

Abstract

A method for conjugating cholesterol to peptide ligands through non-disperse polyethylene glycol (ND-PEG) through a non-hydrolysable linkage is described. The iterative addition of tetraethylene glycol macrocyclic sulfate to cholesterol (Chol) renders a family of highly pure well-defined Chol-PEG compounds with different PEG lengths from 4 up to 20 ethylene oxide units, stably linked through an ether bond. The conjugation of these Chol-PEG compounds to the cyclic (RGDfK) peptide though Lys5 side chains generates different lengths of Chol-PEG-RGD conjugates that retain the oligomer purity of the precursors, as analysis by HRMS and NMR has shown. Other derivatives were synthesized with similar results, such as Chol-PEG-OCH3 and Chol-PEG conjugated to glutathione and Tf1 peptides through maleimide–thiol chemoselective ligation. This method allows the systematic synthesis of highly pure uniform stable Chol-PEGs, circumventing the use of activation groups on each elongation step and thus reducing the number of synthesis steps.
Introduction
Cholesterol (Chol) is an essential component of the plasma membrane of the eukaryotic cell. By improving the physical stability of the membrane, cholesterol plays a crucial role in membrane integrity, modulating membrane permeability and transmembrane signaling.1 Chol is extensively used to stabilize liposomes and other lipid-based nanovesicles (L-NVs) and also reduce the permeability of encapsulated drugs, thereby enhancing the efficiency of drug encapsulation.2−4 Furthermore, its 3β-hydroxyl group offers a chemical point for derivatization, thus allowing the generation of numerous Chol conjugates. In this regard, cholesteryl–polyethylene glycol (Chol-PEG) conjugates are commonly used in the production of PEG-coated liposomes or other nanovesicles to prolong their circulation time in plasma, improving the pharmacokinetics and efficacy of the encapsulated drugs.5,6 Chol-PEG conjugates also behave as solubilizing surfactants that serve to prevent aggregation of the formulated nanovesicles.5 The coating abilities of these conjugates are also used to increase the colloidal stability and aqueous dispersity of metallic nanoparticles, such as gold nanorods.7 Moreover, the amphiphilic character of Chol-PEG conjugates can facilitate their self-assembly by forming micelles, which have been used as delivery systems for poorly water-soluble anticancer agents such as quercetin8 and docetaxel.9 In fact, some drug delivery systems based on Chol-PEG conjugates exploit the capacity of cholesterol moieties to trigger their endocytosis through a lipid-raft-mediated mechanism, minimizing the endo-lysosomal trafficking after internalization.10,11 Cholesterol’s ability to anchor into cell membranes is also exploited for the development of Chol-PEG-based cell membrane imaging probes12−14 and eukaryote15 and bacteria16 cell surface engineering. Additionally, these Chol-PEG moieties are versatile scaffolds that can be modified with specific targeting ligands and used in drug nanoformulations to enhance their capacity to accumulate in specific cells and/or tissues. Recently, Chol-peptide conjugates have demonstrated great potential as hybrid materials with unique properties and pharmacological applications.17
In the literature, Cholesterol and PEGs have been conjugated mainly through an ester bond linkage and, to a lesser extent, by an ether or a carbamate bond.1,5,18 The preferential use of an ester bond is associated with the straightforward introduction of the different-PEG-length moieties onto Chol. However, esters are easily hydrolyzed, thus releasing Chol and altering the behavior of the nanovesicles, for instance, favoring their aggregation. The use of Chol-PEG conjugates linked through a more stable chemical function, like an ether, can overcome these drawbacks and generate nanoformulation systems with a more predictable behavior.19,20 These stable Chol-PEG conjugates are usually synthesized by polymerization processes, and the final molecules contain non-uniform PEG moieties. However, the synthesis of Chol-PEG units linked through an ether bond containing non-disperse PEGs (ND-PEGs) is arduous.21−24 Given the known correlation between erratic biological activities and non-uniform PEG lengths,25−27 the use of ND-PEGs in drug delivery systems has recently attracted a growing interest. Here, we report the synthesis of a family of non-disperse ether-linked Chol-PEGn-OH derivatives (from 4 up to 20 ethylene oxide units) prepared using a recently described methodology28,29 based on the use of tetraethylene glycol macrocyclic sulfate as the electrophile moiety to allow iterative PEG chain elongation. This methodology afforded the desired pure compounds via straightforward work-up procedures, without any chromatographic purification. Additionally, Chol-PEGn-OH moieties were derivatized with targeting peptides, either via carbamate bonds or by a chemoselective thiol maleimide ligation, obtaining Chol-PEGn-peptide conjugates in good yield and excellent purity (up to 99%). In the future, we plan to explore the impact of the PEG lengths of these conjugates on the physicochemical properties of different lipid-based nanovesicles and on their biological behavior, but herein we focus on the development of a methodology for their production.
Results and Discussion
Synthesis of Chol-PEGn
To generate the Chol-PEGn units, we explored two strategies: first, a sequential two-step process based on alcohol tosylation of Chol or Chol-PEG and subsequent reaction with tetraethylene glycol (TEG) and, second, an iterative addition of tetraethylene glycol macrocyclic sulfate to Chol or Chol-PEG hydroxyl groups. We have previously reported the synthesis of Chol-PEG4-RGD starting from Chol.30 The formation of Chol-PEG4-OH (4a) was carried out using a Williamson-type ether synthesis (Scheme 1), achieving good yields through the reaction of cholesteryl p-toluenesulfonate (3a) with TEG using 1,4-dioxane as a solvent under reflux. However, attempts to increase the PEG chain length of 4a following this strategy did not afford the desired PEG-chain elongation. Recently, the synthesis of linear ND-PEGs has been described using oligoethylene glycol (OEG) macrocyclic sulfates as electrophilic moieties to react with PEG terminal hydroxyl groups.28,29 Compared to other reported methods, with this strategy, it is not necessary to work with protecting or activating groups during the elongation process and the number of synthesis steps is reduced. The use of the tetraethylene glycol macrocyclic sulfate as an elongation building block that contains the activation moiety allows us to explore Chol-PEG growth avoiding the use of activating groups such as p-toluensulfonate, which in our hands has resulted in being unsatisfactory to produce Chol-PEGn (n > 4). We then considered applying this methodology to the synthesis of Chol-PEGn structures with a well-defined molecular size. To develop an economically effective synthesis and following our previous work,19,20 we performed the PEG elongation experiments using compound 4a as the starting material and we generated Chol-PEGn derivatives of different PEG lengths applying this new methodology. Chol-PEG4 4a was treated with NaH at 0 °C for 2 h. The resulting alkoxide was reacted with the macrocyclic sulfate 2 at reflux overnight to afford the intermediate Chol-PEG8-sulfate. This intermediate was hydrolyzed with a THF/H2O mixture and H2SO4 at reflux for 5 h to render Chol-PEG8 5 in an 83% yield (Scheme 1). Other methodologies to prepare compound 5 have been reported in the literature,22 but they involve tedious steps of selective monoprotection of oligoethylene glycol moieties. Iterative addition of TEG macrocyclic sulfate 2 following the same procedure resulted in a viable approach to increase the PEG chain length up to PEG20 for the first time with a 57% yield and excellent purity (99% as shown by HPLC, see Table 1 and the Supporting Information) and without the need of any chromatographic purification. The use of 3 equiv of macrocyclic sulfate 2 was necessary to ensure complete reactions and avoid mixtures of Chol-PEGn units with different PEG lengths. We assessed the scalability of the process by preparing Chol-PEG8 5 in a 2 g scale with the same excellent results.
Scheme 1. Synthesis of Chol-PEGn Moieties of Different PEG Lengths via a Macrocyclic Sulfate Strategy.

Table 1. Purity, Polydispersity Index Values (PDI), and Oligomer Purity (OP) of the Chol-PEGn Derivatives (n.s.: Not Synthesized).

|
R = OH |
R = OCH3 |
R = RGD |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| derivatives | compd | purity | PDI/OP | compd | purity | PDI/OP | compd | purity | PDI/OP |
| Chol-PEG4-R | 4a | 99% | 1.0/100% | 9 | 99% | 1.0/100% | 13 | 99% | 1.0/100% |
| Chol-PEG8-R | 5 | 99% | 1.000052/98.7% | 10 | 99% | 1.000039/98.8% | 14 | 97% | 1.000001/99.9% |
| Chol-PEG12-R | 6 | 99% | 1.000007/99.7% | 11 | 99% | 1.000055/98.0% | 15 | 95% | 1.000013/98.4% |
| Chol-PEG16-R | 7 | 99% | 1.000009/99.5% | n.s. | 16 | 90% | 1.000045/96.3% | ||
| Chol-PEG20-R | 8 | 99% | 1.000023/98.3% | n.s. | 17 | 97% | 1.000019/97.6% | ||
Using this approach, we also prepared compound 4b that retains the chirality of the cholesterol β-hydroxyl group (Scheme 1) being the enantiomer of the previously described 4a. In this case, treating Chol 1 with NaH at 0 °C for 2 h followed by a reaction with macrocyclic sulfate 2 at rt was unsuccessful. Remarkably, when the temperature of the ring-opening reaction was increased (up to reflux using THF as a solvent), product 3b was obtained in good yields after 12 h. Acid hydrolysis of this intermediate afforded Chol-PEG4 4b, which was analyzed by HRMS (ESI) to confirm its integrity. To the best of our knowledge, no previous examples using Chol 1 as a nucleophile for the ring-opening reaction of macrocyclic sulfates have been described. The methodology described herein will allow the preparation of even longer Chol-PEG derivatives in a straightforward manner. Chol-PEGn moieties were purified by flash chromatography using hexane/ethyl acetate for Chol-PEG4 and CH2Cl2 with an increasing percentage of ethanol (0 to 80%) for Chol-PEG8 to Chol-PEG20. The purity of all compounds was determined by UPLC-PDA-ELSD-MS with it being higher than 95% in all cases (Table 1). HRMS analysis confirmed the excellent purity and integrity of the final Chol-PEGn products (4a, 5, 6, 7, and 8) and was used to determine their polydispersity indexes (PDIs).
