

Alternative treatments to combat rotavirus infection are a requirement for susceptible communities where vaccines cannot be applied. This demand is urgent for newborn infants, immunocompromised patients, adults traveling to high-risk regions, and even for the livestock industry. Aside from structural and physiological divergences among RV species studied before now, all replicate within cytosolic inclusions termed viroplasms. These inclusions are composed of viral and cellular proteins and viral RNA. Viroplasm-like structures (VLS), composed of RV protein NSP5 with either NSP2 or VP2, are models for investigating viroplasms. In this study, we identified a conserved amino acid in the VP2 protein, L124, necessary for its interaction with NSP5 and the formation of both VLSs and viroplasms. As RV vaccines cover a narrow range of viral strains, the identification of VP2 L124 residue lays the foundations for the design of drugs that specifically block NSP5-VP2 interaction as a broad-spectrum RV antiviral.
KEYWORDS: NSP5, VLS, VP2, dominant negative, hyperphosphorylation, protein interface, replication, rotavirus, viral factories, viroplasms
ABSTRACT
One step of the life cycle common to all rotaviruses (RV) studied so far is the formation of viroplasms, membrane-less cytosolic inclusions providing a microenvironment for early morphogenesis and RNA replication. Viroplasm-like structures (VLS) are simplified viroplasm models consisting of complexes of nonstructural protein 5 (NSP5) with the RV core shell VP2 or NSP2. We identified and characterized the domains required for NSP5-VP2 interaction and VLS formation. VP2 mutations L124A, V865A, and I878A impaired both NSP5 hyperphosphorylation and NSP5/VP2 VLS formation. Moreover, NSP5-VP2 interaction does not depend on NSP5 hyperphosphorylation. The NSP5 tail region is required for VP2 interaction. Notably, VP2 L124A expression acts as a dominant-negative element by disrupting the formation of either VLS or viroplasms and blocking RNA synthesis. In silico analyses revealed that VP2 L124, V865, and I878 are conserved among RV species A to H. Detailed knowledge of the protein interaction interface required for viroplasm formation may facilitate the design of broad-spectrum antivirals to block RV replication.
IMPORTANCE Alternative treatments to combat rotavirus infection are a requirement for susceptible communities where vaccines cannot be applied. This demand is urgent for newborn infants, immunocompromised patients, adults traveling to high-risk regions, and even for the livestock industry. Aside from structural and physiological divergences among RV species studied before now, all replicate within cytosolic inclusions termed viroplasms. These inclusions are composed of viral and cellular proteins and viral RNA. Viroplasm-like structures (VLS), composed of RV protein NSP5 with either NSP2 or VP2, are models for investigating viroplasms. In this study, we identified a conserved amino acid in the VP2 protein, L124, necessary for its interaction with NSP5 and the formation of both VLSs and viroplasms. As RV vaccines cover a narrow range of viral strains, the identification of VP2 L124 residue lays the foundations for the design of drugs that specifically block NSP5-VP2 interaction as a broad-spectrum RV antiviral.
INTRODUCTION
Rotaviruses (RVs) are double-stranded RNA (dsRNA) viruses of the Reoviridae family and are responsible for severe gastroenteritis in infants and young children, killing approximately 128,000 children per year, mainly in developing countries (1). RV virions are organized in three concentric layers surrounding the viral genome. The spike protein VP4 and the glycoprotein VP7 form the outermost layer, while VP6 makes up the intermediate layer. The innermost layer, the core shell, is composed of 120 copies of VP2, organized in twelve asymmetric decamers (2). Each core shell encapsidates the eleven segments of the viral genome as well as the replication complexes composed of the RNA-dependent RNA polymerase (RdRp) VP1 and the guanylyl-methyltransferase VP3. There is evidence that one replication complex is located beneath each 5-fold axis of the twelve VP2 decamers (2, 3). During virus entry the external layer is lost, and a transcriptionally active double-layered particle (DLP) is released into the cytosol (4). The newly released transcripts enable the synthesis of viral proteins necessary for viral replication. Among those proteins, the NTPase/RTPase NSP2 and the phosphoprotein NSP5, together with the structural proteins VP1, VP2, VP3, and VP6 and the nonstructural NSP4, constitute the RV factories termed viroplasms (5, 6). The viroplasms correspond to membrane-less cytosolic electron-dense inclusions where viral genome transcription and replication, as well as the packaging of the newly synthesized pregenomic RNA segments into the viral cores, take place. Interestingly, coexpression of the main viroplasm protein, NSP5, with either NSP2 or VP2 leads to the formation of cytosolic inclusions, named viroplasm-like structures (VLS), that are morphologically similar to viroplasms but unable to yield viral progeny (7–12). VLS serve as a useful simplified model for studying RV viroplasms, since they share physiological traits with viroplasms, including coalescence and perinuclear condensation (10, 13–15). NSP5 is hyperphosphorylated (16, 17) and O-GlcNAc glycosylated (18). While the role of NSP5 phosphorylation in RV replication remains obscure, it has been demonstrated that NSP2 or VP2 is required to enhance the hyperphosphorylation state (19, 20). Moreover, NSP5 is required for viroplasm formation and virus replication, as shown by knockdown experiments using short interfering RNAs (siRNAs) (21, 22). NSP5 has a multifunctional role in the RV life cycle, interacting with NSP6 (12), NSP2 (9), VP1 (23), VP2 (24), and, in an unspecific manner, dsRNA (25), which also is consistent with its predicted partially unfolded nature (26–28). NSP5 was recently described as being covalently sumoylated (29), suggesting that this modification is a prerequirement for the interaction with viral or host components. Taking these studies together, NSP5 can be considered an essential component for RV replication. Likewise, the core shell protein VP2, besides its structural function protecting the viral genome, is also able to activate and regulate the RdRp VP1 protein (30), permitting genome replication. In triple-layered particles and DLPs, VP2 spontaneously oligomerizes by forming an asymmetric decameric structure with VP2 isoforms A and B that converges in the 5-fold axis and cannot dissociate (2, 28, 31–33). Each subunit of the decamer is composed of a primary domain of VP2 (residues ∼100 to 880), forming a thin comma-shaped plate, which in turn also divides into three subdomains: apical, central, and dimer forming. The N-terminal domain (NTD) (residues ∼1 to 100 for type A and ∼1 to 80 for type B) is unfolded and localizes in the decamer beneath the 5-fold axis. VP1, VP3, VP6, and NSP2 (2, 34–36), as well as unspecific single-stranded RNA (ssRNA) (37), have been mapped to bind to VP2, most of them directly associated with the NTD. However, these viral proteins can also make contact with the VP2 scaffold region; for example, VP1 contacts the VP2 region 341–374 (38–40). These interactions are all directly related to the core shell structure and genome replication and transcription. VP2 is also a main component of the viroplasms, as denoted by immunofluorescence of RV-infected cells and its ability to form VLS when coexpressed with NSP5 (7, 10, 41). The VLS induced by VP2 (here termed VLS) are dynamic and able to shift to the perinuclear region by a mechanism that is still under investigation (10). Further evidence indicates that VP2 triggers NSP5 hyperphosphorylation (7). Recently, it was suggested that VP2 has additional roles at early times postinfection because of its interaction with NSP2, preventing its spontaneous oligomerization and sumoylation, which increases the ability of VP2 to interact with other proteins (10, 29). Therefore, we hypothesized that the interaction of these two proteins to form VLS is an essential step for RV replication.
In this study, we show that specific point mutations in VP2 abrogate VLS formation by disrupting NSP5 binding and phosphorylation. Additionally, we demonstrate that NSP5 requires its 18 C-terminal amino acids to interact with VP2 directly. Importantly, VP2 harboring the point mutation L124A disrupts VLS induced by the expression of NSP5-VP2 and NSP5-NSP2. Similarly, in RV-infected cells, VP2 L124A expression results in a reduced number of infected cells and viroplasms with aberrant structures and defective in viral RNA synthesis. Altogether, these data provide the mechanistic explanation that a conserved NSP5-VP2 interaction interface is required for efficient RV replication.
RESULTS
VP2 amino acid regions 103–135 and 840–880 are necessary for VLS formation with NSP5.
To elucidate the nature of the association between NSP5 and VP2, we assessed the capacity of these two proteins to form viroplasm-like structures when coexpressed in the absence of other rotavirus proteins (7). To this end, and as denoted in Fig. 1A, we constructed a series of VP2 mutants by deleting N-terminal and C-terminal regions as well as constructs expressing single regions of the protein based on the published tertiary VP2 structure (42). For the detection of the VP2 deletion mutants, a human influenza virus hemagglutinin (HA) tag was added at the N terminus. All HA-VP2 deletion mutants were verified by immunoblotting for correct expression and size (data not shown). We next performed immunofluorescence analysis of cotransfected cells to assess VLS formation, which was considered positive only when colocalization of both HA-VP2 and NSP5 was observed in cytosolic inclusions. The results, summarized in Fig. 1, show that full-length HA-VP2 (fl) was homogeneously distributed in the cytosol of transfected cells (Fig. 1B, top row, −NSP5). However, when HA-VP2fl was coexpressed with NSP5 (Fig. 1B, top row, +NSP5), it formed cytosolic inclusions that colocalized with NSP5, corresponding to VLSs. In the presence of NSP5, HA-VP2ΔN103 was able to form VLSs, while HA-VP2ΔN135 was not (Fig. 1B). The shortest HA-VP2 C-terminal deletion mutant tested (Fig. 1B), HA-VP2 ΔC840, was also unable to form VLSs when coexpressed with NSP5, suggesting that the amino acid region 840 to 880 is necessary for VLS formation with NSP5 as well. Of note, none of the constructs that tested positive for VLS formation colocalized with ubiquitin when coexpressed with NSP5 (data not shown), suggesting that the observed structures correspond to genuine VLS. These data indicate that amino acids 103 to 135 and 840 to 880 (termed regions 103–135 and 840–880) of VP2 are necessary for VLS formation between VP2 and NSP5. As expected, none of the HA-VP2 single regions formed VLSs when coexpressed with NSP5. A summary of the results of all HA-VP2 deletion mutants tested for VLS formation is shown in the right column of Fig. 1A.
FIG 1.
