

Chikungunya virus is a vector-borne pathogen of global significance. The morbidity associated with chikungunya virus (CHIKV) infection, neurovirulence and adaptability to Aedes albopictus, necessitates a deeper understanding of the interaction of CHIKV with the host immune system. Here, we demonstrate that CHIKV is resistant to neutralization by one of the potent barriers of the innate immune arm, the complement system. Chikungunya virus showed marked resistance to complement despite activation and deposition of complement proteins. Interestingly the C3 component associated with the virion was found to be inactive C3b (iC3b), a key factor implicated in the pathogenesis and disease severity in the mouse model of Ross River virus infection. CHIKV also had an associated unique factor I-like activity that mediated the inactivation of C3b into iC3b. We have unraveled a smart strategy adopted by CHIKV to limit complement which has serious implications in viral dissemination, pathogenesis, and disease.
KEYWORDS: chikungunya virus, complement activation, complement evasion, factor I-like activity
ABSTRACT
Chikungunya virus (CHIKV) is an emerging pathogen capable of causing explosive outbreaks. Prior studies showed that exacerbation in arthritogenic alphavirus-induced pathogenesis is attributed to its interaction with multiple immune components, including the complement system. Viremia concomitant to CHIKV infection makes exposure of the virus to complement unavoidable, yet very little is known about CHIKV-complement interactions. Here, we show that CHIKV activated serum complement to modest levels in a concentration- and time-dependent manner, but the virus effectively resisted complement-mediated neutralization. Heat-inactivated serum from seropositive donors could actively neutralize CHIKV due to the presence of potent anti-CHIKV antibodies. Deposition of key complement components C3 and C4 did not alter the resistance of CHIKV to complement. Further, we identified a factor I-like activity in CHIKV that limited complement by inactivating C3b into inactive C3b (iC3b), the complement component known to significantly contribute to disease severity in vivo, but this activity had no effect on C4b. Inactivation of C3b by CHIKV was largely dependent on the concentration of the soluble host cofactor factor H and the virus concentration. A factor I function-blocking antibody had only a negligible effect on the factor I-like activity associated with CHIKV, suggesting that this activity is independent of host factor I and could be of viral origin. Thus, our findings suggest a complement modulatory action of CHIKV which not only helps the virus to evade human complement but may also have implications in alphavirus-induced arthritogenic symptoms.
IMPORTANCE Chikungunya virus is a vector-borne pathogen of global significance. The morbidity associated with chikungunya virus (CHIKV) infection, neurovirulence and adaptability to Aedes albopictus, necessitates a deeper understanding of the interaction of CHIKV with the host immune system. Here, we demonstrate that CHIKV is resistant to neutralization by one of the potent barriers of the innate immune arm, the complement system. Chikungunya virus showed marked resistance to complement despite activation and deposition of complement proteins. Interestingly the C3 component associated with the virion was found to be inactive C3b (iC3b), a key factor implicated in the pathogenesis and disease severity in the mouse model of Ross River virus infection. CHIKV also had an associated unique factor I-like activity that mediated the inactivation of C3b into iC3b. We have unraveled a smart strategy adopted by CHIKV to limit complement which has serious implications in viral dissemination, pathogenesis, and disease.
INTRODUCTION
The initial defense of the host from a barrage of pathogens is realized primarily by the innate arm of the immune system, which includes the complement system. Complement activation pathways, effected by a highly concerted group of proteins, are multifunctional and not restricted to targeting pathogens (1, 2). Activated complement directly or indirectly plays an important role in limiting viral dissemination, thereby largely influencing the magnitude of pathogenesis and the associated disease outcomes. The effector function of complement occurs through either one or multiple pathways, namely, the classic pathway (CP), the lectin pathway (LP), and the alternative pathway (AP). Unique viral signatures and infection-induced endogenous danger-associated molecular patterns can activate complement pathways, leading to cleavage of the central component C3 into C3a and C3b (3). Complement components such as C3b and C4b are potent opsonins, as they can covalently bind to the pathogen surface or form convertases facilitating further amplification and progression of pathways, resulting in virus neutralization (4). Unlike the Ca2+-dependent CP or LP triggered by the immune complex or the mannose-binding lectin (MBL)-sugar complex, basal levels of activation of AP are maintained and support CP and LP via the amplification loop (5). Viruses, both enveloped and nonenveloped, encompassing diverse families have been shown to activate complement, resulting in virus neutralization. Viruses such as measles virus (MV), mumps virus (MuV), parainfluenza virus 5 (PIV5), dengue virus, influenza A virus, human immunodeficiency virus (HIV), coxsackievirus B3 virus, West Nile virus, and vesicular stomatitis virus (VSV) have been shown to activate one or multiple complement pathways (6–19). Irrespective of the pathway involved, C3 plays a predominant role in the neutralization of many viruses (9, 20, 21). The assembly of the pore-forming unit called membrane attack complex (MAC) is the final step in the complement cascade which supports virolysis, while terminal pathway-independent neutralization has also been frequently reported (9, 22). The role of complement in the neutralization of virus is not limited to lysis or aggregation but is multifactorial, with specific complement components contributing to opsonization, phagocytosis, inflammation, and priming of the adaptive immune responses (23, 24).
The inability to discriminate the self from nonself by the complement system, due to a lack of memory, is circumvented by a highly concerted group of membrane-bound or soluble proteins called regulators of complement activation (RCA). The RCA proteins function either as cofactors by inactivating critical components such as C3b and C4b, e.g., CD46, CD35, factor H, and C4BP, or by accelerating the decay of C3/C5 convertase, e.g., CD55 and CD35 (25). In the race to override complement, viruses have adopted smart strategies, including exploiting RCAs. Recruitment or incorporation of the host’s membrane-associated or soluble RCAs has been reported in the case of many enveloped viruses, including PIV5, VSV, MuV, MV, HIV, and poxviruses (26, 27). It has also been established that certain viruses, such as vaccinia, variola, herpes simplex virus 1, and Kaposi sarcoma-associated herpesvirus, encode mimics that are structurally and functionally similar to RCAs and are known to possess both cofactor and decay accelerating activity (28–33). Preferential recruitment of factor H by Sindbis virus is facilitated by the enrichment of sialic acid on the virus envelope, and the NS1 protein of West-Nile virus acts as a conduit for factor H binding, thus mediating evasion of complement (34, 35). Factor I is a cofactor-dependent complement-specific protease that inactivates C3b/C4b, and recently, the resistance of the Nipah virus (NiV) to human complement was attributed to a factor I-like activity associated with the virus (36). Thus, exploitation of RCAs or other complement regulatory proteins is an interesting ploy adopted by a range of viruses. The overall premise of this study is to investigate the effect of the human complement system on chikungunya virus (CHIKV) and to understand the mechanism of complement modulation, if any, by CHIKV.
Chikungunya virus is an important human pathogen causing significant joint morbidity. The significance of CHIKV as a reemerging global pathogen can be inferred from the magnitude of recent outbreaks that occurred across diverse geographical regions (37, 38). It is a vector-borne alphavirus with a positive-sense single-strand RNA genome and is transmitted by the Aedes aegypti or Aedes albopictus species of mosquitoes (39). Chikungunya and Sindbis virus treated with serum from naive wild-type mice were found to resist mouse complement (40). Much of the information on alphavirus-complement interactions has been obtained from studies using the Ross River virus (RRV). Unlike other viruses, complement was found to support RRV-associated disease pathogenesis. Using C3−/− and MBL−/− mice, it was demonstrated that the MBL pathway and C3 were highly essential for the development of RRV-induced arthrogenic disease (41, 42). The disease symptoms were exacerbated due to the involvement of complement receptor 3 (CR3) and the downstream signaling at the sites of inflammation, which thus promote tissue damage (43). In contrast to RRV, complement had a protective role in Venezuelan equine encephalitis virus (VEEV) infection by promoting peripheral virus clearance and abatement of neuroinvasion and neuropathology (44). Although the effect of mouse complement on CHIKV is known, not much is known about the outcome of CHIKV-human complement interaction. Posttransmission by mosquitoes, dissemination of CHIKV to distal sites occurs via the hematogenous route (45). The presence of complement both locally and in the serum necessitates the adoption of smart strategies by the virus to circumvent complement. Here, we demonstrate that CHIKV can activate complement to modest levels, but this does not result in virus neutralization despite the deposition of complement components C3b and C4b. Neutralization was observed only with serum from seropositive donors due to the presence of anti-CHIKV antibodies. The resistance of CHIKV to human complement is in part due to a factor-I like activity associated with the virion which mediated the inactivation of C3b into inactive C3b (iC3b) with the mere addition of factor H. Our study gains significance, as it offers valuable insights into the mechanism by which CHIKV limits the potency of the host complement.
RESULTS
Chikungunya virus activates complement in normal human serum.