A key parameter for the high quality of the Chol-PEGn platforms is the base selected to generate the alkoxide derivatives, as Tanaka,31 Davis,32 and Bruce25 reported. In our protocol, we used NaH, a strong base that can generate PEG alkoxides that lead to chain scission, but the control of the number of equivalents of NaH and the reaction times were enough to minimize this secondary reaction. The PDIs observed for all the Chol-PEGn synthesized are indicative of highly monodisperse compounds (Table 1).
Synthesis of Chol-PEGn-OCH3
Since PEGs are susceptible to oxidative degradation when exposed to air, most of the PEGs used for coating nanoparticles are CH3 end-capped. Methylation of the Chol-PEGn-OH moieties was achieved by their treatment with NaH at 0 °C for 1 h and then by reaction with CH3I at rt for an extra hour, achieving excellent yields and purities (Table 1).
Synthesis of Chol-PEGn-RGD
We then studied the introduction of a peptide ligand as a targeting unit into these Chol-PEGn-OH moieties. As a proof of concept, we selected the cyclo-(RGDfK) peptide 12, a specific ligand for integrin receptors that has been widely used in drug delivery systems for cancer therapies.33 This peptide has a ζ-NH2 group in the Lys side chain (HZ1,2), which can be easily derivatized and used for conjugation purposes.
The conjugation of cyclo-(RGDfK)19 was performed via a two-step process based on the transformation of the hydroxyl group of Chol-PEGn-OH to the corresponding chloroformate or ester carbonate and subsequent peptide introduction through a carbamate bond. The following hydroxyl derivatizations to ester carbamate were explored: (a) nitrophenyl carbonate by a reaction with 4-nitrophenyl chloroformate and (b) succinimidyl carbonate by direct derivatization with N,N′-disuccinimidyl carbonate. In both the chloroformate and nitrophenyl carbonate derivatizations, the reaction with the peptide produces a side product resulting from the incorporation of a second unit of the peptide to the Chol-PEG4-RGD moiety. In detail, in these reactions, the carboxylic acid of the side chain of the Asp residue of Chol-PEG4-RGD was transformed into a reactive derivative, thus favoring the introduction of a second unit of cyclo-(RGDfK) 12 by means of an amide bond. Although condition b allowed full conversion of Chol-PEG4 into Chol-PEG4-RGD, a more appropriate two-step derivatization strategy was developed (Scheme 2), consisting of the preparation of the chloroformate derivatives by a reaction of Chol-PEGn-OH with a phosgene solution in toluene. These compounds were then smoothly converted into the succinimidyl ester carbonates derivatives by addition of N-hydroxysuccinimide (NHS). Cyclo-(RGDfK) introduction onto these Chol-PEGn derivatives allowed the preparation of Chol-PEGn-RGD compounds (13–17) with a high yield (up to 87%) and purity (up to 99%, see Table 1, Figures 1 and 2, and the Supporting Information).
Scheme 2. Synthesis of Chol-PEGn-RGD Conjugates.

Figure 1.
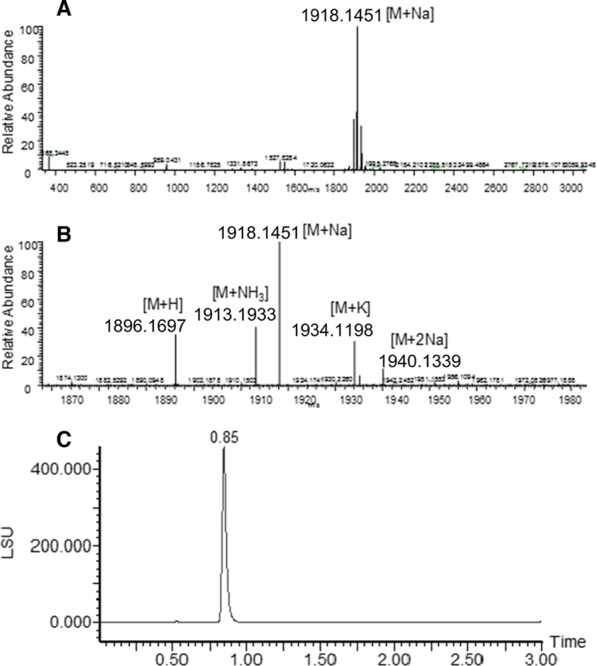
HRMS and UPLC-ELSD of Chol-PEG20-RGD. A: full mass spectrum. B: zoomed-in deconvoluted mass spectrum. C: UPLC-ELSD trace.
Figure 2.

A: 2D TOCSY (amide-aliphatic region) of the RGD peptide 12 (black) superposed to Chol-PEG20-RGD 17 (blue) in H2O/D2O (9/1). Amino acids are labeled. Chemical shift differences of Lys5 protons upon conjugation to the Chol-PEG20 moiety are labeled and indicated with arrows. B: 2D NOESY of Chol-PEG20-RGD 17 in H2O/D2O (aliphatic region). PEG, cholesterol, and RGD resonances are highlighted. C: 2D NOESY of Chol-PEG12-RGD 15 in DMSO. RGD, cholesterol, DMSO, and PEG12 resonances are indicated.
Remarkably, the PDIs of all Chol-PEGn-RGDs showed values similar to those previously determined for their precursors (Table 1 and the Supporting Information).25,31,32 The purification of Chol-PEGn-RGD conjugates when n was 4, 8, and 12 (compounds 13, 14, and 15) was carried out by washing the compound precipitated with H2O to remove the excess c(RGDfK) peptide and CH3CN to eliminate the unreacted Chol-PEGn. Due to the solubility in H2O of Chol-PEG16-RGD (16) and Chol-PEG20-RGD (17), these compounds were purified by solid-phase extraction using a PoraPak Rxn cartridge (Waters) and eluting with H2O with an increasing percentage of ethanol (0 to 100%).
HR-NMR Analysis of the c(RGDfK) Peptide and Chol-PEGn-RGD
The c(RGDfK) peptide was unambiguously assigned using a combination of 2D TOCSY and NOESY experiments both in water and in DMSO solvents. We used 1D 13C NMR to follow the intensity changes at the PEG chain resonances (−OCH2CH2O−) and confirm the PEG length increases in the different Chol-PEGn-RGD conjugates (Figure S52 in the Supporting Information). In these comparisons, the remaining signals of the molecule are the same, corroborating the data achieved by mass spectrometry. Furthermore, we also acquired and assigned 2D TOCSY and 2D NOESY experiments for each Chol-PEGn-RGD derivative. Depending on the solubility of the samples, these experiments were acquired either in DMSO (for derivatives up to PEG16) or in water (for Chol-PEG20). Specific chemical shift changes were observed for the Lys5 side-chain resonances, corroborating that the Chol-PEGn system is attached to the RGD peptide through the Lys5 HZ1 proton. To illustrate these differences, the superposition of the RGD peptide to Chol-PEG20-RGD (17) is shown in Figure 2. The conjugate Chol-PEG12-RGD is also indicated. Other conjugated systems are shown as well in the Supporting Information.
Synthesis of Other Chol-PEG-Peptide Ligands
We also studied the introduction of two other peptide ligands into the Chol-PEG platforms using chemoselective ligation between maleimides and thiols to generate the corresponding conjugates (Scheme 3). For this purpose, we selected two Cys-containing peptides for conjugation: T7, a transferrin-receptor 1 (TfR1) binding peptide3421 modified with a Cys at its N-terminal position, which was synthesized by standard solid-phase peptide synthesis, and a reduced glutathione, a tripeptide that contains a Cys on its sequence, used to permeate the blood–brain barrier through a transporter protein.35
Scheme 3. Synthesis of Chol-PEG4-Maleimide and Conjugation with Blood–Brain Barrier Shuttle Peptides.

Chol-PEG4-maleimide derivative 20 was prepared by a reaction of Chol-PEG4-OH 4a with a phosgene solution in toluene for 2 h and then followed by the addition of the previously synthesized maleimide moiety 19, achieving a yield of 35%. The T7 peptide 21 was conjugated to intermediate 20 by reacting for 36 h in DMF at rt. After solvent evaporation and subsequent addition of acetonitrile, the final product 22 was isolated as a precipitate in a 58% yield. In the case of glutathione conjugation, we used DMSO as a solvent because of the insolubility of the reduced glutathione in DMF. Compound 23 was obtained by precipitation in acetonitrile with a yield of 49%. All compounds were characterized by HRMS, which confirmed their chemical integrity.
Conclusions
Although a large number of methods to generate Chol-PEG conjugates have been described, practically all of them render nonuniform Chol-PEG where both subunits can be linked through degradable (ester) or stable (carbamate or ether) bonds. Generally, the instability and/or the nonuniform character of these conjugates has been associated to reproducibility issues and erratic behavior in in vivo experiments of diverse nanovesicles in different applications. This fact, together with the increased characterization requirements for nanoformulation components, has generated an increasing interest in the development of strategies to produce well-defined hybrid molecules as Chol-PEG conjugates. We have devised an efficient methodology to synthesize highly non-disperse directly linked Chol-PEGn (n = 4–20) via an ether linkage involving iterative addition of tetraethylene glycol macrocyclic sulfate to cholesterol in combination with an accurate control of base equivalents and reaction times. The use of the tetraethylene glycol macrocyclic sulfate as an elongation building block, which contains the sulfate as an activation moiety, circumvents the use of protecting or activation groups on each PEG growth step. Thus, compared to other methods the number of synthesis steps is reduced during the PEG elongation process. This approach allows us to prepare stable Chol-PEGn compounds with n ≥ 6 in a straightforward manner without a purification process during PEG chain growth and permits the methodic synthesis of all Chol-PEGn (n, n + 4, n + 8, ...), allowing us to study systematically the effect of increasing units of ethylene glycol on properties such as solubility or integration capacity onto natural or artificial membranes (nanovesicles). The use of these uniform Chol-PEGn conjugates in different applications such as liposomes or nanovesicle formulations can help fine-tune their physical–chemical properties and behavior in vivo. Furthermore, we have conjugated Chol-PEGn with different peptide ligands preserving its uniform properties and generating highly pure peptide conjugates that were easily isolated by simple precipitation or solid-phase extraction cartridges. Other methodologies purify the protected final compound by gel silica chromatography and then eliminate the protecting groups.