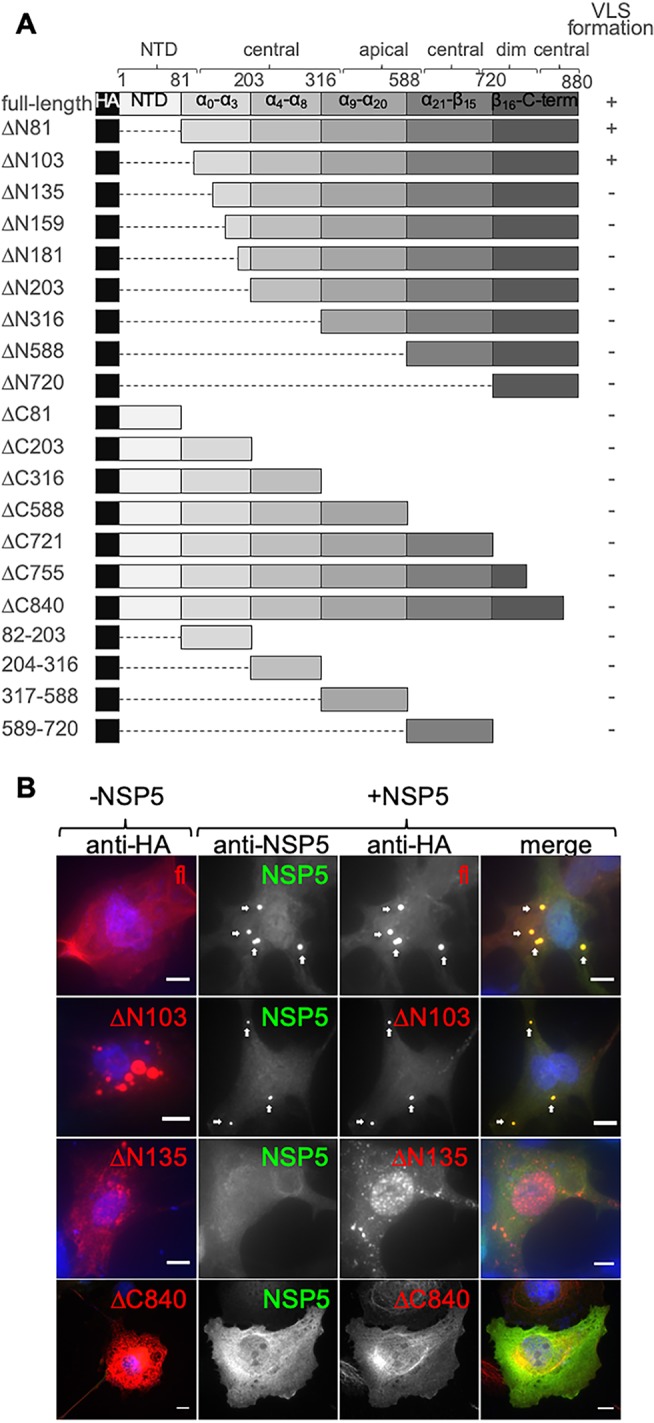
VP2 amino acid regions 103–134 and 840–880 are necessary for VLS formation. (A) Schematic representation of VP2 deletion mutants fused to an HA tag at the N terminus (not to scale). The dashed lines correspond to deleted regions. Positive (+) or negative (−) VLS phenotype is indicated at the right. N-terminal (NTD), central, apical, and dimerization (dim) VP2 tertiary domains are indicated. (B) Representative photomicrographs of MA104 cells expressing the indicated N and C termini of HA-VP2 deletion mutants alone (−NSP5 panel) or together with NSP5 (+NSP5 panel). At 16 hpt, cells were fixed and immunostained for detection of HA-VP2 deletion mutant (anti-HA, red) and NSP5 (anti-NSP5, green). A merged image is shown in the right column. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). White arrows point to VLS. Scale bar is 10 μm.
VP2 amino acids L124, V865, and I878 are required for VLS formation.
We next determined the residues in region 103–135 of VP2 (RV simian strain SA11) that are relevant for VLS formation. For this, we first performed primary sequence alignment of all VP2 proteins from species A to H (Fig. 2A). This analysis revealed that four amino acid residues are highly conserved among all the aligned VP2 proteins, specifically I106, D112, L124, and I127. To analyze their role in VLS formation, we mutated each of these residues to alanine in HA-VP2ΔN103, which is the minimal HA-VP2 deletion mutant able to form VLS when coexpressed with NSP5. The point mutations I106A, D112A, and I127A (data not shown), as well as the parent construct (HA-VP2ΔN103), supported the formation of VLS together with NSP5, while HA-VP2ΔN103 (L124A) did not. VP2 carrying this point mutation (L124A) also was not able to form VLS with NSP5 when introduced into a full-length untagged VP2 (Fig. 2C), while wild-type (wt) VP2 and VP2 D112A did (D112A was chosen as a positive control for NSP5-HA-VP2ΔN103 VLS formation [data not shown]).
FIG 2.

VP2 residues L124, V865, and I878 are required for VLS formation. Alignment of rotavirus VP2 (strain SA11) amino acid region 103–135 (A) and region 840–880 (B). The VP2 amino acid sequences of several representative virus strains are shown. GenBank accession numbers are listed in Materials and Methods. Members of rotavirus families A to H are indicated. Highly conserved amino acids are shown in dark blue. Amino acid residues mutated to alanine are indicated at the top. A minus sign indicates deletion. (C) Immunofluorescence of MA104 cells expressing wt VP2 or point mutations alone (−NSP5 column) or together with NSP5 (+NSP5 panel). At 16 hpt, cells were fixed and VLS were detected by immunostaining of HA-VP2 (anti-HA, red) or VP2 (anti-VP2, green) with NSP5 (anti-NSP5, green or red). A merged image is shown in the right column. Nuclei were stained with DAPI (blue). Scale bar is 10 μm.
Similarly, as for the VP2 region 103–135, we aligned the amino acid region 840–880 of VP2-SA11 using all the sequences deposited in the NCBI database for VP2. This alignment revealed several conserved amino acids in VP2, including M843, L856, V860, V865, F873, and I878, in RV species A to H (Fig. 2B). Interestingly, F850 was highly conserved among RVA, RVC, RVF, and RVD species but absent from RVB, RVG, and RVH species. We mutated each of these residues to alanine in wt VP2 and tested for VLS formation in coexpression with NSP5. As shown in Fig. 2C, wt VP2 and VP2 harboring point mutations M843A, F850A, L856A, V860A, and F873A were able to form VLS. In contrast, VP2 harboring a point mutation in residues V865 and I878 was unable to form VLS. In contrast to wt VP2, the mutants VP2 V865A and I878A do not diffuse homogeneously in the cytosol. However, they did not form aggresomes either (data not shown), suggesting the spontaneous generation of genuine or aberrant core-like structures. All the other point mutations maintained their ability to form VLS. Importantly, as observed in Fig. 3A, cognate NSP5(SA11)-VP2(SA11) pairs, as well as unrelated NSP5(OSU)-VP2(SA11), can form VLS. Meanwhile, VP2L124A impairs VLS either with NSP5(SA11) or NSP5(OSU). Collectively, our data suggest that L124, V865, and I878 of VP2-SA11 are necessary for VLS formation in the presence of NSP5.
FIG 3.

VLS formation and triggering of NSP5 hyperphosphorylation from cognate and noncognate strains are sensitive to VP2 L124A. (A) Immunofluorescence of MA104 cells for detection of VLS composed of NSP5 from simian strain SA11 (top) or NSP5 from porcine strain OSU (bottom) in coexpression with VP2 from simian strain SA11 wt or L124A. At 16 hpt, cells were fixed and immunostained for detection of NSP5 (anti-NSP5, green) and VP2 (anti-VP2, red). A merged image is in the right column of each panel. Nuclei were stained with DAPI (blue). Scale bar is 10 μm. (B) Immunoblotting of lysates from MA104 cells coexpressing NSP5-OSU (lanes 1 to 3) or NSP5-SA11 (lanes 4 to 6) with VP2-SA11 wt (lanes 2 and 5) or L124A (lanes 4 and 6). The membranes were incubated with specific antibodies for the detection of NSP5. Alpha-tubulin was used as a loading control. The red brackets indicate NSP5 hyperphosphorylation. (C) Sequence alignment of NSP5 from simian strain SA11 and porcine strain OSU. The identity between the two proteins corresponds to 94.95%. Noncanonical casein kinase I alpha region and oligomerization tail are labeled in blue and red, respectively.
VP2 residues L124, V865, and I878 are required for triggering NSP5 hyperphosphorylation.
NSP5 is a phosphoprotein that, during RV infection, shows a mobility shift on SDS-PAGE corresponding to a hyperphosphorylated pattern (16, 17). However, when NSP5 is expressed in the absence of other viral proteins, it is detected only as a single band (19). Interestingly, it has been described that the coexpression of NSP5 with NSP2 (19) and VP2 (7) triggers the NSP5 hyperphosphorylation pattern. In order to determine which VP2 regions are involved in triggering the NSP5 hyperphosphorylation, we analyzed cell extracts coexpressing NSP5 in the presence of VP2 constructs with deletions at the N terminus (Fig. 4A) and C terminus (Fig. 4B). As shown in Fig. 4, wt VP2 and VP2ΔN81 (Fig. 4A, lanes 2 and 3, and B, lane 2) triggered NSP5 hyperphosphorylation, while all other VP2 deletion mutants tested did not (Fig. 4A, lanes 4 to 7, and B, lanes 3 to 7), as the NSP5 pattern was comparable to that when NSP5 is expressed alone (Fig. 4A and B, lane 1). Similarly, VP2 proteins harboring L124A, V865A, and I878A point mutations (Fig. 4C, lanes 3 to 5, and 3B, lanes 3 and 6) failed to induce NSP5 hyperphosphorylation as well (Fig. 4C, lane 2, and 3B, lanes 2 and 4). As previously demonstrated (19), the treatment of cellular extracts with lambda-phosphatase confirmed that the NSP5 PAGE mobility shift corresponds to phosphorylation (Fig. 4D). Our results suggest that the VP2 residues involved in VP2-NSP5 VLS formation and in triggering NSP5 hyperphosphorylation are identical.
FIG 4.

VP2 residues L124, V865, and I878 are necessary to trigger NSP5 hyperphosphorylation. Coexpression of VP2 N terminus (lanes 3 to 7) (A) or C terminus (lanes 3 to 7) (B) deletion mutants with NSP5. NSP5 alone (lane 1) or in coexpression with wt VP2 (fl; lane 2) is shown. NSP5 in cellular extracts was detected by immunoblotting. Alpha-tubulin was used as a loading control. (C) Immunoblotting of total cellular lysates expressing NSP5 alone (lane 1) or in coexpression with VP2 wt (lane 2) or point mutations (lanes 3 to 5). NSP5 hyperphosphorylation was detected with anti-NSP5, and VP2 was detected with anti-VP2. Alpha-tubulin was used as a loading control. (D) λ-Phosphatase treatment of NSP5 coexpressed with empty vector (−), wt VP2, or VP2 L124A. NSP5 is visualized by immunoblotting. Untreated (−) and treated (+) samples are indicated. Red brackets indicate the NSP5 mobility shift. The red dots show the NSP5 unphosphorylated isoform. (E) Schematic representation of point mutations in wt NSP5 and NSP5 S67D. S to D modification is indicated in red (44). (F) Immunofluorescence of MA104 cells for detection of VLSs formed with wt or mutant NSP5 with wt VP2 (upper) or VP2 L124A (lower). At 16 hpt, cells were fixed and immunostained for detection of VLS with anti-NSP5 (green) and anti-VP2 (red). Nuclei were stained with DAPI (blue). Scale bar is 10 μm. (G) Immunoblotting of total cellular lysates expressing wt NSP5 (lanes 1 to 3) or NSP5 S67D (lanes 4 to 6) with wt VP2 or VP2 L124A. Where indicated, the membranes were incubated with anti-VP2 or anti-NSP5. The brackets indicate the NSP5 hyperphosphorylation smear. (H) Pulldown assay of cell extracts coexpressing H6-NSP5 (lanes 1 and 2 and lanes 5 and 6) or H6-NSP5 S67D (lanes 3 and 4 and lanes 7 and 8) with wt VP2 (lanes 1, 3, 5, and 7) or VP2 L124A (lanes 2, 4, 6, and 8). Samples were detected by immunoblotting using anti-NSP5 and anti-VP2 antibodies. Input (lanes 1 to 4) and elution fractions are indicated (lanes 5 to 8).
