

HIV-1 Nef mediates evasion of the host immune system by inhibiting MHC-I surface presentation of viral antigens. To achieve this goal, Nef modifies the intracellular trafficking of MHC-I molecules in several ways. Despite being the subject of intense study, the molecular details underlying these modifications are not yet fully understood. Adaptor protein 1 (AP-1) plays an essential role in the Nef-mediated downregulation of MHC-I molecules such as HLA-A in different cell types. However, AP-1 has two functionally distinct variants composed of either γ1 or γ2 subunit isoforms. Because previous studies on the role of AP-1 in MHC-I downregulation by Nef focused on AP-1γ1, an important open question is the participation of AP-1γ2 in this process. Here, we show that AP-1γ2 is also essential for Nef-mediated depletion of surface HLA-A molecules in T cells. Our results indicate that Nef hijacks AP-1γ2 to modify HLA-A intracellular transport, redirecting these proteins to lysosomes for degradation.
KEYWORDS: γ2-adaptin, AP-1γ1, AP-1γ2, MHC-I, HLA-A downregulation, HIV-1, Nef, MVB, lysosome
ABSTRACT
The HIV-1 accessory protein Nef downregulates the cell surface expression of major histocompatibility complex class I (MHC-I) molecules to facilitate virus spreading. The Nef-induced downregulation of MHC-I molecules such as HLA-A requires the clathrin adaptor protein 1 (AP-1) complex. The cooperative interaction of Nef, AP-1, and the cytosolic tail (CT) of HLA-A leads to a redirection of HLA-A targeting from the trans-Golgi network (TGN) to lysosomes for degradation. Although the γ-adaptin subunit of AP-1 has two distinct isoforms (γ1 and γ2), which may form two AP-1 complex variants, so far, only the importance of AP-1γ1 in MHC-I downregulation by Nef has been investigated. Here, we report that the AP-1γ2 isoform also participates in this process. We found that AP-1γ2 forms a complex with Nef and HLA-A2_CT and that this interaction depends on the Y320 residue in HLA-A2_CT and Nef expression. Moreover, Nef targets AP-1γ1 and AP-1γ2 to different compartments in T cells, and the depletion of either AP-1 variant impairs the Nef-mediated reduction of total endogenous HLA-A levels and rescues HLA-A levels on the cell surface. Finally, immunofluorescence and immunoelectron microscopy analyses reveal that the depletion of γ2 in T cells compromises both the Nef-mediated retention of HLA-A molecules in the TGN and targeting to multivesicular bodies/late endosomes. Altogether, these results show that in addition to AP-1γ1, Nef also requires the AP-1γ2 variant for efficient MHC-I downregulation.
IMPORTANCE HIV-1 Nef mediates evasion of the host immune system by inhibiting MHC-I surface presentation of viral antigens. To achieve this goal, Nef modifies the intracellular trafficking of MHC-I molecules in several ways. Despite being the subject of intense study, the molecular details underlying these modifications are not yet fully understood. Adaptor protein 1 (AP-1) plays an essential role in the Nef-mediated downregulation of MHC-I molecules such as HLA-A in different cell types. However, AP-1 has two functionally distinct variants composed of either γ1 or γ2 subunit isoforms. Because previous studies on the role of AP-1 in MHC-I downregulation by Nef focused on AP-1γ1, an important open question is the participation of AP-1γ2 in this process. Here, we show that AP-1γ2 is also essential for Nef-mediated depletion of surface HLA-A molecules in T cells. Our results indicate that Nef hijacks AP-1γ2 to modify HLA-A intracellular transport, redirecting these proteins to lysosomes for degradation.
INTRODUCTION
HIV-1 promotes several changes in the physiology of the host cell in order to promote virus survival and increased replication. The rapid progression of HIV-1 infection in humans and animal models is closely linked to the viral accessory protein Nef (1). Nef facilitates viral particle release, prevents viral antigen presentation, and increases the infectivity of HIV-1 progeny by altering the intracellular trafficking of host transmembrane proteins (2). Specifically, Nef downregulates the surface expression of major histocompatibility complex class I (MHC-I) molecules (3), the CD4 receptor (4), and the host antiretroviral proteins serine incorporator 3 (SERINC3) and SERINC5 (SERINC3/5) (5, 6), frequently targeting these proteins for lysosomal degradation. Nef-mediated downregulation of MHC-I prevents the recognition and lysis of HIV-infected cells by CD8+ cytotoxic T lymphocytes (CTLs) (7), thereby promoting host immune evasion and viral spread (8). Interestingly, these effects of Nef are restricted to MHC-I allotypes HLA-A and HLA-B, not affecting the surface levels of HLA-C and HLA-E. Besides avoiding CTL recognition, this selectivity is thought to preclude the recognition of infected cells by natural killer cells, which target cells expressing low levels of MHC-I (8, 9). The differential sensitivity of HLA molecules to Nef was ascribed to a tyrosine-based (YSQAASS) sequence present in the cytosolic tail (CT) of HLA-A and HLA-B allotypes, which is absent in HLA-C and HLA-E (10).
After loading of a cytosolic peptide in the lumen of the endoplasmic reticulum (ER), mature MHC-I molecules are transported through the secretory pathway and reach the plasma membrane (PM) to present the peptide ligand (11). Adaptor protein 1 (AP-1), a clathrin adaptor complex involved in protein transport between the trans-Golgi network (TGN) and endosomes, is crucial for Nef downregulation of HLA-A (12–15). In HIV-1-infected T cells, Nef cooperatively binds to AP-1 and to the CT of HLA-A (HLA-A_CT) (16–19), inducing conformational changes in AP-1 that promote clathrin-coated vesicle (CCV) biogenesis and cargo loading (20, 21). These events are thought to redirect MHC-I molecules to endosomes and subsequently to multivesicular bodies (MVBs) and lysosomes to be degraded (12, 15).
AP-1, a complex of four distinct subunits (γ, β1, μ1, and σ1), is one of the most studied Nef binding partners. Different isoforms of AP-1 subunits have been described, including two γ (γ1 and γ2), two μ1 (μ1A and μ1B), and three σ1 (σ1A, σ1B, and σ1C) isoforms (22). Several studies using RNA interference demonstrated that the expression of both μ1A and γ1 subunits is required for the downregulation of MHC-I by Nef (12–15, 23). Interestingly, there is growing evidence that some of these isoforms may combine to form functionally distinct AP-1 complexes (24–28). Indeed, we have previously demonstrated that a functional variant of AP-1 that contains γ2 (AP-1γ2), but not the AP-1γ1 complex, is required for efficient downregulation of CD4 by Nef (26). Here, we show that γ2 is present in an AP-1:Nef:HLA-A2_CT tripartite complex and participates in cell surface downregulation and lysosomal targeting of HLA-A molecules mediated by Nef in T cells. A nonredundant role for AP-1γ1 and AP-1γ2 complexes in the cellular depletion of MHC-I molecules induced by Nef is discussed.
RESULTS
γ2-Adaptin forms a tripartite complex with Nef and the HLA-A2 cytosolic tail similarly to γ1-adaptin.
Downregulation of HLA-A2 by Nef requires the formation of a Nef:HLA-A2:AP-1 tripartite complex, including the γ1 subunit of AP-1 (16–19). To test whether γ2 can also participate in a similar tripartite complex, we used a glutathione S-transferase (GST)-HLA-A2_CT-Nef chimeric fusion protein as bait to capture γ2 from the cell lysate in a GST pulldown assay. HLA-A2_CT-Nef was indeed able to capture γ2 as well as another AP-1 subunit, μ1A (Fig. 1). The interactions were specific because HLA-A2_CT-Nef failed to capture the ε subunit of AP-4, which was previously shown not to interact with Nef (29). Importantly, the interaction with either γ2 or γ1 was abolished when the Y320 residue within the HLA-A2_CT, essential for Nef:HLA-A2_CT:AP-1 complex stabilization (16–19), was mutated (Fig. 1), indicating that Nef and HLA-A2_CT interact with AP-1γ2 through the same region used to bind to AP-1γ1.
FIG 1.