This strategy enhances the feasibility to synthesize “a la carte” Chol-PEG conjugates with a range of PEG lengths, linkages, and peptide ligands, thereby modulating the properties of these systems in the formulation of targeted nanosystems for drug delivery. Given the fact that very few very well defined PEG drugs are available, this synthesis makes a significant contribution to expanding the portfolio of Chol-PEGn compounds with potential applications in drug delivery systems, membrane imaging or engineering, and sensors, thus helping them progress into clinic practice. Currently, we are studying the physical–chemical and biological properties of our different-length Chol-PEG conjugates in diverse lipid nanoformulations for drug delivery purposes.
Experimental Section
Synthesis of Chol-PEGn
Tetraethylene Glycol Macrocyclic Sulfate (2)
Compound 2 was synthesized as described previously by Zhang et al.28 Briefly, tetraethylene glycol (10.00 g, 51.49 mmol) was placed in a 2 L round-bottomed flask under a N2 atmosphere and dissolved with anhyd CH2Cl2 (1200 mL), and DMAP (314 mg, 0,05 equiv) and DIEA (42 mL, 4.8 equiv) were added. Then, a solution of thionyl chloride (7.51 mL, 2 equiv) in anhyd CH2Cl2 (250 mL) was added slowly at 0 °C. After 6 h, sat. NaCl (500 mL) was added slowly at 0 °C, and the reaction was stirred overnight. After this period, the two phases were separated, and the aqueous phase was washed with CH2Cl2 (3 × 100 mL). All the organic phases were pulled together, dried over MgSO4, and filtered, and the solvent was evaporated to dryness. The intermediate was purified by flash chromatography using silica gel as the stationary phase and hexane/CH2Cl2/EtOAc as the mobile phase to afford the tetraethylene glycol macrocyclic sulfite intermediate (8.64 g, 70% yield). 1H NMR: (400 MHz, CDCl3) δ: 3.68 (m, 8H), 3.82 (m, 4H), 4.10 (m, 2H), 4.35 (m, 2H). This compound (3.088 g, 12.85 mmol) was suspended in a mixture of CH3CN:CCl4:H2O (1:1:1.5, 122 mL:122 mL:504 mL), and then NaIO4 (5.50 g, 2 equiv) and RuCl3 (13.3 mg, 0.005 equiv) were added. The resulting mixture was stirred for 2 h, and both phases were separated. The aqueous phase was washed with CH2Cl2 (4 × 100 mL). All the organic phases were pulled together, and the solvent was evaporated affording compound 2 (pure, 2.90 g, 75% yield). HPLC-MS (ESI, elution condition 1): tR: 1.52 min, calcd mass for C8H16O7S+: 256.27; found: 257.4 [M + H]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3) δ: 3.67 (m, 8H), 3.84 (m, 4H), 4.48 (m, 4H).
Chol-PEG4 via Williamson-Type Ether (4a)
Into a 100 mL round-bottomed flask, cholesterol 1 (2.06 g, 5.33 mmol) was dissolved in pyridine (14 mL) and 4-toluensulfonyl chloride was added slowly at 0 °C, and the reaction was stirred overnight. Then, pyridine was evaporated under reduced pressure, and the crude was dissolved in 14 mL of chloroform. Subsequently, 80 mL of CH3OH was slowly added until a precipitate was formed. The crude was washed with CH3OH (2 × 25 mL) and CH3CN (25 mL) yielding 2.306 g (82% yield) of compound 3a as a white solid.
Compound 3a (2.306 g, 4.26 mmol) was weighted, and a solution of tetraethylene glycol (13.97 g, 12.42 mL, 71.94 mmol, 17 equiv) in anhyd 1,4-dioxane (35 mL) was added dropwise with an addition funnel. The mixture was refluxed with stirring for 24 h under an Ar atmosphere. Then, the mixture was cooled down to rt, and it was concentrated to dryness. The resulting crude was dissolved in CH2Cl2 (50 mL) and washed with saturated NaHCO3 (2 × 20 mL), H2O (2 × 30 mL), and saturated NaCl (1 × 20 mL). The organic phases were dried over MgSO4 and filtered, and the solvent was evaporated to dryness. The crude was purified by flash chromatography using silica gel as the stationary phase and hexane/EtOAc as the mobile phase to afford 4a (1.91 g, 80% yield) as a yellowish oil. UPLC-MS (ESI, elution condition 7): tR: 1.03 min, calcd mass for C35H63O5+ [M + H]+: 563.46; found: 580.7 [M + H + H2O]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3) δ: 0.67 (s, 3H), 0.85 (d, J = 1.8 Hz, 3H), 0.87 (d, 1.8 Hz, 3H), 0.91 (d, J = 6.5 Hz, 3H), 0.99 (s, 3H), 1.01–1.20 (m, 8H), 1.21–1.41 (m, 5H), 1.42–1.62 (m, 8H), 1.76–2.03 (m, 5H), 2.23 (m, 1H), 2.37 (m, 1H), 3.18 (m, 1H), 3.65 (m, 14H), 3.73 (m, 2H), 5.34 (m, 1H). 13C NMR: (100 MHz, CDCl3) δ: 12.01, 18.87, 19.53, 21.22, 22.71, 22.97, 23.98, 24.45, 28.17, 28.39, 28.46, 32.05, 32.11, 35.94, 36.35, 37.03, 37.39, 39.14, 39.67, 39.95, 42.48, 50.35, 56.31, 56.94, 61.91, 67.39, 70.45, 70.71, 70.73, 70.78, 71.03, 72.80, 79.70, 121.72, 141.10. HRMS (ESI): calculated mass for C35H63O5+ [M + H]+: 563.4670, found: 563.4679; calcd for C70H125O10+ ([2 M + H]+): 1125.9267, found: 1125.9313. The molar oligomer distribution from MS was 100.0% Chol-PEG4, PDI = 1.000000.
Chol-PEG4 via Macrocyclic Sulfate (4b)
In a two-neck 100 mL round-bottomed flask equipped with a refrigerator was placed 0.021 g of NaH 60% dispersed in mineral oil (0.522 mmol) with 8 mL of THF and then added a solution of Chol (0.101 g, 0.26 mmol) in THF (13 mL) under an argon atmosphere at 0 °C. The white suspension was stirred for 1 h at 0 °C. After that, it was allowed to temper until reaching room temperature and, a solution of cyclic sulfate 2 (0.200 g, 0.780 mmol, 3 equiv) in anhyd THF (10 mL) was added. The resulting mixture was stirred for 36 h at reflux and then cooled down, and THF was removed under vacuum. The residue was dissolved in water (50 mL) then H2SO4 (2 mL, 35.49 mmol) was added, and it was refluxed for 5 h. The reaction was quenched with 5% NaHCO3 (35 mL) solution, and Na2CO3 was added until reaching pH 6–7. The solution was extracted with CH2Cl2 (5 × 25 mL). The organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to obtain the desired compound 4b. Chol-PEG4 4b was purified by flash chromatography using silica as the stationary phase and a gradient of EtOH in CH2Cl2 as the mobile phase (0% to 20%) to obtain pure compound 4b (0.074 g, 50% yield) as a white oil. HPLC-MS (ESI, elution condition 1): tR: 1.60 min, calcd mass for C35H63O5+ [M + H]+: 563.46; found: 580.7 [M + H + H2O]+. Chemical purity: 99%.
Chol-PEG8 (5)
The synthesis of compound 5 was performed following the experimental procedure for compound 4b: using a NaH 60% dispersion in mineral oil (0.285 g, 7.13 mmol, 2 equiv) and compounds 4 (2.008 g, 3.56 mmol) and 2 (2.74 g, 1.07 mmol, 3 equiv) and stirring for 12 h at reflux to afford 5. After the reaction was quenched, the solution was extracted with CH2Cl2, and the organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to obtain compound 5, which was purified by flash chromatography using silica as the stationary phase and a gradient of EtOH in CH2Cl2 as the mobile phase (0% to 20%) yielding pure compound 5 (2.2 g, 83% yield) as a white oil. UPLC-MS (ESI, elution condition 7): tR: 0.99 min, calcd mass for C43H79O9+: 739.57; found: 756.08 [M + H + H2O]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3) δ: 0.67 (s, 3H), 0.85 (d, J = 1.8 Hz, 3H), 0.86 (d, J = 1.8 Hz, 3H), 0.91 (d, J = 6.5 Hz, 3H), 0.99 (s, 3H), 1.00–1.19 (m, 8H), 1.22–1.38 (m, 5H), 1.41–1.62 (m, 8H), 1.77–2.03 (m, 6H), 2.21 (m, 1H), 2.36 (ddd, J = 2.3, 4.7, 13.2 Hz, 1H), 3.17 (tt, J = 0, 4.5, 11.3 Hz, 1H), 3.65 (m, 30H), 3.72 (m, 2H), 5.33 (m, 1H). 13C NMR: (100 MHz, CDCl3) δ: 12.01, 18.87, 19.54, 21.22, 22.71, 22.97, 23.98, 24.45, 28.17, 28.39, 28.52, 32.06, 32.11, 35.94, 36.35, 37.03, 37.40, 39.22, 39.67, 39.95, 42.48, 50.35, 56.31, 56.94, 61.90, 67.44, 70.49, 70.70, 70.73, 70.76, 70.78, 71.04, 72.71, 79.64, 121.68, 141.14. HRMS (ESI): calculated mass for C43H79O9+ [M + H]+: 739.57186; found: 739.57365. The molar oligomer distribution from MS was 98.7% Chol-PEG8, 1.16% Chol-PEG7, and 0.07% Chol-PEG6; PDI = 1.000052.