Chikungunya virus being a hematogenous virus, we first investigated if it can activate human complement in the fluid phase. To determine the effect of virus concentration on complement activation, 2-fold dilutions of sucrose-gradient-purified CHIKV starting with 2.5 μg to 0.07 μg were incubated with normal human serum (NHS; 1:20) for 45 min at 37°C. The levels of C3a generated, an established complement activation marker, were determined using Western blotting. Background levels of C3a, similar to the serum-only controls, were seen at a CHIKV concentration of 0.07 μg, with a gradual increase observed at 0.15 and 0.31 μg, suggesting concentration dependency. The levels of C3a generated saturated at concentrations above 0.6 μg (Fig. 1A). The data presented are a representation of three independent experiments. Complement activation by CHIKV was also time dependent. Upon incubation of a fixed concentration of CHIKV (1.25 μg) with NHS (1:20) for various time points, negligible levels of C3a were observed at 1 min, followed by a gradual increase starting at 5 min, with the highest levels being observed at 45 min (Fig. 1B, lanes 8 to 13). Only background levels of C3a could be detected in the corresponding serum-only control (Fig. 1B, lanes 2 to 7). Zymosan treated with NHS served as the positive control, where robust levels of C3a were generated. Complement activation levels observed with CHIKV never equaled the levels achieved with the Zymosan control. Thus, CHIKV was found to activate human complement in solution; however, increased virus concentration and the prolonged incubation period required for saturation of C3a indicated that CHIKV supported complement activation to modest levels.
FIG 1.
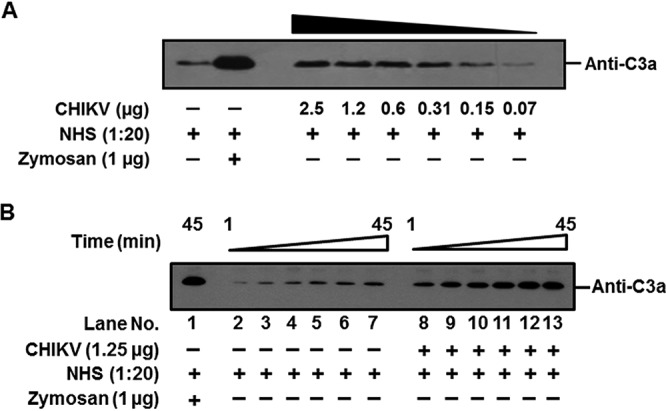
Activation of human complement by CHIKV is concentration and time dependent. Complement activation and subsequent generation of C3a upon incubation of CHIKV with NHS are shown. Serum treated with zymosan served as a positive control, while the negative control was set up by incubating serum with PBS. (A) Levels of C3a generated in the serum when incubated with various concentrations of CHIKV for 45 min. CHIKV activated complement in a concentration-dependent manner. (B) Levels of C3a generated in the serum at different time points (1, 5, 10, 20, 30, and 45 min) when incubated with a set concentration of CHIKV. A gradual increase in the levels of C3a occurred with the increase in time (lanes 8–13); background levels of C3a were detected at 1 min, with marked saturation occurring midway. The serum-only control incubated for the indicated time period showed only basal levels of C3a (lanes 2–7), which were significantly lower than for the samples containing both CHIKV and NHS. The Western blots are representative of three independent experiments.
CHIKV is resistant to complement-mediated neutralization.
It was previously demonstrated that CHIKV is less sensitive to neutralization by mouse complement (40). Having observed that CHIKV is a poor activator of the human complement, we hypothesized that unlike highly susceptible enveloped viruses, CHIKV would be less sensitive to the human complement. Complement-dependent neutralization assays were performed by incubating CHIKV with different dilutions of NHS or heat-inactivated NHS (HI-NHS) for 1 h at 37°C, and the remaining infectivity was determined by performing a plaque assay on monolayers of Vero cells. The virus was found to actively resist neutralization at all the dilutions tested. Compared to the input, virus-only control, only a 10 to 25% reduction in the number of plaques was seen when the serum concentration was high (1:5 to 1:40; Fig. 2A). Although the reduction in numbers showed statistical significance when the minimal essential medium (MEM) control and NHS- or HI-NHS-treated CHIKV were compared, the relative difference between NHS- and HI-NHS-treated CHIKV samples was not significant. Because the reduction in plaque numbers was not specific to NHS treatment and was close enough to HI-NHS-treated samples, a role of serum factors other than complement may be attributed. It should be pointed out here that each data point is the mean ± standard error of the mean (SEM) of values from experiments carried out with seven independent donor serum samples. Thus, our findings support the hypothesis that CHIKV is resistant to human complement. To further substantiate the resistance of CHIKV to NHS, a comparative analysis was carried out between CHIKV and Chandipura virus (CHPV). We had earlier observed that the neurotrophic rhabdovirus CHPV, known to cause encephalitis in humans, is readily neutralized by human complement (46). A marked reduction in the number of plaques was seen in the case of CHPV compared to CHIKV upon treatment with NHS at all serum dilutions tested (Fig. 2B). As observed earlier, a minor reduction in the plaque numbers was observed when CHIKV was treated with either NHS or HI-NHS, while treatment with HI-NHS had no substantial effect on the infectivity of CHPV. These observations confirm that CHIKV is highly refractory to complement-mediated neutralization in vitro.
FIG 2.

Chikungunya virus is less sensitive to serum complement factors but is readily neutralized by anti-CHIKV antibody in seropositive donor sera. Complement-dependent neutralization of CHIKV and CHPV was assessed by incubating equal amounts of PFU of the virus with MEM or a 2-fold dilution of NHS or HI-NHS for 1 h at 37°C followed by plaque assay. (A) Effect of NHS/HI-NHS on CHIKV. A reduction in the number of plaques can be observed with both the NHS- and HI-NHS-treated samples compared to the CHIKV-alone control. No significant differences were observed within the NHS- and HI-NHS-treated groups at every dilution tested except at the highest serum concentration of 1:5. The data represent the mean ± SEM of all data points obtained from 7 individual donor serum samples. The symbols in the figure indicate significance as follows: #, P ≤ 0.0001; ^, P ≤ 0.01; *, P ≤ 0.05; ns, nonsignificance. (B) Comparison of susceptibility of CHIKV and CHPV to NHS. Chandipura virus was highly sensitive to NHS compared to CHIKV, with neutralization observed in all serum concentrations tested. Heat-inactivated serum (1:10) was not capable of limiting both CHIKV and CHPV. The data are the mean ± SEM of three independent experiments performed in duplicate. ***, P ≤ 0.0001; **, P ≤ 0.01; ns, nonsignificance. (C) Effect of HI sera from both seropositive and seronegative donors on CHIKV. Equal PFU of CHIKV were incubated with HI-sera from seropositive/seronegative donors, and the remaining viral infectivity was determined using a plaque assay. Heat-resistant factors in seropositive donors and not seronegative serum neutralized CHIKV. The data represent the mean ± SEM of 6 independent experiments. ***, P ≤ 0.001. (D) ELISA to detect anti-CHIKV antibodies in seropositive donors. Purified CHIKV adsorbed to ELISA plates was incubated with 2-fold serial dilutions of HI-donor sera, and the bound anti-CHIKV antibody was estimated colorimetrically at 405 nm. Binding was observed only with the serum from the seropositive donor and positive control and not from the seronegative serum. Serum from a seronegative donor (D8) and immune sera from rabbits immunized with UV-inactivated CHIKV (P) served as the negative and positive controls, respectively. The data are the mean ± SEM of 3 independent experiments.
Since both NHS and HI-NHS had no significant effect on CHIKV, we wanted to determine the effect of serum from seropositive donors on the virus. Serum samples from 6 individuals with a clinical history of CHIKV infection and one naive donor (donor 8 [D8]) were heat-inactivated to inactivate complement and were tested for their neutralizing effect on CHIKV. Based on the prior standardization, 100 PFU of CHIKV was incubated with the donor serum at a dilution of 1:2,500 for 1 h at 37°C, and the remaining infectious particles were determined by plaque assay (Fig. 2C). Compared to the MEM-treated control, a marked reduction in plaque numbers was observed when CHIKV was treated with HI sera from seropositive donors. The number of plaques equaled that of the control upon treatment with HI serum from the seronegative donor (D8). The neutralization pattern among the individuals varied, with donors D92 and D145 showing >75% neutralization, while the remaining donors had a neutralization range between 50 and 60%. The differences in the neutralization patterns of the seropositive donors were consistent with existing variation in the antibody profile against CHIKV (47).
