

Abstract
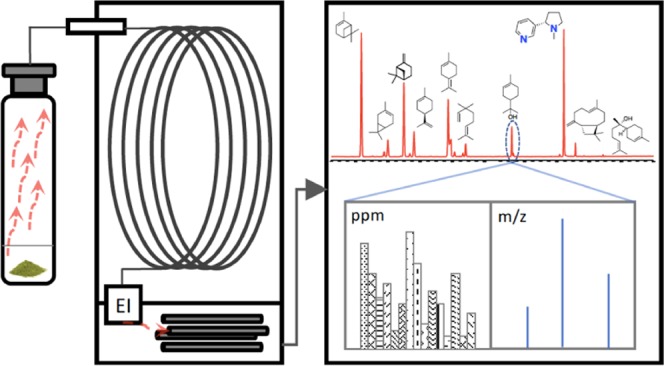
Plants are the main sources of many high-value bioactive terpenoids used in the medical, fragrance, and food industries. Increasing demand for these bioactive plants and their derivative products (e.g., cannabis and extracts thereof) requires robust approaches to verify feedstock, identify product adulteration, and ensure product safety. Reported here are single-laboratory validation details for a robust testing method to quantitate select terpenes and terpenoids in dry plant materials and terpenoid-containing vaping liquids (e.g., a derivative product) using high-temperature headspace gas chromatography–mass spectrometry, with glycerol used as a headspace solvent. Validated method recoveries were 75–103%, with excellent repeatability (relative standard deviation (RSD) < 5%) and intermediate precision (RSD < 12%). The use of high-temperature headspace (180 °C) permitted terpene and terpenoid profiles to be monitored at temperatures consistent with vaping conditions.
Introduction
Medicinal and psychoactive plants have been used throughout human history, with bioactive small molecules, also known as natural products or specialized metabolites, being the key components that elicit biological effects. Although significant progress in biotechnology and chemical synthesis of these valuable molecules has been made, plants remain the main source for most of them.1,2 Reliable and robust authentication procedures are therefore needed to help commercial suppliers of natural product-containing plants and their derived products validate the integrity of their supply chain, while at the same time providing regulatory agencies with tools to evaluate product authenticity and safety.3,4 This has become a more pressing issue as the market for bioactive plants and their extracts are fast expanding, particularly following the legalization of recreational cannabis (Cannabis sativa L.) consumption in jurisdictions around the world.
Of all the natural products found in plants, terpenoids constitute the largest, most functionally and structurally diverse class with more than 50 000 compounds identified to date.5 Some terpenes and terpenoids have been reported to possess anticancer, pesticidal, antimicrobial, anti-inflammatory, and immuno-modulatory properties.6−9 While these bioactive properties are of immense academic and industrial interest, terpenes and terpenoids are still best known for their flavor and aroma profiles that contribute to consumer preferences (e.g., essential oils, food flavoring, perfume, etc.). Such preferences are becoming apparent in the way consumers select the cannabis they are purchasing for recreational purposes as well.10,11 In addition to their distinct sensory attributes, terpenes and terpenoids may exert some “entourage” neurological effects on the cannabis user experience,12,13 all of which make their accurate quantitation a critical analytical objective.
In cannabis, terpene and terpenoid profiles include many mono- and sesquiterpenoids (Figure 1) at different abundance levels, in addition to the well-known psychoactive Δ9-tetrahydrocannabinol (THC) and the bioactive cannabidiol (CBD).14,15 Regardless of different opinions on the medicinal value of cannabis, existing evidence suggests that it should be subjected to rigorous quality and safety tests before entering the market, as is required for other bioactive plant and natural health products.16,17 Given their potential bioactivity, such characterization should include terpenoid quantitation.
Figure 1.

Select terpenes and terpenoids naturally occurring in important medicinal and recreational plants such as cannabis and included in this work.
In addition to their natural occurrence in plant materials, terpenoids are found in e-cigarette vaping liquids (e-juices) and supplements where they impart a wide range of flavors (i.e., piney, sweet, succulent, bitter, citrusy, etc.). Although adding flavors is prohibited in conventional cigarette products in Canada, the United States, and the European Union, at the time of manuscript preparation, this is not the case for e-juices. Such flavorants appeal to users despite the possible health implications of vaping.18 This is of acute public concern given that vaping has long been advertised as a safer alternative to smoking and has particularly attracted younger consumers. With recent vaping-related deaths and subsequent legislative proposals to ban or at least discourage flavorant-added e-juices,19,20 it is important that flavorants, including terpenoids, in vaping liquids should be analyzed quantitatively for quality control purposes. Such efforts will support an understanding of the potential safety impacts that they may have.
Analysis of terpenes and terpenoids in plants and other products generally involves organic solvent extraction of plant materials followed by direct injection and analysis by gas chromatography (GC) coupled with flame ionization or mass spectrometry (MS) detectors.21−23 While effective at yielding an exhaustive chemical profile from the plant materials’ complex organic matrix, several extraction steps using organic solvents and sample concentration may be required.24 In addition, complex sample matrices could be problematic for direct injection due to coextracted matrix interferences—particularly for gas chromatography–mass spectrometry (GC–MS) where the coextracted low-volatility macromolecules can interfere with chromatography and introduce contamination in injection ports.
Headspace sampling techniques are frequently employed, as only compounds with suitable vapor pressures are analyzed.25−27 Furthermore, the resulting gas phase phytochemicals may better reflect those compounds that users of vaporized products are exposed to. In addition, much of the sensory attributes to these products are related to terpenes and terpenoids—such users do not employ vigorous, exhaustive extraction with organic solvents prior to product use. This is especially true of materials that are highly processed or intended for inhalation after combustion or high-temperature vaporization. Headspace solid-phase microextraction (SPME) methods have been developed for analyzing terpenoids in liquid and solid plant products,23,28,29 but variation in chemical selectivity depending on the chemistry of the fiber can make comprehensive terpenoid quantitation difficult. Full evaporative transfer (FET) is an alternate approach that has been recently reported for the quantitation of 93 terpenes and terpenoids in cannabis.27 While FET does not suffer from the challenges of SPME fiber chemistry, care must be taken when using FET, as it could be problematic if nontrivial sample amounts (e.g., >5 mg) are used; higher sample quantities are likely required to ensure representative results, even after thorough processing.23,30
Here, we present the use of GC–MS with high-temperature headspace sampling for the quantitation of select terpenes and terpenoids in plant samples and e-juices. The reported method is simple, sensitive, and specific, and should be broadly amenable to the quantitation of all mono- and sesquiterpenoids due to the high-temperature incubation utilized. To demonstrate high-temperature headspace GC–MS as an effective tool to analyze various matrices and analyte classes, method validation data are presented for cannabis terpenoid mixtures and e-juice matrices in addition to plant materials.