Chol-PEG12 (6)
The synthesis of compound 6 was performed following the experimental procedure for compound 5: using a NaH 60% dispersion in mineral oil (0.023 g, 0.576 mmol, 2 equiv) and compounds 5 (0.214 g, 0.290 mmol) and 2 (0.223 g, 0.870 mmol, 3 equiv) and stirring for 12 h at reflux to afford 6 with quantitative yields. After the reaction was quenched, the solution was extracted with CH2Cl2, and the organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to obtain compound 6, which was purified by flash chromatography using silica as the stationary phase and a gradient of EtOH in CH2Cl2 as the mobile phase (0% to 40%), yielding pure compound 6 (0.171 g, 65% yield) as a white oil. UPLC-MS (ESI, elution condition 7): tR: 0.91 min, calcd mass for C51H95O13+: 915.67, found: 932.9 [M + H + H2O]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3) δ: 0.66 (s, 3H), 0.84 (d, J = 1.8 Hz, 3H), 0.86 (d, J = 1.8 Hz, 3H), 0.90 (d, J = 6.5 Hz, 3H), 0.98 (s, 3H), 1.00–1.19 (m, 8H), 1.20–1.40 (m, 5H), 1.40–1.61 (m, 8H), 1.74–2.04 (m, 6H), 2.19 (m, 1H), 2.35 (ddd, J = 2.2, 4.8, 13.2 Hz, 1H), 3.16 (tt, J = 0, 4.5, 11.3 Hz, 1H), 3.63 (m, 46H), 3.71 (m, 2H), 5.32 (m, 1H). 13C NMR: (100 MHz, CDCl3) δ: 11.98, 18.84, 19.51, 21.19, 22.68, 22.94, 23.94, 24.41, 28.13, 28.35, 28.49, 32.02, 32.07, 35.90, 36.31, 36.99, 37.37, 39.19, 39.64, 39.91, 42.44, 50.31, 56.28, 56.90, 61.83, 67.41, 70.44, 70.65, 70.68, 70.69, 70.72, 71.01, 72.65, 79.60, 121.64, 141.10. HRMS (ESI): calculated mass for C51H95O13+ [M + H]+: 915.67672, found: 915.67816; calcd for C51H94O13Na+ [M + Na]+: 937.65867, found: 937.66114. The molar oligomer distribution from MS was 99.7% Chol-PEG12 and 0.31% Chol-PEG11; PDI = 1.000007.
Chol-PEG16 (7)
The synthesis of compound 7 was performed following the experimental procedure for compound 5: using a NaH 60% dispersion in mineral oil (0.028 g, 0.70 mmol, 2 equiv.) and compounds 6 (0.322 g, 0.352 mmol) and 2 (0.270 g, 1.05 mmol, 3 equiv) and stirring for 12 h at reflux to afford 7. After the reaction was quenched, the solution was extracted with CH2Cl2, and the organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to obtain compound 7, which was purified by flash chromatography using silica as the stationary phase and a gradient of EtOH in CH2Cl2 as the mobile phase (0% to 60%) affording pure compound 7 (0.168 g, 44% yield) as a white oil. UPLC-MS (ESI, elution condition 7): tR: 0.86 min, calcd mass for C59H111O17+: 1091.78; found: 1108.9 [M + H + H2O]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3) δ: 0.66 (s, 3H), 0.84 (d, J = 1.7 Hz, 3H), 0.86 (d, J = 1.7 Hz, 3H), 0.90 (d, J = 6.5 Hz, 3H), 0.98 (s, 3H), 1.00–1.19 (m, 8H), 1.22–1.37 (m, 5H), 1.42–1.62 (m, 8H), 1.77–2.03 (m, 6H), 2.20 (m, 1H), 2.35 (m, 1H), 3.16 (m, 2H), 3.62 (m, 62H), 3.71 (m, 2H), 5.32 (m, 1H). 13C NMR: (100 MHz, CDCl3) δ: 11.98, 18.83, 19.50, 21.18, 22.68, 22.93, 23.94, 24.41, 28.12, 28.35, 28.47, 32.01, 32.06, 35.89, 36.30, 36.99, 37.36, 39.18, 39.63, 39.90, 42.44, 50.30, 56.27, 56.89, 61.76, 61.80, 67.40, 70.35, 70.39, 70.57, 70.68, 71.00, 72.68, 79.60, 121.64, 141.08. HRMS (ESI): calculated mass for C59H111O17+ [M + H]+: 1091.78158, found: 1091.78214; calcd for C59H114O17N+ ([M + NH3]+): 1108.8081, found: 1108.8092; calcd for C59H110O17K+ ([M + K]+): 1129.7375, found: 1129.7394. The molar oligomer distribution from MS was 99.5% Chol-PEG16, 0.43% Chol-PEG15, and 0.04% Chol-PEG14; PDI = 1.000009.
Chol-PEG20 (8)
The synthesis of compound 8 was performed following the experimental procedure for compound 5: using a NaH 60% dispersion in mineral oil (0.007 g, 0.170 mmol, 2 equiv) and compounds 7 (0.093 g, 0.0852 mmol) and 2 (0.066 g, 0.258 mmol, 3 equiv) and stirring for 12 h at reflux to afford 7. After the reaction was quenched, the solution was extracted with CH2Cl2, and the organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to obtain compound 8, which was purified by flash chromatography using silica as the stationary phase and a gradient of EtOH in CH2Cl2 as the mobile phase (0% to 80%), yielding pure compound 8 (0.062 g, 57% yield) as a white oil. UPLC-MS (ESI, elution condition 7): tR: 0.85 min, calcd mass for C67H127O21+: 1267.88; found: 1284.8 [M + H + H2O]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3) δ: 0.66 (s, 3H), 0.84 (d, J = 1.8 Hz, 3H), 0.86 (d, J = 1.8 Hz, 3H), 0.90 (d, J = 6.5 Hz, 3H), 0.98 (s, 3H), 1.00–1.19 (m, 8H), 1.20–1.40 (m, 5H), 1.40–1.61 (m, 7H), 1.75–2.06 (m, 7H), 2.19 (m, 1H), 2.35 (ddd, J = 2.1, 4.6, 13.3 Hz, 1H), 3.16 (m, 1H), 3.63 (m, 78H), 3.71 (m, 2H), 5.32 (m, 1H). 13C NMR: (100 MHz, CDCl3) δ: 12.01, 18.87, 19.53, 21.22, 22.71, 22.96, 23.97, 24.44, 28.16, 28.38, 28.50, 32.05, 32.10, 35.93, 36.34, 37.02, 37.39, 39.19, 39.66, 39.94, 42.47, 50.33, 56.30, 56.93, 61.77, 67.42, 70.33, 70.58, 70.61, 70.63, 70.69, 70.73, 71.02, 72.74, 79.64, 121.68, 141.12. HRMS (ESI): calculated mass for C67H127O21+ [M + H]+: 1267.8864; found: 1267.8944. Calcd for C67H126O21Na+ [M + Na]+: 1289.8684; found: 1289.8724. The molar oligomer distribution from MS was 98.3% Chol-PEG20, 1.63% Chol-PEG19, and 0.08% Chol-PEG18; PDI = 1.000023.
Synthesis of Chol-PEGn-OCH3
Chol-PEG4-OCH3 (9)
In a two-neck 50 mL round-bottomed flask was placed a suspension of NaH (0.048 g, 60% dispersed in mineral oil, 1.2 mmol, 2.5 equiv) in THF (5 mL) and added a solution of 4 (0.270 g, 0.480 mmol) in THF (5 mL) under an argon atmosphere at 0 °C. The white suspension was stirred for 2 h at 0 °C and then once rt was reached, 0.060 mL of CH3I (0.964 mmol, 2 equiv) was added. The resulting mixture was stirred for 4 h, and then H2O was added. The resulting residue was dissolved in ethyl acetate (50 mL) and washed with H2O (3 × 25 mL). Layers were separated, dried over MgSO4, and organic phase was evaporated to afford compound 9, which was purified by flash chromatography using silica as the stationary phase and a gradient of EtOAc in hexane as the mobile phase (0% to 100%) yielding pure compound 9 (0.151 g, 55% yield) as a yellow oil with 99% purity. HPLC-MS (ESI, elution condition 1): tR: 1.63 min, calcd mass for C36H65O5+: 576.48; found: 593.8 [M + H + H2O]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3, 298 K) δ: 0.67 (s, 3H), 0.85 (d, J = 1.8 Hz, 3H), 0.86 (d, J = 1.8 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.99 (s, 3H), 1.00–1.19 (m, 8H), 1.19–1.40 (m, 5H), 1.41–1.61 (m, 8H), 1.76–2.04 (m, 5H), 2.20 (m, 1H), 2.36 (m, 1H), 3.17 (m, 1H), 3.37 (s, 3H), 3.54 (m, 2H), 3.64 (m, 14H), 5.33 (m, 1H). 13C NMR: (100 MHz, CDCl3, 298 K) δ: 12.00, 18.86, 19.52, 21.21, 22.70, 22.96, 23.97, 24.43, 28.15, 28.37, 28.50, 32.04, 32.09, 35.92, 36.33, 37.01, 37.39, 39.21, 39.66, 39.93, 42.46, 50.33, 56.30, 56.92, 59.17, 67.43, 70.66, 70.72, 70.74, 70.75, 71.03, 72.08, 79.63, 121.66, 141.12. HRMS (ESI): calculated mass for C36H65O5 [M + H]+: 577.4826; found: 577.4839. The molar oligomer distribution from MS was 100.0% Chol-PEG4-OCH3; PDI = 1.0.