In order to validate the presence or absence of anti-CHIKV antibodies in the donor serum, a CHIKV-specific enzyme-linked immunosorbent assay (ELISA) was carried out by coating 125 ng of gradient-purified CHIKV. Then, 2-fold dilutions of HI-serum were incubated with the adsorbed virus, and the bound antibodies were detected using horseradish peroxidase (HRP)-labeled secondary antibodies. Antibodies in the serum from seropositive donors reacted to bound CHIKV saturating at the highest concentration (1/100 and 1/200). Variation in absorbance was observed between individuals, which is clearly evident between the dilutions 1/200 and 1/6,400 (Fig. 2D). As expected, antibodies from donor D8 did not bind to CHIKV; however, antibodies in the hyper-immune sera from a rabbit immunized with UV-inactivated CHIKV were highly reactive to the viral antigens, with absorbance at 405 nm being >2 at the highest concentration. Taken together, these findings make it clear that serum components, including complement in the naive serum, are incapable of neutralizing CHIKV, which is reversed only when CHIKV-specific antibodies are present as in the case of serum from seropositive donors.
Complement activation by CHIKV results in deposition of complement components C3b and C4b.
Deposition of activated complement components, including C3b and C4b, is an important step in the progression of the complement cascade and largely dictates the susceptibility of viruses to complement. We had earlier demonstrated that despite complement activation by CHIKV (Fig. 1), neutralization did not ensue (Fig. 2A and B). We sought to understand if the block in neutralization is due to the lack of deposition of complement components. An ultracentrifugation approach was first employed, wherein gradient-purified CHIKV was incubated with either MEM or NHS at a dilution of 1:2 for 1 h at 37°C and layered on a 15 to 60% sucrose gradient and was centrifuged at 150,000 × g for 5 h at 4°C. Fractions of equal volumes collected from the bottom of the tubes were analyzed with Western blotting to detect CHIKV proteins and the complement components C3 and C4. Of the total 17 fractions analyzed, viral antigens were detected between fractions 6 and 8 (Fig. 3A, top panel) in the CHIKV+MEM sample. Treatment with NHS had no effect in the migration pattern of viruses, as viral antigens were present in identical fractions 6 to 8, similar to the CHIKV+MEM samples (Fig. 3A, second panel from top). In the case of enveloped viruses such as PIV5 or mumps virus, differential shifts in migration have been reported which corresponded to their mechanism of neutralization, viz., aggregation or lysis (9). In the case of CHIKV, it is interesting to note that no such differences existed. All the fractions from the CHIKV+NHS sample were also probed to detect complement proteins C3 and C4. Complement component C3 was found to comigrate with the viral fractions (6–8), but C3 reactivity was also observed in the bottom fractions 3 to 5. The remaining unbound C3 components were detected at the top of the gradient between fractions 13 and 17 (Fig. 3A, third panel from top). The association of C4 components to CHIKV and migration also followed a pattern similar to that of C3. The peak fractions (6–8) showed reactivity to C4 antibodies with signal also detected in fractions 4, 5, and 9. Maximum reactivity to C4 antibody was observed in the fractions at the top of the gradient, similar to C3 (Fig. 3A, bottom panel). These results indicate that complement components comigrate with the virus particles through the sucrose gradient, suggesting deposition of the complement proteins. This deposition is limited, as the larger fraction of the complement proteins was found unbound at the top of the sucrose gradient.
FIG 3.

Exposure of CHIKV to NHS results in deposition of complement components on the virus. (A) Western blot analysis of sucrose density gradient fractions of NHS- or MEM-treated CHIKV. Purified CHIKV was incubated with NHS or MEM and subjected to ultracentrifugation over a preequilibrated 15 to 60% linear sucrose gradient. Fractions of equal volume collected from the bottom of the tube were analyzed for CHIKV antigens and the associated complement components. Complement components C3/C4 comigrated with CHIKV in a linear sucrose gradient when subjected to high-speed ultracentrifugation. Note that fraction 1 is from the bottom, while fraction 17 is the topmost of the gradient as indicated. The migration pattern of viral antigens in both MEM- (top) and NHS-treated CHIKV (2nd from top) was identical to the reactivity observed between fractions 6 to 8. Components of complement proteins C3 (3rd from top) and C4 (bottom) could be seen migrating with CHIKV antigens, but excess or unbound C3/C4 in the CHIKV + NHS sample appeared in the top fractions. P indicates purified CHIKV (top two blots, extreme left lane), C3 (third from the top), and C4 (bottom). The Western blot data comprise a representative image of three independent experiments. (B) Visualization of the C3/C4 component bound to CHIKV by electron microscopy. Chikungunya virus was adsorbed on EM grids and treated with NHS. Bound complement components (C3/C4) to CHIKV were detected after multiple washes followed by probing with anti-human C3 (middle panel, magnification 68,000×) or anti-human C4 (bottom panel, magnification 98,000×) antibody followed by anti-CHIKV antisera and nanogold-conjugated secondary antibodies. The controls included secondary only (top left, magnification 68,000×) and particles probed for viral antigens only (top right, magnification 120,000×). Viral antigens are highlighted by 6-nm (closed triangles) and C3/C4 by 12-nm (arrows) gold secondary antibodies, respectively, with the scale bar indicating 0.025 μm. (C) Pie chart depicting the percentage of particles staining positive for C3 deposition in comparison to C3-negative particles.
To further confirm deposition of C3 and C4 components on CHIKV, electron microscopy (EM) experiments were carried out. Gradient-purified CHIKV was adsorbed to carbon-coated gold EM grids, incubated with NHS (1:10), and probed for C3/C4 deposition. Treatment with NHS resulted in deposition of both C3 and C4 components on CHIKV. Anti-CHIKV antibodies bound to the particles were detected readily, as observed by the marked staining of the 6-nm nano-gold anti-rabbit antibodies (Fig. 3B, top-right panel). Upon probing with anti-C3/C4 antibodies, positive reactivity to both the proteins was observed as evident from the 12-nm gold secondary antibody binding to reactive C3 (Fig. 3B, middle two panels) and C4 antibodies (Fig. 3B, bottom two panels). Thus, the electron microscopic observations further confirm that vital complement components, C3 and C4, are deposited on the virion post-complement activation by CHIKV. However, the overall number of particles which showed positive reactivity to C3 deposition was lower. Particles in 14 independent fields were chosen and checked for positive reactivity to anti-C3 antibodies. Out of the 165 particles analyzed, only 35 showed staining for C3, which accounted for only 21% positivity (Fig. 3C). These data suggest that the activation of complement by CHIKV resulted in deposition of complement proteins on the virus particles, but the limited deposition did not support virus neutralization.
The C3 component comigrating with the viral fractions lacks the α′ subunit.
Having observed that C3 deposition occurred on the virion without affecting the virus integrity, as no neutralization was observed, it was intriguing to identify the form of C3 component associated with the virion. Upon initial activation, the native C3 is cleaved into the active C3b, which can bind covalently to pathogens. However, unprecedented complement activation can generate significant levels of C3b and C4b, which is inactivated readily by the combined action of a complement-specific serine protease, factor I, and cofactors. Cofactors such as factor H and CD46 support the factor I-mediated inactivation of C3b into iC3b. The 102-kDa α′ subunit of C3b is cleaved into 68- and 43-kDa fragments with the release of C3f as depicted in the stick diagram (Fig. 4A). A closer analysis of the peak viral fractions in the CHIKV+NHS samples (for gradient samples see Fig. 3A) with Western blotting revealed that the species of C3 associated with the virion lacked the intact α′ subunit. This clearly resembled iC3b, as both the 68- and 43-kDa subunits were also detected in the peak fraction (Fig. 4B, lane 4). This led to the hypothesis that CHIKV harbors an associated factor that can mediate the inactivation of C3b into iC3b. A nonspecific signal between the 68- and 43-kDa bands was also observed, the identity of which is not clear. However, based on the molecular weight, the bands corresponded to that of the envelope proteins of CHIKV.
FIG 4.

An inactive form of C3b is associated with CHIKV. (A) Schematic diagram depicting C3b and the first order of inactivation to iC3b. C3b comprises a 102-kDa α′ subunit and a 75-kDa β subunit linked by an intermolecular disulfide bond. Inactivation of C3b by a specific serine protease factor I in the presence of cofactors such as factor H, CD35, or CD46 results in the first-step cleavage of the C3b α′ subunit into 68- and 43-kDa fragments with the release of a 2-kDa fragment called C3f. The resulting C3b component is called iC3b. The closed balloon indicates the position of the thioester bond, and the solid vertical arrows indicate the site of first-order cleavage by factor I. (B) Western blot analysis of the peak fractions to identify the species of C3b associated with CHIKV. Blots containing the peak fractions 6 to 8 (lanes 3 to 5), purified C3b (lane 1), and iC3b (lane 2) were probed with anti-C3 antibody. The samples in lanes 3, 4, and 5 lacked the α′-subunit of C3b (note lane 1) and corresponded to iC3b (note lane 2) characterized by the presence of the 68- and 43-kDa fragments, suggesting the association of inactive C3b (iC3b). The image is representative of 3 independent experiments.
A factor I-like activity associated with CHIKV mediates the inactivation of C3b into iC3b and not that of C4b.