NSP5 S67D cannot restore VLS formation with VP2 L124A.
As previously reported, NSP5 hyperphosphorylation is triggered by phosphorylation on serine 67 when in the presence of NSP2 (19) or VP2 (7) due to a mechanism involving casein kinase I (CKI) alpha (43, 44). Interestingly, when serine 67 (Fig. 4E) is mutated to an aspartic acid, which mimics a phosphorylated serine, NSP5 gets hyperphosphorylated, similar to what occurs RV infection (44). We therefore investigated whether NSP5 S67D would allow VLS formation with the VP2 point mutation L124A. For this, we compared VLS formation of wt NSP5 and NSP5 S67D with wt VP2 (Fig. 4F, upper) or VP2 L124A (Fig. 4F, lower). As expected, wt NSP5 or NSP5 S67D forms VLS with wt VP2 but not with VP2 L124A. To test whether the phosphorylation state of NSP5 S67D was impaired when coexpressed with VP2 L124A (Fig. 4G), we inspected the NSP5 hyperphosphorylation patterns of both wt NSP5 and NSP5 S67D when coexpressed with wt VP2 (Fig. 4G, lanes 2 and 5) or VP2 L124A (Fig. 4G, lanes 3 and 6). wt NSP5, as anticipated, had an increased mobility shift in the presence of wt VP2 but not in the presence of VP2 L124A, which showed a pattern comparable to that observed when wt NSP5 is expressed alone. Surprisingly, NSP5 S67D showed a more substantial shift with wt VP2 (Fig. 4G, lane 2), while with VP2 L124A a single NSP5 band was detected, similar to that observed when wt NSP5 was expressed alone (Fig. 4G, lane 3). Since VP2 triggered NSP5 hyperphosphorylation, we hypothesized that the hypophosphorylation of NSP5 in the presence of VP2 L124A is impaired because of the lack of interaction between the two proteins. To address this (Fig. 4H), we pulled down cellular extracts coexpressing hexahistidine (H6)-tagged wt NSP5 (H6-NSP5) or NSP5 S67D (H6-NSP5/S67D) with wt VP2 or VP2 L124A. The results showed that independent of the NSP5 genotype (wt or S67D), VP2 L124 did not bind NSP5 while wt VP2 did. Collectively, these data suggest that NSP5 and VP2 associate to form VLS independent of the NSP5 phosphorylation state.
VP2 L124 and I878 are necessary to bind NSP5.
To determine if VP2 and NSP5 requirements for VLS formation correlate with their ability to associate, we performed a pulldown assay by adding an H6 tag at the N terminus of NSP5 (H6-NSP5), followed by coexpression with wt VP2 or VP2 L124A, VP2 V865A, or VP2 I878A point mutations (Fig. 5A). As expected, wt VP2 associated with NSP5, as evident by the presence of wt VP2 in the eluted fraction (Fig. 5A, lane 3). In contrast, VP2 L124A was not detected in the elution (Fig. 5A, lanes 6), suggesting no association with H6-NSP5. Also, VP2 V865A got eluted from the pulldown assay (Fig. 5A, lane 9). Meanwhile, VP2 I878 weakly eluted, suggesting no or weak association with NSP5 (Fig. 5A, lane 12). Interestingly, when pulling down H6-NSP5 with wt VP2, the NSP5 eluted fraction showed a higher mobility shift in PAGE (Fig. 5A, lane 3), which represents the NSP5 hyperphosphorylated fraction. The H6-NSP5 eluted fraction obtained by coexpression with VP2 L124A showed a unique band that correlated with the unphosphorylated NSP5 isoform.
FIG 5.

NSP5 tail and VP2 residues L124 and I878 are required for their association. (A) Immunoblotting of nickel resin pulldown cellular extracts coexpressing histidine-tagged NSP5 (H6-NSP5) with wt VP2 or harboring the indicated point mutation (ce, cellular extract input; ft, flowthrough; el, column eluate). (B) Schematic representation of histidine-tagged (H6)-NSP5 deletion mutants used in panels C, D, and E. Amino acid residues delimiting the NSP5 regions are labeled at the top. Images are not to scale. (C) Immunoblotting of pulldown lysates from cells coexpressing H6-NSP5 (lanes 1 to 3) or H6-NSP5ΔT (lanes 4 to 6) with wt VP2. (D) Immunoblotting of pulldown lysates from cells coexpressing wt VP2 with H6-Δ1Δ3 (lanes 1 to 3) or H6-Δ1Δ3ΔT (lanes 4 to 6). (E) Immunoblotting of pulldown lysates from cells coexpressing VP2 L124A (lanes 1 to 3) or wt VP2 (lanes 4 to 6) with H6-Δ1Δ3. When indicated, the membranes were incubated with anti-VP2 or anti-NSP5 antibody. (F) Schematic representation of H6-EGFP fused to NSP5 regions used in the assay shown in panel E. Images are not to scale. (G) Plot showing the wt VP2 binding to H6-EGFP fused to NSP5 C-terminal regions. Data are represented as means ± standard deviations (SD) from four independent experiments. *, P < 0.05 by Student's t test.
NSP5 amino acid residues 180 to 198 are necessary for the association with VP2.
It was previously described that the NSP5 tail (T), corresponding to residues 180 to 198, is needed for its dimerization (9, 12, 44) and to associate with NSP2 (9, 35), NSP6 (12), and VP1 (23). Using a pulldown assay, we tested (Fig. 5B and C) the capacity of an H6-tagged, tail-deleted NSP5 (H6-NSP5ΔT) to bind to VP2. The results show that H6-NSP5 can bind VP2 while H6-NSP5ΔT did not. Likewise, H6-NSP5Δ1Δ3, an NSP5 deletion mutant known to allow self-dimerization (44), pulled down VP2, whereas the same NSP5 deletion mutant but without the tail region (H6-NSP5Δ1Δ3ΔT) did not (Fig. 5B and D). Interestingly, H6-NSP5Δ1Δ3 was not able to pull down VP2 L124A (Fig. 5B and E). To confirm these results, we constructed an H6-tagged enhanced green fluorescent protein (EGFP) fused at its C terminus with the NSP5 tail (H6-EGFP-T) or region 4 (amino acid residues 134 to 180; H6-EGFP-4). The fusion constructs were coexpressed with wt VP2 and pulled down with nickel resin to detect an association with VP2. As shown in Fig. 5E and F, H6-EGFP-T can bind VP2 while H6-EGFP and H6-EGFP-4 cannot. Collectively, these results indicate that the NSP5 tail is required for the association with VP2.
VP2 and NSP5 are protein-protein interaction partners.
The next experiment was performed to determine whether VP2 and NSP5 proteins can interact directly. For this, we employed the recently developed tripartite split-GFP association assay (45). As shown in Fig. 6A, GFP beta strands 10 and 11 are fused to bait (NSP5 or NSP5-T) and prey (wt VP2 or VP2 point mutations), respectively. The detector fragment GFP1-9OPT is added separately. When protein interaction occurs (Fig. 6Ai), GFP10 and GFP11 are tethered and spontaneously associate with the GFP1–9 fragment to form a full-length GFP, which fluoresces green. If the bait and prey do not interact (Fig. 6Aii), GFP10 and GFP11 are not tethered, and the assembly of GFP10, GFP11, and GFP1–9 does not occur (45); therefore, no green fluorescence is detected by flow cytometry. As expected, neither GFP10-NSP5 nor GFP11-VP2 produced green fluorescence when expressed together with GFP11 or GFP10, respectively (Fig. 6B and data not shown). In contrast, coexpression of GFP10-NSP5 with GFP11-VP2 produced green fluorescent cells showing a direct interaction between wt NSP5 and wt VP2. Consistent with the data shown in Fig. 5A, coexpression of GFP10-NSP5 with either GFP11-VP2 L124A or GFP11-VP2 I878A did not generate green fluorescent cells, thereby confirming their incapacity to interact. In contrast, GFP10-NSP5, together with GFP11-VP2 V865A, as well as GFP10-(T)NSP5 together with GFP11-VP2, produced green fluorescent cells. These data confirmed that the NSP5 tail is necessary for the interaction with VP2. We next investigated whether VP2 L124A would bind to the wt VP2 and, thus, perturb its interaction with NSP5. To address this question (Fig. 6C), we expressed the protein-protein interaction (PPI) partners GFP10-NSP5 and GFP11-VP2 together with an increasing amount of GFP11-VP2 L124A. As anticipated, the PPI partner NSP5-VP2 yielded a large number of green fluorescent cells that were reduced upon the addition of VP2 L124A in a dose-dependent manner, suggesting a reduction in the binding between NSP5 and wt VP2.
FIG 6.