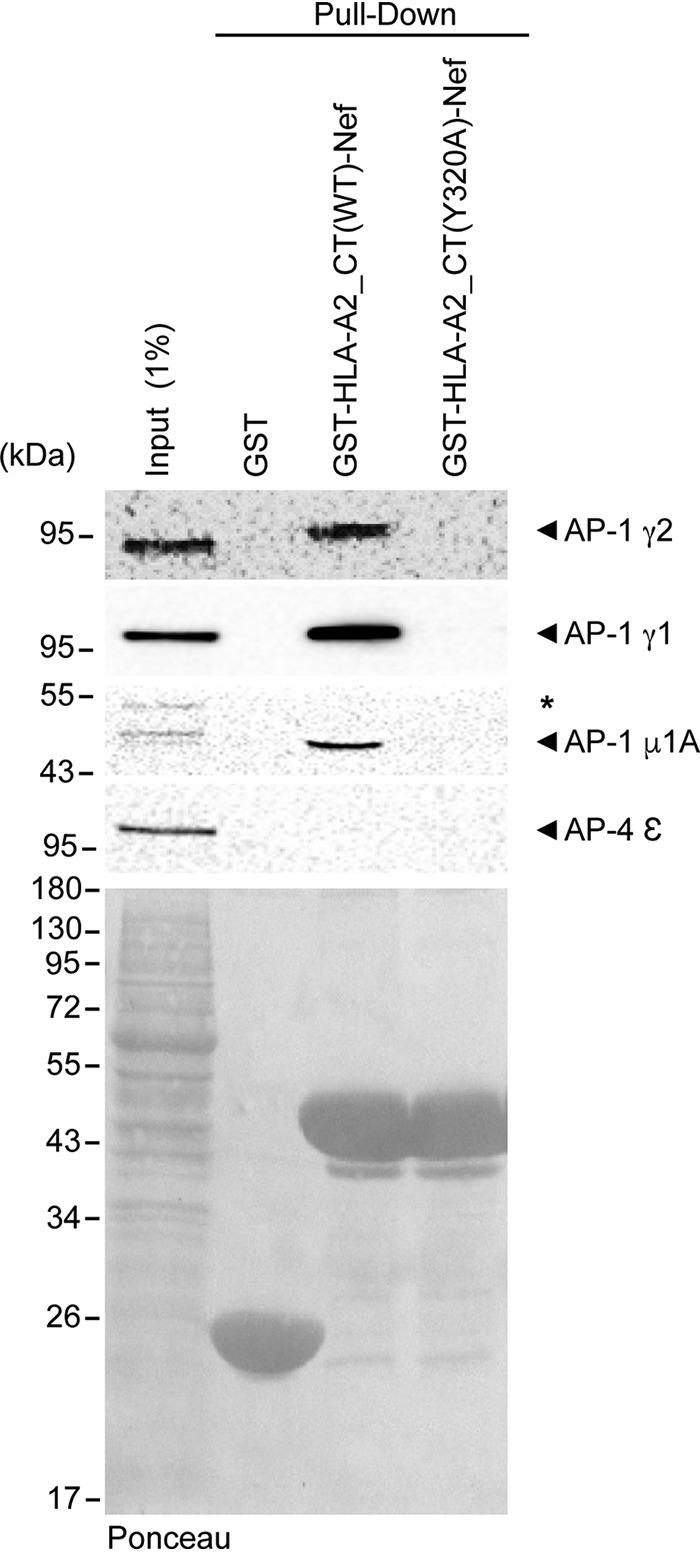
AP-1γ2 interacts with HLA-A2_CT-Nef through the noncanonical tyrosine motif of HLA-A2_CT. Immunoblots for AP-1γ2, AP-1γ1, AP-1μ1A, and AP-4ε indicate that the AP-1 subunits γ2, γ1, and μ1A interact with a chimera of Nef and the cytosolic tail of HLA [GST-HLA-A2_CT(WT)-Nef] and that this interaction is inhibited by a point mutation in the Tyr320 residue of HLA-A2 [GST-HLA-A2_CT(Y320A)-Nef]. AP-4ε was used as a negative control for the interaction. GST-HLA-A2_CT(WT)-Nef and GST-HLA-A2_CT(Y320A)-Nef were produced in E. coli, immobilized onto glutathione beads, and incubated with cleared HeLa cell lysates. One percent of the total protein lysate incubated with the beads was used as the input. As a loading control, the nitrocellulose membrane was stained with Ponceau S (bottom).
AP-1γ2 is involved in Nef-induced HLA-A downregulation and subcellular redistribution.
The fact that AP-1γ2 may form a tripartite complex with Nef and HLA-A2 prompted us to test whether this AP-1 variant is involved in the Nef-mediated downregulation of endogenous HLA-A in T cells. To address this question, we transduced A3.01 T cells, which endogenously express HLA-A1 and HLA-A31 allotypes, with a retrovirus to express either green fluorescent protein (GFP) alone or GFP and Nef (GFP/Nef) and selected a pool of GFP-expressing cells using a cell sorter. These results showed a partial (∼1.9-fold) but significant reduction in the surface levels of endogenous HLA-A molecules in cells expressing Nef (Fig. 2A) (30). To test whether the intensity of HLA-A surface downregulation observed in our system is relevant in the context of infection, we infected Rev-CEM GFP T cells with wild-type (WT) and Nef-defective (ΔNef) HIV-1 NL4-3. These cells express GFP driven by the HIV-1 long terminal repeat (LTR) and, like A3.01 cells, endogenously express HLA-A1 and HLA-A31 allotypes. The results showed that surface levels of endogenous HLA-A in cells infected with the WT virus were ∼1.5-fold lower than those in cells infected with Nef-deleted virus (Fig. 2B). This effect is comparable to that of Nef expression alone in A3.01 T cells (Fig. 2A).
FIG 2.

AP-1γ2 and AP-1γ1 are required for efficient Nef-mediated depletion of surface and total levels of HLA-A in T cells. (A) A3.01 GFP T cells or A3.01 Nef/GFP T cells were surface labeled with anti-HLA-A antibody and analyzed by FACS analysis. The bar graph represents the fold downmodulation of HLA-A levels in Nef cells, which was calculated by dividing the mean fluorescence intensity (MFI) of HLA-A in A3.01 GFP by the MFI of HLA-A in A3.01 Nef/GFP cells. The data represent the means ± SEM (n = 3). (B) Rev-CEM GFP reporter cells were infected with HIV-1 NL4-3 WT or HIV NL4-3 ΔNef, cultured, processed, and analyzed by FACS analysis to verify the surface expression of HLA-A in GFP-expressing cells. The bar graph represents the fold downmodulation of HLA-A levels in cells infected with HIV-1 WT. Fold downmodulation was calculated by dividing the MFI of HLA-A in cells infected with HIV-1 ΔNef by the MFI of HLA-A in cells infected with HIV-1 WT. The data represent the mean ± SEM (n = 3). (C) A3.01 GFP T cells or A3.01 Nef/GFP T cells were transduced with lentivirus to express control shRNA or shRNA against AP-1γ1 or AP-1γ2, and after 120 h, cell surface HLA-A levels were analyzed by FACS analysis. (D) Bar graph representing the fold downmodulation of HLA-A levels under each condition. Fold downmodulation was calculated as described above for panel A. The data represent the means ± SEM (n = 3). **, P < 0.005; ****, P < 0.0005 (two-tailed paired t test); ns, not significant. (E) Cells were treated as described above for panel C but this time incubated for either 96 h or 120 h after shRNA transduction, as indicated. Cell lysates were subjected to SDS-PAGE and immunoblotting against HLA-A, Nef, AP-1γ1, AP-1γ2, and β-actin. (F) HLA-A, Nef, AP-1γ1, and AP-1γ2 signal intensities under each condition in panel E were determined using Image Lab software (Bio-Rad) and are expressed as a percentage of each protein signal from A3.01 GFP T cells infected with the shRNA control. The data represent the means ± SEM (n = 3). *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ns, not significant (one-way analysis of variance [ANOVA] with Bonferroni corrections).
Next, we used short hairpin RNA (shRNA) to deplete AP-1γ2 or AP-1γ1 levels in either A3.01 Nef/GFP or A3.01 GFP cells. As expected, under control conditions (control shRNA), Nef decreased the surface level of HLA-A (Fig. 2C and D), a process that requires AP-1γ1 (14, 15) (Fig. 2C and D), and similarly, AP-1γ2 depletion reduced this effect (Fig. 2C and D). Although at the time of the experiment, a fraction of the cells in the pool (∼50%) lost Nef/GFP expression, a significant reduction in total levels of HLA-A was detected in total lysates of Nef cells (Fig. 2E and F). This effect was compromised by AP-1γ2 knockdown (KD) (as well as by γ1 knockdown) (Fig. 2E and F).
The depletion of the AP-1μ1A subunit of AP-1, known to be present in both AP-1γ1- and AP-1γ2-containing variants of AP-1 (29), led to the codepletion of AP-1γ1 and AP-1γ2, as previously observed (26). This treatment resulted in the full recovery of HLA-A surface levels in cells expressing Nef (Fig. 3A and B), similarly to the individual depletion of AP-1γ1 or AP-1γ2 (Fig. 2B and C). In contrast, knockdown of AP-1μ1A did not fully revert the reduction in the total levels of HLA-A induced by Nef (Fig. 3C and D), as observed for the isolated depletion of AP-1γ1 or AP-1γ2 (Fig. 2E and F). Together, these results indicate that the effect of AP-1γ1 and AP-1γ2 KD is not additive in inhibiting Nef-mediated depletion.
FIG 3.