Results and Discussion
Method Development and Optimization
The chromatographic gradient was optimized to resolve all 30 mono- and sesquiterpenoids of interest using the selected ion monitoring (SIM) mode (Table S1; Figures 1 and 2). Using this thermal program, the stable working conditions for the system were optimized by establishing analyte recoveries and response repeatability. This was evaluated using a range of stock solvents, headspace carrier liquids, and vial incubation and loop/sample path temperatures (with 10–12 replicate injections under all conditions evaluated).
Figure 2.

Sample selected ion monitoring (SIM) chromatogram for the select terpenes and terpenoids under investigation. Numbers represent structures depicted in Figure 1. IS: internal standard, (±)-linalool-d3.
The first headspace liquid examined was brine, as this is an inexpensive and easily prepared solution that should increase the equilibrium vapor pressure of terpenes. Terpenoid standards were prepared in hexanes at 1 μg/mL and fortified to 5 ng/mL in 5 mL of brine. These samples were analyzed with a 5:1 split ratio and vial incubation and sample loop temperatures of 85 °C. The intention with this approach was to leverage the ability of hexane to quantitatively extract terpenes from plant materials with the reduction of coextraction interferences that headspace analysis would provide. Analyte responses showed low calculated relative standard deviations (RSD; ≤ 5%) for the low-boiling analytes (mostly nonoxygenated monoterpenes, compounds 1–13, Figure 1), and larger variations (up to 20% RSD) for the higher boiling components (oxygenated monoterpenoids and sesquiterpenoids, compounds 14–30, Figure 1). To improve method precision, different sample path temperatures (95–125 °C) were evaluated, as the higher sample path temperatures might prevent condensation of analytes. In addition, higher septum purge flows (10 and 20 mL/min) were also evaluated for possible flow dynamic effects within the high-temperature headspace sampler. Results showed that an offset of 10–20 °C between vial incubation and sample path temperatures and a purge flow of 20 mL/min improved the precision of instrumental replicates (n = 10). At 5 mL/min purge flow, a 100 °C sample path temperature produced all RSD values below 20%, with all but three below 10%. When higher septum purge flows were applied, all RSDs were less than 10% at 100 and 125 °C sample path temperatures (Table 1). Neither increasing septum purge flow beyond 20 mL/min nor changing needle purge flow levels improved the RSDs further. A substantial gap between vial incubation and sample path temperatures (85 and 190 °C) did not improve repeatability (Table 1).
Table 1. Calculated Absolute Response Precision (Relative Standard Deviation; RSD) of Analytes in Brine Incubated at 85 °C and Analyzed with Different Sample Path Temperatures and Septum Purge Flowsa.
| RSD
(%) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| septum purge flow (mL/min) | 5 |
10 |
20 |
|||||||
| ID | sample path temperature (°C) | 85b | 95b | 100c | 125b | 100c | 125c | 100c | 125c | 190c |
| 1 | α-pinene | 2.6 | 4.1 | 4.9 | 2.0 | 4.3 | 3.9 | 2.4 | 1.6 | 6.3 |
| 2 | camphene | 3.5 | 4.5 | 5.1 | 3.8 | 4.9 | 4.5 | 2.4 | 1.6 | 6.7 |
| 3 | β-myrcene | 5.5 | 5.9 | 5.6 | 7.6 | 5.9 | 5.9 | 3.5 | 2.5 | 8.1 |
| 4 | (−)-β-pinene | 5.4 | 5.5 | 5.6 | 7.0 | 5.7 | 5.5 | 3.2 | 2.3 | 7.6 |
| 5 | δ-3-carene | 3.7 | 4.0 | 4.9 | 4.5 | 4.7 | 4.6 | 2.7 | 1.5 | 6.4 |
| 6 | α-terpinene | 9.4 | 6.3 | 6.2 | 9.6 | 6.0 | 6.0 | 3.3 | 2.8 | 8.2 |
| 7 | cis-β-ocimene | 3.4 | 3.8 | 5.3 | 3.7 | 4.7 | 4.2 | 2.5 | 1.9 | 7.7 |
| 8 | (+)-limonene | 3.0 | 4.0 | 5.0 | 2.6 | 4.1 | 3.9 | 2.5 | 1.6 | 6.5 |
| 9 | p-cymene | 7.4 | 5.1 | 5.7 | 6.8 | 5.4 | 5.1 | 2.8 | 2.2 | 6.8 |
| 10 | trans-β-ocimene | 5.5 | 4.8 | 5.3 | 5.9 | 5.2 | 4.9 | 3.1 | 2.1 | 7.6 |
| 11 | 8-cineole (eucalyptol) | 4.6 | 4.6 | 5.0 | 4.8 | 4.8 | 4.1 | 2.7 | 1.3 | 7.6 |
| 12 | γ-terpinene | 3.7 | 3.5 | 4.8 | 3.5 | 4.2 | 4.1 | 2.8 | 1.6 | 6.4 |
| 13 | terpinolene | 8.2 | 4.9 | 5.8 | 8.7 | 6.0 | 5.3 | 2.9 | 2.3 | 7.6 |
| 14 | cis-linalool oxide | 15 | 9.1 | 7.4 | 15 | 8.6 | 7.1 | 3.7 | 3.8 | 9.3 |
| 15 | trans-linalool oxide | 12 | 7.5 | 6.5 | 9.3 | 6.6 | 5.2 | 2.9 | 2.9 | 8.0 |
| 16 | linalool | 11 | 5.6 | 6.6 | 7.0 | 5.6 | 4.9 | 2.9 | 2.0 | 10 |
| 17 | (−)-isopulegol | 10 | 6.1 | 7.4 | 7.2 | 6.1 | 5.1 | 3.2 | 2.3 | 9.3 |
| 18 | α-terpineol | 14 | 8.7 | 7.2 | 13 | 7.4 | 6.6 | 3.4 | 3.1 | 11 |
| 19 | citronellol | 19 | 9.6 | 16 | 36 | 12 | 7.5 | 6.2 | 5.1 | 9.0 |
| 20 | nerol | 7.1 | 9.3 | 13 | 24 | 10 | 7.0 | 4.5 | 4.6 | 11 |
| 21 | geraniol | 20 | 11 | 17 | 41 | 14 | 8.0 | 7.4 | 7.0 | 9.6 |
| 22 | β-damascenone | 4.3 | 5.4 | 5.9 | 3.5 | 4.6 | 4.8 | 2.6 | 2.2 | 11 |
| 23 | β-caryophyllene | 6.1 | 4.7 | 5.6 | 6.1 | 4.9 | 5.1 | 2.5 | 2.4 | 7.9 |
| 24 | β-damascone | 7.8 | 6.9 | 6.6 | 9.1 | 6.0 | 6.5 | 3.6 | 2.8 | 11 |
| 25 | α-humulene | 6.3 | 5.3 | 5.8 | 7.6 | 5.5 | 5.3 | 2.8 | 2.5 | 7.8 |
| 26 | cis-nerolidol | 11 | 8.7 | 8.7 | 10 | 6.5 | 6.3 | 4.6 | 6.0 | 14 |
| 27 | trans-nerolidol | 13 | 11 | 9.9 | 13 | 8.1 | 7.0 | 6.0 | 7.1 | 15 |
| 28 | (−)-guaiol | 14 | 9.3 | 8.6 | 15 | 7.6 | 7.0 | 4.4 | 5.4 | 15 |
| 29 | (−)-caryophyllene oxide | 14 | 37 | 9.0 | 19 | 7.8 | 27 | 5.5 | 6.2 | 4.9 |
| 30 | (−)-α-bisabolol | 14 | 13 | 10 | 14 | 7.6 | 6.8 | 8.2 | 8.9 | 15 |
Values were calculated from 10–12 replicates.