C3b and C4b cofactor assays were reconstituted with purified C3b, factor H or C4b, C4BP, factor I, and CHIKV. In the presence of the cofactor factor H, the serine protease factor I mediates the cleavage of the α′ subunit of C3b into the 68- and 43-kDa fragments, resulting in C3b inactivation (Fig. 5A, lane 2). Viruses such as PIV5 and VSV have been shown to recruit the host CD46, and the mere addition of purified factor I to the substrate C3b and the CD46 harboring virus was sufficient to inactivate C3b into iC3b. In order to check if this is same in the case of CHIKV, C3b was incubated with the virus and only factor I; however, no inactivation was observed even after 6 h of incubation (Fig. 5A, lane 5). Interestingly, in the absence of factor I, the mere addition of the cofactor factor H and CHIKV to C3b resulted in the cleavage of the α′ subunit of C3b (Fig. 5A, lane 6). This suggested that CHIKV harbors a factor I-like activity capable of inactivating C3b into iC3b.
FIG 5.

Chikungunya virus supports factor H-mediated cleavage of C3b- and not C4BP-dependent inactivation of C4b. (A) A C3b cofactor activity assay was carried out by incubating the indicated purified proteins, including gradient-purified CHIKV, and subjecting the samples to SDS-PAGE followed by GelCode blue staining. The virus alone without the addition of the enzyme factor I mediated the cleavage of the C3b-α′ with the cofactor factor H, indicated by the closed arrowhead (lane 6), similar to the control (lane 2). CHIKV by itself (lane 4) or upon supplementation of factor I (lane 5) was incapable of inactivating C3b. (B) A C4b cofactor activity assay was performed with the proteins and CHIKV as indicated in the figure, followed by SDS-PAGE analysis. C4b inactivation into C4c and C4d can be observed only in the control lane with C4BP and factor I (lane 2). Unlike the C3b inactivation by CHIKV, C4b was found to be intact in both the samples where either factor I (lane 5) or C4BP (lane 6) was supplemented. The gel images are representative of two independent experiments. The labels En and Ca in the gels indicate the positions of the CHIKV envelop and nucleocapsid proteins, respectively, while the closed arrows point to the 68-kDa fragment generated upon cleavage of the α′ subunit of C3b.
Yet another function of factor I is that it can cleave C4b, another important complement activation product, into C4c and C4d in the presence of the cofactor C4BP (Fig. 5B, lane 2). We wanted to determine if the CHIKV-associated factor I-like activity can support C4b inactivation similarly to C3b. Incubation of C4b with C4BP and CHIKV did not result in the cleavage of the α′ subunit of C4b (Fig. 5B, lane 6); similarly, addition of factor I and not the cofactor also did not have any effect on C4b (Fig. 5B, lane 5). Thus, the factor I-like activity supports only the inactivation of C3b into iC3b and not that of C4b into C4c and C4d.
Inactivation of C3b by CHIKV is dependent on the concentration of the virus, factor H, and time.
Having observed that the factor I-like activity associated with CHIKV can inactivate C3b, in-depth analysis of C3b inactivation with respect to factor H and CHIKV concentration and time was carried out. In order to test the effects of CHIKV concentration on C3b-α′ cleavage, while C3b (3 μg) and factor H (1 μg) were kept constant, the concentration of CHIKV was varied (2.5, 5, 10, 12.5, 15, 17.5, and 20 μg). As observed earlier, incubation of CHIKV with C3b and only the cofactor, factor H, without the protease factor I resulted in cleavage of the α′ subunit into the 68- and 43-kDa fragments. This inactivation was highly dependent on the concentration of virus. As the concentration of CHIKV was increased, the level of 68-kDa fragment generated also increased (Fig. 6A, lanes 4 to 10). C3b cleavage occurred even at the lowest concentration of CHIKV (2.5 μg), and a gradual increase in the cleaved product was seen as the concentration was increased. The function of factor I is indispensable for cofactor activity as may be observed in the control (Fig. 6A, lane 2), wherein C3b cleavage occurred only in the presence of both the cofactor (factor H) and the enzyme (factor I). The 68-kDa fragment generated was taken as a measure of C3b inactivation, and densitometry analysis of this fragment clearly showed a dependency on CHIKV concentration (Fig. 6B).
FIG 6.

Chikungunya virus harbors a factor I-like activity that supports C3b inactivation into iC3b. C3b cofactor activity was set up with purified CHIKV, C3b, factor H, and factor I. After the indicated incubation at 37°C, the entire assay volume was loaded onto a 12% gel, subjected to SDS-PAGE, and stained with GelCode blue. C3b α′ cleavage was assessed by varying either the concentration of purified CHIKV (A; lanes 4 to 10), the duration of incubation (C; lanes 4 to 10), or the concentration of factor H (E, lanes 5 to 10). Controls included C3b with factor I (lane 1; A, C, E) and virus alone (lane 3; A, C, E). The positive control (lane 2; A, C, E) included purified C3b, factor H, and factor I incubated for 6 h at 37°C. Note the disappearance of the α′ subunit and generation of 68-and 43-kDa fragments. The labels En and Ca in the gels indicate the positions of the CHIKV envelop and nucleocapsid proteins, respectively, while the closed arrows point to the 68-kDa fragment generated upon cleavage of the α′ subunit of C3b. (B, D, F) Densitometry analysis of the 68-kDa fragment of C3b generated upon its inactivation by either varying CHIKV concentration (A), time (C), or factor H (E). The analysis is based on three independent experiments on images acquired using the VersaDoc MP 5000 and Image Lab software.
Similarly, time course experiments were performed by incubating a fixed concentration of CHIKV (10 μg), factor H (2.5 μg), and C3b (3 μg) for the indicated time points (0.5, 1, 2, 3, 4, 5, and 6 h). The 68-kDa fragment generated was minimal at the shortest time of incubation (0.5 and 1 h). However, an increase in levels was observed as time progressed (Fig. 6C). Densitometry analysis of three independent experiments further confirmed that increased incubation periods resulted in greater levels of C3b inactivation by CHIKV (Fig. 6D).
Our earlier data showed that CHIKV did not possess a cofactor-like activity (Fig. 5A, lane 5); however, it required a host cofactor, factor H, to inactivate C3b. In order to check if the concentration of the cofactor, factor H, also contributed to the degree of inactivation, the C3b cofactor activity assay was carried out with increasing concentrations of factor H (0.5, 1, 2.5, 5, 7.5, and 10 μg). Minimal 68-kDa fragment generation was observed when 0.5 μg of factor H was used; however, a gradual increase in the generation of cleavage product was observed with an increase in factor H concentration (Fig. 6E). The increase in the degree of C3b-α′ cleavage was proportional to the factor H concentration, as can be observed in the plot of densitometry analysis of three independent experiments (Fig. 6F). Greater than 50% decay in C3b-α′ was evident at a factor H concentration of 5 μg. Thus, the concentration of factor H is critical for the inactivation of C3b by the factor I-like protease in CHIKV.
The factor I-like activity associated with CHIKV is not due to the host factor I.
The cofactor activity assays clearly show that CHIKV harbors a factor I-like activity associated with the virus. Recruitment of host membrane-bound complement regulators such as CD46 and CD55 and soluble regulators such as factor H have been reported in viruses. We wanted to address if the C3b inactivation by CHIKV is due to the recruitment of host factor I or if it is of viral origin. Gradient-purified CHIKV was subjected to Western blotting and probed with an antibody against human factor I. Reactivity to the antibody was observed only in the factor I control lane. No signal was observed in the CHIKV-only lanes even at the highest concentration of 10 μg (Fig. 7A). To further confirm that CHIKV does not harbor host factor I, ELISA was performed by coating 2-fold dilutions of gradient-purified CHIKV starting from 10 to 0.078 μg, while bovine serum albumin (BSA) served as the control. The bound antibodies were detected by probing with anti-factor I antibody, and the absorbance was measured at 405 nm. Factor I served as the positive control, and the initial concentration taken for the assay was 1 μg. No reactivity to factor I antibody was observed in the wells coated with CHIKV or BSA, with absorbance matching only background levels, while at the highest concentration of factor I (1 μg), the absorbance was found to be 1.5, followed by a concentration-dependent reduction in absorbance (Fig. 7B).
FIG 7.