Chol-PEG8-OCH3 (10)
In a two-neck 50 mL round-bottomed flask was placed a suspension of NaH (0.013 g, 60% dispersed in mineral oil, 0.32 mmol, 5 equiv) in THF (5 mL) and added a solution of 5 (0.048 g, 0.065 mmol) in THF (5 mL) under an argon atmosphere at 0 °C. The white suspension was stirred for 2 h at 0 °C, and then once having reached room temperature, 0.008 mL of CH3I (0.130 mmol, 2 equiv) was added. The resulting mixture was stirred for 4 h, and then H2O was added. The resulting residue was dissolved in ethyl acetate (20 mL) and washed with H2O (3 × 20 mL). Layers were separated and dried over MgSO4, and the organic phase was evaporated to afford compound 10, which was purified by flash chromatography using silica as the stationary phase and a gradient of EtOAc in hexane as the mobile phase (0% to 100%) yielding pure compound 10 (0.034 g, 69% yield) as a yellow oil with 99% purity. HPLC-MS (ESI, elution condition 1): tR: 1.70 min, calcd mass for C44H81O9+: 753.58; found: 770.8 [M + H + H2O]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3, 298 K) δ: 0.66 (s, 3H), 0.84 (d, J = 1.7 Hz, 3H), 0.86 (d, J = 1.7 Hz, 3H), 0.90 (d, J = 6.5 Hz, 3H), 0.98 (s, 3H), 1.00–1.19 (m, 8H), 1.19–1.40 (m, 5H), 1.41–1.61 (m, 8H), 1.75–2.03 (m, 5H), 2.19 (m, 1H), 2.35 (m, 1H), 3.16 (m, 1H), 3.37 (s, 3H), 3.54 (m, 2H), 3.64 (m, 30H), 5.33 (m, 1H). 13C NMR: (100 MHz, CDCl3, 298 K) δ: 11.99, 18.85, 19.52, 22.69, 22.95, 23.95, 28.14, 28.36, 28.49, 32.03, 32.08, 35.91, 36.32, 37.00, 37.38, 39.20, 39.65, 39.92, 42.46, 50.32, 56.29, 56.91, 59.17, 67.42, 70.65, 70.72, 70.74, 71.02, 72.07, 79.62, 121.66, 141.11. HRMS (ESI): calculated mass for C44H81O9+ [M + H]+: 753.5875; found: 753.5889. The molar oligomer distribution from MS was 98.8% Chol-PEG8-OCH3 and 1.16% Chol-PEG7-OCH3; PDI = 1.000039.
Chol-PEG12-OCH3 (11)
In a two-neck 50 mL round-bottomed flask was placed a suspension of NaH (0.008 g, 60% dispersed in mineral oil, 0.20 mmol, 5 equiv.) in THF (5 mL) and added a solution of 6 (0.037 g, 0.040 mmol) in THF (5 mL) under an argon atmosphere at 0 °C. The white suspension was stirred for 2 h at 0 °C, and then once having reached room temperature, 0.005 mL of CH3I (0.080 mmol, 2 equiv) was added. The resulting mixture was stirred for 4 h, and then H2O was added. The resulting residue was dissolved in ethyl acetate (20 mL) and washed with H2O (3 × 20 mL). Layers were separated and dried over MgSO4, and the organic phase was evaporated to afford compound 11, which was purified by flash chromatography using silica as the stationary phase and a gradient of EtOAc in hexane as the mobile phase (0% to 100%) yielding pure compound 11 (0.027 g, 71% yield) as a yellow oil with 99% purity. HPLC-MS (ESI, elution condition 1): tR: 1.63 min, calcd mass for C52H97O13+: 929.69; found: 946.9 [M + H + H2O]+. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3, 298 K) δ: 0.67 (s, 3H), 0.85 (d, J = 1.8 Hz, 3H), 0.87 (d, J = 1.8 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.99 (s, 3H), 1.00–1.17 (m, 8H), 1.20–1.41 (m, 5H), 1.41–1.60 (m, 8H), 1.76–2.04 (m, 5H), 2.20 (m, 1H), 2.36 (m, 1H), 3.17 (m, 1H), 3.37 (s, 3H), 3.54 (m, 2H), 3.64 (m, 46H), 5.33 (m, 1H). 13C NMR: (100 MHz, CDCl3, 298 K) δ: 12.00, 14.26, 18.86, 19.53, 21.22, 22.70, 22.96, 23.97, 24.44, 28.16, 28.38, 28.51, 29.84, 32.05, 32.10, 35.93, 36.34, 37.02, 37.39, 39.21, 39.66, 39.94, 42.47, 50.34, 56.30, 56.93, 59.17, 67.43, 70.66, 70.71, 70.75, 71.03, 72.08, 79.63, 121.67, 141.12. HRMS (ESI): calculated mass for C52H97O13+ [M + H]+: 929.69237; found: 929.69326. Calcd for C52H100O13N+ [M + NH4]+: 946.71947; found: 946.72068. Calcd for C52H96O13K+ [M + K]+: 967.54880; found: 967.65017. The molar oligomer distribution from MS was 98.0% Chol-PEG12-OCH3, 1.86% Chol-PEG11-OCH3, and 0.16% Chol-PEG10-OCH3; PDI = 1.000055.
Synthesis of Chol-PEGn-RGD
Cyclo-(RGDfK) Peptide (12)
The synthesis was carried out manually in a filter funnel with a pore size-1 fritted glass disc that was modified with a PTFE key using 2-chlorotritil resin (10.62 g, 0.68 mmol/g loading). First, the amino acid, Gly (3.75 g), was introduced in CH2Cl2 (4 mL) using N,N-disopropylethylamine (DIEA, 10.71 mL, 5 equiv) and left for 8 h. Then, capping with CH3OH (5 mL) and DIEA (1.071 mL, 0.5 equiv) was done for 30 min and consequently washing with CH2Cl2, DMF, and CH2Cl2. The cleavage of the peptide was carried out by treatment with a CH2Cl2:TFA (99:1 v/v) mixture (15–20 mL/g of resin) for 5 min and repeated five times affording the protected peptide. Cyclization was carried out as described by Dai et al.36 with some modifications. The crude was dissolved in ethyl acetate (1.3 L), and 4.86 mL of Et3N (20 equiv) and 4.2 mL of T3P (8 equiv.) were added and stirred for 2 h. The protecting groups from the cyclic peptide were eliminated by treatment with a mixture of 40 mL of TFA:TIPS:H2O (95:2.5:2.5 v/v/v) for 3 h. The final peptide was precipitated by addition of cold diethyl ether and centrifuged, and the supernatant was discarded affording the cyclo-(RGDfK) (3.44 g, 58%) without purification. HPLC-MS (ESI, elution condition 4): tR: 2.47 min, calculated mass for C27H41N9O7+ [M + H]+: 603.68; found: 604.3, chemical purity: 97%.
Chol-PEG4-RGD (13)
In a 50 mL round-bottomed flask, Chol-PEG4 4 (0.362 g, 0.643 mmol, 1 equiv) was dissolved in anhyd CH2Cl2 (8 mL) and cooled to −5 °C. Then, 0.68 mL of a phosgene solution of 20 wt % in toluene (2 equiv) was added, and the reaction was left for 1 h at this temperature. After this time, extra phosgene solution (0.34 mL, 1equiv) was added, and the mixture was stirred an additional 1 h. When the starting material was consumed (monitored by TLC), the solvent was evaporated to dryness and co-evaporated two times with toluene to remove the residual phosgene. The crude was then dissolved in anhyd CH2Cl2 (10 mL) and cooled to 0 °C, and NHS (0.089 g, 0.773 mmol, 1.2 equiv) was dissolved in CH3CN (2 mL); DCM (4 mL) and DIEA (0.44 mL, 4 equiv) were added, and the solution was stirred for 1 h at room temperature. Then, solvents were removed under reduced pressure, and CH2Cl2 (10 mL) was added. This organic phase was washed with 5% aqueous NaHCO3 (2 × 20 mL), and the combined organic phases were dried with MgSO4 and evaporated to dryness. The crude was used without any further purification in the peptide incorporation reaction. In a 50 mL round-bottomed flask, the cyclo-(RGDfK) peptide (0.446 g, 0.536 mmol, 1 equiv) was dissolved in anhyd DMF (7 mL) and DIEA (0.09 mL, 1 equiv.), and a solution of a succinimidyl ester carbonate derivative of compound 4 (0.453 g, 0.643 mmol. 1.2 equiv) in anhyd DMF (9 mL) was added. The reaction was stirred for 30 h at room temperature. Then, the solvent was removed to dryness, and CH3CN (10 mL) was added; a white precipitate slowly appeared. The suspension was sonicated for 20 min and filtered in a glass filter funnel with a pore size-4 fritted disc. The final compound was washed with CH3CN (3 × 15 mL), H2O (5 × 15 mL), and CH3CN (3 × 15 mL) and dried overnight in the desiccator. The final product 13 (0.554 g, 87% yield) was obtained as an off-white solid. UPLC-MS (ESI, elution condition 7): tR: 1.52 min, calculated mass for C63H102O13N9: 1192.75916; found: 1192.8 [M + H]+. Chemical purity: 99%. HPLC-MS (ESI, elution condition 6): tR: 17.97 min, calculated mass for C63H102O13N9: 1192.75916; found: 1192.8 [M + H]+. Chemical purity: 99%. HRMS (ESI, elution condition 6): calculated mass for C63H102O13N9+ [M + H]+: 1192.75916; found: 1192.75932. Calcd for C63H102O13N9K+ [M + H + K]2+: 615.86116; found: 615.85870. The molar oligomer distribution from MS was 100.0% Chol-PEG4-RGD, PDI = 1.000000.