n = 10.
n = 12.
Additional headspace liquids were explored, including dimethyl sulfoxide (DMSO), dimethylformamide (DMF), and glycerol. It was hypothesized that their higher boiling points compared to water might improve the equilibrium vapor pressure of terpenes with lower inherent volatility (i.e., higher boiling terpenes). Also, the ability to use high temperatures with glycerol would facilitate investigations into profiling terpenoids across a wider range of incubation temperatures (vide infra). Analysis of terpenes and terpenoids fortified in DMSO and DMF at various incubation temperatures up to 175 and 140 °C, respectively, revealed low signal-to-noise ratios for most analytes, and nondetects for many others (data not shown). For this reason, further evaluations with these solvents were not explored. Conversely, when glycerol was used, the most abundant raw responses for higher boiling terpenes were observed (relative to brine), likely due to the utilized vial incubation temperature of 180 °C and a sample path temperature of 200 °C. Of the headspace liquids evaluated, glycerol was unique in permitting the exploration of very high incubation temperatures due to its boiling point of 290 °C, versus 189 and 153 °C for DMSO and DMF, respectively. This facilitated the better transfer of analytes to the gas phase, and improved precision.
It was interesting to note that analyte recoveries for calibrators and spike-recovery samples using glycerol as the headspace solvent were dependent on the solvent used to prepare the terpenoid stock solutions. The low-boiling solvent hexanes (68 °C) and the high-boiling solvent tetradecane (254 °C) were used to investigate this effect. Analyses of samples fortified with terpenoids in the absence of plant material (i.e., a calibration sample) in hexane-based stocks showed that glycerol samples displayed higher absolute responses for low-boiling terpenoids (compounds 1–13, Figure 1), but lower absolute responses (as low as 10–20%) for high-boiling terpenoids (compounds 14–30, Figure 1) compared to brine samples. When the analogous tetradecane-based terpenoid stocks were used, most analytes displayed higher responses with glycerol compared to brine (Figure S1). This suggested that the solvents used to prepare stock solutions increased the partial pressure in the headspace such that the amount of analyte in the headspace decreased when the low-boiling hexane was used. It is postulated that the higher boiling point of tetradecane mitigated this effect. When the plant material was also present in the headspace vials, higher absolute responses relative to the data obtained in brine were observed for the majority of terpenoids when glycerol was used regardless of terpenoid stock solvents (hexanes or tetradecane). The reason for this effect is currently being explored.
Based on these investigations, terpenoid stock solutions for calibration samples and spike-recovery tests were prepared in tetradecane and glycerol was selected as the optimal headspace carrier liquid.
Method Validation for Plant Tissue Analysis
The developed method was evaluated for the dynamic range, repeatability (n = 5), intermediate precision (n = 17, over three days), carry-over, method detection limit (MDL), method reporting limit (MRL), and spike recovery. Calibration functions for all 30 terpenes and terpenoids were determined from 25 to 4000 ng/mL (equivalent to 2.5–400 ppm in plant material; dynamic range = 2.2). The split ratio can be adjusted to accommodate different expected terpenoid concentrations or to permit the evaluation of different quantities of plant material (i.e., not 50 mg). However, the issue of suitable sample quantity to overcome potential homogeneity issues needs to be considered; erring on the side of more material sampled for a given analysis is generally the prudent approach, with the authors suggesting 50 mg as a minimum quantity. Across a range of 25–4000 ng/mL, all calibration functions had correlation coefficients >0.99 and the accuracy of all calibration concentrations were ± 20% (data not shown). The calibration functions were consistent across three days of method validation and subsequent sample analyses. Additionally, all analytes presented with <0.1% carry-over when a method blank was analyzed immediately after a high concentration calibration sample.
Stinging nettle was selected as the matrix for method development, and validation as the dry tissues of this plant contain nominal incurred terpenoids, which made it an ideal blank matrix with which spike-recovery studies could be conducted (Figure S2). This was necessary for the method development reported herein, as the intended matrices for this method (i.e., cannabis and industrial hemp) have high concentrations of many terpenoids, such that performing spike-recovery studies would be problematic due to limitations in the dynamic range. Using dry, ground stinging nettle tissue, terpenoid spike recoveries of at least 70% were achieved, with 27 out of 30 terpenes and terpenoids reaching 80–108% at 10 ppm (Table 2). At the evaluated mid and high spike-recovery concentrations (125 and 400 ppm, respectively), the spike-recovery data ranged from 82 to 104% for all analytes, except for (−)-caryophyllene oxide, which has a high concentration spike recovery of 129% (Table 2). MDLs for all analytes were less than 4 ppm in the plant (40 ppm in the vial), and MRLs were verified at 10 ppm in the plant (100 pm in the vial) for 50 mg of plant material (Table 2). Lower MRLs could be achieved for many analytes, but given the expected concentrations in cannabis and industrial hemp, it was not necessary to fully optimize this statistically verified reporting limit. All of the method validation data reported herein demonstrate the suitability of the method for the intended goal of quantitating terpenoids.
