty B. Acetaminophen-induced liver injury in obesity and nonalcoholic fatty liver disease. Liver Int. 2014;34(7):e171–e179. doi: 10.1111/liv.12514. [DOI] [PubMed] [Google Scholar]
- 4.Craig DG, Bates CM, Davidson JS, Martin KG, Hayes PC, Simpson KJ. Overdose pattern and outcome in paracetamol-induced acute severe hepatotoxicity. Br J Clin Pharmacol. 2011;71(2):273–282. doi: 10.1111/j.1365-2125.2010.03819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong HY, Medrano N, Borobia AM, Ruiz JA, Martinez AM, Martin J, et al. Hepatotoxicity induced by acute and chronic paracetamol overdose in children: where do we stand? World J Pediatr. 2017;13(1):76–83. doi: 10.1007/s12519-016-0046-6. [DOI] [PubMed] [Google Scholar]
- 6.Schnell RC, Bozigian HP, Davies MH, Merrick BA, Johnson KL. Circadian rhythm in acetaminophen toxicity: role of nonprotein sulfhydryls. Toxicol Appl Pharmacol. 1983;71(3):353–361. doi: 10.1016/0041-008X(83)90022-4. [DOI] [PubMed] [Google Scholar]
- 7.Schnell RC, Bozigian HP, Davies MH, Merrick BA, Park KS, McMillan DA. Factors influencing circadian rhythms in acetaminophen lethality. Pharmacology. 1984;29(3):149–157. doi: 10.1159/000138005. [DOI] [PubMed] [Google Scholar]
- 8.Vermeulen NP, Bessems JG, Van de Straat R. Molecular aspects of paracetamol-induced hepatotoxicity and its mechanism-based prevention. Drug Metab Rev. 1992;24(3):367–407. doi: 10.3109/03602539208996298. [DOI] [PubMed] [Google Scholar]
- 9.Claridge LC, Eksteen B, Smith A, Shah T, Holt AP. Acute liver failure after administration of paracetamol at the maximum recommended daily dose in adults. Bmj. 2010;341:c6764. doi: 10.1136/bmj.c6764. [DOI] [PubMed] [Google Scholar]
- 10.Jaeschke H, Knight TR, Bajt ML. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett. 2003;144(3):279–288. doi: 10.1016/S0378-4274(03)00239-X. [DOI] [PubMed] [Google Scholar]
- 11.Hinson JA, Reid AB, McCullough SS, James LP. Acetaminophen-induced hepatotoxicity: role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab Rev. 2004;36(3–4):805–822. doi: 10.1081/DMR-200033494. [DOI] [PubMed] [Google Scholar]
- 12.Boxill GC, Nash CB, Wheeler AG. Comparative pharmacological and toxicological evaluation of N-acetyl-p-aminophenol, salicylamide, and acetylsalicylic acid. J Am Pharm Assoc Am Pharm Assoc. 1958;47(7):479–487. doi: 10.1002/jps.3030470706. [DOI] [PubMed] [Google Scholar]
- 13.Boyd EM, Bereczky GM. Liver necrosis from paracetamol. Br J Pharmacol Chemother. 1966;26(3):606–614. doi: 10.1111/j.1476-5381.1966.tb01841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eder H. Chronic toxicity studies on phenacetin, N-acetyl-P-aminophenol (Napa) and acetylsalicylic acid on cats. Acta Pharmacol Toxicol (Copenh) 1964;21:197–204. doi: 10.1111/j.1600-0773.1964.tb01784.x. [DOI] [PubMed] [Google Scholar]
- 15.Potter WZ, Thorgeirsson SS, Jollow DJ, Mitchell JR. Acetaminophen-induced hepatic necrosis. V. Correlation of hepatic necrosis, covalent binding and glutathione depletion in hamsters. Pharmacology. 1974;12(3):129–143. doi: 10.1159/000136531. [DOI] [PubMed] [Google Scholar]
- 16.McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol. 2012;264(3):387–394. doi: 10.1016/j.taap.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaeschke H, Xie Y, McGill MR. Acetaminophen-induced liver injury: from animal models to humans. J Clin Transl Hepatol. 2014;2(3):153–161. doi: 10.14218/JCTH.2014.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chia R, Achilli F, Festing MF, Fisher EM. The origins and uses of mouse outbred stocks. Nat Genet. 2005;37(11):1181–1186. doi: 10.1038/ng1665. [DOI] [PubMed] [Google Scholar]