The factor I-like activity associated with CHIKV is not due to human factor I. (A) Western blotting to check if host factor I is bound to CHIKV. No reactivity to anti-human factor I antibody was observed in the lanes containing 0.1, 1, or 10 μg of CHIKV, while robust signal was seen in the factor I (FI) control lane (top panel). A positive reactivity to anti-E1 antibody correlating to the levels of CHIKV is evident from the bottom panel. The image is a representation of three independent experiments. (B) ELISA to detect bound host factor I. Various concentrations of gradient-purified CHIKV, factor I, or BSA as indicated in the x axis of the graph were coated and probed with anti-human factor I to detect bound factor I if any. The dose-dependent response can be seen in the case of the positive control factor I with background levels of binding in the case of CHIKV-and BSA-immobilized wells. The data are the mean ± SEM of 3 independent experiments. The # symbol indicates that the initial concentration of human factor I begins with 1 μg. (C) Factor I inhibition assay. C3b cofactor activity was reconstituted with purified CHIKV, C3b, factor H, or factor I in either the presence or absence of a human factor I function-blocking antibody. The effects were assessed using SDS-PAGE followed by GelCode blue staining. The function-blocking antibody was found to completely inhibit the C3b-α′ cleavage by factor I (lane 5) compared to the no-antibody control (lane 4). The antibody did not have any effect on the factor I-like activity associated with CHIKV (lane 7). The labels En and Ca indicate the position of the envelope and nucleocapsid protein, respectively, while HC and LC indicate the heavy and the light chain of the blocking antibody, respectively. (D) The experiments were carried out in triplicate, and the percent reduction in the proteolytic activity of factor I or CHIKV in the presence or absence of factor I functional blocking antibody was assessed by performing a densitometry analysis of the 68-kDa fragment generated using Image Lab software. The data are the mean ± SEM with. ***, P ≤ 0.01.
Although the biochemical assays showed that the host factor I is not associated with CHIKV, we wanted to further confirm this by carrying out a functional assay. A well-established factor I function-blocking antibody was used in the cofactor activity assays. This antibody specifically blocks the protease activity of factor I responsible for cleavage of C3b-α′. As expected, while factor I effectively mediated C3b cleavage into iC3b (Fig. 7C, lane 4), incubation with anti-factor I-blocking antibody inhibited this function, resulting in an intact C3b-α′ (Fig. 7C, lane 5). Interestingly, the antibody had only a minimal effect on factor I-like activity associated with CHIKV, with levels of the 68-kDa fragment generated being similar in both instances, either with or without antibody treatment (Fig. 7C, lanes 6 to 7). Densitometry analysis of the 68-kDa fragment generated from three independent experiments showed more than 95% inhibition of human factor I activity with negligible levels of inhibition of the CHIKV-associated factor I-like activity (Fig. 7D). The relative difference between the antibody-treated samples showed a high degree of significance. These results clearly suggest that the factor I-like activity associated with CHIKV could be of viral origin but not of host-derived factor I.
With all of the preceding data taken together, we have confirmed earlier findings that CHIKV is resistant to complement, with the addition that this resistance occurs despite complement activation and deposition of the critical complement components C3b and C4b. This resistance can be attributed at least in part to the inherent virion-associated factor I-like activity capable of inactivating C3b into iC3b.
DISCUSSION
Facing the innate immune barrier, including the complement system, is an unavoidable circumstance faced by most viruses. The systemic presence of complement apart from blood signifies its role in limiting viruses, irrespective of the route of infection (48, 49). The severity of clinical symptoms during CHIKV infection is directly related to viremia, which raises the important question of the nature of the interaction of CHIKV with the human complement system. A role for complement in the severity of disease symptoms involving alphaviruses has been elegantly demonstrated in mouse models using the Ross River virus (41–43, 50); however, the direct effect of human complement on CHIKV remained unexplored.
Enveloped viruses, including VSV, dengue virus, MuV, PIV5, and WNV, activate complement (7–11, 17–19). We observed that CHIKV also activated complement. Our observation that CHIKV activates complement at a higher concentration (2.5 μg) and a prolonged incubation period (45 min) suggests that CHIKV is a poor activator of complement. This strikingly contrasts with complement activation by other viruses, including NiV, wherein just 0.01 μg of the virus was sufficient to activate complement to saturating levels within 2 min (36).
The progress of the complement cascade upon activation by viruses often results in virus neutralization and may involve one or multiple complement pathways. In the fluid phase, deposition of complement proteins on the virus can lead to virus aggregation (e.g., in PIV5) or virolysis (e.g., in MuV and VSV), depending upon the extent of the pathway involved (9, 51). Exceptions include NiV, where both wild-type (WT) NiV and pseudotypes bearing NiV glycoproteins, despite being potent activators of complement, were resistant to neutralization by complement (36, 52). Limited complement activation indicated that CHIKV possessed mechanisms to overcome complement. Treatment of CHIKV with both NHS and HI-NHS showed marked resistance to neutralization. Even at the highest concentrations of serum treatment (1:5, 1:10), only a modest reduction in plaques was observed; however, a role for complement in this reduction can be ruled out, as the percent reduction was comparable in NHS and HI-NHS. Our findings are very much in line with earlier observations that CHIKV and Sindbis virus are less sensitive to mouse complement (40). The significance of this resistance by CHIKV is further highlighted by the potent reduction in viral titer observed in the case of CHPV treated with NHS, where >90% neutralization occurred within 1 h. Complete resistance to neutralization by complement is not a common feature and thus holds great significance for the viruses, including CHIKV, in establishing productive infection and overall pathogenesis.
Remarkable differences have been reported in the mechanism of complement activation and resultant virus neutralization among paramyxoviruses, which include CP-mediated neutralization of measles, AP-dependent neutralization of PIV5 and MuV, and antibody dependency in the case of measles virus and HPIV2, while NiV was completely resistant to complement (9, 36, 53–55). Antibodies, which are either natural, specific, or cross-reactive to a specific virus, have significant roles in dictating the mechanism of virus neutralization. While serum components, including antibodies and complement in normal human serum, had no effect on CHIKV, a reversal of this effect was observed when the serum of seropositive donors to CHIKV was used. The donor serum used in this study is from a South Indian cohort in an area which saw widespread infection during the 2008 outbreak (56), yet the prevalence of anti-CHIKV neutralizing or cross-reacting antibodies was negligible among the normal donors. Chikungunya virus treated with serum from seropositive donors was readily neutralized, although to various degrees, compared to that of the MEM- or D8-NHS-treated samples (Fig. 2C). NHS, HI-NHS, and antibodies purified from seropositive donors were all capable of neutralizing CHIKV, which suggested that anti-CHIKV antibodies in these samples were the neutralizing factor (data not shown). Chikungunya virus-specific ELISA further confirmed the presence of anti-CHIKV antibodies only in the seropositive and not in the seronegative donor serum. Antibody-dependent enhancement in complement-mediated virus neutralization was reported earlier in viruses such as measles, VSV, etc. (55, 57); however, not much improvement in complement involvement was observed even after employing subneutralizing concentrations of anti-CHIKV antibodies (data not shown). The sensitivity of CHIKV to only virus-specific antibodies from seropositive donors and not to NHS further confirmed the earlier findings on resistance to complement.
As mentioned earlier, in most viruses, complement activation results in deposition of active C3b and C4b on viral surfaces. This covalent interaction paves the way for further amplification, leading to multiple effector functions, including opsonization, aggregation, and assembly of MAC followed by lysis (24). For example, virolysis post-complement deposition has been reported in the case of MuV and VSV, but viral aggregation was observed with PIV5 (9, 58). A comparison between two paramyxoviruses, PIV5 and NiV, showed that while significant levels of C3b were found to be deposited on PIV5 exposed to NHS, negligible levels were seen on NiV (36). Western blot analysis of sucrose gradient fractions of CHIKV treated with NHS subjected to ultracentrifugation showed comigration of C3 and C4 components with the virus, which was further validated by electron microscopy. Although deposition of complement components C3 and C4 occurred, the levels of complement deposition were significantly less. Electron microscopic analysis of C3 deposition on CHIKV in over 15 independent fields in the EM grids showed just 21% positive reactivity to anti-C3 antibodies. Similar experiments reported earlier with PIV5 showed marked enrichment of C3 moieties on virus exposed to NHS (36). The rate of deposition of C3 components on CHIKV had striking similarities to that of Nipah virus, where a marked reduction in complement deposition was observed.
In-depth analysis of the species of C3 comigrating with the virion showed that the associated C3 component is iC3b and not C3b. The accumulation of active complement components in sites affected by CHIKV infection is clinically significant, as it contributes to the severity of the disease symptoms. In the RRV model, C3 component deposition was observed in disease sites, including the ankles, quadriceps, and skeletal muscle in WT C57BL/6 mice and RAG1−/− mice and not in C3−/− or MBL−/− mice (41, 42). This is pathologically significant because in vivo studies with RRV demonstrated that iC3b was found in all the affected tissue samples tested. Even though iC3b is an inactivation product generated by the cofactor activity of RCAs such as factor H or CD46 with factor I, the binding of this ligand to complement receptor 3 (CR3) leads to downstream signaling, leading to proinflammatory responses. This is supported by yet another RRV study, where it was found that the generation of iC3b and the resultant engagement with CR3 exacerbated disease symptoms in wild-type mice compared to CR3−/− mice (43).