AP-1μ1A is required for efficient Nef-mediated depletion of surface and total levels of HLA-A. (A and B) A3.01 GFP T cells or A3.01 Nef/GFP T cells were transduced with lentivirus to express control shRNA or shRNA against AP-1μ1A, and after 120 h, cell surface HLA-A levels were analyzed by FACS analysis. (B) Bar graphs representing the fold downmodulation of HLA-A levels under each condition. Fold downmodulation was calculated by dividing the MFI of HLA-A in A3.01 GFP cells by the MFI of HLA-A in A3.01 Nef/GFP cells. The data represent the means ± SEM (n = 3). ***, P < 0.0005 (two-tailed paired t test). (C) Cells were treated as described above for panels A and B, and cell lysates were subjected to SDS-PAGE and immunoblotting against HLA-A, Nef, AP-1γ1, AP-1γ2, AP-1μ1A, and β-actin. (D) HLA-A, Nef, AP-1γ1, AP-1γ2, and AP-1μ1A signal intensities under each condition in panel C were determined using Image Lab software (Bio-Rad) and are expressed as a percentage of each protein signal from A3.01 GFP T cells infected with control shRNA. The data represent the means ± SEM (n = 3). *, P < 0.05; ***, P < 0.0005; ns, not significant (one-way ANOVA with Bonferroni corrections).
HLA-A downregulation from the cell surface correlates with a redistribution to cytoplasmic vesicular structures (Fig. 4A, B, E, and F), where HLA-A partially colocalizes with either AP-1γ1 (Fig. 4B and C and Table 1) or AP-1γ2 (Fig. 4F and G and Table 1). Knockdown of AP-1γ1 leads to the recovery of HLA-A labeling at the cell surface (Fig. 4D), as does AP-1γ2 knockdown (Fig. 4H). Taken together, these results suggest that the expression of both γ-adaptin isoforms is required for efficient HLA-A downregulation by Nef.
FIG 4.

Depletion of either AP-1γ1 or AP-1γ2 compromises Nef-induced redistribution of HLA-A in T cells. (A and B) A3.01 GFP T cells (A) and A3.01 Nef/GFP T cells (B) were transduced with lentivirus encoding control shRNA, and after 120 h, cells were fixed, permeabilized, and stained for HLA-A (red channel) and AP-1γ1 (green channel). (C) Bar graph showing the mean Manders’ colocalization coefficients of the HLA-A signal overlapping the AP-1γ1 signal ± SEM (n ≥ 6). ***, P < 0.0005; n.s., not significant (two-tailed paired t test). (D) A3.01 Nef/GFP T cells expressing shRNA for AP-1γ1 for 120 h were subjected to immunofluorescence as described above for panels A and B. (E and F) Cells were treated as described above for panels A and B, fixed, permeabilized, and stained with an antibody that recognizes HLA-A, -B, and -C (red channel) and AP-1γ2 (green channel). (G) Bar graph showing the mean Manders’ colocalization coefficients of the HLA-A/B/C signal overlapping the AP-1γ2 signal ± SEM (n ≥ 6). ***, P < 0.0005 (two-tailed paired t test). (H) A3.01 Nef/GFP T cells expressing shRNA for AP-1γ2 for 120 h were subjected to immunofluorescence as described above for panels E and F. Bars, 7 μm.
TABLE 1.
Manders’ colocalization coefficients determined in this studya
| Proteins | Mean Manders’ coefficient ± SEM (no. of cells) |
|
|---|---|---|
| tM1 | tM2 | |
| HA-HLA-A2 and TGN46 in HeLa cells (siRNA control) | 0.09 ± 0.009 (5) | 0.21 ± 0.07 (5) |
| HA-HLA-A2 and TGN46 in HeLa cells expressing Nef (siRNA control) | 0.39 ± 0.02 (8) | 0.45 ± 0.03 (8) |
| HA-HLA-A2 and TGN46 in HeLa cells expressing Nef (siRNA AP-1γ1) | 0.16 ± 0.01 (6) | 0.19 ± 0.03 (6) |
| HA-HLA-A2 and TGN46 in HeLa cells expressing Nef (siRNA AP-1γ2) | 0.16 ± 0.006 (8) | 0.26 ± 0.02 (8) |
| HLA-A and AP-1γ1 in A3.01 GFP T cells (shRNA control) | 0.17 ± 0.02 (6) | 0.24 ± 0.01 (6) |
| HLA-A and AP-1γ1 in A3.01 Nef/GFP T cells (shRNA control) | 0.77 ± 0.005 (7) | 0.63 ± 0.01 (7) |
| HLA-A/B/C and AP-1γ2 in A3.01 GFP T cells (shRNA control) | 0.20 ± 0.02 (6) | 0.20 ± 0.02 (6) |
| HLA-A/B/C and AP-1γ2 in A3.01 Nef/GFP T cells (shRNA control) | 0.73 ± 0.03 (7) | 0.45 ± 0.02 (7) |
| HLA-A and TGN46 in A3.01 GFP T cells (shRNA control) | 0.31 ± 0.02 (5) | 0.44 ± 0.03 (5) |
| HLA-A and TGN46 in A3.01 Nef/GFP T cells (shRNA control) | 0.64 ± 0.01 (5) | 0.64 ± 0.01 (5) |
| HLA-A and TGN46 in A3.01 Nef/GFP T cells (shRNA AP-1γ1) | 0.33 ± 0.02 (5) | 0.44 ± 0.01 (5) |
| HLA-A and TGN46 in A3.01 Nef/GFP T cells (shRNA AP-1γ2) | 0.38 ± 0.01 (5) | 0.45 ± 0.02 (5) |
| AP-1γ2 and AP-1γ1 in GFP T cells | 0.57 ± 0.02 (6) | 0.79 ± 0.01 (6) |
| AP-1γ2 and AP-1γ1 in Nef/GFP T cells | 0.28 ± 0.02 (6) | 0.65 ± 0.03 (6) |
The colocalization threshold plug-in for ImageJ software (55) was used for the quantification of colocalization. For the determination of single-channel-specific Manders’ overlap coefficients, tM1 is defined as the total intensity of pixels from channel 1 (green) for which the intensity in channel 2 (red) is above zero relative to the total intensity in channel 1, and tM2 is defined as the total intensity of pixels from channel 2 for which the intensity in channel 1 is above zero relative to the total intensity in channel 2. The coefficients were calculated using an automatically determined threshold value, for both channels, according to an algorithm described previously (56), where a value of 1 represents total colocalization. Shown are the means ± standard errors of the means (SEM) for tM1 and tM2, indicating the extent of the overlap between the signals of the indicated proteins. Data for each pairwise comparison were obtained from a series of z-slices (with 0.3-μm intervals) for at least five cells from three independent experiments.
AP-1γ2 is involved in the efficient targeting of HLA-A to MVBs in T cells by Nef.
Previous work showed that Nef requires γ1 and μ1A subunits to perturb the transport of newly synthesized HLA-A molecules to the PM, leading to TGN retention and targeting to MVBs/lysosomes for degradation (12–15). The requirement for AP-1γ2 in Nef-mediated cell surface depletion of HLA-A by Nef (Fig. 2) prompted us to test the involvement of AP-1γ2 in the subcellular redistribution of HLA-A by Nef. Indeed, the reduction in cell surface staining of HLA-A in T cells induced by Nef was accompanied by a strong accumulation of HLA-A in the juxtanuclear region. Under these conditions, HLA-A staining overlapped that of TGN46, a widely used marker for the trans-Golgi network (31) (Fig. 5A, B, and E and Table 1). Importantly, Nef-induced colocalization of HLA-A with TGN46 was clearly diminished upon the depletion of either AP-1γ1 or AP-1γ2 (Fig. 5C to E), indicating that the expression of both complexes is required for efficient intracellular retention of HLA-A in the TGN. Additionally, immunoelectron microscopy (immuno-EM) analysis of T cells expressing Nef revealed that AP-1γ2 knockdown decreases HLA-A detection in MVBs/late endosomes (LE) and increases HLA-A staining on the cell surface (Fig. 6A to C). Thus, the lack of AP-1γ2 antagonized the effect of Nef, which targeted HLA-A to MVBs and lysosomes for degradation (Fig. 6A to C) (12, 15). This indicates that HLA-A targeting to the endolysosomal degradation pathway by Nef requires a variant of AP-1 that contains the γ2 subunit isoform.
FIG 5.

Knockdown of AP-1γ2 or AP-1γ1 impairs the Nef-mediated retention of HLA-A in the TGN. (A to D) A3.01 GFP T cells (A) and A3.01 Nef/GFP T cells (B to D) were transduced with lentivirus encoding control shRNA (A and B), shRNA against AP-1γ1 (C), or shRNA against AP-1γ2 (D), and after 120 h, cells were fixed, permeabilized, and stained for HLA-A (red channel) and TGN46 (green channel). (E) Bar graph showing the mean Manders’ colocalization coefficients of the HLA-A signal overlapping the TGN46 signal ± SEM (n ≥ 5). ***, P < 0.0005; n.s., not significant (two-tailed paired t test). Bars, 7 μm.
FIG 6.