Table 2. Method Validation for Quantitating Terpenoids in Plant Materials by Headspace GC–MSa.
| limit
(ppm) |
recovery
(%) |
RSD
(%) |
||||||
|---|---|---|---|---|---|---|---|---|
| ID | analyte | MDL | MRL | low (10 ppm) | mid (125 ppm) | high (350 ppm) | intraday | interday |
| 1 | α-pinene | 0.95 | 10.0 | 84.2 | 86.4 | 83.3 | 1.34 | 8.70 |
| 2 | camphene | 0.87 | 10.0 | 83.3 | 86.2 | 85.1 | 0.85 | 7.33 |
| 3 | β-myrcene | 1.13 | 10.0 | 82.8 | 89.1 | 87.9 | 1.19 | 4.77 |
| 4 | (−)-β-pinene | 1.03 | 10.0 | 80.6 | 88.5 | 86.4 | 1.07 | 4.12 |
| 5 | δ-3-carene | 1.28 | 10.0 | 83.4 | 89.0 | 85.5 | 0.90 | 3.41 |
| 6 | α-terpinene | 1.72 | 10.0 | 99.5 | 93.6 | 84.8 | 1.90 | 11.2 |
| 7 | cis-β-ocimene | 1.57 | 10.0 | 74.2 | 97.4 | 88.5 | 1.95 | 7.60 |
| 8 | (+)-limonene | 1.37 | 10.0 | 80.4 | 91.5 | 85.2 | 1.25 | 6.38 |
| 9 | p-cymene | 1.19 | 10.0 | 70.2 | 81.8 | 82.8 | 1.30 | 6.92 |
| 10 | trans-β-ocimene | 1.36 | 10.0 | 90.5 | 95.6 | 89.3 | 1.07 | 6.36 |
| 11 | 8-cineole (eucalyptol) | 0.93 | 10.0 | 81.8 | 95.1 | 92.7 | 1.39 | 8.71 |
| 12 | γ-terpinene | 0.94 | 10.0 | 84.0 | 101 | 89.1 | 0.90 | 6.02 |
| 13 | terpinolene | 1.39 | 10.0 | 93.4 | 87.5 | 85.9 | 2.11 | 5.77 |
| 14 | cis-linalool oxide | 2.10 | 10.0 | 93.3 | 96.9 | 91.6 | 3.65 | 6.76 |
| 15 | trans-linalool oxide | 1.32 | 10.0 | 95.3 | 102 | 102 | 2.32 | 5.76 |
| 16 | linalool | 1.09 | 10.0 | 97.4 | 103 | 99.0 | 1.24 | 2.29 |
| 17 | (−)-isopulegol | 1.19 | 10.0 | 94.9 | 95.0 | 101 | 1.81 | 4.79 |
| 18 | α-terpineol | 1.41 | 10.0 | 91.5 | 96.1 | 102 | 2.58 | 6.21 |
| 19 | citronellol | 1.30 | 10.0 | 79.5 | 90.4 | 101 | 1.76 | 4.91 |
| 20 | nerol | 3.15 | 10.0 | 104 | 98.0 | 96.6 | 2.13 | 9.41 |
| 21 | geraniol | 2.01 | 10.0 | 79.3 | 93.5 | 90.8 | 3.74 | 5.67 |
| 22 | β-damascenone | 1.15 | 10.0 | 96.5 | 95.9 | 91.5 | 2.14 | 3.15 |
| 23 | β-caryophyllene | 2.43 | 10.0 | 110 | 99.9 | 85.3 | 1.87 | 5.10 |
| 24 | β-damascone | 0.98 | 10.0 | 88.5 | 90.1 | 91.5 | 2.02 | 6.41 |
| 25 | α-humulene | 2.49 | 10.0 | 87.6 | 98.2 | 84.8 | 2.38 | 5.61 |
| 26 | cis-nerolidol | 2.02 | 10.0 | 86.9 | 86.8 | 94.0 | 2.38 | 8.10 |
| 27 | trans-nerolidol | 2.54 | 10.0 | 90.1 | 87.8 | 95.3 | 1.50 | 4.29 |
| 28 | (−)-guaiol | 1.77 | 10.0 | 95.0 | 92.5 | 99.4 | 2.20 | 4.75 |
| 29 | (−)-caryophyllene oxide | 3.10 | 10.0 | 108 | 104 | 129 | 3.46 | 6.05 |
| 30 | (−)-α-bisabolol | 2.42 | 10.0 | 87.5 | 87.1 | 93.0 | 2.83 | 5.80 |
Concentrations are presented based on the plant weight. MDL: method detection limit. MRL: method reporting limit. The calibration ranges were from 2.5 to 400 ppm (25–4000 ppb in the vial), except for nerol and (−)-caryophyllene oxide (5–400 ppm in samples or 50–4000 ppb in the vial).
As a relatively new market segment, there is currently a dearth of proficiency testing and/or certified reference materials for cannabis and industrial hemp. This is especially true for terpenoids that, unlike cannabinoids or pesticides, are not faced with any regulatory restrictions regarding their absolute concentrations. The absence of these materials makes it challenging to demonstrate the accuracy of an analytical method for a broad range of terpenoids—a new program exists in the United States for hemp flower, but the number of terpene values provided is limited to ten. To provide a better assessment of method performance for all terpenes and terpenoids evaluated herein, a cross-validation approach was utilized to verify method accuracy. Since the presence of incurred terpenoids was not a concern for the cross-validation, hops (Humulus lupulus L. flower), a close phylogenetic relative of the family Cannabaceae (i.e., cannabis), was used.
The method used for the cross-validation is an ISO/IEC 17025 accredited method for quantitating terpenes in plant material based on a hexane extraction, followed by direct analysis of the organic extract using GC–tandem mass spectrometry (method validation parameters were consistent with those reported herein for the high-temperature headspace method; Table S5). In addition to being accredited, this method has also passed several rounds of proficiency testing for terpenes in hemp oil. While not an exact matrix match, these results do provide strong support for the accuracy of the method as a benchmark. When comparing analytes above the respective MRLs for each method, a correlation matrix was obtained that demonstrated the accuracy of the high-temperature headspace method developed herein (Figure 3A).
Figure 3.

Cross-validation of terpene/terpenoid quantitation between headspace GC–MS and liquid-injection GC–tandem mass spectrometry (GC–MS/MS). (A) Dry hop tissue (unit: ppm). (B) Commercial cannabis terpene mixtures (unit: mg/mL).
Method Validation For e-Juices and Related Vaping Products
The original focus of this method development project was on the quantitation of terpenes in plant material. Paralleling our method development efforts in plant material were reports coming out about the safety of e-cigarettes and related vaping products intended to be inhaled after high-temperature vaporization.18,20,31 E-juices are generally formulated in a mixture of glycerol and propylene glycol, with other common ingredients, including nicotine, caffeine, and flavoring agents (i.e., terpenes).32 Given our finding that glycerol was a suitable matrix for the high-temperature headspace quantitation of terpenes, it was decided that the scope of this method should be extended to include the quantitation of terpenes in e-juices and related products. Understanding the terpene concentrations is likely to become a critical regulatory issue, as it has been documented that flavoring agents can decompose during consumer use of e-cigarettes to produce potentially harmful breakdown products.18,33−35 Since nicotine is frequently included in e-juice formulations, it was also included in the expansion of the analytical scope of the method reported herein (Figure 4).