Since CHIKV was resistant to complement-mediated neutralization, and iC3b, not C3b, was associated with the virion, we hypothesized that CHIKV has an inherent mechanism to overcome complement. Enveloped viruses such as PIV5, MuV, and VSV have been shown to incorporate CD46, a membrane-associated RCA in the virus envelope during budding. The associated CD46 was found to be functionally active and capable of mediating the inactivation of C3b into iC3b, thus providing a selective survival advantage over complement compared to viruses lacking CD46 (51, 59). Poxviruses, including vaccinia and variola, also have been shown to encode proteins capable of mimicking RCA cofactor activity by inactivating both C3b and C4b into iC3b and C4c (28, 30, 60). Unlike the examples mentioned above, interestingly, incubation of CHIKV with C3b and factor I did not result in inactivation of C3b into iC3b (Fig. 5A, lane 5). Although it was not investigated if, in fact, CD46 was associated with the virion, the functional assay proved beyond a doubt that the presence or absence of CD46 was immaterial, as it did not support C3b inactivation. However, addition of factor H and C3b to gradient-purified CHIKV without factor I was sufficient to convert C3b into iC3b, suggesting that CHIKV possessed a factor I-like activity. Recruitment of factor I or possessing its activity is not a common feature among pathogens; earlier, a factor I-like activity was reported in NiV, supporting the inactivation of C3b into iC3b, similar to in CHIKV. The factor I activity associated with NiV was more potent than that with CHIKV since the cofactor activities of both factor H and CD35 were supported by NiV. CD35-mediated inactivation of C3b by factor I involves two steps. The first step is conversion of C3b into iC3b, followed by conversion of iC3b into C3c and C3d, with NiV supporting only the first cleavage (36). The inactivation of C3b into iC3b mediated by CHIKV depended on the concentration of virus and factor H and the time of incubation. C3b cleavage into iC3b is a unique function of factor I, and the activity associated with CHIKV could be due to a host factor I recruited by the virus or may be of viral origin. Factor I function-blocking experiments with a specific inhibiting antibody showed that the antibody could block the function of factor I and not that of the CHIKV derived factor I-like activity. This suggests that the activity is of viral origin. Identification of the precise CHIKV signature needs further investigation. In summary, the factor I-like activity associated with CHIKV may be considered one of the weapons in the virus arsenal to target complement.
As discussed earlier, the role of complement in the overall pathogenesis, including the arthritogenic symptoms, both mild and severe, has been well established. Being a virus adapted to the hematogenous route of spread, our confirmation that CHIKV is resistant to human serum addresses the fundamental question of the survival and dissemination of CHIKV in serum. In addition, the identification of a factor I-like activity associated with CHIKV highlights a plausible mechanism of complement evasion and, thereby, iC3b generation, a key factor implicated in Alphavirus-specific disease manifestations. These findings, besides providing invaluable insights into the nature of interaction of CHIKV with human complement, offer a rationale to develop new strategies for improving therapeutic interventions.
MATERIALS AND METHODS
Ethics statement.
All human ethical guidelines were followed with regard to the use of human samples for this study. The study protocol “IHEC/1/2018/14” was reviewed and approved by the institutional human ethics committee of the Rajiv Gandhi Centre for Biotechnology (RGCB) as per the guidelines of the government of India Ministry of Health and Family Welfare (the Drugs and Cosmetics Act, 1940, and the Drugs and Cosmetics Rules, 1945). Only adult human samples (serum) were used in this study, and all the participants gave written informed consent. Animal experiments were performed after approval was obtained for the study protocol “IAEC/559/JOHN/2016” from the institutional animal ethics committee of RGCB as per the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Department of Animal Husbandry and Dairying, Ministry of Agriculture and Farmers Welfare, government of India. Approval was obtained from the institutional biosafety committee of RGCB for use of both chikungunya virus and Chandipura virus.
Cells and viruses.
Vero cells were grown and maintained in minimal essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM/liter glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified CO2 incubator (5%). Both CHIKV (reference identifiers [National Institute of Virology, Pune, India] NIV_No634029, M/K-No M-16229) and CHPV (NIV_No-653514, M/K-No M-1461) were kindly provided by the director of the National Institute of Virology, Pune, India. The viruses used in the study were validated by partial sequencing of the E2 gene of CHIKV and the N, M, and G genes of CHPV in a 3730XL DNA analyzer (Applied Biosystems, CA). Viruses were grown and titrated using a plaque assay in Vero cells. Briefly, plaque assays were carried out by infecting a monolayer of Vero cells with CHIKV suspended in MEM supplemented with 10% BSA fraction V and incubated for 2 h at 37°C. Postinoculation, the monolayer was washed with phosphate-buffered saline (PBS) and overlaid with 2% of 1,500 cP carboxymethyl cellulose (Sigma, MO) in Dulbecco modified Eagle medium (DMEM) (Millipore) supplemented with 2% FBS, penicillin, streptomycin, and 5 mM HEPES, incubated for 72 h, and fixed with 3.7% formaldehyde. Plaques were enumerated after staining with 0.2% crystal violet in 20% ethanol.
Chikungunya virus was purified as described earlier (61) with slight modifications. Briefly, Vero cells were infected with CHIKV at a multiplicity of infection (MOI) of 1. After 48 h, the culture supernatant was collected and clarified by centrifugation at 3,000 × g for 10 min, and the virus in the supernatant was precipitated with 40% polyethylene glycol 8000 (Sigma, MO) to a final concentration of 8% by gently stirring for 12 h at 4°C. The precipitate was pelleted by centrifugation at 8,000 × g for 30 min and resuspended in ice-cold NTE buffer (150 mM NaCl, 50 mM Tris-HCl, 5 mM EDTA, pH 8.0). The suspension was then layered on top of a 30 to 60% sucrose gradient followed by ultracentrifugation at 90,000 × g using a Beckman Coulter Optima XL-100k centrifuge (Beckman, CA) for 12 h at 4°C in an SW28 rotor. The opaque virus band at the interphase of 30% and 60% sucrose gradient was collected, diluted with NTE buffer, and pelleted by further ultracentrifugation at 9,000 × g for 6 h at 4°C using an SW41Ti rotor over a 20% sucrose cushion. The pelleted virus was resuspended in MEM, aliquoted, and stored at –80°C.
Sera, complement reagents, proteins, and antibodies.
Blood was collected from human donors with either a history of chikungunya virus infection or no such history after consent was obtained per IHEC/1/2018/14. Serum was separated from the blood as described earlier (62). Briefly, the collected blood was allowed to clot at room temperature (RT) for 30 min and was clot retracted and centrifuged at 3,000 × g for 10 min at RT. The serum obtained was subjected to a second high-speed centrifugation at 20,000 × g for 5 min before being aliquoted and frozen at –80°C. Purified human complement proteins C3b, factor H, factor I, and antibodies against human C3, C4, factor H, and C3a were procured from Complement Technologies (Tyler, TX). Rabbit anti-human IgG-HRP was from Santa Cruz Biotechnology (catalog number sc-2769). Anti-CHIKV polyclonal antibodies were raised in-house in a New Zealand white rabbit using one prime and two boost schemes after immunizing it with UV-inactivated CHIKV mixed with Freund’s complete and incomplete adjuvant (Sigma-Aldrich, CA). The secondary antibody used in this study included anti-human IgG-HRP (Santa Cruz, CA), anti-rabbit and -mouse-HRP (Bio-Rad, CA), and 6- and 12-nm gold-labeled anti-mouse, -rabbit, or -goat antibodies (Jackson ImmunoResearch, PA).
Activation and neutralization assays.
Concentration- and time-dependent activation of complement by CHIKV was determined by analyzing the levels of C3a generated by Western blotting. Various concentrations of purified CHIKV (0.07, 0.15, 0.31, 0.6, 1.2, and 2.5 μg) were incubated with a set dilution (1:20) of NHS for 45 min at 37°C to study the effect of CHIKV concentration. For the time course activation assay, a set concentration of CHIKV (1.2 μg) was incubated for various time periods (1, 5, 10, 20, 30, and 45 min) with NHS (1:20) at 37°C. SDS-PAGE followed by Western blotting was performed on the samples to detect the levels of C3a generated by probing the blots with anti-C3a antibody (1:5,000) and anti-rabbit HRP (1:10,000). The blots were developed using the SuperSignal West Pico chemiluminescence substrate (Pierce, IL). Neutralization assays were carried out by incubating 100 PFU of CHIKV/CHPV with a 2-fold serial dilution of NHS or HI-NHS (1:5 to 1:80) from a single or multiple donors for 1 h at 37°C. Virus incubated with MEM alone served as the control. Following incubation, the remaining infectivity in the samples was determined by plaque assays as described earlier. An antibody-dependent neutralization assay was performed using 1:2,500 diluted HI serum samples from 6 seropositive donors (D2, D3, D7, D92, D95, and D145) and one seronegative donor (D8), and the remaining virus was determined using plaque assays.
ELISA and Western blotting.