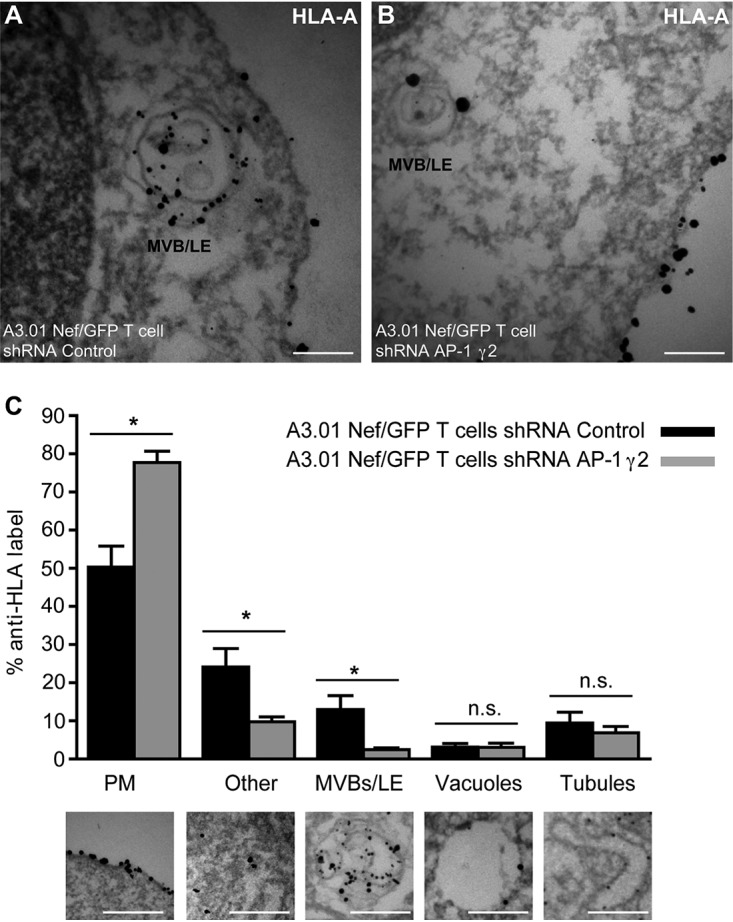
Knockdown of AP-1γ2 impairs the redistribution of HLA-A to late endosomes by Nef. (A and B) A3.01 Nef/GFP T cells were transduced with lentivirus encoding either control shRNA (A) or shRNA for AP-1γ2 (B), and after 72 h, the cells were fixed, immunogold labeled for HLA-A, and processed for immuno-EM. Bars, 0.5 μm. (C) Quantification of HLA-A staining. The bar graph indicates the percentages of the total numbers of gold particles counted and associated with the indicated compartments. The plasma membrane (PM), cytoplasm, MVBs/late endosomes (LE), vacuoles, and tubules are defined below the graph. Gold particles associated with unidentifiable membranes and other cytoplasmic structures are categorized as “other.” Bars, 0.5 μm. P values were calculated using Student’s t test (*, P < 0.05; n.s., not significant).
AP-1γ1 and AP-1γ2 are recruited to partially distinct membrane structures upon Nef expression in T cells.
In the above-described experiments, the staining of AP-1γ1 in A3.01 T cells was mostly concentrated at the juxtanuclear region, while AP-1γ2 staining was also abundant in peripheral structures (Fig. 4). Colabeling of AP-1γ1 and AP-1γ2 confirmed that these proteins display a partially distinct pattern of distribution in A3.01 GFP and A3.01 Nef/GFP T cells (Fig. 7A to C). To gain further insights into the partitioning of AP-1γ1 and AP-1γ2 within the endomembrane system and their behavior upon Nef expression, we performed subcellular fractionation of postnuclear supernatants of GFP and GFP/Nef A3.01 T cells by sedimentation on glycerol gradients (Fig. 7D and E). In GFP cells, HLA-A was found at the bottom of the gradient in fractions corresponding to the plasma membrane (fractions 11 and 12) and also in intermediate fractions enriched with the endosomal markers early endosome antigen 1 (EEA1) and hepatocyte-responsive serum phosphoprotein (HRS) (fractions 6 and 7). Also, in GFP cells, AP-1γ2 was found at the top and middle of the gradient (fractions 2 to 7), in fractions corresponding to the cytosol (fraction 2, GFP), the TGN (fractions 4 to 6, TGN46), and endosomes (fractions 6 to 8, EEA1 and HRS). The distribution of AP-1γ1 was similar to that of AP-1γ2, with the exception that AP-1γ1 was also detected in bottom fractions enriched with the recycling endosome marker transferrin receptor (TfR) (Fig. 7D). In Nef-expressing cells, HLA-A shifted from the bottom to the middle fractions enriched in TGN and endosome proteins. Also, in Nef cells, AP-1γ1 and AP-1γ2 were clearly displaced from lighter fractions corresponding to the cytosol and transport intermediates and were strongly recruited to heavier membrane fractions (Fig. 7E). Interestingly, under these conditions, AP-1γ2 appears to segregate from AP-1γ1 and concentrates in a fraction that is enriched in EEA1 and HRS, which contain a low proportion of AP-1γ1. Therefore, Nef stabilizes the association of AP-1γ1 and AP-1γ2 with membranes of distinct compartments.
FIG 7.

Subcellular fractionation reveals that Nef induces the accumulation of AP-1γ2 in membranes positive for HLA-A and the early endosome markers. (A and B) A3.01 GFP (A) and A3.01 Nef/GFP (B) cells were fixed, permeabilized, and costained for AP-1γ1 (red channel) and AP-1γ2 (green channel). Bars, 5 μm. (C) Graph representing Manders’ overlap coefficients between AP-1γ1 and AP-1γ2 signals in cells treated as described above for panels A and B. P values were calculated using Student’s t test (*, P < 0.05; ***, P < 0.0005). (D and E) A3.01 GFP or A3.01 Nef/GFP cells were subjected to subcellular fractionation on 10% to 30% glycerol gradients. After ultracentrifugation, fractions were collected from the top (lane 1) to the bottom (lane 12), and the proteins from each fraction were precipitated and subjected to SDS-PAGE and immunoblotting against HLA-A, TGN46 AP-1γ1, AP-1γ2, AP-1μ1A, HRS, EEA1, TfR, Nef, and GFP. Note that Nef induces the accumulation of AP-1γ2 in membrane fractions containing HLA-A, TGN46, Nef, and the early endosome markers EEA1 and HRS (a component of the ESCRT-0 machinery).
AP-1γ2 is required for Nef-mediated retention of HLA-A in the trans-Golgi network of HeLa cells.
To test the requirement for AP-1γ2 in MHC-I downregulation by Nef in a different system, we generated a HeLa cell line constitutively expressing hemagglutinin (HA)-tagged HLA-A2 (HeLa HLA-A2) and used small interfering RNA (siRNA) to specifically deplete either AP-1γ2 or AP-1γ1 levels (Fig. 8). Nef expression did not change the steady-state levels of HA-HLA-A2 in cell extracts of HeLa HLA-A2 cells (Fig. 8A), corroborating previously reported observations that Nef-mediated degradation of MHC-I is weak in HeLa cells (32). However, the surface levels of HA-HLA-A2 were reduced in Nef-expressing cells (Fig. 8B), and this reduction was compromised upon the depletion of either AP-1γ1 or AP-1γ2 (Fig. 8B). As observed for T cells (Fig. 5), Nef expression led to a strong accumulation of HA-HLA-A2 at the TGN (Fig. 8C and D and Table 1), a phenotype that was attenuated upon the depletion of either AP-1γ1 or AP-1γ2 (Fig. 8E and F and Table 1). These results confirm that the expression of both complexes is independently required for the efficient intracellular retention of HLA-A2 at the TGN.
FIG 8.

Knockdown of AP-1γ1 or AP-1γ2 compromises the Nef-induced redistribution of HLA-A2 from the cell surface to the TGN in HeLa cells. (A) HeLa cells constitutively expressing HA-HLA-A2 were transfected twice with control siRNA, siRNA for AP-1γ1, or siRNA for AP-1γ2, and after 24 h, cells were transfected with an empty vector (pCI) or pCI-Nef WT. After 24 h, cells were incubated at 26°C for 16 h and then subjected to SDS-PAGE and immunoblotting for HA, Nef AP-1γ1, AP-1γ2, and β-actin. (B) Cells were treated as described above for panel A and transfected with pNef-WT.IRES.GFP or pIRES.GFP, and surface HA-HLA-A2 levels were analyzed by FACS analysis. (C to F) HeLa HA-HLA-A2 cells were transfected twice with control siRNA (C and D), siRNA for AP-1γ1 (E), or siRNA for AP-1γ2 (F), and after 24 h cells, were transfected with an empty vector (C) or pCI-Nef WT (D to F). After 24 h, cells were incubated at 26°C for 16 h, fixed, permeabilized, and stained for HA-HLA-A2 (red channel) and TGN46 (green channel). Bars, 10 μm. (G) Bar graph showing the mean Manders’ colocalization coefficients for the HA-HLA-A2 signal overlapping the TGN46 signal ± SEM (n ≥ 5). **, P < 0.005; ***, P < 0.0005; n.s., not significant (two-tailed paired t test).