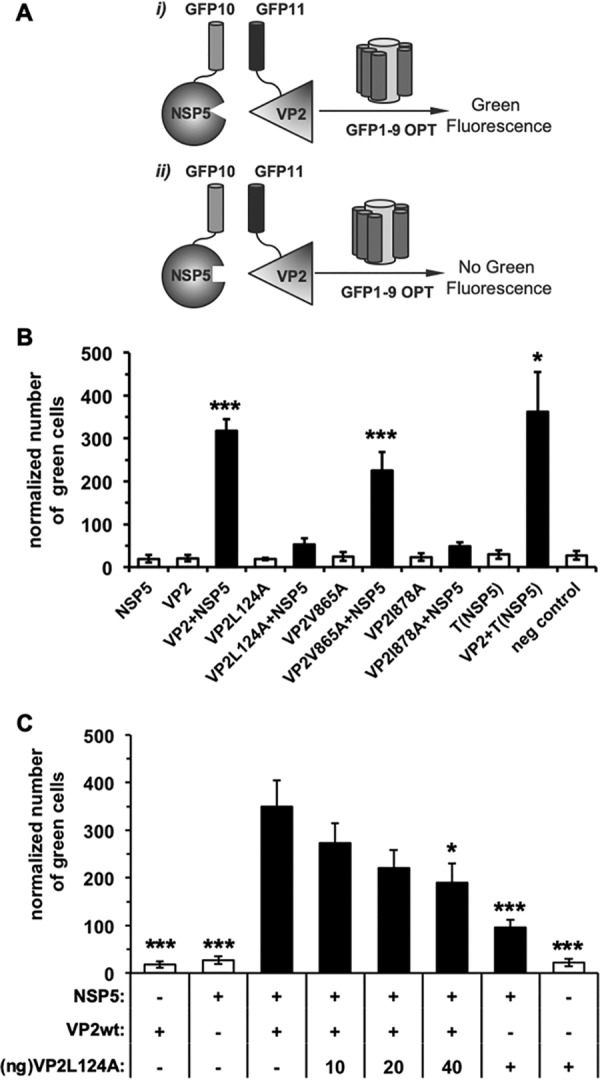
NSP5 and VP2 interact directly. (A) Schematic representation of NSP5-VP2 interaction sensor base in the tripartite split-GFP association. GFP beta strands 10 and 11 are fused to bait, NSP5 or T-NSP5, prey, wt VP2, or point mutations. The detector fragment, GFP1–9, is added separately. (i) When protein interaction occurs, GFP10 and GFP11 are tethered and spontaneously associate with the GFP1–9 fragment to form a full-length GFP, which fluoresces green. (ii) If the bait and prey do not interact, GFP10 and GFP11 are not tethered and entropy is too high to allow complementation with GFP1–9; therefore, no green fluorescence is detected. (B) The NSP5-VP2 direct binding plot shows the normalized number of green fluorescent cells for the indicated bait-prey pairs. The samples showing a single partner were expressed together with GFP10 or GFP11. (C) Competition assay plot for NSP5 binding between wt VP2 and VP2 L124A. In panels B and C, data show means ± standard errors of the means (SEM) from six independent experiments. By two-tailed, nonparametric Mann-Whitney test, P < 0.001 (***) and P < 0.05 (*) compared to NSP5-VP2 green fluorescence.
VP2 L124A hinders the formation of VLS(NSP5+HA-VP2) and VLS(NSP5+NSP2).
Since VP2 L124A impedes the direct binding of NSP5 and VP2 (Fig. 6), we hypothesized that increased amounts of VP2 L124A would disrupt VLS(NSP5+HA-VP2). To investigate this possibility, we coexpressed NSP5 and HA-VP2 to permit the formation of VLS(NSP5+HA-VP2). As shown in Fig. 1B (and data not shown), VLS(NSP5+HA-VP2) can be readily detected as yellow fluorescent cytosolic inclusions upon staining with anti-NSP5 (red) and anti-HA (green) antibodies. With increasing amounts of wt VP2, the color of the VLS shifted to red (Fig. 7A), reflecting competition between wt VP2 and HA-VP2. With increasing amounts of VP2 L124A, the cytosolic inclusions became disrupted (Fig. 7B). Using the same approach, we then tested whether overexpression of VP2 L124A hinders VLS(NSP5+NSP2) formation. Coexpression of NSP5 and NSP2 resulted in the formation of VLS(NSP5+NSP2), readily detected as yellow fluorescent cytosolic inclusions upon staining with anti-NSP5 (red) and anti-NSP2 (green) antibodies; VP2 (anti-VP2, green) was not detected (Fig. 7C, upper). Upon coexpression of NSP5, NSP2, and wt VP2, all three proteins colocalized in the cytosolic VLS (Fig. 8C, middle). However, when coexpressing NSP5, NSP2, and VP2 L124A, unstructured VLSs were detected (Fig. 7C, lower). In support of this result (data not shown), a drastic reduction (P < 0.001) in the number of cells showing VLS(NSP5+NSP2) is observed in coexpression with VP2 L124A (12.8%) compared to that of wt VP2 (38.3%). Additionally, 100% of VLS(NSP5+NSP2) formed with wt VP2 showed a globular morphology that is in contrast to the reduced number (36%) of globular structures observed among VLS(NSP5+NSP2) elements formed with VP2 L124A (data not shown). Accordingly, VP2 L124A expression blocked the formation of both VLS(NSP5+VP2) and VLS(NSP5+NSP2).
FIG 7.

VP2 L124A disrupts VLS formation. For immunofluorescence of VLS formation, MA104 cells expressed NSP5 with HA-VP2 plus increasing amounts (0.25, 0.5, and 1 μg) of either wt VP2 (A) or VP2 L124A (B). At 16 hpt, cells were fixed and immunostained for VLS by detecting NSP5 (anti-NSP5, red) and HA-VP2 (anti-HA, green). Nuclei were stained with DAPI (blue). White arrowheads denote impaired VLS. Scale bar is 10 μm. (C) Immunofluorescence of VLS(NSP5+NSP2) alone (upper), with wt VP2 (middle), or with VP2 L124A (lower). At 16 hpt, MA104 cells were fixed and immunostained for the VLS with anti-NSP5 (red) and anti-NSP2 or anti-VP2 (green). Nuclei were stained with DAPI (blue). A merged image is shown at the right of each panel. The white arrowheads point to misshaped VLS. Scale bar is 10 μm.
FIG 8.

Viroplasms are damaged by VP2 L124A expression. (A) Immunoblotting of lysates from BSR-T7 cells expressing wt VP2 (lane 2) and VP2 L124A (lane 3) at 48 hpt. OSU virus particles were used as a positive control (lane 1). (B) Plot of the percentage of cells showing viroplasms when expressing either wt VP2 or VP2 L124A. A cell was considered viroplasm positive when anti-NSP5 cytosolic inclusions were detected. Data are represented as means ± SD from three independent experiments. ***, P < 0.001 by two-tailed unpaired Student's t test, n > 1,500 cells per experimental point. (C) Representative immunofluorescence images comparing viroplasm morphologies (5 hpi) (anti-NSP5, green) of cells expressing VP2 L124A (upper, frames i, ii, iii, and iv) or wt VP2 (lower, frames v and vi). Nuclei were stained with DAPI (blue). Scale bar is 10 μm. (D) Confocal immunofluorescence of RV proteins of cells expressing wt VP2 (left) or VP2 L124A (right). At 24 hpt, BSR-T7 cells were RV infected (MOI, 75 VFU/cell). At 5 hpi, cells were fixed and immunostained for the detection of NSP5 (anti-NSP5, green or red), VP2 (anti-VP2, green), NSP2 (anti-NSP2, green), VP6 (anti-VP6, red), NSP3 (anti-NSP3, red), and VP6, VP7, and VP4 (anti-RV, green). Nuclei were stained with DAPI (blue). Scale bar is 5 μm.
VP2 L124A expression affects viroplasm morphology.
As VLS are a simplified model to study viroplasms, we interrogated whether overexpression of VP2 L124A could also compromise viroplasm formation. For this purpose, we overexpressed either wt VP2 or VP2 L124A in BSR-T7 cells (Fig. 8A), followed by RV infection, and quantified cells showing viroplasms under these two conditions. As shown in Fig. 8B, the number of cells with viroplasms was significantly reduced upon coexpression of VP2 L124A compared to that of cells expressing wt VP2. Interestingly, most viroplasms in cells expressing VP2 L124A presented an atypical filamentous (Fig. 8Ci and ii) or diffuse and juxtanuclear polarized (Fig. 8Ciii and iv) morphology. In contrast, viroplasms formed in the presence of wt VP2 cells showed a characteristic globular morphology (Fig. 8Cv and vi). Since viroplasms are composed of several host and viral proteins, as well as ssRNAs and dsRNAs, we investigated if the localization of the viral proteins diverges in viroplasms of cells expressing VP2 L124A. For this purpose, RV-infected BSR-T7 cells expressing wt VP2 or VP2 L124A were stained with specific antibodies detecting viroplasm proteins, such as NSP5, VP2, VP6, and NSP3 (Fig. 8D and data not shown). As expected (Fig. 8D, left, and data not shown), the viroplasms formed in the presence of wt VP2 expression exhibit delineated globular structures when detected for the tested proteins. However, in the cells expressing VP2 L124A, the viral protein components of viroplasms, including NSP2 and VP6, showed a diffuse pattern (Fig. 8D, right, and data not shown). For NSP3 and the capsid proteins (VP6, VP7, and VP4, detected by anti-RV antibody), even amyloid-like structures were observed that surrounded aberrant viroplasm structures. Collectively, our data indicate that viroplasm morphology is sensitive to VP2 L124A expression.
VP2 L124A expression precludes transcription and replication.
Since VP2 L124A expression disrupts viroplasm morphology, we inspected if other functions of viroplasms, such as viral transcription and replication, are also inhibited. The synthesis of RV plus-strand ssRNA, as a template for dsRNA generation, is uniquely dependent on RdRp-VP1 (46). We assessed RV transcription by treating RV-infected BSR-T7 cells expressing wt VP2 or VP2 L124A at 4 h postinfection (hpi) with actinomycin D for 30 min (Fig. 9A). Of note, actinomycin D has been shown not to interfere with the RdRp activity of VP1 (47). The cells then were fed with 5-bromouridine 5′-triphosphate sodium salt (BrUTP) for 1 h, and the incorporation signal was monitored by immunofluorescence using a fluorescence-labeled anti-bromodeoxyuridine (BrdU) antibody. The viroplasms were stained with an anti-NSP5 antibody. Cells not treated with BrUTP were used as a control. As expected (46), cells expressing wt VP2 incorporated BrUTP in viroplasms (Fig. 9A, upper row), indicating RV viral transcription, while cells expressing VP2 L124A did not (Fig. 9A, lower row). To confirm our result, we stained RV-infected cells expressing wt VP2 or VP2 L124A with an anti-dsRNA antibody (clone J2) that can recognize dsRNA with a size larger than 40 bp (48). In noninfected cells (Fig. 9B, −RV), punctuated anti-dsRNA signal was found distributed in the whole cytoplasm. Upon RV infection (Fig. 9B, +RV), most of the dsRNA signal localized to viroplasms in both nontransfected and wt VP2-expressing cells. These results are also confirmed by intensity profiles among the signals of both viroplasms and dsRNA (Fig. 9C). Such a pattern, however, is not repeated in VP2 L124A-expressing cells, where no or mild localization of anti-dsRNA signal in viroplasm and no coincidence of these signals in the intensity profile plot was observed (Fig. 9C). Thus, our results suggest that RV RNA synthesis is at least inefficient in VP2 L124A cells.
FIG 9.