Chol-PEG8-RGD (14)
The synthesis of compound 14 was performed following the experimental procedure described previously for compound 13: using 1.044 g of compound 5 (1.413 mmol) as a starting material, 1.49 mL of phosgene solution 20 wt% in toluene (2 equiv), 0.195 g of NHS (1.695 mmol, 1.2 equiv), and 0.96 mL of DIEA (4 equiv) to generate the corresponding succinimidyl ester carbonate derivative that was used without any further purification in the peptide incorporation reaction. Peptide incorporation was carried out by conjugation of 0.978 g (1 equiv) of cyclo-(RGDfK) with the succinimidyl ester carbonate derivative of compound 5 and DIEA (0.24 mL, 1 equiv). After completion of the reaction, compound 14 was isolated following the separation procedure described previously for compound 13, affording pure compound 14 (1.417 g, 88% yield) as an off-white solid. UPLC-MS (ESI, elution condition 7): tR: 1.56 min, calculated mass for C71H118O17N9+ [M + H]+: 1368.86; found: 1369.0. Chemical purity: 97%. HPLC-MS (ESI, elution condition 6): tR: 17.68 min, calculated mass for C71H118O17N9+ [M + H]+: 1368.86; found: 1369.0. Chemical purity: 97%. HRMS (ESI): calculated mass for C71H118O17N9+ [M + H]+: 1368.86402; found: 1368.87080. The molar oligomer distribution from MS was 99.9% Chol-PEG8-RGD and 0.12% Chol-PEG7-RGD, PDI = 1.00000124.
Chol-PEG12-RGD (15)
The synthesis of compound 15 was performed following the experimental procedure described previously for compound 13: using 0.115 g of compound 6 (0.126 mmol) as a starting material, 0.132 mL of phosgene solution 20 wt% in toluene (0.251 mmol, 2 equiv), 0.017 g of NHS (0.148 mmol, 1.2 equiv), and 0.09 mL of DIEA (4 equiv) to generate the corresponding succinimidyl ester carbonate derivative that was used without any further purification in the peptide incorporation reaction. Peptide incorporation was carried out by conjugation of 0.063 g of cyclo-(RGDfK) (0.104 mmol, 1 equiv) with the succinimidyl ester carbonate derivative of compound 6 and DIEA (0.018 mL, 1 equiv). After completion of the reaction, compound 15 was isolated following the separation procedure described previously for compound 13, affording pure compound 15 (0.097 g, 60% yield) as a white oil. UPLC-MS (ES, elution condition 7): tR: 1.58 min, calculated mass for C79H134O21N9+ [M + H]+: 1544.96; found: 1545.0. Chemical purity: 95%. HPLC-MS (ES, elution condition 6): tR: 17.62 min, calculated mass for C79H134O21N9+ [M + H]+: 1544.96; found: 1545.0. Chemical purity: 95%. HRMS (ESI): calculated mass for C79H134O21N9+ [M + H]+: 1544.96888; found: 1544.97002. The molar oligomer distribution from MS was 98.4% Chol-PEG12-RGD and 1.59% Chol-PEG11-RGD; PDI = 1.00001275.
Chol-PEG16-RGD (16)
The synthesis of compound 16 was performed following the experimental procedure described previously for compound 13: using 0.114 g of compound 7 (0.104 mmol) as a starting material, 0.110 mL of phosgene solution 20 wt% in toluene (0.208 mmol, 2 equiv), 0.014 g of NHS (0.122 mmol, 1.2 equiv), and 0.071 mL of DIEA (4 equiv) to generate the corresponding succinimidyl ester carbonate derivative that was used without any further purification in the peptide incorporation reaction. Peptide incorporation was carried out by conjugation of 0.053 g of cyclo-(RGDfK) (0.088 mmol, 1 equiv) with the succinimidyl ester carbonate derivative of compound 7 and DIEA (0.010 mL, 1 equiv) to afford 16. The final crude compound was dissolved in H2O, purified by solid-phase extraction using a PoraPak Rxn cartridge (Waters) and eluting with H2O with an increasing percentage of EtOH (0 to 100%), and lyophilized. The final compound 16 was obtained as a white oil (0.075 g, 42% yield). UPLC-MS (ESI, elution condition 7): tR: 1.59 min, calculated mass for C87H149O25N9+ [M]+: 1721.5; found: 1722.2. Chemical purity: 90%. HPLC-MS (ESI, elution condition 6): tR: 18.22 min, calculated mass for C87H149O25N9+ [M]+: 1721.5; found: 1722.2. Chemical purity: 90%. HRMS (ESI): calculated mass for C87H149O25N9+ [M]+: 1720.06591; found: 1720.06576. Calcd for C87H152O25N10+ [M + NH3]+: 1737.09246; found: 1737.08824. The molar oligomer distribution from MS was 96.3% Chol-PEG16-RGD, 2.56% Chol-PEG15-RGD, and 1.16% Chol-PEG14-RGD; PDI = 1.00004543.
Chol-PEG20-RGD (17)
The synthesis of compound 17 was performed following the experimental procedure described previously for compound 13: using 0.051 g of compound 8 (0.0402 mmol) as a starting material, 0.042 mL of phosgene solution 20 wt% in toluene (0.0804 mmol, 2 equiv), 0.006 g of NHS (0.052 mmol, 1.2 equiv), and 0.03 mL of DIEA (4 equiv) to generate the corresponding succinimidyl ester carbonate derivative that was used without any further purification in the peptide incorporation reaction. Peptide incorporation was carried out by conjugation of 0.028 g of cyclo-(RGDfK) (1 equiv) with the succinimidyl ester carbonate derivative of compound 8 (1.2 equiv) and DIEA (0.007 mL, 1 equiv) to afford 17. This crude compound was dissolved in water, purified by solid-phase extraction using a PoraPak Rxn cartridge (Waters) and eluting with H2O with an increasing percentage of EtOH (0 to 100%), and lyophilized. The final product 17 was obtained as a foamy solid (0.017 g, 22% yield). UPLC-MS (ESI, elution condition 7): tR: 1.58 min, calculated mass for C95H165O29N9+ [M + H]+: 1896.17; found: 1897.7. Chemical purity: 97%. HPLC-MS (ESI, elution condition 6): tR: 18.32 min, calculated mass for C95H165O29N9+ [M + H]+: 1896.17; found: 1897.7. Chemical purity: 97%. HRMS (ESI): calculated mass for C95H165O29N9+ [M + H]+: 1896.1713; found: 1896.1697. Calcd for C95H168O29N10+ [M + NH3]+: 1913.1979; found: 1913.1933. Calcd for C95H164O29N9Na+ [M + Na]+: 1918.1533; found: 1918.1451. Calcd for C95H164O29N9K+ [M + K]+: 1934.1272; found: 1934.1198. The molar oligomer distribution from MS was 97.6% Chol-PEG20-RGD, 2.03% Chol-PEG19-RGD, and 0.41% Chol-PEG18-RGD; PDI = 1.00001936.
Synthesis of Chol-PEG4-Peptide Ligands
N-(2-Boc-aminoethyl)-3-maleimidopropanamide (18)
A solution of N-boc-ethylenediamine (522 mg, 3.26 mmol, 1.1 equiv) in CH2Cl2 (4 mL) was added to a suspension of 3-maleimidopropionic acid (500 mg, 2.96 mmol, 1.0 equiv), EDC·HCl (680 mg, 3.55 mmol, 1.2 equiv), and HOBt·H2O (543 mg, 3.55 mmol, 1.2 equiv) in CH2Cl2 (20 mL). The resulting mixture was stirred at room temperature overnight (16 h). After this time, the crude was washed with saturated NaHCO3 (3 × 25 mL), 0.5% w/v citric acid (3 × 25 mL), and brine (1 × 25 mL). The organic phase was dried over MgSO4 and evaporated. The resulting crude was purified by flash chromatography using silica as the stationary phase and a gradient of CH3OH in CH2Cl2 as the mobile phase (0 to 5%), affording compound 18 (610 mg, 1.96 mmol, 66% yield). HPLC-MS (ESI, elution condition 3): tR: 1.85 min, calculated mass for calculated mass for C14H21N3O5+ [M + H]+: 311.1; found: 312.0. Chemical purity: 99%. 1H NMR (400 MHz, CDCl3, 298 K) δ: 1.44 (s, 9H), 2.51 (t, J = 7.1 Hz, 2H), 3.28–3.20 (m, 2H), 3.37–3.29 (m, 2H), 3.84 (t, J = 7.1 Hz, 2H), 4.98 (bs, NH), 6.34 (bs, NH), 6.70 (s, 2H). 13C NMR (101 MHz, CDCl3, 298 K) δ: 170.64, 170.39, 157.05, 134.35, 79.89, 40.90, 40.24, 34.89, 34.43, 28.50. MS: calculated exact mass for C14H21N3O5+: 311.1; found by HPLC-MS (ESI): 312.0 [M + H].
N-(2-Aminoethyl)-3-maleimidopropanamide Trifluoroacetate Salt (19)
Compound 18 (93.0 mg, 299 μmol, 1.0 equiv) was dissolved in a solution of 40% TFA in CH2Cl2 (5 mL, v/v), and the resulting mixture was stirred for 1 h at room temperature. Then, the solvent was evaporated to dryness to afford compound 19 (97.0 mg, 298 μmol, 99% yield). HPLC-MS (ESI, elution condition 3): tR: 0.30 min, calculated exact mass for C9H13N3O3+ [M + H]+: 211.1, found 211.9 [M + H], chemical purity: 99%. 1H NMR (400 MHz, D2O, 298 K) δ: 6.87 (s, 2H), 3.81 (t, J = 6.5 Hz, 2H), 3.46 (t, J = 6.0 Hz, 2H), 3.12 (t, J = 6.1 Hz, 2H), 2.54 (t, J = 6.5 Hz, 2H). 13C NMR (101 MHz, D2O, 298 K) δ: 174.65, 172.62, 134.40, 38.96, 36.73, 34.55, 34.24.