- 19.Kim YC, Lee SJ. Temporal variation in hepatotoxicity and metabolism of acetaminophen in mice. Toxicology. 1998;128(1):53–61. doi: 10.1016/S0300-483X(98)00046-8. [DOI] [PubMed] [Google Scholar]
- 20.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev. 2012;44(1):88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee YH, Kim SH, Lee S, Kim KM, Jung JC, Son TG, et al. Antioxidant Effect of Barley Sprout Extract via Enhancement of Nuclear Factor-Erythroid 2 Related Factor 2 Activity and Glutathione Synthesis. Nutrients. 2017;9(11):1252. doi: 10.3390/nu9111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reitman S, Frankel S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am J Clin Pathol. 1957;28(1):56–63. doi: 10.1093/ajcp/28.1.56. [DOI] [PubMed] [Google Scholar]
- 23.Kim SH, Kwon DY, Kwak JH, Lee S, Lee YH, Yun J, et al. Tunicamycin-Induced ER Stress is Accompanied with Oxidative Stress via Abrogation of Sulfur Amino Acids Metabolism in the Liver. Int J Mol Sci. 2018;19(12):4114. doi: 10.3390/ijms19124114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patten CJ, Thomas PE, Guy RL, Lee M, Gonzalez FJ, Guengerich FP, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol. 1993;6(4):511–518. doi: 10.1021/tx00034a019. [DOI] [PubMed] [Google Scholar]
- 25.Kim SJ, Lee MY, Kwon DY, Kim SY, Kim YC. Alteration in metabolism and toxicity of acetaminophen upon repeated administration in rats. J Pharmacol Sci. 2009;111(2):175–181. doi: 10.1254/jphs.09151FP. [DOI] [PubMed] [Google Scholar]
- 26.Jung YS. Metabolism of sulfur-containing amino acids in the liver: a link between hepatic injury and recovery. Biol Pharm Bull. 2015;38(7):971–974. doi: 10.1248/bpb.b15-00244. [DOI] [PubMed] [Google Scholar]
- 27.Meister A, Tate SS. Glutathione and related gamma-glutamyl compounds: biosynthesis and utilization. Annu Rev Biochem. 1976;45:559–604. doi: 10.1146/annurev.bi.45.070176.003015. [DOI] [PubMed] [Google Scholar]
- 28.Laine JE, Auriola S, Pasanen M, Juvonen RO. Acetaminophen bioactivation by human cytochrome P450 enzymes and animal microsomes. Xenobiotica. 2009;39(1):11–21. doi: 10.1080/00498250802512830. [DOI] [PubMed] [Google Scholar]
- 29.Du K, Ramachandran A, Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol. 2016;10:148–156. doi: 10.1016/j.redox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muldrew KL, James LP, Coop L, McCullough SS, Hendrickson HP, Hinson JA, et al. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab Dispos. 2002;30(4):446–451. doi: 10.1124/dmd.30.4.446. [DOI] [PubMed] [Google Scholar]
- 31.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010;196:369–405. doi: 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal VK, McHutchison JG, Hoofnagle JH. Drug-induced liver injury N. important elements for the diagnosis of drug-induced liver injury. Clin Gastroenterol Hepatol. 2010;8(5):463–470. doi: 10.1016/j.cgh.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee WM. Etiologies of acute liver failure. Semin Liver Dis. 2008;28(2):142–152. doi: 10.1055/s-2008-1073114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed in this study are available from the corresponding author on reasonable request.