DISCUSSION
Here, we demonstrate that the γ2 subunit of AP-1, which has previously been shown to be involved in Nef-induced CD4 downregulation (26), is also required for the downregulation of MHC-I molecules by Nef. Our results revealed that AP-1γ2 interacts with Nef and HLA-A2 to form a tripartite complex involving the critical tyrosine residue (Y320) in the cytosolic tail of HLA-A2, as previously demonstrated for AP-1γ1 (16, 18–20). This interaction is essential to remove HLA-A from the cell surface and to target it to lysosomes for degradation. Overall, the data indicate that AP-1γ2 is an important nexus in the immune evasion mechanism mediated by Nef in HIV-infected cells.
Biochemical and structural data show that Nef cooperatively binds to HLA-A2_CT and AP-1, forming a tripartite complex (16–19) in a conformation that is compatible with clathrin-coated vesicle (CCV) biogenesis and cargo loading (21). The γ1 and μ1A subunits of AP-1 integrate this tripartite complex (16, 20, 21, 33) and are essential for efficient MHC-I downregulation by Nef (12–15, 23). Whereas our GST pulldown result confirmed this notion, it also revealed a novel Nef:AP-1:HLA-A2_CT tripartite complex, containing γ2 instead of γ1 (Fig. 1). Moreover, these data suggest that the two Nef:AP-1:HLA-A2_CT complexes, containing either γ1 or γ2, coexist in Nef-expressing cells. Previous studies showed that the Y320 residue within the Y320SQA motif of HLA-A2_CT is required for Nef:AP-1γ1:HLA-A2_CT complex formation (16–19). In the context of this complex, the Y320SQA motif functions as a tyrosine-based sorting signal, despite the absence of an essential hydrophobic residue in position 323, making HLA-A2_CT able to bind μ1A (18). Likewise, our data show that a 320Y/A substitution abolished γ2 binding to the GST-Nef:HLA-A2_CT chimera, indicating that the Y320SQA motif in HLA-A2_CT is also crucial for Nef:AP-1γ2:HLA-A2_CT complex formation (Fig. 1).
Nef-dependent downregulation of MHC-I molecules requires the expression of both μ1A and γ1 subunits of AP-1 (12–15, 23). Our data show that the depletion of either AP-1γ2 or AP-1γ1 impaired the Nef-induced decrease of cell surface HLA-A in T cells (Fig. 2). Therefore, Nef hijacks both AP-1 complex variants to disrupt HLA-A transport to the PM. The specific requirement for AP-1γ1 and AP-1γ2 in the Nef-mediated cell surface downregulation of HLA-A2 was also confirmed in HeLa cells (Fig. 8). Nef’s ability to reduce the total levels of HLA-A in T cells was sensitive but not fully inhibited by either the individual (Fig. 2) or simultaneous (Fig. 3) depletion of AP-1γ2 and AP-1γ1. A possible interpretation is that these AP-1 variants are involved in distinct but interdependent steps of the same pathway of Nef-mediated degradation of HLA and that AP-1-independent pathways may also operate, as previously discussed (34).
Previous studies have shown that different subunit isoforms may combine to form distinct AP-1 complexes, whose variants exert different functions in intracellular trafficking. Specifically, σ1A and σ1B differentially coordinate endosomal maturation in brain tissues (27), whereas the μ1A and μ1B subunit isoforms increase the repertoire of proteins sorted by AP-1 by recognizing distinct cargoes (25). Our group reported that the Nef-mediated downregulation of CD4 requires the function of AP-1γ2 but not AP-1γ1 (26). Similarly, knockdown of either γ1 or γ2 leads to distinct phenotypes in zebrafish development (28). Here, however, we describe that the specific depletion of either γ1 or γ2 compromises HLA-A downregulation by Nef, via a process in which two different AP-1 subunits interact with the same molecules but with nonredundant functions.
The processes of CD4 and MHC-I downregulation by Nef are mechanistically distinct (2). In HLA-A downregulation, Nef retains newly synthesized HLA-A molecules in the TGN and endosomes, preventing their delivery to the PM and targeting them to MVBs/lysosomes for degradation (32, 35). In contrast, the downregulation of CD4 initiates with Nef-induced endocytosis of CD4 via AP-2/clathrin pits (36–40), followed by delivery to early endosomes and subsequently to MVBs and lysosomes (12, 41, 42). Despite the differences in the initial steps, the downregulation of both CD4 and HLA-A molecules culminates with their lysosomal delivery (12). Considering our previous report describing the requirement for AP-1γ2 in CD4 downregulation (26) and the data shown here, we propose that AP-1γ2 is a common host factor used by Nef to target both CD4 and MHC-I molecules to lysosomes. Supporting this notion, our microscopy analysis shows that AP-1γ2 depletion in T cells or HeLa cells expressing Nef decreased HLA-A retention at the TGN (Fig. 5 and 8) and its targeting to MVBs/LE (Fig. 6), while increasing its presence at the PM, compared to control T cells expressing Nef (Fig. 2 and 5). The involvement of AP-1γ2 in lysosomal protein targeting has been proposed previously (43, 44). The depletion of AP-1γ2 compromises the targeting of the epidermal growth factor (EGF):EGF receptor (EGFr) complex to the canonical MVB pathway (26, 44), and AP-1γ2 may interact with subunits of the endosomal sorting complexes required for transport (ESCRTs) (44). Moreover, the depletion of AP-1γ2 prevents the lysosomal clearance of the HIV-1 Gag protein, leading to an increase of HIV-1 particle release (44). Here, we show that Nef induces the accumulation of AP-1γ2 in membrane fractions that contain HLA-A, Nef, and endosome markers such as HRS (a subunit of ESCRT-0, involved in MVB biogenesis) and a low proportion of AP-1γ1 (Fig. 7). We propose that Nef predominately targets HLA-A molecules in the TGN via the γ1-containing form of AP-1 and uses the γ2-containing variant of AP-1 to target HLA-A in endosomes, thus retaining these molecules in a pathway that leads to lysosomes.
In sum, our results demonstrate that HLA-A2 downregulation by HIV-1 Nef in T cells requires the two functional variants of the AP-1 complex. We postulate that AP-1γ2 retains and/or sorts cargo in the MVB/lysosome route and that Nef interacts with AP-1γ2 to target both HLA-A and CD4 to the degradative pathway. It remains to be determined why Nef interaction with AP-1γ1 is not needed for CD4 downregulation (14, 26), a full understanding of which may require the structural characterization of these complexes. In fact, a recent high-resolution structural analysis of the tripartite complex formed by Nef, AP-1, and the cytosolic tail of either HLA-A or tetherin showed that the actual function of the Nef:AP-1 interaction is allosterically modulated by posttranslational modifications and the type of cargo involved (21). In terms of HIV pathogenesis, targeting AP-1γ1 at the TGN and AP-1γ2 in endosomes to prevent PM delivery of newly synthesized HLA-A molecules is critical, since MHC molecules may be charged with virus antigens in the ER after infection. On the other hand, newly synthesized CD4 molecules are targeted for degradation directly from the ER by another HIV-1 accessory protein, Vpu (45, 46). For the early viral protein Nef, the priority seems to be targeting preexisting CD4 molecules at the PM for internalization via AP-2 to avoid superinfection and facilitate virus progeny release (2) and then delivering these molecules for lysosomal degradation using molecules such as AP-1γ2, β-COP, and Alix (12, 26, 42). Therefore, AP-1γ2 may serve as an “Achilles’ heel” that could be exploited to interfere with these two important functions of Nef. Moreover, considering the key role of AP-1γ2 in Nef-mediated targeting of HLA-A and CD4 to the lysosome pathway, it would be important to investigate whether AP-1γ2 plays a role in the downregulation of the antiviral protein SERINC3/5 and other host targets of Nef.
MATERIALS AND METHODS
Expression plasmids.
The pNL4-3 Nef.IRES.GFP and pCIneoNL4-3 Nef plasmids were described previously (47). The HIV-1 NL4-3 (pNL4-3) (catalog no. 114) and the HIV-1 NL4-3 ΔNef (catalog no. 12755) infectious molecular clones were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (48, 49). To obtain a construct encoding the human HLA-A2 heavy chain with an N-terminal HA epitope tag (pHA-HLA-A2), two cDNA fragments were amplified and inserted into pCIneo (Promega) using the EcoRI/SalI sites. One fragment encodes the mature sequence of the HLA-A2 heavy chain flanked by XbaI and SalI sites. The other fragment encodes the signal peptide of HLA-A2 flanked by an EcoRI site and an HA epitope sequence followed by an XbaI site. For GST pulldown experiments, a chimera composed of GST-HLA-A2_CT-Nef (16) was generated as follows. A sequence encoding an HLA-A2-Nef fusion protein was obtained by PCR amplification of the full-length HIV-1 Nef sequence using a forward primer encoding residues 314 to 341 of the mature HLA-A2 protein. This PCR fragment was then cloned as an EcoRI/SalI insert into pGEX5.1 (GE Healthcare). GST-HLA-A2_CT(Y320A)-Nef was generated by site-directed mutagenesis. All open reading frames were verified by nucleotide sequence analysis.