Chol-PEG4-Mal (20)
A volume of 0.22 mL of a solution of phosgene 20 wt % in toluene (422 μmol, 2.0 equiv) was added to compound 4 (119 mg, 211 μmol, 1.0 equiv) dissolved in dry CH2Cl2 (4 mL) at −5 °C, and the resulting mixture was stirred for 2 h at this temperature. Then, the solvent was evaporated to dryness and co-evaporated with toluene (2 × 5 mL). A cooled solution (0 °C) of the crude in dry DMF (2 mL) was added to a solution of compound 19 (77.0 mg, 237 μmol, 1.1 equiv) in dry DMF (3 mL) and DIEA (60 μL, 353 μmol, 1.7 equiv.), and the resulting mixture was stirred for 1 h at 0 °C. After this time, the crude was evaporated to dryness and purified by flash chromatography using silica and a gradient of CH3OH in CH2Cl2 as the mobile phase (0 to 5%), affording compound 20 (58.7 mg, 73.4 μmol, 35% yield). HPLC-MS (ESI, elution condition 2): tR: 1.85 min, calculated mass for calculated mass for C45H74O9N3+ [M + H]+: 800.5; found: 817.5 [M + H + H2O]. Chemical purity: 99%. 1H NMR: (400 MHz, CDCl3, 298 K) δ: 0.67 (s, 3H), 0.85 (d, 1.8 Hz, 3H), 0.87 (d, 1.8 Hz, 3H), 0.91 (d, 6.5 Hz, 3H), 0.98 (s, 3H), 1.01–1.19 (m, 8H), 1.20–1.38 (m, 5H), 1.40–1.63 (m, 8H), 1.77–2.05 (m, 5H), 2.20 (m, 1H), 2.35 (m, 1H), 2.50 (t, J = 7.0 Hz, 2H), 3.17 (m, 1H), 3.28 (m, 2H), 3.34 (m, 2H), 3.65 (m, 14H), 3.83 (m, 2H), 4.21 (t, J = 4.5 Hz), 5.33 (m, 1H), 5.44 (bs, 1H), 6.45 (bs, 1H), 6.70 (s, 2H). 13C NMR: (100 MHz, CDCl3, 298 K) δ: 12.01, 18.87, 19.53, 21.21, 22.71, 22.96, 23.97, 24.44, 28.16, 28.38, 28.49, 32.04, 32.09, 34.50, 34.90, 35.93, 36.34, 37.01, 37.37, 39.18, 39.66, 39.93, 40.36, 40.66, 42.47, 50.33, 56.30, 56.92, 64.23, 67.38, 69.64, 70.66, 70.71, 70.76, 71.03, 79.69, 121.78, 134.38, 141.01, 157.32, 170.69. HRMS (ESI): calculated mass for C45H74O9N3+ [M + H]+: 800.5425; found: 800.5412. Calcd for C45H73O9N3Na+ [M + Na]+: 822.5244; found: 822.5222.
Transferrin Peptide Ligand Tf1 (21)
The synthesis was carried out manually in a disposable polypropylene syringe fitted with a polyethylene porous disc using the Fmoc-Rink amide aminomethyl polystyrene resin (0.74 mmol/g loading). The cleavage and protecting-group elimination of the peptide were carried out by treatment with a TFA:H2O:TIPS (95:2.5:2.5 v/v/v) mixture (15–20 mL/g of resin) for 3 h at room temperature. Then, the volume of the cleavage mixture was reduced by evaporation (approximately to 1/3), and the peptide was precipitated by addition of cold diethyl ether and centrifuged, and the supernatant was discarded. The resulting crude was purified by semipreparative reversed-phase HPLC, affording the transferrin receptor-binding peptide Tf1 as pure (781.3 mg, 73% yield). HPLC-MS (ESI, elution condition 5): tR: 1.32 min, calculated mass for C44H66O16N9S+ [M + H]+: 995.18; found: 996.4, chemical purity: 99%.
Chol-PEG4-Tf1 (22)
Compound 20 (58.7 mg, 73.4 μmol, 1.0 equiv) and Tf1 transferrin peptide ligand 21 (117 mg, 80.6 μmol, 1.1 equiv) were dissolved in DMF (2 mL), and the resulting mixture was stirred at room temperature for 36 h. Then, the solvent was evaporated to dryness, and 4 mL of CH3CN was added to the resulting crude then the mixture was sonicated for 5 min. The precipitate formed was filtered by vacuum and washed with CH3CN (3 × 5 mL), H2O (3 × 5 mL), and finally again CH3CN (3 × 5 mL). The resulting product 22 was dried by vacuum overnight (76.3 mg, 42.5 μmol, 58% yield). HPLC-MS (ESI, elution condition 6): tR: 13.50 min, calculated mass for C89H140O18N19S+ [M + 2H]2+: 897.5; found: 898.2, C89H141O18N19S+ [M + 3H]3+: 598.6; found: 599.3. Chemical purity: 99%. HRMS (ESI): calculated mass for C89H139O18N19S+ [M + H]+: 1794.02662; found: 1794.02218.
Chol-PEG4-Glutathione (23)
A mixture of 20 (61.7 mg, 77.1 μmol, 1.0 equiv) and reduced glutathione (26.1 mg, 84.8 μmol, 1.1 equiv) was suspended in DMSO (2 mL), and the crude was stirred at room temperature for 48 h under an Ar atmosphere. After this time, H2O (30 mL) is added, and a precipitate is formed. The solid formed was filtered by vacuum and washed with H2O (3 × 5 mL). The resulting product 23 was dried by vacuum overnight (41.8 mg, 37.7 μmol, 49% yield). HPLC-MS (ESI, elution condition 6): tR: 18.43 min, calculated mass for C55H91O15N6S+ [M + H]+: 1107.62; found: 1107.6. Chemical purity: 98%. HRMS (ESI): calculated mass for C55H91O15N6S+ [M + H]+: 1107.62576; found: 1107.62330.
Acknowledgments
We thank M. Díaz and M. Vilaseca (Mass Spectrometry Core Facility, IRB Barcelona) for the support with the MS data and the Peptide Synthesis Unit (U3) of Nanbiosis ICTS. This work was supported by the Spanish Government (no. RTC-2014-2207-1, Centers of Excellence Severo Ochoa), CIBER-BBN (Lipocell, no. CB06-01-0074), European Community Horizon 2020 (Smart4Fabry, no. 720942), IRB Barcelona, BBVA foundation (M.J.M.), and Generalitat de Catalunya (nos. 2017-SGR-1439 and 2017-SGR-50 and the CERCA Programme). M.J.M. is an ICREA Programme Investigator.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00130.
General methods: description and characterization data (1H NMR, 13C NMR, HRMS, and UPLC-PDA-ELSD-MS) for all Chol-PEG derivatives and 2D TOCSY and 2D NOESY NMR spectra for all Chol-PEGn-RGD (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version.
The authors declare no competing financial interest.
Supplementary Material
References
- Ercole F.; Whittaker M. R.; Quinn J. F.; Davis T. P. Cholesterol Modified Self-Assemblies and Their Application to Nanomedicine. Biomacromolecules 2015, 16, 1886–1914. 10.1021/acs.biomac.5b00550. [DOI] [PubMed] [Google Scholar]
- Grimaldi N.; Andrade F.; Segovia N.; Ferrer-Tasies L.; Sala S.; Veciana J.; Ventosa N. Lipid-Based Nanovesicles for Nanomedicine. Chem. Soc. Rev. 2016, 45, 6520–6545. 10.1039/C6CS00409A. [DOI] [PubMed] [Google Scholar]
- Briuglia M. L.; Rotella C.; McFarlane A.; Lamprou D. A. Influence of Cholesterol on Liposome Stability and on in Vitro Drug Release. Drug Deliv. Transl. Res. 2015, 5, 231–242. 10.1007/s13346-015-0220-8. [DOI] [PubMed] [Google Scholar]
- Sawant R. R.; Torchilin V. P. Liposomes as ‘Smart’ Pharmaceutical Nanocarriers. Soft Matter 2010, 6, 4026–4044. 10.1039/b923535n. [DOI] [Google Scholar]
- He Z. Y.; Chu B. Y.; Wei X. W.; Li J.; Edwards C. K. 3rd; Song X. R.; He G.; Xie Y. M.; Wei Y. Q.; Qian Z. Y. Recent Development of Poly(Ethylene Glycol)-Cholesterol Conjugates as Drug Delivery Systems. Int. J. Pharm. 2014, 469, 168–178. 10.1016/j.ijpharm.2014.04.056. [DOI] [PubMed] [Google Scholar]
- Chen H.-H.; Lu I.-L.; Liu T.-I.; Tsai Y.-C.; Chiang W.-H.; Lin S.-C.; Chiu H.-C. Indocyanine Green/Doxorubicin-Encapsulated Functionalized Nanoparticles for Effective Combination Therapy Against Human MDR Breast Cancer. Colloids Surf., B 2019, 177, 294–305. 10.1016/j.colsurfb.2019.02.001. [DOI] [PubMed] [Google Scholar]
- Mahmoud N. N.; Sabbah D. A.; Abu-Dabah R.; Abuarqoub D.; Abdallah M.; Ameerah H. I.; Khalil E. A. RSC Adv. 2019, 9, 12718–12731. 10.1039/C9RA01041F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; He Z.; Yu S.; Li S.; Ma Q.; Yu Y.; Zhang J.; Li R.; Zheng Y.; He G.; Song X. Micelles Based on Methoxy Poly(Ethylene Glycol)- Cholesterol Conjugate for Controlled and Targeted Drug Delivery of a Poorly Water Soluble Drug. J. Biomed. Nanotechnol. 2012, 8, 809–817. 10.1166/jbn.2012.1433. [DOI] [PubMed] [Google Scholar]
- Yu Y.; He Y.; Xu B.; He Z.; Zhang Y.; Chen Y.; Yang Y.; Xie Y.; Zheng Y.; He G.; Song X. Self-Assembled Methoxy Poly(Ethylene Glycol)-Cholesterol Micelles for Hydrophobic Drug Delivery. J. Pharm. Sci. 2013, 102, 1054–1062. 10.1002/jps.23418. [DOI] [PubMed] [Google Scholar]