Figure 4.

Extracted ion chromatogram from the analysis of a commercial e-juice for select terpenes, terpenoids, and nicotine in the same injection. Terpene/terpenoid and nicotine peaks were extracted with m/z 93 and 84, respectively. Numbers represent structures depicted in Figure 1.
A mixture of glycerol and propylene glycol (1:1 ratio) was used as a surrogate for typical e-juice matrices to develop the method for terpene/terpenoid and nicotine quantitation. Using this mixture, terpenoid spike recoveries of at least 93% were obtained (Table 3). At the evaluated mid and high spike-recovery concentrations in e-juice (75 and 250 ppm for terpenoids, and 7.5 and 25 mg/mL for nicotine), the spike-recovery data ranged from 83 to 104% for all analytes. MDLs for all terpenoids and nicotine were less than 3 ppm for terpenoids and 0.1 mg/mL for nicotine, and MRLs were verified at 10 ppm for terpenoids and 0.5 mg/mL for nicotine (Table 3). These method performance parameters are all suitable for the intended scope, with typical nicotine concentrations in e-juices ranging mostly from 1 to 30 mg/mL.36,37
Table 3. Method Validation for Quantitating Terpenoids and Nicotine in e-Juices by Headspace GC–MSa.
| limitb |
recovery
(%)c |
RSD
(%) |
||||||
|---|---|---|---|---|---|---|---|---|
| ID | analyte | MDL | MRL | low | mid | high | intraday | interday |
| 1 | α-pinene | 0.04 | 5.00 | 105 | 94.6 | 101 | 2.64 | 4.86 |
| 2 | camphene | 0.05 | 5.00 | 104 | 94.2 | 99.9 | 2.16 | 4.35 |
| 3 | β-myrcene | 0.13 | 5.00 | 99.7 | 95.9 | 99.1 | 1.78 | 2.84 |
| 4 | (−)-β-pinene | 0.22 | 5.00 | 105 | 99.0 | 104 | 2.24 | 4.04 |
| 5 | δ-3-carene | 0.16 | 5.00 | 105 | 98.8 | 104 | 2.08 | 3.97 |
| 6 | α-terpinene | 0.13 | 5.00 | 101 | 94.7 | 99.6 | 1.91 | 4.01 |
| 7 | cis-β-ocimene | 0.77 | 5.00 | 102 | 97.2 | 99.0 | 2.44 | 2.40 |
| 8 | (+)-limonene | 0.12 | 5.00 | 98.7 | 94.8 | 99.2 | 2.36 | 3.58 |
| 9 | p-cymene | 0.10 | 5.00 | 100 | 94.2 | 99.6 | 2.59 | 4.15 |
| 10 | trans-β-ocimene | 0.62 | 5.00 | 99.0 | 94.3 | 98.5 | 2.36 | 3.49 |
| 11 | 8-cineole (eucalyptol) | 0.18 | 5.00 | 101 | 95.3 | 98.6 | 1.81 | 2.91 |
| 12 | γ-terpinene | 0.16 | 5.00 | 101 | 94.4 | 99.3 | 2.26 | 3.82 |
| 13 | terpinolene | 0.33 | 5.00 | 105 | 97.8 | 102 | 2.21 | 3.66 |
| 14 | cis-linalool oxide | 0.59 | 5.00 | 104 | 96.5 | 99.3 | 3.01 | 2.78 |
| 15 | trans-linalool oxide | 1.83 | 5.00 | 105 | 102 | 104 | 3.02 | 2.49 |
| 16 | linalool | 1.35 | 10.0 | 101 | 95.7 | 97.0 | 2.65 | 2.18 |
| 17 | (−)-isopulegol | 0.35 | 5.00 | 101 | 96.2 | 98.2 | 3.18 | 2.52 |
| 18 | α-terpineol | 0.53 | 5.00 | 97.3 | 93.2 | 96.7 | 3.81 | 3.65 |
| 19 | citronellol | 2.54 | 10.0 | 97.8 | 95.3 | 94.7 | 2.69 | 2.83 |
| 20 | nerol | 0.69 | 5.00 | 96.5 | 96.1 | 99.4 | 3.61 | 3.49 |
| 21 | geraniol | 1.66 | 10.0 | 104 | 91.3 | 96.0 | 4.72 | 4.61 |
| 22 | β-damascenone | 0.67 | 5.00 | 98.3 | 103 | 103 | 1.74 | 1.82 |
| 23 | β-caryophyllene | 0.50 | 5.00 | 103 | 95.5 | 101 | 1.87 | 4.31 |
| 24 | β-damascone | 0.63 | 5.00 | 95.7 | 94.2 | 95.6 | 1.54 | 2.30 |
| 25 | α-humulene | 0.74 | 5.00 | 105 | 96.5 | 101 | 1.56 | 3.86 |
| 26 | cis-nerolidol | 1.58 | 10.0 | 100 | 96.2 | 98.3 | 2.13 | 2.11 |
| 27 | trans-nerolidol | 0.41 | 5.00 | 100 | 93.7 | 96.9 | 1.97 | 2.95 |
| 28 | (−)-guaiol | 0.39 | 5.00 | 94.6 | 89.0 | 93.3 | 0.92 | 3.58 |
| 29 | (−)-caryophyllene oxide | 0.33 | 5.00 | 97.8 | 97.8 | 97.3 | 2.08 | 2.14 |
| 30 | (−)-α-bisabolol | 0.96 | 5.00 | 101 | 96.0 | 97.5 | 1.34 | 2.15 |
| 31 | nicotine | 0.05 | 0.50 | 94.3 | 101 | 102 | 3.53 | 2.71 |
Concentrations are presented based on e-juice volume. MDL: method detection limit. MRL: method reporting limit. The calibration ranges for terpenoids were from 1 to 300 ppm (10–3000 ppb in the vial), except for trans-β-ocimene, linalool, citronellol, and geraniol (5–300 ppm, or 50–3000 ppb in the vial). The calibration range for nicotine was from 0.25 to 30 mg/mL (2.5–300 μg/mL in the vial).
Unit: ppm for terpenoids, and mg/mL for nicotine.
Concentration: Low-, mid-, and high-level concentrations for terpenoids are 10, 75, and 250 ppm, respectively. Low-, mid-, and high-level concentrations for nicotine are 1, 7.5, and 25 mg/mL, respectively.