The presence of anti-CHIKV antibodies in donor sera was determined using ELISA, carried out in Nunc MaxiSorp flat bottom 96-well plates (Thermo Fisher Scientific) coated with sucrose gradient-purified CHIKV (125 ng) followed by overnight incubation at 4°C. The wells were blocked with 2% skim milk in PBS for 2 h at 37°C, washed with PBS containing 0.05% Tween, and incubated with 2-fold serially diluted donor sera (1:100 to 1:224,800) for 1 h at 37°C. Hyperimmune serum obtained from a rabbit was used as a positive control, while serum from a seronegative donor served as the negative control. HRP-conjugated anti-human or anti-rabbit secondary antibodies were used at a dilution of 1:5,000 for 1 h at 37°C.
In order to determine if human factor I is associated with the virion, an ELISA was carried out by coating 2-fold dilutions of gradient-purified CHIKV or BSA beginning with 10 μg to 0.007 μg. Two-fold dilutions of factor I were used as positive controls, but the starting dilution used for coating was 1 μg. The coated samples were probed with anti-human factor I antibody at a dilution of 1:2,500 followed by anti-goat HRP conjugated secondary antibody.
For both the ELISA protocols described above, absorbance was measured at 405 nm 20 min after the addition of ABTS [2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt] substrate (Roche, IN) in 0.1 M Na-citrate.
Ultracentrifugation and Western blotting.
Sucrose gradient-purified CHIKV (50 μg) was mixed with an equal volume (1:1) of NHS or MEM alone and incubated for 1 h at 37°C. Following incubation, the reaction mixture was layered on top of 15 to 60% preequilibrated sucrose gradient and subjected to ultracentrifugation at 100,000 × g for 5 h at 4°C using an SW41Ti rotor. Fractions of approximately 500 μl were collected from the bottom of the tube and analyzed using SDS-PAGE and Western blotting using anti-CHIKV antibodies (1:2,500). The blots were further stripped, blocked, and probed with goat polyclonal anti-human C3/C4 (1:5,000). Virus alone or purified complement proteins were used as markers for comparison of their respective signals from the sucrose gradient fractions being analyzed. Antibody binding was detected using the Pierce West Pico chemiluminescent substrate.
The presence or absence of factor I in gradient-purified CHIKV was determined using Western blotting by taking a range of virus concentration (0.1 to 10 μg) with purified factor I serving as the control. The blots were probed with anti-human factor I antibody (1:5,000 dilution) and mouse anti-CHIKV E1 antibody (EastCoast Bio, ME) at a dilution of 1:2,500 followed by the corresponding secondary labeled antibodies. Detection was carried out with a chemiluminescent substrate as described above.
Factor I cofactor activity assay.
In vitro C3b/C4b cofactor activity assays were set up as described earlier (36) with slight modifications. Briefly, the purified complement protein C3b (3 μg) or C4b (3 μg) was incubated with gradient-purified CHIKV (10 μg) either alone or in the presence or absence of the serine protease factor I (100 ng) and the respective complement regulator factor H (2.5 μg) or C4BP (2.5 μg) as indicated. Positive controls included a combination of C3b, factor H, and factor I or C4b, C4BP, and factor I for the C3b or C4b cofactor activity assays, respectively. Concentration-dependent C3b cofactor activity assays were set up by incubating C3b with various concentrations of either CHIKV (with a constant 2.5 μg of factor H) or factor H (with a constant 10 μg of CHIKV) as indicated. Time course experiments, on the other hand, were carried out by keeping the concentration of C3b (3 μg), factor H (2.5 μg), and CHIKV (10 μg) constant but by varying the time as indicated. In the case of the cofactor activity inhibition assays, factor I (100 ng) or CHIKV (10 μg) was incubated first with 5 μg of the function-blocking anti-human factor I antibody (A247; Quidel, CA) followed by the addition of other components of the reaction. The assay and the analysis were continued as described above.
In all cases, the final volume of the reaction was made up to 20 μl with PBS containing Ca2+ and Mg2+, and all incubations were carried out at 37°C for 6 h unless otherwise indicated. After SDS-PAGE followed by GelCode blue staining (Thermo Fisher Scientific, IL), the images of the gels were acquired using a VersaDoc MP 5000 imaging system. The extent of C3b inactivation was estimated by carrying out a densitometry analysis of the 68-kDa fragment generated using the Image Lab Software (Bio-Rad, CA).
Electron microscopy.
Complement component (C3 and C4) deposition on CHIKV was detected by incubating purified virus with equal volumes (1:1) of NHS (1:10) or PBS for 1 h at 37°C. Postincubation, 10 μl of the sample was loaded onto gold Formvar carbon support film on square grids (FCF200-Au; Electron Microscopy Sciences, PA) and allowed to adsorb at RT for 30 min. After adsorption, the grids were fixed with 2.5% glutaraldehyde and blocked with 1% BSA for 30 min at RT. Deposition was detected using a 2-step dual-staining protocol where the grids were initially probed with goat polyclonal anti-human C3/C4 antibody (1:10) for 1 h before being probed with rabbit polyclonal anti-CHIKV antisera (1:10) for another 1 h. For the immunodetection of CHIKV antigens and associated complement proteins, grids were washed with PBS and incubated with a mixture of 12- and 6-nm gold particle-conjugated anti-goat and anti-rabbit secondary antibodies, respectively (Jackson ImmunoResearch Laboratories, PA) at a dilution of 1:20. The grids were negatively stained with 2% phosphotungstic acid (pH 6.6), and images were acquired using a 1011 transmission electron microscope (TEM) (Jeol, MA) using the Digital Micrograph software (Gatan, CA).
Statistical analysis.
GraphPad Prism software was used to analyze and plot the data. The analysis of statistical significance was performed using Student’s t test wherever required.
ACKNOWLEDGMENTS
We thank M. Radhakrishna Pillai, Director of RGCB, for his constant support. D. T. Mourya, Director of NIV, and A. Basu are duly acknowledged for providing the viruses used in this study. We thank Sabu Thomas and Arumugam Rajavelu and Reji Varghese and Nandagopal (IISER, Trivandrum) and Arvind Sahu (NCCS-Pune) for their critical comments and thoughtful suggestions during the discussions. We also acknowledge the RGCB Transmission Electron Microscopy and Image Processing Core and Animal Research Facility and the respective personnel, Gopi Krishnan and Vishnu Sunil Jaikumar, for their excellent services rendered.
This work was supported by a Department of Biotechnology-Ramalingaswami fellowship, BT/RLF/Re-entry/29/2012, a Ministry of Science and Technology grant, and the Department of Science and Technology-Science and Engineering Research Board (DST-SERB) SERB-ECR/2015/000261 (to J.B.J.) and CSIR, India, for fellowship 09/716(0164)/2015-EMR-I (to J.N.).
We declare no conflict of interest.