Recombinant protein expression and GST pulldown assays.
All GST recombinant proteins were expressed in Escherichia coli BL21 Rosetta(DE3) cells from a pGEX5.1 vector (GE Healthcare) for 16 h at 22°C after induction with 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After this period, cells were centrifuged for 5 min at 4°C at 3,000 × g, and the pellet was resuspended in ice-cold lysis buffer 1 (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM EDTA, 10 mM dithiothreitol [DTT]), supplemented with 500 μg/ml lysozyme, disrupted by sonication, supplemented with 1% Nonidet P-40, and centrifuged for 20 min at 4°C. The cleared cell lysate was incubated with glutathione-Sepharose 4B beads (GE Healthcare) for 2 h at 4°C under orbital rotation. After this period, beads were washed with lysis buffer 2 (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 10% [vol/vol] glycerol, 5 mM EDTA, and 1% [vol/vol] Triton X-100 supplemented with a cocktail of protease inhibitors [Sigma-Aldrich, St. Louis, MO, USA]) and subsequently incubated with cleared HeLa cells lysed overnight at 4°C under orbital rotation. Following this incubation, the beads were washed with lysis buffer 2, resuspended in sample buffer (4% SDS, 160 mM Tris-HCl [pH 6.8], 20% glycerol, 100 mM DTT, and 0.005% bromphenol blue), and incubated for 5 min at 90°C. The samples were then subjected to SDS-PAGE (10%) and immunoblotting.
Cell lines and cell culture.
HeLa ATCC CCL2 cells (American Type Culture Collection, Manassas, VA) and HEK 293T Peak cells (50) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM l-glutamine, and 10% fetal bovine serum (FBS) (Life Technologies) at 37°C with 5% CO2. The human A3.01 CD4+ T cell line was obtained from the NIH AIDS Research and Reference Reagent Program (Germantown, MD), originally deposited by Thomas Folks (51). A3.01 T cells expressing Nef and GFP (A3.01 Nef/GFP) or GFP alone (A3.01 GFP) were obtained by transduction with a bicistronic internal ribosome entry site (IRES)-based retroviral vector as previously described (30). A3.01 T cells were grown in RPMI 1640 medium (Life Technologies, Carlsbad, CA) and supplemented as described above. To obtain a HeLa cell line constitutively expressing HA-HLA-A2, cells were transfected with the pHA-HLA_A2 plasmid, and positive clones were selected with 500 μg/ml Geneticin (Life Technologies), followed by immunofluorescence analysis. The Rev-CEM cell line (catalog no. 11467), which expresses GFP upon HIV-1 infection driven by the HIV-1 NL4-3 LTR, was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (52). These cells were grown in RPMI 1640 medium (Life Technologies, Carlsbad, CA) and supplemented as described above.
HLA typing.
HLA-A typing was performed using sequence-specific oligonucleotides of a commercial kit (Micro SSP DNA typing tray; One Lambda, Canoga Park, CA), according to the manufacturer’s instructions.
Subcellular fractionation.
Subcellular fractionation on glycerol gradients was performed as described previously (53). Briefly, 10 million A3.01 Nef/GFP or A3.01 GFP cells were washed 2 times with cold phosphate-buffered saline (PBS) and once with cold STE buffer (250 mM sucrose, 20 mM Tris-HCl [pH 7.4], and 1 mM EDTA with protease inhibitors) and resuspended in 800 μl of ice-cold STE buffer without sucrose. Next, the cells were homogenized by 20 passages through a 25-gauge needle and centrifuged at 1,000 × g for 2 min at 4°C. The supernatant (750 μl) was layered on top of a 2%-stepwise, 10 to 30% (wt/vol) glycerol gradient (13.75 ml in 200 mM Tris-HCl [pH 7.4] and 1 mM EDTA on a 0.5-ml 80% sucrose cushion) and centrifuged at 29,000 rpm for 2 h at 4°C in a Thermo TH660 instrument. Fractions of 1.25 ml were collected from the top, precipitated with trichloroacetic acid (TCA), resuspended in sample buffer, and analyzed by SDS-PAGE and Western blotting.
Plasmids and small interfering RNA transfections in HeLa cells.
HeLa HA-HLA-A2 cells were treated twice with control siRNA (Mission siRNA universal negative control, catalog no. SIC001; Sigma-Aldrich), siRNA for AP-1γ1 (5′-GGAAGAGCCUAUUCAGGUA-3′) (Sigma-Aldrich), or siRNA for AP-1γ2 (5′-AAACCCUGCUUUGCUGUUAA-3′) (Sigma-Aldrich), with 48-h intervals, by using Oligofectamine reagent (Thermo Scientific). One day after the second round of siRNA transfection, cells were transfected with Nef or control plasmids by using Lipofectamine 2000 (Thermo Scientific). After 24 h of incubation at 37°C, cells were further incubated at 26°C with 5% CO2 for 16 h, before they were analyzed by immunofluorescence, flow cytometry (fluorescence-activate cell sorter [FACS] analysis), or immunoblotting.
HIV-1 production and infection.
HIV-1 production was performed in HEK 293T cells, which were transfected with pNL4-3 WT or ΔNef with 25-kDa linear polyethylenimine (PEI) transfection reagent (Polysciences Inc., Warrington, PA). At 48 h posttransfection, the virus-containing media were collected and used to infect CD4+ T cells (Rev-CEM cell line, in which GFP expression is controlled by HIV-1 infection) by spinoculation at 1,200 × g for 2 h at 23°C. The cells were cultured for 5 days and processed for flow cytometry analyses to assess HLA-A surface expression (see below). The infected cells were assessed by GFP fluorescence.
Virus production and transduction of T cells.
For knockdown experiments in T cells using shRNA, Peak cells were transfected with 0.5 μg of control shRNA (Mission TRC2 pLKO.5-puro nonmammalian shRNA control plasmid, catalog no. SHC202; Sigma-Aldrich) or shRNA against AP-1γ1 (Mission TRC2 pLKO.5-puro, catalog no. TRCN0000065152; Sigma-Aldrich), AP-1γ2 (Mission TRC2 pLKO.5-puro, catalog no. TRCN0000233143), or AP-1μ1A (Mission TRC2 pLKO.5-puro, catalog no. TRCN000006508) together with 0.375 μg of psPAX2 (catalog no. 12259; Addgene) and 0.125 μg of pMD2.G (catalog no. 12259; Addgene) by using 8 μl of 25-kDa linear PEI (1-mg/ml stock solution) for each well of a 6-well plate. After 15 h posttransfection, the culture medium was removed, and 1 ml of RPMI medium supplemented with 30% FBS (without antibiotics) was added to each well. The virus supernatants were subsequently collected for 2 days and cleared by centrifugation for 10 min at 2,000 × g at 4°C. After centrifugation, 1 ml of the viral supernatant was added to 1 × 106 A3.01 Nef/GFP or A3.01 GFP T cells in a 12-well plate. Cells were incubated for 120 h under puromycin selection before the assays were performed.
Antibodies.
The rabbit polyclonal antibody to human HLA-A, used in most experiments, was purchased from Proteintech (Chicago, IL). In immunofluorescence assays, in combination with the rabbit anti-AP-1γ2 antibody, a mouse anti-HLA class I antibody that recognises HLA-A, -B, and -C loci (W6/32, catalog no. ab23755; Abcam, Cambridge, MA) was used. Rabbit polyclonal anti-HRS (catalog no. ab155539; Abcam, Cambridge, MA), mouse monoclonal anti-TfR (catalog no. 136800; Invitrogen), and rabbit antiserum to GFP, a generous gift of R. Hegde (MRC, Cambridge, UK), were used in the immunoblot analyses. Mouse monoclonal anti-EEA1 (catalog no. 610456; BD Biosciences, San Jose, CA) was used in the immunofluorescence and Western blot assays. Mouse monoclonal anti-β-actin (C4, catalog no. sc-47778; Santa Cruz Biotechnology) and rabbit polyclonal anti-μ1A-adaptin (catalog no. AB111135; Abcam, Cambridge, MA) antibodies were used in immunoblot assays. Mouse monoclonal anti-AP-1γ1 (clone 88, adaptin-γ, catalog no. 610386; BD Biosciences, San Jose, CA) and rabbit anti-AP-1γ2 (catalog no. HPA004106; Sigma-Aldrich) antibodies were used in the immunoblot and immunofluorescence assays. Sheep polyclonal antibody to TGN46 (AbD Serotec, Oxford, UK) and mouse monoclonal antibody to the HA epitope (catalog no. H3663; Sigma-Aldrich) were used in the immunofluorescence and Western blot assays. Mouse monoclonal anti-AP-4ε antibody (catalog no. 612018; BD Biosciences, San Jose, CA) was used in the immunoblot assay. Rabbit antiserum to HIV-1 Nef, used for immunoblot experiments, was obtained from the NIH AIDS Research and Reference Reagent Program (54). Horseradish peroxidase (HRP)-conjugated donkey anti-mouse immunoglobulin G (IgG) and donkey anti-rabbit IgG were obtained from GE Healthcare. Secondary antibodies conjugated to Alexa fluorophores were purchased from Thermo Scientific, and anti-rabbit nanogold-conjugated secondary antibody used for immunoelectron microscopy experiments was purchased from Nanoprobes.