VP2 L124A expression decreases viral transcription and replication of viroplasms. (A) At 24 hpt, for expression of wt VP2 (upper row) and VP2 L124A (lower row), BSR-T7 cells were RV infected (MOI, 75 VFU/cell). At 4 hpi, cells were treated with 10 μM actinomycin D for 30 min and then transfected for 1 h with 1 mM BrUTP (+BrUTP) or not (−BrUTP) before fixation. Cells were immunostained for BrUTP incorporation (anti-BrdU, green) and viroplasm detection (anti-NSP5, red). Nuclei were stained with DAPI (blue). Scale bar is 5 μm. (B) Confocal immunofluorescence of BSR-T7 cells alone, expressing wt VP2 or VP2 L124A, followed at 24 hpt by RV infection (MOI, 150 VFU/cell). At 5 hpi, cells were fixed and immunostained for detection of dsRNA (anti-dsRNA, red) and viroplasms (anti-NSP5, green). Nuclei were stained with DAPI (blue). N.T., nontransfected cells; a.u., arbitrary units. Scale bar is 5 μm. (C) Intensity profile plot of dsRNA (red line) and viroplasms (green line) of the indicated linear region of interest of cells from panel B.
DISCUSSION
In RV viroplasms several processes take place, including viral transcription, packaging of the viral pregenomic RNA into newly synthesized viral cores, and RNA replication, followed by the addition of the second layer of proteins. None of these processes have been directly demonstrated as a whole in the viroplasms, and all the current evidence is based on in vitro data, providing models such as the packaging of the virus genome (49). The fact that rotavirus encodes only a small number of proteins, eleven or twelve (NSP6 is present only in certain RV strains [50–53]), supports the hypothesis that at least some viral proteins have multifunctional roles in the replication cycle. A clear example is VP2, which is well characterized as the RV core shell encapsidating the viral genome (2, 32, 33, 54, 55), acting as a cofactor of RdRp VP1 (28, 30, 39) and anchoring VP6 to form DLPs (34, 55). Aside from its structural functions, VP2 has other roles during the RV life cycle, such as permitting perinuclear motion of viroplasms (10) or being associated with the RV-induced cell cycle arrest (56). Another exciting aspect is its role as the main building block for viroplasm formation. In this sense, the coexpression of NSP5 with either NSP2 or VP2 forms VLS, denoting the role of VP2 in viroplasm formation (7, 10, 41). In the present study, we fully characterized the interaction between NSP5 and VP2 proteins and the importance of this interaction for VLS formation. In this context, we identified three amino acids in VP2 of simian RV strains SA11, L124, V865, and I878 that are highly conserved among cognate proteins of all RV species present in the NCBI data bank. As observed in the calculated three-dimensional model for VP2-SA11, V865 and I878 (data not shown) are positioned behind the beta-strand β25 at the VP2 dimerization region (2, 57). Accordingly, these hydrophobic residues, when modified to alanine, can directly abrogate VP2 dimerization and VLS formation, indicating that VP2 dimerization is a requirement for VLS formation in concomitant association with NSP5. A particular example corresponds to VP2 point mutations V865A and I878, which associate with NSP5, as demonstrated by binding assays, but support neither VLS formation nor NSP5 hyperphosphorylation. Moreover, VP2 L124A impairs VLS formation and NSP5 hyperphosphorylation in NSP5 from RV strain OSU or SA11. This outcome was not surprising, since both strains have 95% identity to each other (Fig. 3C), where the NSP5 tails (region 180–198) and the CKI noncanonical phosphorylation site for S67D are identically conserved.
Interestingly, the residue L124 faces toward the VP2 inner central domains (data not shown), and its modification to alanine affects VLS formation, the direct association with NSP5, and the NSP5 phosphorylation state. This suggests that the VP2-NSP5 interaction interface impedes core assembly to favor viroplasm formation and allow the packaging of genome segments. We show that NSP5 can associate with VP2 through its tail region (residues 180 to 198). This finding is not surprising, since the role of the NSP5 tail has previously been described to play a role in association with other viral proteins, including NSP2 (9, 35), NSP6 (12), and VP1 (23), and even as a self-oligomerization domain (12, 44).
Collectively, our data show that NSP5-VP2 interaction and VLS formation are immediate prior steps to the NSP5 hyperphosphorylation. In support of this viewpoint, we provide data showing that (i) a hyperphosphorylated NSP5 (NSP5 S67D) is not sufficient to form VLS with VP2 L124A, (ii) NSP5 S67D does not bind VP2 L124A, (iii) VP2 L124A does not hyperphosphorylate wt NSP5, and (iv) NSP5 S67A forms VLS with wt VP2 (7). Our conclusion then provides additional insights into the molecular mechanism previously proposed (44, 58) for the formation of viroplasms and subsequent NSP5 hyperphosphorylation. This mechanistic interpretation is consistent with previous observations that viroplasm formation requires a phosphorylation cascade triggered by NSP2, which depends on CK1-alpha (43, 44, 59). It also is consistent with the fact that NSP5 phosphorylation on S67 by CK1-alpha is a step after the interaction with VP2 (7 and this study) and NSP2 (19, 44, 58). The phosphorylation of other serines and threonines of NSP5 by host cell kinases (8, 43, 60) will follow, in agreement with a model previously described (44, 58). Our data also suggest that the NSP5-VP2 interaction relies on conserved L124 of VP2.
As described above, NSP2 and VP2 can both trigger NSP5 hyperphosphorylation. We also provide evidence that VP2-NSP5 interaction, similar to NSP2-NSP5 interaction, provides a scaffold for viroplasm formation. Impairment of the NSP5-VP2 interaction by VP2 L124A, for example, abolishes the formation of VLS and viroplasms. This observation is consistent with the previous model for viroplasm formation (44, 58, 59), whereas NSP5 hyperphosphorylation is an initial step followed by the cytosolic inclusion formation in association with both NSP2 (58) and VP2 (this study).
A dominant-negative designation refers to a mutation in a protein that adversely affects the wild-type phenotype, for example, by disrupting a functional domain but retaining its dimerization property. Therefore, the dominant-negative protein can dimerize with the wild-type protein within the same cell, decreasing the activity of the wild-type protein (61). We provide strong evidence that VP2 L124 works as a dominant-negative protein for the formation of both VLS and viroplasms. In this sense, we have shown that (i) VP2 L124 disrupts both VLS(NSP5+VP2) and VLS(NSP5+NSP2), (ii) NSP5-VP2 interaction, in the split tripartite EGFP assay, is reduced in a dose-dependent manner with VP2 L124, and (iii) NSP5 and VP2 L124A neither form VLS nor bind to each other. Thus, we conclude that VP2 L124A necessarily dimerizes with wt VP2, displacing NSP5-wt VP2 interaction. It is well described that NSP2 binds VP2 at several amino acid regions (residues 34 to 86, 514 to 518, 664 to 674, and 836 to 845) (35). Hence, VP2 L124A also is dominant negative over NSP2, because VP2 L124A preserves NSP2 binding sites but did not allow VLS formation. Moreover, the NSP2-VP2 interaction seems stronger than the NSP5-VP2 interaction. We provide evidence for the idea that VP2 L124A can disrupt viroplasms by showing that the expression of VP2 L124A interferes with RV replication by damaging viroplasms. Specifically, viroplasms upon VP2 L124A expression form aberrant inclusions and delocalize viroplasmic viral proteins in diffuse cytosolic patterns or amyloid-like structures. Moreover, these aberrant viroplasms are decreased in viral transcription (plus-strand ssRNA synthesis) and dsRNA synthesis. Hence, VP2 L124A works as a bona fide dominant-negative factor for the assembly and formation of viroplasms. Accordingly, a point mutation on VP2 L124 to alanine on RV, and possible rescue by a reverse genetic technique (62), would undoubtedly provide new insights into the regulation of the RV life cycle. However, several attempts to rescue a recombinant RV harboring VP2 L124A were performed without success, in contrast to the case of the control recombinant RV that retains wt VP2 (data not shown), reinforcing the relevance of this motif in the RV replication cycle.
We believe this study could lay the foundations for the designing of inhibitors that specifically bind at the interaction site between NSP5 and VP2.
MATERIALS AND METHODS
Cells and viruses.
MA104 cells (embryonic rhesus monkey kidney; ATCC CRL-2378) and HEK293T cells (human embryonic kidney; ATCC CRL-573) were cultured in complete Dulbecco’s modified Eagle’s medium (cDMEM; Gibco BRL) supplemented with 10% fetal calf serum (FCS) (AMIMED; BioConcept, Switzerland) and penicillin (100 U/ml)–streptomycin (100 μg/ml) (Gibco, Life Technologies). BSR-T7 cells (baby hamster kidney; CVCL-RW96), which express T7 DNA-directed RNA polymerase, were cultured in DMEM supplemented with 10% FCS, 1 mg/ml Geneticin (G418; Thermo Fisher Scientific), and penicillin (100 U/ml)–streptomycin (100 μg/ml). The T7 RNA polymerase recombinant vaccinia virus (strain vvT7.3) was amplified as previously described (63).
Rotavirus porcine OSU strain (G5;P [9]) was propagated in MA104 cells, as described previously (64, 65). Virus titer was determined as described previously by Eichwald et al. (10) and expressed as viroplasm-forming units (VFU) per milliliter.
Antibodies.
Guinea pig anti-NSP5, guinea pig anti-NSP2, guinea pig anti-VP2, and mouse scFV anti-NSP5 clone 1F2 were described previously (8, 19, 23, 66). Guinea pig anti-VP6 was described previously (10, 67). Mouse monoclonal anti-VP6 (clone 2F) was a gift from N. Mattion (CEVAN, Buenos Aires, Argentina). Rabbit anti-NSP3 was kindly provided by Susana López (UNAM, Cuernavaca, Mexico). Mouse monoclonal antibody (MAb) anti-VP2 (clone3E8) was kindly provided by Harry Greenberg (Stanford University, CA, USA). Mouse MAb anti-HA (clone HA-7) and mouse monoclonal anti-glyceraldehyde dehydrogenase (GAPDH) (clone GAPDH-71.1) were obtained from Sigma-Aldrich. Mouse MAb anti-dsRNA (clone J2) was purchased from SCICONS J2, English & Scientific Consulting, Hungary. Goat anti-mouse immunoglobulin G (IgG) (H+L) conjugated to Alexa 594 and goat anti-guinea pig IgG conjugated to Alexa 488 were obtained from Molecular Probes, Invitrogen, USA. Goat polyclonal anti-mouse IgG F(ab')-peroxidase was purchased from Sigma-Aldrich. Rabbit polyclonal anti-guinea pig IgG-peroxidase was purchased from Dako Cytomation, Denmark.
Plasmid construction.