- Jia H. R.; Zhu Y. X.; Xu K. F.; Liu X.; Wu F. G. Plasma membrane-anchorable photosensitizing nanomicelles for lipid raft-responsive and light-controllable intracellular drug delivery. J. Controlled Release 2018, 286, 103–113. 10.1016/j.jconrel.2018.07.027. [DOI] [PubMed] [Google Scholar]
- Xu K.-F.; Jia H.-R.; Zhu Y.-X.; Liu X.; Gao G.; Li Y.-H.; Wu F.-G. Cholesterol-Modified Dendrimers for Constructing a Tumor Microoenvironment-Responsive Drug Delivery System. ACS Biomater. Sci. Eng. 2019, 5, 6072–6081. 10.1021/acsbiomaterials.9b01386. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zhang X.; Wang H.-Y.; Chen Z.; Wu F.-G. Subcellullar Fate of a Fluorescence Cholesterol-Poly (Ethylen Glycol) Conjugate: An Excellent Plasma Membrane Imaging Agent. Langmuir 2016, 32, 10126–10135. 10.1021/acs.langmuir.6b02288. [DOI] [PubMed] [Google Scholar]
- Wang H.-Y.; Wua X.-W.; Jia H.-R.; Li C.; Lin F.; Chen Z.; Wu F.-G. Universal Cell Surface Imaging for Mammalian, Fungal and Bacterial Cells. ACS Biomater. Sci. Eng. 2016, 2, 987–997. 10.1021/acsbiomaterials.6b00130. [DOI] [PubMed] [Google Scholar]
- Jia H.-R.; Zhu Y.-X.; Xu K.-F.; Pan G.-Y.; Liu X.; Qiao L.; Wu F.-G. Efficient cell surface labelling of life zebrafish embryos: wash-free fluorescence imaging for cellular dynamics tracking and nanotoxicity evaluation. Chem. Sci. 2019, 10, 4062–4068. 10.1039/C8SC04884C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabbilisetty P.; Boron M.; Nie H.; Ozhegov E.; Sun X.-L. Chemical Reactive Anchoring Lipids with Different Performance for Cell Surface Re-engineering Application. ACS Omega 2018, 3, 1589–1599. 10.1021/acsomega.7b01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H.-R.; Zhu Y.-X.; Chen Z.; Wu F.-G. Cholesterol-Assisted Bacterial Cell Surface Engineering for Photodynamic Activation of Gram-Positive and Gram-Negative Bacteria. ACS Appl. Mater. Interfaces 2017, 9, 15943–15951. 10.1021/acsami.7b02562. [DOI] [PubMed] [Google Scholar]
- Mozhdehi D.; Luginbuhl K. M.; Dzuricky M.; Costa S. A.; Xiong S.; Huang F. C.; Lewis M. M.; Zelenetz S. R.; Colby C. D.; Chilkoti A. Genetically Encoded Cholesterol-Modified Polypeptides. J. Am. Chem. Soc. 2019, 141, 945–951. 10.1021/jacs.8b10687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Wang R.; Wei J.; Cheng C.; Zheng Y.; Pan Y.; He X.; Ding M.; Tan H.; Fu Q. Conformation-Directed Micelle-to-Vesicle Transition of Cholesterol-Decorated Polypeptide Triggered by Oxidation. J. Am. Chem. Soc. 2018, 140, 6604–6610. 10.1021/jacs.8b01873. [DOI] [PubMed] [Google Scholar]
- Cabrera I.; Elizondo E.; Esteban O.; Corchero J. L.; Melgarejo M.; Pulido D.; Córdoba A.; Moreno E.; Unzueta U.; Vazquez E.; Abasolo I.; Schwartz S. Jr.; Villaverde A.; Albercio F.; Royo M.; García-Parajo M. F.; Ventosa N.; Veciana J. Nanovesicle-Bioactive Conjugates to be used as Nanomedicines, Prepared by a One-Step Scalable Method Using CO2 -Expanded Solvents. Nano Lett. 2013, 13, 3766–3774. 10.1021/nl4017072. [DOI] [PubMed] [Google Scholar]
- Cabrera I.; Abasolo I.; Corchero J. L.; Elizondo E.; Gil P. R.; Moreno E.; Faraudo J.; Sala S.; Bueno D.; González-Mira E.; Rivas M.; Melgarejo M.; Pulido D.; Albericio F.; Royo M.; Villaverde A.; García-Parajo M. F.; Schwartz S. Jr.; Ventosa N.; Veciana J. α-Galactosidase-A Loaded-Nanoliposomes with Enhanced Enzymatic Activity and Intracellular Penetration. Adv. Healthcare Mater. 2016, 5, 829–840. 10.1002/adhm.201500746. [DOI] [PubMed] [Google Scholar]
- Barragan-Montero V.; Winum J. Y.; Molès J. P.; Juan E.; Clavel C.; Montero J. L. Synthesis and Properties of Isocannabinoid and Cholesterol Derivatized Rhamnosurfactants: Application to Liposomal Targeting of Keratinocytes and Skin. Eur. J. Med. Chem. 2005, 40, 1022–1029. 10.1016/j.ejmech.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Schmidt F.; Spoerner M.; Kalbitzer H. R.; König B. Synthesis of New Water-Soluble Cholesterol Derivatives. Synth. Commun. 2011, 41, 2876–2887. 10.1080/00397911.2010.515362. [DOI] [Google Scholar]
- Xie F.; Yao N.; Qin Y.; Zhang Q.; Chen H.; Yuan M.; Tang J.; Li X.; Fan W.; Zhang Q.; Wu Y.; Hai L.; He Q. Investigation of Glucose-Modifed Liposomes Using Polyethylene Glycols with Different Chain Lengths as the Linkers for Brain Targeting. Int. J. Nanomed. 2012, 7, 163–175. 10.2147/IJN.S23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanal A.; Fang S. Solid Phase Stepwise Synthesis of Polyethylene Glycols. Chem. – Eur. J. 2017, 23, 15133–15142. 10.1002/chem.201703004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranski K.; Andreev Y. G.; Bruce P. G. Synthesis of Poly(Ethylene Oxide) Approaching Monodispersity. Angew. Chem., Int. Ed. 2014, 53, 6411–6413. 10.1002/anie.201403436. [DOI] [PubMed] [Google Scholar]
- Székely G.; Schaepertoens M.; Gaffney P. R. J.; Livingston A. G. Beyond PEG2000: Synthesis and Functionalisation of Monodisperse Pegylated Homostars and Clickable Bivalent Polyethyleneglycols. Chem. – Eur. J. 2014, 20, 10038–10051. 10.1002/chem.201402186. [DOI] [PubMed] [Google Scholar]
- Segal M.; Avinery R.; Buzhor M.; Shaharabani R.; Harnoy A. J.; Tirosh E.; Beck R.; Amir R. J. Molecular Precision and Enzymatic Degradation: From Readily to Undegradable Polymeric Micelles by Minor Structural Changes. J. Am. Chem. Soc. 2017, 139, 803–810. 10.1021/jacs.6b10624. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Li X.; Shi Q.; Li Y.; Xia G.; Chen L.; Yang Z.; Jiang Z.-X. Highly Efficient Synthesis of Monodisperse Poly (Ethylene Glycols) and Derivatives through Macrocyclization of Oligo (Ethylene Glycols). Angew. Chem., Int. Ed. 2015, 54, 3763–3767. 10.1002/anie.201410309. [DOI] [PubMed] [Google Scholar]
- Wan Z.; Li Y.; Bo S.; Gao M.; Wang X.; Zeng K.; Tao X.; Li X.; Yang Z.; Jiang Z.-X. Amide Bond-Containing Monodisperse Polyethylene Glycols beyond 10,000 Da. Org. Biomol. Chem. 2016, 14, 7912–7919. 10.1039/C6OB01286H. [DOI] [PubMed] [Google Scholar]
- Ventosa L.; Veciana J.; Cabrera I.; Elizondo E.; Melgarejo M.; Royo M.; Albericio F.; Pulido D.; Sala S.; Corchero J. L.; Schwartz S.; Abasolo I.; Villaverde A. P.. Functionalized Liposomes Useful for the Delivery of Bioactive Compounds. WO2014/001509A1.
- Ahmed S. A.; Tanaka M. Synthesis of Oligo(Ethylene Glycol) toward 44-Mer. J. Org. Chem. 2006, 71, 9884–9886. 10.1021/jo0617464. [DOI] [PubMed] [Google Scholar]
- French A. C.; Thompson A. L.; Davis B. G. High-Purity Discrete PEG-Oligomer Crystals Allow Structural Insight. Angew. Chem., Int. Ed. 2009, 48, 1248–1252. 10.1002/anie.200804623. [DOI] [PubMed] [Google Scholar]
- Miura Y.; Takenaka T.; Toh K.; Wu S.; Nishihara H.; Kano M. R.; Ino Y.; Nomoto T.; Matsumoto Y.; Koyama H.; Cabral H.; Nishiyama N.; Kataoka K. Cyclic RGD-Linked Polymeric Micelles for Targeted Delivery of Platinum Anticancer Drugs to Glioblastoma through the Blood-Brain Tumor Barrier. ACS Nano 2013, 7, 8583–8592. 10.1021/nn402662d. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Engler J. A.; Collawn J. F.; Moore B. A. Receptor Mediated Uptake of Peptides That Bind the Human Transferrin Receptor. Eur. J. Biochem. 2001, 268, 2004–2012. 10.1046/j.1432-1327.2001.02073.x. [DOI] [PubMed] [Google Scholar]
- Gaillard P. J.; Appeldoorn C. C.; Rip J.; Dorland R.; van der Pol S. M.; Kooij G.; de Vries H. E.; Reijerkerk A. Enhanced Brain Delivery of Liposomal Methylprednisolone Improved Therapeutic Efficacy in a Model of Neuroinflammation. J. Controlled Release 2012, 164, 364–369. 10.1016/j.jconrel.2012.06.022. [DOI] [PubMed] [Google Scholar]
- Dai X. D.; Su Z.; Liu J. O. An Improved Synthesis of a Selective Alpha(v)Beta(3)-Integrin Antagonist Cyclo(-RGDfK-). Tetrahedron Lett. 2000, 41, 6295–6298. 10.1016/S0040-4039(00)01060-1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.