Immunofluorescence and confocal microscopy.
HeLa cells were grown on coverslips, whereas A3.01 T cells were incubated on coverslips coated with Biobond tissue adhesive (Electron Microscopy Sciences, Hatfield, PA) for adhesion at a concentration of 2 × 105 cells/ml in PBS. Cells were then fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.01% saponin in blocking buffer (1% pork skin gelatin in PBS). Subsequently, the cells were incubated with specific primary antibodies diluted in blocking solution, washed with PBS, and incubated with appropriate secondary antibodies conjugated to Alexa fluorophores. The coverslips were then mounted on slides using Fluoromount G (Electron Microscopy Sciences, Hatfield, PA, USA) and imaged using a Leica TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany) or a Zeiss confocal laser scanning microscope (LSM780; Zeiss, Jena, Germany). The obtained images were analyzed using Fiji/ImageJ software using z-stacks of at least five cells under each condition from three independent experiments for Manders’ coefficient calculation.
SDS-PAGE and immunoblot analysis.
The samples were equalized for total protein concentration, and the protein extracts were submitted to SDS-PAGE and electrotransferred to nitrocellulose membrane (Millipore, Bedford, MA, USA). The membranes were then blocked with PBS-T (PBS, 05% Tween 20) and 5% nonfat dry milk, followed by individual incubation with primary and secondary antibodies conjugated to HRP. Proteins were detected by using an enhanced chemiluminescence (ECL) solution and visualized with ChemiDoc imaging systems (GE Life Science, Chicago, IL, USA).
FACS analysis.
To assess surface HLA-A, unfixed cells were incubated at 4°C with rabbit polyclonal antibody to human HLA-A, followed by incubation with F(ab′)2 goat anti-rabbit IgG(H+L) cross-adsorbed Alexa Fluor 647 secondary antibody (catalog no. A-21246; Thermo Scientific), and prepared for FACS analysis. FACS data were acquired using the levels of Alexa Fluor 647 fluorescence in cells expressing GFP with a FACSDiva flow cytometer (BD Biosciences). FlowJo software (TreeStar, Ashland, OR) was used for data analyses.
Electron microscopy analysis.
A3.01 T cells expressing Nef/GFP were transduced with a retrovirus encoding either a control shRNA or AP-1γ2 shRNA, as described above. Cells were fixed with 4% (wt/vol) PFA in PBS for 15 min, rinsed with 50 mM glycine in PBS for 15 min, and blocked with 1% (wt/vol) bovine serum albumin in PBS for 30 min. Cells were then permeabilized with 0.05% (wt/vol) saponin and 1% (wt/vol) bovine serum albumin in PBS for 30 min, incubated with antibody to HLA-A (Proteintech, Chicago, IL) for 1 h at room temperature, rinsed, and subsequently incubated with nanogold-conjugated secondary antibodies (Nanoprobes, Yaphank, NY) for 1 h at room temperature. Cells were then fixed with glutaraldehyde (2.5% [vol/vol] in 0.1 M cacodylate buffer) for 1 h, rinsed, treated with a gold enhancement mixture (Nanoprobes, Yaphank, NY) for 6 min, and postfixed in reduced osmium prior to embedding in Embed 812 (EM Sciences). All steps described above were done upon orbital agitation. Sections of 70 to 100 nm were stained with lead citrate prior to imaging with a JEOL 100cx transmission electron microscope (JEOL, Tokyo, Japan). A quantitative analysis was done with 1,093 gold particles of control shRNA samples and 1,227 gold particles of AP-1γ2 shRNA samples counted from pictures taken from two grids under each sample condition. The cellular distribution of the HLA-A signal was determined as gold particles associated with the plasma membrane, cytoplasm, multivesicular bodies (MVBs)/LE, vacuoles, and tubules. The gold particles scored were expressed as a percentage of the total number of gold particles counted for each picture.
ACKNOWLEDGMENTS
We thank J. Bonifacino and the NIH AIDS Research and Reference Reagent Program for the kind donation of reagents, J. A. Maulin and M. D. S. Ferreira (FMRP-USP Electron Microscopy Facility) for help with TEM experiments, and E. Tozatto (LIAREC-FCFRP-USP) for help with confocal microscopy. We are grateful to D. M. Jorge (CPV-FMRP-USP) for analysis of HLA-A sequences and L. Jesus da Costa (UFRJ) for helpful discussions.
The present study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (São Paulo Research Foundation) grants (18/00297-7 and 14/02438-6) and by Fundação de Apoio ao Ensino, Pesquisa e Assistência do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (FAEPA), grants to L.L.P.D. L.A.T. was supported by a doctoral fellowship, and R.M.S. and C.S.C. were supported by master’s fellowships from FAPESP (16/18207-9, 16/05945-1, and 18/08966-5, respectively). J.V.D.C. was supported by a postdoctoral fellowship (2015/26667-7) from FAPESP. L.L.P.D. is the recipient of a long-standing investigator scholarship from CNPq.
REFERENCES
- 1.Sugden SM, Bego MG, Pham TN, Cohen EA. 2016. Remodeling of the host cell plasma membrane by HIV-1 Nef and Vpu: a strategy to ensure viral fitness and persistence. Viruses 8:67. doi: 10.3390/v8030067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pereira EA, daSilva LL. 2016. HIV-1 Nef: taking control of protein trafficking. Traffic 17:976–996. doi: 10.1111/tra.12412. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz O, Maréchal V, Le Gall S, Lemonnier F, Heard JM. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med 2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 4.Garcia JV, Miller AD. 1991. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350:508–511. doi: 10.1038/350508a0. [DOI] [PubMed] [Google Scholar]
- 5.Usami Y, Wu Y, Gottlinger HG. 2015. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526:218–223. doi: 10.1038/nature15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, Santoni FA, Pizzato M. 2015. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526:212–217. doi: 10.1038/nature15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 8.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671. doi: 10.1016/S1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 9.Specht A, DeGottardi MQ, Schindler M, Hahn B, Evans DT, Kirchhoff F. 2008. Selective downmodulation of HLA-A and -B by Nef alleles from different groups of primate lentiviruses. Virology 373:229–237. doi: 10.1016/j.virol.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 10.Williams M, Roeth JF, Kasper MR, Fleis RI, Przybycin CG, Collins KL. 2002. Direct binding of human immunodeficiency virus type 1 Nef to the major histocompatibility complex class I (MHC-I) cytoplasmic tail disrupts MHC-I trafficking. J Virol 76:12173–12184. doi: 10.1128/jvi.76.23.12173-12184.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donaldson JG, Williams DB. 2009. Intracellular assembly and trafficking of MHC class I molecules. Traffic 10:1745–1752. doi: 10.1111/j.1600-0854.2009.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaefer MR, Wonderlich ER, Roeth JF, Leonard JA, Collins KL. 2008. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog 4:e1000131. doi: 10.1371/journal.ppat.1000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roeth JF, Williams M, Kasper MR, Filzen TM, Collins KL. 2004. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J Cell Biol 167:903–913. doi: 10.1083/jcb.200407031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonard JA, Filzen T, Carter CC, Schaefer M, Collins KL. 2011. HIV-1 Nef disrupts intracellular trafficking of major histocompatibility complex class I, CD4, CD8, and CD28 by distinct pathways that share common elements. J Virol 85:6867–6881. doi: 10.1128/JVI.00229-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lubben NB, Sahlender DA, Motley AM, Lehner PJ, Benaroch P, Robinson MS. 2007. HIV-1 Nef-induced down-regulation of MHC class I requires AP-1 and clathrin but not PACS-1 and is impeded by AP-2. Mol Biol Cell 18:3351–3365. doi: 10.1091/mbc.e07-03-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noviello CM, Benichou S, Guatelli JC. 2008. Cooperative binding of the class I major histocompatibility complex cytoplasmic domain and human immunodeficiency virus type 1 Nef to the endosomal AP-1 complex via its mu subunit. J Virol 82:1249–1258. doi: 10.1128/JVI.00660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh RK, Lau D, Noviello CM, Ghosh P, Guatelli JC. 2009. An MHC-I cytoplasmic domain/HIV-1 Nef fusion protein binds directly to the mu subunit of the AP-1 endosomal coat complex. PLoS One 4:e8364. doi: 10.1371/journal.pone.0008364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia X, Singh R, Homann S, Yang H, Guatelli J, Xiong Y. 2012. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat Struct Mol Biol 19:701–706. doi: 10.1038/nsmb.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wonderlich ER, Williams M, Collins KL. 2008. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J Biol Chem 283:3011–3022. doi: 10.1074/jbc.M707760200. [DOI] [PubMed] [Google Scholar]