Using this method, the terpenoids of interest were quantitated in commercially available terpenoid distillates and nicotine-containing e-juices. Owing, again, to the lack of suitable proficiency testing and/or certified reference materials, a cross-validation approach (using the same method as reported for cross-validation of terpene quantitation in plant material) was utilized to evaluate method accuracy for the terpenoid distillates. The data produced were in excellent agreement (Figure 3B), demonstrating the applicability of this method for the quantitation of terpenoids in e-juices and other glycerol-based vaporization products.
In addition to terpenoids, the method reported here is suitable for the analysis of nicotine (Figure 4, Table 3)—a bioactive compound for which concerns about the combined neurological impact with cannabis38,39 has been raised. In 2018, the Canadian Task Force on Cannabis Legalization and Regulation also recommended that mixed products, including those containing both cannabis and nicotine and/or caffeine, should be prohibited.40 In this context, this method could be used as a regulatory tool to screen for nicotine, while at the same time quantitating (or profiling) the terpenoids present.
Conclusions
The headspace GC–MS analysis method presented here facilitates the precise quantitation of select terpenes and terpenoids in plant materials and vaping liquids with minimal sample preparation. Sample matrices (plant tissues and liquid extracts) that do not affect method performance and critical nonterpenoid components of e-juices, such as nicotine, can also be included in the analysis.
Materials and Methods
General Details
For dry plant tissue, the sample was homogenized using a commercial coffee grinder. Samples and standards were weighed on a VWR-164AC analytical balance (VWR) and analyzed using a TriPlus 500 GC Headspace Autosampler and a TRACE 1300 GC coupled with an ISQ 7000 Single Quadrupole MS System (Thermo Fisher).
Where cross-validation was required, samples were also analyzed using a TriPlus RSH Autosampler, TRACE 1310 GC, and a TSQ 9000 Triple Quadrupole GC–MS/MS system equipped with an advanced electron ionization (AEI) source (Thermo Fisher).
Plant Materials, e-Juices, Reagents, and Chemicals
Dry stinging nettle (Urtica dioica L.) was purchased from Westpoint Naturals (British Columbia, Canada). Cannabis terpenoid mixtures were purchased from Vapeur Terp (California), and e-juices were products of Canada Vape Lab (Ontario, Canada), Illusions Vape (Ontario, Canada), and Premium Labs (British Columbia, Canada).
Anhydrous, ACS-grade sodium chloride, high-performance liquid chromatography (HPLC)-grade solvents (water, glycerol, DMSO, DMF, hexanes, and tetradecane), and nicotine were purchased from Sigma-Aldrich (Ontario, Canada) and used as received. Cannabis terpenoid standards were acquired from Restek (California): Cannabis Terpenes Standard 1 contains 2500 μg/mL (−)-α-bisabolol, camphene, δ-3-carene, (−)-β-caryophyllene, geraniol, (−)-guaiol, α-humulene, p-cymene, (−)-isopulegol, (+)-limonene, linalool, β-myrcene, nerolidol (cis and trans), β-ocimene (cis and trans), α-pinene, (−)-β-pinene, α-terpinene, γ-terpinene, and terpinolene; Cannabis Terpenes Standard 2 contains 2500 μg/mL (−)-caryophyllene oxide, and 1,8-cineole (eucalyptol). In addition, analytical standards of linalool oxides, citronellol, nerol, β-damascone, β-damascenone, and α-terpineol were purchased from Sigma-Aldrich and used as received. Mixed terpenoid and nicotine calibration samples (5–800 μg/mL) were prepared in tetradecane and stored at −20 °C for up to three months (Tables S2 and S3). We used (±)-linalool-d3 (CDN Isotopes, Quebec, Canada) as an internal standard. A 400 μg/mL linalool-d3 stock solution was prepared in tetradecane and stored at −20 °C for up to 12 months.
GC–MS Analysis
Water, brine (saturated NaCl solution [stored under ambient conditions]), DMSO, DMF, or glycerol was used as the headspace carrier solvent. Ground plant materials (50 mg), e-juices (50 μL), and cannabis terpenoid mixtures (25 μL) were added to a 5 mL of headspace carrier solvent in 20 mL headspace sampling vials (Chromatographic Specialties, Ontario, Canada) sealed with an 18 mm (diameter) × 1.524 mm (thickness) polytetrafluoroethylene (PTFE)/silicone gas-tight septa (Supelco, Pennsylvania). Vials were incubated and agitated at the “fast” setting for 15 min at temperatures ranging from 85 to 220 °C. Vial pressure was set to 100 kPa with an equilibration time of 12 s. The samples were then injected using a 1 mL loop with an equilibration time of 30 s and an injection time of 30 s, with sample loop temperatures ranging from 85 to 220 °C.
All samples were analyzed using a 30 m × 0.25 mm × 1.4 μm TG-624SilMS capillary GC column (Thermo Fisher). Helium (99.999%) was used as carrier gas at 1.5 mL/min. The inlet temperature was set at 125 °C with a septum purge flow of 20 mL/min and a split ratio of 100:1 (unless otherwise noted). The GC oven temperature gradient started at 60 °C for 30 s, followed by a ramp of 50 °C/min to 130 °C and 3-min hold, another increase to 140 °C at 5 °C/min, and finally to 280 °C at 22 °C/min and held for 3 min. The total gradient run time was 16.5 min. The temperatures of the MS transfer line, electron impact source, and ISQ transfer line were 250, 300, and 300 °C, respectively. Data were acquired in either scan or selected ion monitoring (SIM) modes (Table S1) and analyzed using the Chromeleon Chromatography Data System (Thermo Fisher; version 7).
For cross-validation purposes, 0.2 g ground plant materials were extracted with 20 mL hexanes in a 50 mL conical tube by shaking for 1 min, followed by incubation in an ultrasonic bath for 10 min and subsequent 10 s vortex. The sample was then centrifuged for 5 min at 3000g, and the clear supernatant was diluted 1000-fold for liquid-injection GC–MS/MS analysis. The GC–MS/MS instrument included a TSQ 9000 AEI triple quadrupole MS coupled with a TRACE 1310 GC. One microliter of the sample was injected by a TriPlusTM RHS autosampler at an inlet temperature of 220 °C and chromatographed with helium (99.999%) as the carrier gas (1.5 mL/min) on a TG-624SilMS capillary column (30 m × 0.25 mm ID 1.4 μm). The same oven temperature gradient, as described above, was used, and the split flow was 6 mL/min. MS transfer line and ion source temperatures were 250 and 300 °C, respectively. Data were acquired in selected reaction monitoring (SRM) mode (Table S2)
Method Validation
Method validation in plant tissue was performed using plant material fortified with select terpenes and terpenoids (vide infra). Each headspace vial included 5 mL glycerol, 25 μL internal standard (400 μg/mL linalool-d3), 25 μL of the fortification (or solvent as method blank), and 50 mg ground, dry stinging nettle where applicable.