The plasmids pcDNA-NSP5 from porcine strain OSU (G5;P [9]), pcDNA-NSP5 from simian strain SA11-4F (G3;P6 [1]), pcDNA-NSP2 from simian strain SA11-4F (G3;P6 [1]), and pcDNA-VP2 from simian strain SA11-4F (G3;P6 [1]) were previously described (11, 19, 21, 23). The plasmids pCI-HA-VP2 full-length, pCI-HA-VP2ΔN81, pCI-HA-VP2ΔN103, pCI-HA-VP2ΔN135, pCI-HA-VP2ΔN159, pCI-HA-VP2ΔN181, pCI-HA-VP2ΔN203, pCI-HA-VP2ΔN316, pCI-HA-VP2ΔN588, pCI-HA-VP2ΔN720, pCI-HA-VP2ΔC81, pCI-HA-VP2ΔC203, pCI-HA-VP2ΔC316, pCI-HA-VP2ΔC588, pCI-HA-VP2ΔC721, pCI-HA-VP2ΔC755, pCI-HA-VP2ΔC840, pCI-HA-VP2(82-203), pCI-HA-VP2(204-316), pCI-HA-VP2(317-588), and pCI-HA-VP2(589-720) were obtained by PCR amplification of pcDNA-VP2 (23) using specific primers to insert flanking XhoI/HA-tag and EcoRI sites, followed by ligation into those sites in pCI-Neo (Promega). Similarly, pCI-VP2ΔN81, pCI-VP2ΔN203, pCI-VP2ΔN316, pCI-VP2ΔN588, pCI-VP2ΔN720, pCI-VP2ΔC81, pCI-VP2ΔC203, pCI-VP2ΔC316, pCI-VP2ΔC588, and pCI-VP2ΔC720 were obtained by PCR amplification of pcDNA-VP2 using specific primers to insert XhoI and EcoRI sites, followed by ligation into those sites in pCI-Neo.
The constructs pCI-H6-NSP5, pCI-H6-NSP5 S67D, pCI-H6-EGFP, pCI-H6-EGFP-T, and pCI-H6-EGFP-dom4 were obtained from PCR amplification of pcDNA-NSP5 (11), pcDNA-NSP5 S67D (44), pEGFP-N1 (Clontech), p(1-EGFP-T) (9), and p(EGFP-4) (9) using specific primers to insert XhoI/hexahistidine tag and NotI sites, following by ligation on those sites in pCI-Neo (Promega). The construct pcDNA-H6-Δ1Δ3 was previously described (8). The constructs pCI-H6-NSP5ΔT and pCI-H6-Δ1Δ3ΔT were obtained by PCR amplification of pcDNA-H6-NSP5 and pcDNA-H6-Δ1Δ3 using specific primers for the insertion of an XhoI restriction site at the 5′ end and the reverse oligonucleotide primer 5′-GATCGCGGCCGCTTAGAAGCACCTTTTCTTATATTT-3′ for the deletion of NSP5 tail and insertion of Stop codon/NotI site at the 3′ end. The PCR fragment was ligated between XhoI/NotI sites in pCI-Neo (Promega). The plasmids pCI-mCherry and pcDNA3.1tn(+)-GFP1-9OPT were previously described (45, 68). The construct p10L-NSP5 was obtained by PCR amplification of pcDNA-NSP5 using specific primers to insert PacI and XhoI sites, followed by ligation in-frame in pcDNA3.1tn(+)GFP10+long linker (45). The construct p10L-NSP5 (T) was obtained by annealing of the oligonucleotides 5′-TAACATTGCACTAAGAATGAGGATGAAGCAAGTCGCAATGCAATTGATCGAAGATTTGTAAC-3′ and 5′-TCGAGTTACAAATCTTCGATCAATTGCATTGCGACTTGCTTCATCCTCATTCTTAGTGCAATGTTAAT-3′, followed by ligation into PacI and XhoI sites of pcDNA3.1tn(+)-GFP10+long linker (45). The construct p11L-VP2 was obtained by PCR amplification of pcDNA-VP2 using specific primers to insert PacI and EcoRI sites, following by ligation on those sites in pcDNA3.1tn(+)-linker+GFP11(45).
pCAG-D1R (Addgene plasmid number 89160) and pCAG-D12L (Addgene plasmid number 89161) were a gift from Takeshi Kobayashi (62).
Version of constructs pCDNA-VP2, pCI-HA-VP2(ΔN103), and p11L-VP2, harboring the VP2 point mutations I106A, D112A, L124A, R125A, I127A, M843A, F850A, L856A, V860A, F865A, F873A, F878A, V865A, and I878A, were built by insertion of point mutations using the QuikChange site-directed mutagenesis kit and protocol (Agilent). All of the oligonucleotides were obtained from Microsynth AG, Switzerland, and are available upon request.
Immunofluorescence.
For VLS detection, 1 × 105 MA104 cells per well were seeded in 24-well multiwell plates over a coverslip. Cells infected with vvT7.3 (3 PFU/cell) were transfected for 15 h posttransfection (hpt) in an empirically determined ratio of 2:1 for NSP5 and VP2, using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. For viroplasm detection, transfected BSR-T7 cells were infected for 5 h at a multiplicity of infection (MOI) of 75 VFU/cell. At the indicated time postinfection, cells were fixed in 2% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature and processed as described by Eichwald et al. (10). When indicated, images were acquired using a fluorescence microscope (DMI6000B; Leica) or a confocal laser scanning microscope (CLSM) (DM550Q; Leica). Data were analyzed with the Leica Application Suite (Mannheim, Germany) and Image J (version 2.0.0-RC-69/1.52i; https://imagej.nih.gov/ij/). Images were prepared for publication using PowerPoint (Microsoft) and Adobe Photoshop software. The intensity profile plots were obtained using Image J, ROI manager, multiplot algorithm.
Pulldown assay.
MA104 cells were infected with vvT7.3 (MOI, 3 PFU/cell), followed by transfection at a ratio of 1:1 for two DNA plasmids using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. At 16 hpt, cells were processed as described previously (69).
Hyperphosphorylation assay.
The hyperphosphorylation assay was performed as described by Eichwald et al. (44). Briefly, 2 × 105 cells in 12-well multiwell plates were infected with T7 RNA polymerase recombinant vaccinia virus (vvT7.3) (63) (MOI, 3 PFU/cell), followed by transfection with 2 μg of total plasmid DNA (1 μg of the substrate and 1 μg activator) and 3 μl Lipofectamine 2000 transfection reagent (ThermoFisher Scientific). At 16 hpt, cells were lysed in 30 μl of TNN lysis buffer (100 mM Tris-HCl, pH 8.0, 250 mM NaCl, 0.5% Nonidet P-40, and cOmplete protease inhibitor cocktail [Roche, Switzerland]) for 10 min at 4°C. Samples were harvested and centrifuged at 17,000 × g for 7 min at 4°C. The supernatant was analyzed by immunoblotting as described previously (10).
Lambda-phosphatase assay.
Cellular extracts were treated with lambda phosphatase, as described by Eichwald et al. (44). For analysis, samples were loaded in SDS-polyacrylamide and analyzed by immunoblotting for the detection of NSP5 (10).
Protein-protein interaction tripartite split-GFP assay.
The split-GFP assay was performed as previously described by Cabantous et al. (45). Briefly, 5 × 104 per well of HEK293T cells were seeded in 48-well multiwell plates. Cells were cotransfected by mixing 20 ng pCI-mCherry, 60 ng pcDNA3.1tn(+) GFP1-9 OPT, 60 ng p10L-NSP5, and 60 ng p11L-VP2 using with 1 μl Lipofectamine 2000 (Thermo Fisher Scientific) in 40 μl Opti-MEM (Thermo Fisher Scientific), followed by 20 min of incubation at room temperature. The transfection mixture was added to the cells held under 160 μl of DMEM supplemented with 10% FCS. At 24 hpt, cells were released with trypsin-EDTA, harvested in cDMEM, and centrifuged at low speed for 2 min. Cellular pellets were resuspended in FC buffer (5 mM EDTA, pH 8.0, 2% FCS in PBS), filtered in cell-strained snap-cap tubes (BD Falcon), and immediately acquired in a Gallios flow cytometer (Beckman Coulter, Inc.). In total, 10,000 events were acquired and excited with 488-nm and 561-nm lasers, followed by recording with default filters of 525/50 nm (GFP, green) and 620/30 nm (mCherry, red), respectively. Data were analyzed by normalizing the number of green cells to 10,000 events on the positive red population using Kaluza flow analysis software. The statistical analysis and plot were performed using Microsoft Excel for MAC 2011, version 14.7.3, and Prism 8 (GraphPad Software, Inc.).
Dominant-negative assay for viroplasm formation.
BSR-T7 cells (2 × 105) seeded in 24-well plates over coverslip were transfected with 1 μg pcDNA-VP2 or pcDNA-VP2 L124A, 0.1 μg pCAG-D1R, and 0.1 μg pCAG-D12L using Lipofectamine 2000 according to the manufacturer’s instructions. At 24 hpt, cells were infected with porcine rotavirus strain OSU at the indicated MOI. The virus was adsorbed for 1 h at 4°C, followed by incubation at 37°C. At 5 hpi, cells were fixed with 2% paraformaldehyde in PBS for 10 min at room temperature and processed for immunofluorescence as described by Eichwald et al. (10). The number of cells containing viroplasms and the total number of cells per field was counted for at least 40 fields using a fluorescence microscope. The percentage of viroplasms, plot, and statistical analysis were performed with Microsoft Excel 2011 for MAC, version 14.7.3, and Prism 8 (GraphPad Software, Inc.).
Detection of RNA synthesis in viroplasm.
BSR-T7 cells (2 × 105), seeded over coverslips in 24-well plates, were transfected for expression of wt VP2 and VP2 L124A, as described above. At 24 hpt, cells were infected with porcine RV strain OSU at an MOI of 75 VFU/cell. The virus was adsorbed for 1 h at 4°C, followed by incubation at 37°C. BrUTP (5-bromouridine 5′-triphosphate sodium salt; Sigma-Aldrich) incorporation was performed as described previously by Silvestri et al. (46), with some modifications. Briefly, cells were pretreated for 30 min with 10 μM actinomycin D (GeneTex) in 500 μl serum-free medium starting at 4 hpi. At 4.5 hpi, cells were transfected with a mixture containing 1 mM BrUTP, 10 μM actinomycin D, and 20 μl of Lipofectamine 2000 transfection reagent (ThermoFisher Scientific) in 500 μl Opti-MEM reduced serum medium (ThermoFisher Scientific) and incubated for 1 h at 37°C and 5% CO2. The cells were immediately fixed in 2% paraformaldehyde in PBS for 10 min at room temperature and processed for immunofluorescence by staining with Alexa Fluor 488 anti-BrdU clone 3D4 (diluted 1:50; BioLegend) and guinea pig anti-NSP5 (1:200), followed by a secondary antibody conjugated to rhodamine isothiocyanate (RITC). Images were subsequently acquired with a CLSM (DM550Q; Leica) and images analyzed using Imaris File Converter 9.0.0 and Image J (version 2.0.0-RC-69/1.52P; http://imagej.net/Contributors). Images were prepared for publication using PowerPoint (Microsoft) and Adobe Photoshop software.
Sequence alignments.