- 20.daSilva LLP, Mardones GA. 2018. HIV/SIV-Nef: pas de trois choreographies to evade immunity. Trends Microbiol 26:889–891. doi: 10.1016/j.tim.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Morris KL, Buffalo CZ, Sturzel CM, Heusinger E, Kirchhoff F, Ren X, Hurley JH. 2018. HIV-1 Nefs are cargo-sensitive AP-1 trimerization switches in tetherin downregulation. Cell 174:659–671. doi: 10.1016/j.cell.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson MS, Bonifacino JS. 2001. Adaptor-related proteins. Curr Opin Cell Biol 13:444–453. doi: 10.1016/s0955-0674(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 23.Yi L, Rosales T, Rose JJ, Chowdhury B, Knutson JR, Venkatesan S. 2010. HIV-1 Nef binds a subpopulation of MHC-I throughout its trafficking itinerary and down-regulates MHC-I by perturbing both anterograde and retrograde trafficking. J Biol Chem 285:30884–30905. doi: 10.1074/jbc.M110.135947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonifacino JS. 2014. Adaptor proteins involved in polarized sorting. J Cell Biol 204:7–17. doi: 10.1083/jcb.201310021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo X, Mattera R, Ren X, Chen Y, Retamal C, Gonzalez A, Bonifacino JS. 2013. The adaptor protein-1 mu1B subunit expands the repertoire of basolateral sorting signal recognition in epithelial cells. Dev Cell 27:353–366. doi: 10.1016/j.devcel.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tavares LA, da Silva EM, da Silva-Januario ME, Januario YC, de Cavalho JV, Czernisz ES, Mardones GA, daSilva LL. 2017. CD4 downregulation by the HIV-1 protein Nef reveals distinct roles for the gamma1 and gamma2 subunits of the AP-1 complex in protein trafficking. J Cell Sci 130:429–443. doi: 10.1242/jcs.192104. [DOI] [PubMed] [Google Scholar]
- 27.Candiello E, Mishra R, Schmidt B, Jahn O, Schu P. 2017. Differential regulation of synaptic AP-2/clathrin vesicle uncoating in synaptic plasticity. Sci Rep 7:15781. doi: 10.1038/s41598-017-16055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zizioli D, Geumann C, Kratzke M, Mishra R, Borsani G, Finazzi D, Candiello E, Schu P. 2017. Gamma2 and gamma1AP-1 complexes: different essential functions and regulatory mechanisms in clathrin-dependent protein sorting. Eur J Cell Biol 96:356–368. doi: 10.1016/j.ejcb.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Mattera R, Boehm M, Chaudhuri R, Prabhu Y, Bonifacino JS. 2011. Conservation and diversification of dileucine signal recognition by adaptor protein (AP) complex variants. J Biol Chem 286:2022–2030. doi: 10.1074/jbc.M110.197178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Carvalho JV, de Castro RO, da Silva EZ, Silveira PP, da Silva-Januário ME, Arruda E, Jamur MC, Oliver C, Aguiar RS, daSilva LL. 2014. Nef neutralizes the ability of exosomes from CD4+ T cells to act as decoys during HIV-1 infection. PLoS One 9:e113691. doi: 10.1371/journal.pone.0113691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfeffer SR. 2011. Entry at the trans-face of the Golgi. Cold Spring Harb Perspect Biol 3:a005272. doi: 10.1101/cshperspect.a005272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kasper MR, Roeth JF, Williams M, Filzen TM, Fleis RI, Collins KL. 2005. HIV-1 Nef disrupts antigen presentation early in the secretory pathway. J Biol Chem 280:12840–12848. doi: 10.1074/jbc.M413538200. [DOI] [PubMed] [Google Scholar]
- 33.Shen QT, Ren X, Zhang R, Lee IH, Hurley JH. 2015. HIV-1 Nef hijacks clathrin coats by stabilizing AP-1:Arf1 polygons. Science 350:aac5137. doi: 10.1126/science.aac5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pawlak EN, Dikeakos JD. 2015. HIV-1 Nef: a master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim Biophys Acta 1850:733–741. doi: 10.1016/j.bbagen.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Kasper MR, Collins KL. 2003. Nef-mediated disruption of HLA-A2 transport to the cell surface in T cells. J Virol 77:3041–3049. doi: 10.1128/jvi.77.5.3041-3049.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burtey A, Rappoport JZ, Bouchet J, Basmaciogullari S, Guatelli J, Simon SM, Benichou S, Benmerah A. 2007. Dynamic interaction of HIV-1 Nef with the clathrin-mediated endocytic pathway at the plasma membrane. Traffic 8:61–76. doi: 10.1111/j.1600-0854.2006.00512.x. [DOI] [PubMed] [Google Scholar]
- 37.Chaudhuri R, Mattera R, Lindwasser OW, Robinson MS, Bonifacino JS. 2009. A basic patch on alpha-adaptin is required for binding of human immunodeficiency virus type 1 Nef and cooperative assembly of a CD4-Nef-AP-2 complex. J Virol 83:2518–2530. doi: 10.1128/JVI.02227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. 2007. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol 81:3877–3890. doi: 10.1128/JVI.02725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren X, Park SY, Bonifacino JS, Hurley JH. 2014. How HIV-1 Nef hijacks the AP-2 clathrin adaptor to downregulate CD4. Elife 3:e01754. doi: 10.7554/eLife.01754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greenberg ME, Bronson S, Lock M, Neumann M, Pavlakis GN, Skowronski J. 1997. Co-localization of HIV-1 Nef with the AP-2 adaptor protein complex correlates with Nef-induced CD4 down-regulation. EMBO J 16:6964–6976. doi: 10.1093/emboj/16.23.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.daSilva LL, Sougrat R, Burgos PV, Janvier K, Mattera R, Bonifacino JS. 2009. Human immunodeficiency virus type 1 Nef protein targets CD4 to the multivesicular body pathway. J Virol 83:6578–6590. doi: 10.1128/JVI.00548-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amorim NA, da Silva EM, de Castro RO, da Silva-Januário ME, Mendonça LM, Bonifacino JS, da Costa LJ, daSilva LL. 2014. Interaction of HIV-1 Nef protein with the host protein Alix promotes lysosomal targeting of CD4 receptor. J Biol Chem 289:27744–27756. doi: 10.1074/jbc.M114.560193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rost M, Döring T, Prange R. 2008. γ2-Adaptin, a ubiquitin-interacting adaptor, is a substrate to coupled ubiquitination by the ubiquitin ligase Nedd4 and functions in the endosomal pathway. J Biol Chem 283:32119–32130. doi: 10.1074/jbc.M802632200. [DOI] [PubMed] [Google Scholar]
- 44.Döring T, Gotthardt K, Stieler J, Prange R. 2010. γ2-Adaptin is functioning in the late endosomal sorting pathway and interacts with ESCRT-I and -III subunits. Biochim Biophys Acta 1803:1252–1264. doi: 10.1016/j.bbamcr.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 45.Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol 66:7193–7200. doi: 10.1128/JVI.66.12.7193-7200.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magadán JG, Pérez-Victoria FJ, Sougrat R, Ye Y, Strebel K, Bonifacino JS. 2010. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog 6:e1000869. doi: 10.1371/journal.ppat.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindwasser OW, Smith WJ, Chaudhuri R, Yang P, Hurley JH, Bonifacino JS. 2008. A diacidic motif in human immunodeficiency virus type 1 Nef is a novel determinant of binding to AP-2. J Virol 82:1166–1174. doi: 10.1128/JVI.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. doi: 10.1128/JVI.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwartz O, Marechal V, Danos O, Heard JM. 1995. Human immunodeficiency virus type 1 Nef increases the efficiency of reverse transcription in the infected cell. J Virol 69:4053–4059. doi: 10.1128/JVI.69.7.4053-4059.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guiraldelli MF, Berenstein EH, Grodzki AC, Siraganian RP, Jamur MC, Oliver C. 2008. The low affinity IgG receptor Fc gamma RIIB contributes to the binding of the mast cell specific antibody, mAb BGD6. Mol Immunol 45:2411–2418. doi: 10.1016/j.molimm.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 51.Folks T, Powell DM, Lightfoote MM, Benn S, Martin MA, Fauci AS. 1986. Induction of HTLV-III/LAV from a nonvirus-producing T-cell line: implications for latency. Science 231:600–602. doi: 10.1126/science.3003906. [DOI] [PubMed] [Google Scholar]
- 52.Gervaix A, West D, Leoni LM, Richman DD, Wong-Staal F, Corbeil J. 1997. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc Natl Acad Sci U S A 94:4653–4658. doi: 10.1073/pnas.94.9.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez-Victoria FJ, Mardones GA, Bonifacino JS. 2008. Requirement of the human GARP complex for mannose 6-phosphate-receptor-dependent sorting of cathepsin D to lysosomes. Mol Biol Cell 19:2350–2362. doi: 10.1091/mbc.e07-11-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shugars DC, Smith MS, Glueck DH, Nantermet PV, Seillier-Moiseiwitsch F, Swanstrom R. 1993. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J Virol 67:4639–4650. doi: 10.1128/JVI.67.8.4639-4650.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abramoff MD, Magelhaes PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int 11:36–42. [Google Scholar]
- 56.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. 2004. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J 86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]