Nine calibration levels were prepared at final concentrations of 25–4000 ng/mL in the vial (Table S3). Calibration functions for all analytes were fit to linear or quadratic curves, with inverse concentration weighting applied to all analytes. The dynamic range was calculated as the logarithm of the high calibration concentration divided by the low calibration concentration for each analyte. Method detection limits (MDL) were evaluated by determining the minimum measured concentration of an analyte at 25–200 ng/mL in the vial (2.5–20 ppm in plant sample; n = 7 over three days) that could be reported as distinct from the method blank with 99% confidence. The method reporting limit (MRL) for each analyte was verified using precision and accuracy specifications as outlined by the United States Environmental Protection Agency.41 MRLs were established at a fortification concentration of 25–200 ng/mL in the vial (n = 7). Carry-over was assessed by comparing the raw area response of each analyte in the high calibration sample (4000 ng/mL in the vial) to that of a method blank analyzed immediately after—an area response ratio < 0.1% in the method blank was accepted as suitable carry-over. Repeatability (n = 5; 1250 ng/mL in the vial or 125 ppm in the plant sample), intermediate precision (n = 17 over 3 days; 1250 ng/mL in the vial or 125 ppm in the plant sample), and method recoveries (n = 5; 3500 ng/mL in the vial or 350 ppm in the plant sample) were also evaluated.
For vaping liquid (e-juice) method validation, each headspace vial included 5 mL glycerol, 25 μL internal standard (400 μg/mL linalool-d3), 25 μL of the fortification (or solvent as method blank), and 50 μL glycerol:propylene glycol (1:1 v/v) as a surrogate vaping liquid where applicable. The calibration range included nine levels of 10–3000 ng/mL and 1–300 mg/mL for terpenoids and nicotine, respectively (Table S4). MDLs and MRLs were evaluated by identifying the minimum concentration of an analyte at 10–100 ng/mL (terpenoid) or 1–10 mg/mL (nicotine) in the vial (1–10 ppm terpenoids or 0.1–1 mg/mL nicotine in e-juice). Repeatability and intermediate precision were established at fortification concentrations of 750 ng/mL terpenoids and 75 mg/mL nicotine in the vial (or 75 ppm terpenoids and 7.5 mg/mL nicotine in surrogate e-juice). All other parameters and settings were identical to those of the plant tissue method validation (vide supra).
Acknowledgments
This work was supported by a Mitacs Elevate Fellowship to TDN.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00384.
Molecular ions used for SIM analysis of terpenes/terpenoids and nicotine in headspace GC–MS; molecular precursor and product ions used for SRM analysis of terpenes/terpenoids in GC–MS/MS; calibration schemes; method validation by GC–MS/MS; relative recoveries of select terpenes/terpenoids with different headspace liquids; SIM analysis of the surrogate matrix (stinging nettle) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Pickens L. B.; Tang Y.; Chooi Y.-H. Metabolic Engineering for the production of natural products. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 211–236. 10.1146/annurev-chembioeng-061010-114209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright G. D. Unlocking the potential of natural products in drug discovery. Microb. Biotechnol. 2019, 12, 55–57. 10.1111/1751-7915.13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid U.; Elkabetz M.; Zamir C.; Cohen K.; Larkov O.; Aly R. Authenticity assessment of natural fruit flavour compounds in foods and beverages by auto-HS–SPME stereoselective GC–MS. Flavour Fragrance J. 2010, 25, 20–27. 10.1002/ffj.1953. [DOI] [Google Scholar]
- Davies N. W.; Larkman T.; Marriott P. J.; Khan I. A. Determination of enantiomeric distribution of terpenes for quality assessment of Australian tea tree oil. J. Agric. Food Chem. 2016, 64, 4817–4819. 10.1021/acs.jafc.6b01803. [DOI] [PubMed] [Google Scholar]
- Christianson D. W. Roots of biosynthetic diversity. Science 2007, 316, 60–61. 10.1126/science.1141630. [DOI] [PubMed] [Google Scholar]
- Croteau R.; Kutchan T. M.; Lewis N. G. In Natural products (secondary metabolites). Biochemistry & Molecular Biology of Plants; Buchanan B.; Gruissem W.; Jones R. L., Eds.; Wiley: Rockville, 2000; pp 1250–1318. [Google Scholar]
- Ajikumar P. K.; Tyo K.; Carlsen S.; Mucha O.; Phon T. H.; Stephanopoulos G. Terpenoids: Opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol. Pharmaceutics 2008, 5, 167–190. 10.1021/mp700151b. [DOI] [PubMed] [Google Scholar]
- Huang M.; Lu J. J.; Huang M. Q.; Bao J. L.; Chen X. P.; Wang Y. T. Terpenoids: natural products for cancer therapy. Expert Opin. Invest. Drugs 2012, 21, 1801–1818. 10.1517/13543784.2012.727395. [DOI] [PubMed] [Google Scholar]
- Abdel-Rahman F. H.; Alaniz N. M.; Saleh M. A. Nematicidal activity of terpenoids. J. Environ. Sci. Health, Part B 2013, 48, 16–22. 10.1080/03601234.2012.716686. [DOI] [PubMed] [Google Scholar]
- Caputi L.; Aprea E. Use of terpenoids as natural flavouring compounds in food industry. Recent Pat. Recent Pat. Food, Nutr. Agric. 2011, 3, 9–16. 10.2174/2212798411103010009. [DOI] [PubMed] [Google Scholar]
- Fischedick J. T. Identification of terpenoid chemotypes among high (−)-trans-Δ9-tetrahydrocannabinol-producing Cannabis sativa L. cultivars. Cannabis Cannabinoid Res. 2017, 2, 34–47. 10.1089/can.2016.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsch J.; Leonti M.; Raduner S.; Racz I.; Chen J. Z.; Xie X. Q.; Altmann K. H.; Karsak M.; Zimmer A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 9099–9104. 10.1073/pnas.0803601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahham S. S.; Tabana Y. M.; Iqbal M. A.; Ahamed M. B. K.; Ezzat M. O.; Majid A. S. A.; Majid A. M. S. A. The anticancer, antioxidant and antimicrobial properties of the sesquiterpene β-caryophyllene from the essential oil of Aquilaria crassna. Molecules 2015, 20, 11808–11829. 10.3390/molecules200711808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo E. B. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. 10.1111/j.1476-5381.2011.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth J. K.; Bohlmann J. Terpenes in Cannabis sativa – from plant genome to humans. Plant Sci. 2019, 284, 67–72. 10.1016/j.plantsci.2019.03.022. [DOI] [PubMed] [Google Scholar]
- Zeng Z.; Chau F. T.; Chan H. Y.; Cheung C. Y.; Lau T. Y.; Wei S.; Mok D. K. W.; Chan C. O.; Liang Y. Recent advances in the compound-oriented and pattern-oriented approaches to the quality control of herbal medicines. Chin. Med. 2008, 3, 9 10.1186/1749-8546-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Health Canada. Quality of Natural Health Products Guide – Version 3.1. 2015.