To identify highly conserved amino acids among cogent proteins of all RV species, an alignment for two VP2 (simian strain SA11) regions (amino acids 103 to 135 and 840 to 880) of all VP2 available in the protein depository of the NCBI was performed using multiple-sequence alignment with Clustal W (70) combined with MAFFT software (71). The obtained alignments were further processed using Jalview software (72). The following selected sequences from rotavirus species (A to H) are represented in the alignments (species, strain, host, and GenBank version accession number listed for each): RVA_K9 (canine) (EU708924), RVA_SA11_lab (simian [si]) (MK184911), RVA_SA11-H96 (si) (NC_011506), RVA_Vanderbilt_VU08-09-24 (human [hu]) (JF491072), RVA_Vanderbilt_VU08-09-25 (hu) (JF491082), RVA_Vanderbilt_VU05-06-15 (hu) (JF490598), RVA_Vanderbilt_VU06-07-29 (hu) (JF490852), RVA_Wa (hu) (X14942), RVA_PO-13 (avian [av]) (AB009630), RVA_EB (murine) (HQ540508), RVA_02V0002G3 (av) (FJ169854), RVA_Cowden (porcine [po]) (M74217), RVA_OSU (po) (ADE44253), RVB_WH-1 (hu) (AY539859), RVB_ADRV (hu) (M91433), RVB_Bang373 (hu) (NC_021545), RVB_IDIRg2 (rabbit) (U00673), RVB_RUBV226 (bovine) (GQ358717), RVC_Bristol (hu) (NC_007546), RVD_05V0049/DEU/2003 (av) (YP_003896047), RVF_03V0568/DEU/2003 (av) (NC_021626), RVG_HK18 (av) (KC876011), RVG_03V0567/DEU/2003 (av) (NC_021580), RVH_MRC_DPRU1575 (po) (KT962028), and RVH_J19 (hu) (DQ113898).
ACKNOWLEDGMENTS
We are grateful to Luca Murer and Jovan Pavlovic for providing the tripartite split-GFP plasmids. We thank Ohad Medalia for his support in the calculated models of VP2. We also thank Guido Papa and Oscar Burrone for their help with RV reverse genetics. We thank Claudio Aguilar and Jakub Kubacki for their valuable comments and criticisms of the manuscript.
This work was supported by a private donation of the late Robert Wyler to M.A. (F-52601-10-01) and the University of Zurich. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We have no conflicts of interest to declare.
REFERENCES
- 1.Troeger C, Khalil IA, Rao PC, Cao S, Blacker BF, Ahmed T, Armah G, Bines JE, Brewer TG, Colombara DV, Kang G, Kirkpatrick BD, Kirkwood CD, Mwenda JM, Parashar UD, Petri WA, Riddle MS, Steele AD, Thompson RL, Walson JL, Sanders JW, Mokdad AH, Murray CJL, Hay SI, Reiner RC. 2018. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 years. JAMA Pediatr 172:958–965. doi: 10.1001/jamapediatrics.2018.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McClain B, Settembre E, Temple BR, Bellamy AR, Harrison SC. 2010. X-ray crystal structure of the rotavirus inner capsid particle at 3.8 A resolution. J Mol Biol 397:587–599. doi: 10.1016/j.jmb.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Periz J, Celma C, Jing B, Pinkney JN, Roy P, Kapanidis AN. 2013. Rotavirus mRNAS are released by transcript-specific channels in the double-layered viral capsid. Proc Natl Acad Sci U S A 110:12042–12047. doi: 10.1073/pnas.1220345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trask SD, McDonald SM, Patton JT. 2012. Structural insights into the coupling of virion assembly and rotavirus replication. Nat Rev Microbiol 10:165–177. doi: 10.1038/nrmicro2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berkova Z, Crawford SE, Trugnan G, Yoshimori T, Morris AP, Estes MK. 2006. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J Virol 80:6061–6071. doi: 10.1128/JVI.02167-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patton JT, Silvestri LS, Tortorici MA, Vasquez-Del Carpio R, Taraporewala ZF. 2006. Rotavirus genome replication and morphogenesis: role of the viroplasm. Curr Top Microbiol Immunol 309:169–187. doi: 10.1007/3-540-30773-7_6. [DOI] [PubMed] [Google Scholar]
- 7.Contin R, Arnoldi F, Campagna M, Burrone OR. 2010. Rotavirus NSP5 orchestrates recruitment of viroplasmic proteins. J Gen Virol 91:1782–1793. doi: 10.1099/vir.0.019133-0. [DOI] [PubMed] [Google Scholar]
- 8.Eichwald C, Vascotto F, Fabbretti E, Burrone OR. 2002. Rotavirus NSP5: mapping phosphorylation sites and kinase activation and viroplasm localization domains. J Virol 76:3461–3470. doi: 10.1128/jvi.76.7.3461-3470.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eichwald C, Rodriguez JF, Burrone OR. 2004. Characterization of rotavirus NSP2/NSP5 interactions and the dynamics of viroplasm formation. J Gen Virol 85:625–634. doi: 10.1099/vir.0.19611-0. [DOI] [PubMed] [Google Scholar]
- 10.Eichwald C, Arnoldi F, Laimbacher AS, Schraner EM, Fraefel C, Wild P, Burrone OR, Ackermann M. 2012. Rotavirus viroplasm fusion and perinuclear localization are dynamic processes requiring stabilized microtubules. PLoS One 7:e47947. doi: 10.1371/journal.pone.0047947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabbretti E, Afrikanova I, Vascotto F, Burrone OR. 1999. Two non-structural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. J Gen Virol 80(Part 2):333–339. doi: 10.1099/0022-1317-80-2-333. [DOI] [PubMed] [Google Scholar]
- 12.Torres-Vega MA, González RA, Duarte M, Poncet D, López S, Arias CF. 2000. The C-terminal domain of rotavirus NSP5 is essential for its multimerization, hyperphosphorylation and interaction with NSP6. J Gen Virol 81:821–830. doi: 10.1099/0022-1317-81-3-821. [DOI] [PubMed] [Google Scholar]
- 13.Cabral-Romero C, Padilla-Noriega L. 2006. Association of rotavirus viroplasms with microtubules through NSP2 and NSP5. Mem Inst Oswaldo Cruz 101:603–611. doi: 10.1590/s0074-02762006000600006. [DOI] [PubMed] [Google Scholar]
- 14.Criglar JM, Hu L, Crawford SE, Hyser JM, Broughman JR, Prasad BV, Estes MK. 2014. A novel form of rotavirus NSP2 and phosphorylation-dependent NSP2-NSP5 interactions are associated with viroplasm assembly. J Virol 88:786–798. doi: 10.1128/JVI.03022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin D, Duarte M, Lepault J, Poncet D. 2010. Sequestration of free tubulin molecules by the viral protein NSP2 induces microtubule depolymerization during rotavirus infection. J Virol 84:2522–2532. doi: 10.1128/JVI.01883-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Afrikanova I, Miozzo MC, Giambiagi S, Burrone O. 1996. Phosphorylation generates different forms of rotavirus NSP5. J Gen Virol 77(Part 9):2059–2065. doi: 10.1099/0022-1317-77-9-2059. [DOI] [PubMed] [Google Scholar]
- 17.Poncet D, Lindenbaum P, L'Haridon R, Cohen J. 1997. In vivo and in vitro phosphorylation of rotavirus NSP5 correlates with its localization in viroplasms. J Virol 71:34–41. doi: 10.1128/JVI.71.1.34-41.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.González SA, Burrone OR. 1991. Rotavirus NS26 is modified by addition of single O-linked residues of N-acetylglucosamine. Virology 182:8–16. doi: 10.1016/0042-6822(91)90642-o. [DOI] [PubMed] [Google Scholar]
- 19.Afrikanova I, Fabbretti E, Miozzo MC, Burrone OR. 1998. Rotavirus NSP5 phosphorylation is up-regulated by interaction with NSP2. J Gen Virol 79:2679–2686. doi: 10.1099/0022-1317-79-11-2679. [DOI] [PubMed] [Google Scholar]
- 20.Contin R, Arnoldi F, Mano M, Burrone OR. 2011. Rotavirus replication requires a functional proteasome for effective assembly of viroplasms. J Virol 85:2781–2792. doi: 10.1128/JVI.01631-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campagna M, Eichwald C, Vascotto F, Burrone OR. 2005. RNA interference of rotavirus segment 11 mRNA reveals the essential role of NSP5 in the virus replicative cycle. J Gen Virol 86:1481–1487. doi: 10.1099/vir.0.80598-0. [DOI] [PubMed] [Google Scholar]
- 22.López T, Rojas M, Ayala-Bretón C, López S, Arias CF. 2005. Reduced expression of the rotavirus NSP5 gene has a pleiotropic effect on virus replication. J Gen Virol 86:1609–1617. doi: 10.1099/vir.0.80827-0. [DOI] [PubMed] [Google Scholar]
- 23.Arnoldi F, Campagna M, Eichwald C, Desselberger U, Burrone OR. 2007. Interaction of rotavirus polymerase VP1 with nonstructural protein NSP5 is stronger than that with NSP2. J Virol 81:2128–2137. doi: 10.1128/JVI.01494-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berois M, Sapin C, Erk I, Poncet D, Cohen J. 2003. Rotavirus nonstructural protein NSP5 interacts with major core protein VP2. J Virol 77:1757–1763. doi: 10.1128/jvi.77.3.1757-1763.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vende P, Taraporewala ZF, Patton JT. 2002. RNA-binding activity of the rotavirus phosphoprotein NSP5 includes affinity for double-stranded RNA. J Virol 76:5291–5299. doi: 10.1128/jvi.76.10.5291-5299.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang X, Jayaram H, Kumar M, Ludtke SJ, Estes MK, Prasad BV. 2006. Cryoelectron microscopy structures of rotavirus NSP2-NSP5 and NSP2-RNA complexes: implications for genome replication. J Virol 80:10829–10835. doi: 10.1128/JVI.01347-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin D, Charpilienne A, Parent A, Boussac A, D'Autreaux B, Poupon J, Poncet D. 2013. The rotavirus nonstructural protein NSP5 coordinates a [2Fe-2S] iron-sulfur cluster that modulates interaction to RNA. FASEB J 27:1074–1083. doi: 10.1096/fj.12-217182. [DOI] [PubMed] [Google Scholar]