- Erythropel H. C.; Jabba S. V.; DeWinter T. M.; Mendizabal M.; Anastas P. T.; Jordt S. E.; Zimmerman J. B. Formation of flavorant-propylene glycol adducts with novel toxicological properties in chemically unstable e-cigarette liquids. Nicotine Tob. Res. 2019, 21, 1248–1258. 10.1093/ntr/nty192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckell J.; Marti J.; Sindelar J. L. Should flavors be banned in combustible and electronic cigarettes? Evidence on adult smokers and recent quitters from a discrete choice experiment. Tob. Control 2019, 28, 168–175. 10.1136/tobaccocontrol-2017-054165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H. Scientists chase cause of mysterious vaping illness. Nature 2019, 574, 303–304. 10.1038/d41586-019-03033-1. [DOI] [PubMed] [Google Scholar]
- McPartland J. M.; Russo E. B. Cannabis and cannabis extracts. J. Cannabis Ther. 2001, 1, 103–132. 10.1300/J175v01n03_08. [DOI] [Google Scholar]
- Piñeiro Z.; Palma M.; Barroso C. G. Determination of terpenoids in wines by solid phase extraction and gas chromatography. Anal. Chim. Acta 2004, 513, 209–214. 10.1016/j.aca.2003.12.044. [DOI] [Google Scholar]
- Jiang Z.; Kempinski C.; Chappell J. Extraction and analysis of terpenes/terpenoids. Curr. Protoc. Plant Biol. 2016, 1, 345–358. 10.1002/cppb.20024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Centre for Ecotoxicology and Toxicology of Chemicals. ECETOC Technical Report No. 117: Understanding the Relationship Between Extraction Technique and Bioavailability. 2013.
- Mills G. A.; Walker V. Headspace solid-phase microextraction procedures for gas chromatographic analysis of biological fluids and materials. J. Chromatogr. A 2000, 902, 267–287. 10.1016/S0021-9673(00)00767-6. [DOI] [PubMed] [Google Scholar]
- Ebeler S. E.Gas chromatographic analysis of wines: current applications and future trends. In Gas Chromatography; Pool C. F., Ed.; Elsevier, 2012. [Google Scholar]
- Shapira A.; Berman P.; Futoran K.; Guberman O.; Meiri D. Tandem mass spectrometric quantification of 93 terpenoids in Cannabis using static headspace injections. Anal. Chem. 2019, 91, 11425–11432. 10.1021/acs.analchem.9b02844. [DOI] [PubMed] [Google Scholar]
- Bouvier-Brown N. C.; Holzinger R.; Palitzsch K.; Goldstein A. H. Quantifying sesquiterpene and oxygenated terpene emissions from live vegetation using solid-phase microextraction fibers. J. Chromatogr. A 2007, 1161, 113–120. 10.1016/j.chroma.2007.05.094. [DOI] [PubMed] [Google Scholar]
- Dziadas M.; Jeleń H. H. Analysis of terpenes in white wines using SPE-SPME-GC/MS approach. Anal. Chim. Acta 2010, 677, 43–49. 10.1016/j.aca.2010.06.035. [DOI] [PubMed] [Google Scholar]
- Jorge T. F.; Mata A. T.; António C. Mass Spectrometry as a quantitative tool in plant metabolomics. Philos. Trans. R. Soc., A 2016, 374, 20150370 10.1098/rsta.2015.0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basáñez T.; Majmundar A.; Cruz T. B.; Allem J. P.; Unger J. B. E-cigarettes are being marketed as “vitamin delivery” devices. Am. J. Public Health 2019, 109, 194–196. 10.2105/AJPH.2018.304804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Academies of Sciences, Engineering, and Medicine of the United States of America. Public health consequences of e-cigarettes 2018.
- Klager S.; Vallarino J.; MacNaughton P.; Christiani D. C.; Lu Q.; Allen J. G. Flavoring chemicals and aldehydes in e-cigarette emissions. Environ. Sci. Technol. 2017, 51, 10806–10813. 10.1021/acs.est.7b02205. [DOI] [PubMed] [Google Scholar]
- Bitzer Z. T.; Goel R.; Reilly S. M.; Elias R. J.; Silakov A.; Foulds J.; Muscat J.; Richie J. P. Effect of flavoring chemicals on free radical formation in electronic cigarette aerosols. Free Radical Biol. Med. 2018, 120, 72–79. 10.1016/j.freeradbiomed.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son Y.; Mishin V.; Laskin J. D.; Mainelis G.; Wackowski O. A.; Delnevo C.; Schwander S.; Khlystov A.; Samburova V.; Meng Q. Hydroxyl radicals in e-cigarette vapor and e-vapor oxidative potentials under different vaping patterns. Chem. Res. Toxicol. 2019, 32, 1087–1095. 10.1021/acs.chemrestox.8b00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter J. F.; Zäther E.; Svensson S. Analysis of refill liquids for electronic cigarettes. Addiction 2013, 108, 1671–1679. 10.1111/add.12235. [DOI] [PubMed] [Google Scholar]
- Cameron J. M.; Howell D. N.; White J. R.; Andrenyak D. M.; Layton M. E.; Roll J. M. Variable and potentially fatal amounts of nicotine in e-cigarette nicotine solutions. Tob. Control 2014, 23, 77–78. 10.1136/tobaccocontrol-2012-050604. [DOI] [PubMed] [Google Scholar]
- Gray T. R.; Eiden R. D.; Leonard K. E.; Connors G. J.; Shisler S.; Huestis M. A. Identifying prenatal cannabis exposure and effects of concurrent tobacco exposure on neonatal growth. Clin. Chem. 2010, 56, 1442–1450. 10.1373/clinchem.2010.147876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbey F. M.; McQueeny T.; Kadamangudi S.; Bice C.; Ketcherside A. Combined effects of marijuana and nicotine on memory performance and hippocampal volume. Behav. Brain Res. 2015, 293, 46–53. 10.1016/j.bbr.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulations amending the Cannabis Regulations (new classes of cannabis):SOR/2019-206. Can. Gaz. 2018, 153, 3558–3728. [Google Scholar]
- United States Environmental Protection Agency. Definition and Procedure for the Determination of Themethod Detection Limit—Revision 2. 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.