- 28.McDonald SM, Patton JT. 2011. Rotavirus VP2 core shell regions critical for viral polymerase activation. J Virol 85:3095–3105. doi: 10.1128/JVI.02360-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campagna M, Marcos-Villar L, Arnoldi F, de la Cruz-Herrera CF, Gallego P, González-Santamaría J, González D, Lopitz-Otsoa F, Rodriguez MS, Burrone OR, Rivas C. 2013. Rotavirus viroplasm proteins interact with the cellular SUMOylation system: implications for viroplasm-like structure formation. J Virol 87:807–817. doi: 10.1128/JVI.01578-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patton JT, Jones MT, Kalbach AN, He YW, Xiaobo J. 1997. Rotavirus RNA polymerase requires the core shell protein to synthesize the double-stranded RNA genome. J Virol 71:9618–9626. doi: 10.1128/JVI.71.12.9618-9626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawton JA, Zeng CQ, Mukherjee SK, Cohen J, Estes MK, Prasad BV. 1997. Three-dimensional structural analysis of recombinant rotavirus-like particles with intact and amino-terminal-deleted VP2: implications for the architecture of the VP2 capsid layer. J Virol 71:7353–7360. doi: 10.1128/JVI.71.10.7353-7360.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Baker ML, Jiang W, Estes MK, Prasad BV. 2009. Rotavirus architecture at subnanometer resolution. J Virol 83:1754–1766. doi: 10.1128/JVI.01855-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Settembre E, Xu C, Dormitzer PR, Bellamy R, Harrison SC, Grigorieff N. 2008. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc Natl Acad Sci U S A 105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charpilienne A, Lepault J, Rey F, Cohen J. 2002. Identification of rotavirus VP6 residues located at the interface with VP2 that are essential for capsid assembly and transcriptase activity. J Virol 76:7822–7831. doi: 10.1128/jvi.76.15.7822-7831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Viskovska M, Anish R, Hu L, Chow DC, Hurwitz AM, Brown NG, Palzkill T, Estes MK, Prasad BV. 2014. Probing the sites of interactions of rotaviral proteins involved in replication. J Virol 88:12866–12881. doi: 10.1128/JVI.02251-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng CQ, Estes MK, Charpilienne A, Cohen J. 1998. The N terminus of rotavirus VP2 is necessary for encapsidation of VP1 and VP3. J Virol 72:201–208. doi: 10.1128/JVI.72.1.201-208.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labbé M, Baudoux P, Charpilienne A, Poncet D, Cohen J. 1994. Identification of the nucleic acid binding domain of the rotavirus VP2 protein. J Gen Virol 75:3423–3430. doi: 10.1099/0022-1317-75-12-3423. [DOI] [PubMed] [Google Scholar]
- 38.Ding K, Celma CC, Zhang X, Chang T, Shen W, Atanasov I, Roy P, Zhou ZH. 2019. In situ structures of rotavirus polymerase in action and mechanism of mRNA transcription and release. Nat Commun 10:2216. doi: 10.1038/s41467-019-10236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Estrozi LF, Settembre EC, Goret G, McClain B, Zhang X, Chen JZ, Grigorieff N, Harrison SC. 2013. Location of the dsRNA-dependent polymerase, VP1, in rotavirus particles. J Mol Biol 425:124–132. doi: 10.1016/j.jmb.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jenni S, Salgado EN, Herrmann T, Li Z, Grant T, Grigorieff N, Trapani S, Estrozi LF, Harrison SC. 2019. In situ structure of rotavirus VP1 RNA-dependent RNA polymerase. J Mol Biol 431:3124–3138. doi: 10.1016/j.jmb.2019.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laimbacher AS, Esteban LE, Castello AA, Abdusetir Cerfoglio JC, Argüelles MH, Glikmann G, D'Antuono A, Mattion N, Berois M, Arbiza J, Hilbe M, Schraner EM, Seyffert M, Dresch C, Epstein AL, Ackermann M, Fraefel C. 2012. HSV-1 amplicon vectors launch the production of heterologous rotavirus-like particles and induce rotavirus-specific immune responses in mice. Mol Ther 20:1810–1820. doi: 10.1038/mt.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boudreaux CE, Vile DC, Gilmore BL, Tanner JR, Kelly DF, McDonald SM. 2013. Rotavirus core shell subdomains involved in polymerase encapsidation into virus-like particles. J Gen Virol 94:1818–1826. doi: 10.1099/vir.0.052951-0. [DOI] [PubMed] [Google Scholar]
- 43.Campagna M, Budini M, Arnoldi F, Desselberger U, Allende JE, Burrone OR. 2007. Impaired hyperphosphorylation of rotavirus NSP5 in cells depleted of casein kinase 1alpha is associated with the formation of viroplasms with altered morphology and a moderate decrease in virus replication. J Gen Virol 88:2800–2810. doi: 10.1099/vir.0.82922-0. [DOI] [PubMed] [Google Scholar]
- 44.Eichwald C, Jacob G, Muszynski B, Allende JE, Burrone OR. 2004. Uncoupling substrate and activation functions of rotavirus NSP5: phosphorylation of Ser-67 by casein kinase 1 is essential for hyperphosphorylation. Proc Natl Acad Sci U S A 101:16304–16309. doi: 10.1073/pnas.0406691101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cabantous S, Nguyen HB, Pedelacq JD, Koraïchi F, Chaudhary A, Ganguly K, Lockard MA, Favre G, Terwilliger TC, Waldo GS. 2013. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci Rep 3:2854. doi: 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silvestri LS, Taraporewala ZF, Patton JT. 2004. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J Virol 78:7763–7774. doi: 10.1128/JVI.78.14.7763-7774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spencer E, Arias ML. 1981. In vitro transcription catalyzed by heat-treated human rotavirus. J Virol 40:1–10. doi: 10.1128/JVI.40.1.1-10.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schönborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, Lukacs N. 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res 19:2993–3000. doi: 10.1093/nar/19.11.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald SM, Patton JT. 2011. Assortment and packaging of the segmented rotavirus genome. Trends Microbiol 19:136–144. doi: 10.1016/j.tim.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmed K, Nakagomi T, Nakagomi O. 2005. Isolation and molecular characterization of a naturally occurring non-structural protein 5 (NSP5) gene reassortant of group A rotavirus of serotype G2P[4] with a long RNA pattern. J Med Virol 77:323–330. doi: 10.1002/jmv.20451. [DOI] [PubMed] [Google Scholar]
- 51.Kojima K, Taniguchi K, Urasawa T, Urasawa S. 1996. Sequence analysis of normal and rearranged NSP5 genes from human rotavirus strains isolated in nature: implications for the occurrence of the rearrangement at the step of plus strand synthesis. Virology 224:446–452. doi: 10.1006/viro.1996.0551. [DOI] [PubMed] [Google Scholar]
- 52.Komoto S, Kanai Y, Fukuda S, Kugita M, Kawagishi T, Ito N, Sugiyama M, Matsuura Y, Kobayashi T, Taniguchi K. 2017. Reverse genetics system demonstrates that rotavirus nonstructural protein NSP6 is not essential for viral replication in cell culture. J Virol 91:e00695-17. doi: 10.1128/JVI.00695-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H, Taniguchi K, Urasawa T, Urasawa S. 1998. Serological and genomic characterization of human rotaviruses detected in China. J Med Virol 55:168–176. doi:. [DOI] [PubMed] [Google Scholar]
- 54.Prasad BV, Wang GJ, Clerx JP, Chiu W. 1988. Three-dimensional structure of rotavirus. J Mol Biol 199:269–275. doi: 10.1016/0022-2836(88)90313-0. [DOI] [PubMed] [Google Scholar]
- 55.Yeager M, Dryden KA, Olson NH, Greenberg HB, Baker TS. 1990. Three-dimensional structure of rhesus rotavirus by cryoelectron microscopy and image reconstruction. J Cell Biol 110:2133–2144. doi: 10.1083/jcb.110.6.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glück S, Buttafuoco A, Meier AF, Arnoldi F, Vogt B, Schraner EM, Ackermann M, Eichwald C. 2017. Rotavirus replication is correlated with S/G2 interphase arrest of the host cell cycle. PLoS One 12:e0179607. doi: 10.1371/journal.pone.0179607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. 2011. Atomic model of an infectious rotavirus particle. EMBO J 30:408–416. doi: 10.1038/emboj.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Papa G, Venditti L, Arnoldi F, Schraner EM, Potgieter C, Borodavka A, Eichwald C, Burrone OR, Papa G, Venditti L, Arnoldi F, Schraner EM, Potgieter C, Borodavka A, Eichwald C, Burrone OR. 2019. Recombinant rotaviruses rescued by reverse genetics reveal the role of NSP5 hyperphosphorylation in the assembly of viral factories. J Virol 94:e01110-19. doi: 10.1128/JVI.01110-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Criglar JM, Anish R, Hu L, Crawford SE, Sankaran B, Prasad BVV, Estes MK. 2018. Phosphorylation cascade regulates the formation and maturation of rotaviral replication factories. Proc Natl Acad Sci U S A 115:E12015–E12023. doi: 10.1073/pnas.1717944115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sotelo PH, Schümann M, Krause E, Chnaiderman J. 2010. Analysis of rotavirus non-structural protein NSP5 by mass spectrometry reveals a complex phosphorylation pattern. Virus Res 149:104–108. doi: 10.1016/j.virusres.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 61.Sheppard D. 1994. Dominant negative mutants: tools for the study of protein function in vitro and in vivo. Am J Respir Cell Mol Biol 11:1–6. doi: 10.1165/ajrcmb.11.1.8018332. [DOI] [PubMed] [Google Scholar]
- 62.Kanai Y, Komoto S, Kawagishi T, Nouda R, Nagasawa N, Onishi M, Matsuura Y, Taniguchi K, Kobayashi T. 2017. Entirely plasmid-based reverse genetics system for rotaviruses. Proc Natl Acad Sci U S A 114:2349–2354. doi: 10.1073/pnas.1618424114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A 83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arnold M, Patton JT, McDonald SM. 2009. Culturing, storage, and quantification of rotaviruses. Curr Protoc Microbiol Chapter 15:Unit 15C.3. doi: 10.1002/9780471729259.mc15c03s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Estes MK, Graham DY, Gerba CP, Smith EM. 1979. Simian rotavirus SA11 replication in cell cultures. J Virol 31:810–815. doi: 10.1128/JVI.31.3.810-815.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Petris G, Bestagno M, Arnoldi F, Burrone OR. 2014. New tags for recombinant protein detection and O-glycosylation reporters. PLoS One 9:e96700. doi: 10.1371/journal.pone.0096700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eichwald C, De Lorenzo G, Schraner EM, Papa G, Bollati M, Swuec P, de Rosa M, Milani M, Mastrangelo E, Ackermann M, Burrone OR, Arnoldi F. 2018. Identification of a small molecule that compromises the structural integrity of viroplasms and rotavirus double-layered particles. J Virol 92:e01943-17. doi: 10.1128/JVI.01943-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eichwald C, Ackermann M, Nibert ML. 2018. The dynamics of both filamentous and globular mammalian reovirus viral factories rely on the microtubule network. Virology 518:77–86. doi: 10.1016/j.virol.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 69.Eichwald C, Kim J, Nibert ML. 2017. Dissection of mammalian orthoreovirus μ2 reveals a self-associative domain required for binding to microtubules but not to factory matrix protein μNS. PLoS One 12:e0184356. doi: 10.1371/journal.pone.0184356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 71.Katoh K, Rozewicki J, Yamada KD. 2019. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]