REFERENCES
- 1.Walport MJ. 2001. Complement. First of two parts. N Engl J Med 344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 2.Walport MJ. 2001. Complement. Second of two parts. N Engl J Med 344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 3.Bajic G, Degn SE, Thiel S, Andersen GR. 2015. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J 34:2735–2757. doi: 10.15252/embj.201591881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernet J, Mullick J, Singh AK, Sahu A. 2003. Viral mimicry of the complement system. J Biosci 28:249–264. doi: 10.1007/bf02970145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elvington A, Atkinson C, Zhu H, Yu J, Takahashi K, Stahl GL, Kindy MS, Tomlinson S. 2012. The alternative complement pathway propagates inflammation and injury in murine ischemic stroke. J Immunol 189:4640–4647. doi: 10.4049/jimmunol.1201904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charlesworth JA, Pussell BA, Roy LP, Robertson MR, Beveridge J. 1976. Measles infection. Involvement of the complement system. Clin Exp Immunol 24:401–406. [PMC free article] [PubMed] [Google Scholar]
- 7.Sissons JG, Cooper NR, Oldstone MB. 1979. Alternative complement pathway-mediated lysis of measles virus infected cells: induction by IgG antibody bound to individual viral glycoproteins and comparative efficacy of F(ab′)2 and Fab′ fragments. J Immunol 123:2144–2149. [PubMed] [Google Scholar]
- 8.Hirsch RL, Wolinsky JS, Winkelstein JA. 1986. Activation of the alternative complement pathway by mumps infected cells: relationship to viral neuraminidase activity. Arch Virol 87:181–190. doi: 10.1007/bf01315298. [DOI] [PubMed] [Google Scholar]
- 9.Johnson JB, Capraro GA, Parks GD. 2008. Differential mechanisms of complement-mediated neutralization of the closely related paramyxoviruses simian virus 5 and mumps virus. Virology 376:112–123. doi: 10.1016/j.virol.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bokisch VA, Top FH Jr, Russell PK, Dixon FJ, Muller-Eberhard HJ. 1973. The potential pathogenic role of complement in dengue hemorrhagic shock syndrome. N Engl J Med 289:996–1000. doi: 10.1056/NEJM197311082891902. [DOI] [PubMed] [Google Scholar]
- 11.Avirutnan P, Hauhart RE, Marovich MA, Garred P, Atkinson JP, Diamond MS. 2011. Complement-mediated neutralization of dengue virus requires mannose-binding lectin. mBio 2:e00276-11. doi: 10.1128/mBio.00276-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anders EM, Hartley CA, Reading PC, Ezekowitz RA. 1994. Complement-dependent neutralization of influenza virus by a serum mannose-binding lectin. J Gen Virol 75:615–622. doi: 10.1099/0022-1317-75-3-615. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien KB, Morrison TE, Dundore DY, Heise MT, Schultz-Cherry S. 2011. A protective role for complement C3 protein during pandemic 2009 H1N1 and H5N1 influenza A virus infection. PLoS One 6:e17377. doi: 10.1371/journal.pone.0017377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rattan A, Pawar SD, Nawadkar R, Kulkarni N, Lal G, Mullick J, Sahu A. 2017. Synergy between the classical and alternative pathways of complement is essential for conferring effective protection against the pandemic influenza A(H1N1) 2009 virus infection. PLoS Pathog 13:e1006248. doi: 10.1371/journal.ppat.1006248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoiber H, Clivio A, Dierich MP. 1997. Role of complement in HIV infection. Annu Rev Immunol 15:649–674. doi: 10.1146/annurev.immunol.15.1.649. [DOI] [PubMed] [Google Scholar]
- 16.Anderson DR, Carthy CM, Wilson JE, Yang D, Devine DV, McManus BM. 1997. Complement component 3 interactions with coxsackievirus B3 capsid proteins: innate immunity and the rapid formation of splenic antiviral germinal centers. J Virol 71:8841–8845. doi: 10.1128/JVI.71.11.8841-8845.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehlhop E, Diamond MS. 2006. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J Exp Med 203:1371–1381. doi: 10.1084/jem.20052388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehlhop E, Whitby K, Oliphant T, Marri A, Engle M, Diamond MS. 2005. Complement activation is required for induction of a protective antibody response against West Nile virus infection. J Virol 79:7466–7477. doi: 10.1128/JVI.79.12.7466-7477.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mills BJ, Cooper NR. 1978. Antibody-independent neutralization of vesicular stomatitis virus by human complement. I. Complement requirements. J Immunol 121:1549–1557. [PubMed] [Google Scholar]
- 20.Aasa-Chapman MM, Holuigue S, Aubin K, Wong M, Jones NA, Cornforth D, Pellegrino P, Newton P, Williams I, Borrow P, McKnight A. 2005. Detection of antibody-dependent complement-mediated inactivation of both autologous and heterologous virus in primary human immunodeficiency virus type 1 infection. J Virol 79:2823–2830. doi: 10.1128/JVI.79.5.2823-2830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beebe DP, Cooper NR. 1981. Neutralization of vesicular stomatitis virus (VSV) by human complement requires a natural IgM antibody present in human serum. J Immunol 126:1562–1568. [PubMed] [Google Scholar]
- 22.Mehlhop E, Fuchs A, Engle M, Diamond MS. 2009. Complement modulates pathogenesis and antibody-dependent neutralization of West Nile virus infection through a C5-independent mechanism. Virology 393:11–15. doi: 10.1016/j.virol.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoermer KA, Morrison TE. 2011. Complement and viral pathogenesis. Virology 411:362–373. doi: 10.1016/j.virol.2010.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. 2015. Complement system part II: role in immunity. Front Immunol 6:257. doi: 10.3389/fimmu.2015.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. 2015. Complement system part I: molecular mechanisms of activation and regulation. Front Immunol 6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. 2017. Complement evasion strategies of viruses: an overview. Front Microbiol 8:1117. doi: 10.3389/fmicb.2017.01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Favoreel HW, Van de Walle GR, Nauwynck HJ, Pensaert MB. 2003. Virus complement evasion strategies. J Gen Virol 84:1–15. doi: 10.1099/vir.0.18709-0. [DOI] [PubMed] [Google Scholar]
- 28.Kotwal GJ, Moss B. 1988. Vaccinia virus encodes a secretory polypeptide structurally related to complement control proteins. Nature 335:176–178. doi: 10.1038/335176a0. [DOI] [PubMed] [Google Scholar]
- 29.Bernet J, Mullick J, Panse Y, Parab PB, Sahu A. 2004. Kinetic analysis of the interactions between vaccinia virus complement control protein and human complement proteins C3b and C4b. J Virol 78:9446–9457. doi: 10.1128/JVI.78.17.9446-9457.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosengard AM, Liu Y, Nie Z, Jimenez R. 2002. Variola virus immune evasion design: expression of a highly efficient inhibitor of human complement. Proc Natl Acad Sci U S A 99:8808–8813. doi: 10.1073/pnas.112220499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spiller OB, Blackbourn DJ, Mark L, Proctor DG, Blom AM. 2003. Functional activity of the complement regulator encoded by Kaposi’s sarcoma-associated herpesvirus. J Biol Chem 278:9283–9289. doi: 10.1074/jbc.m211579200. [DOI] [PubMed] [Google Scholar]
- 32.Mullick J, Bernet J, Singh AK, Lambris JD, Sahu A. 2003. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) open reading frame 4 protein (kaposica) is a functional homolog of complement control proteins. J Virol 77:3878–3881. doi: 10.1128/jvi.77.6.3878-3881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huemer HP, Wang Y, Garred P, Koistinen V, Oppermann S. 1993. Herpes simplex virus glycoprotein C: molecular mimicry of complement regulatory proteins by a viral protein. Immunology 79:639–647. [PMC free article] [PubMed] [Google Scholar]
- 34.Hirsch RL, Griffin DE, Winkelstein JA. 1981. Host modification of Sindbis virus sialic acid content influences alternative complement pathway activation and virus clearance. J Immunol 127:1740–1743. [PubMed] [Google Scholar]
- 35.Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, Atkinson JP, Diamond MS. 2006. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc Natl Acad Sci U S A 103:19111–19116. doi: 10.1073/pnas.0605668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson JB, Borisevich V, Rockx B, Parks GD. 2015. A novel factor I activity in Nipah virus inhibits human complement pathways through cleavage of C3b. J Virol 89:989–998. doi: 10.1128/JVI.02427-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrison TE. 2014. Reemergence of chikungunya virus. J Virol 88:11644–11647. doi: 10.1128/JVI.01432-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wahid B, Ali A, Rafique S, Idrees M. 2017. Global expansion of chikungunya virus: mapping the 64-year history. Int J Infect Dis 58:69–76. doi: 10.1016/j.ijid.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz O, Albert ML. 2010. Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol 8:491–500. doi: 10.1038/nrmicro2368. [DOI] [PubMed] [Google Scholar]
- 40.Fuchs A, Lin TY, Beasley DW, Stover CM, Schwaeble WJ, Pierson TC, Diamond MS. 2010. Direct complement restriction of flavivirus infection requires glycan recognition by mannose-binding lectin. Cell Host Microbe 8:186–195. doi: 10.1016/j.chom.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrison TE, Fraser RJ, Smith PN, Mahalingam S, Heise MT. 2007. Complement contributes to inflammatory tissue destruction in a mouse model of Ross River virus-induced disease. J Virol 81:5132–5143. doi: 10.1128/JVI.02799-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gunn BM, Morrison TE, Whitmore AC, Blevins LK, Hueston L, Fraser RJ, Herrero LJ, Ramirez R, Smith PN, Mahalingam S, Heise MT. 2012. Mannose binding lectin is required for alphavirus-induced arthritis/myositis. PLoS Pathog 8:e1002586. doi: 10.1371/journal.ppat.1002586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morrison TE, Simmons JD, Heise MT. 2008. Complement receptor 3 promotes severe Ross River virus-induced disease. J Virol 82:11263–11272. doi: 10.1128/JVI.01352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brooke CB, Schafer A, Matsushima GK, White LJ, Johnston RE. 2012. Early activation of the host complement system is required to restrict central nervous system invasion and limit neuropathology during Venezuelan equine encephalitis virus infection. J Gen Virol 93:797–806. doi: 10.1099/vir.0.038281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva LA, Dermody TS. 2017. Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J Clin Invest 127:737–749. doi: 10.1172/JCI84417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kunnakkadan U, Nag J, Kumar NA, Mukesh RK, Suma SM, Johnson JB. 2019. Complement mediated neutralization of a potent neurotropic human pathogen, Chandipura virus, is dependent on C1q. J Virol 93:e00994-19. doi: 10.1128/JVI.00994-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pierro A, Rossini G, Gaibani P, Finarelli AC, Moro ML, Landini MP, Sambri V. 2015. Persistence of anti-chikungunya virus-specific antibodies in a cohort of patients followed from the acute phase of infection after the 2007 outbreak in Italy. New Microbes New Infect 7:23–25. doi: 10.1016/j.nmni.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, Whitmore A, Heise MT, Baric RS. 2018. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 9:e01753-18. doi: 10.1128/mBio.01753-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suresh M, Molina H, Salvato MS, Mastellos D, Lambris JD, Sandor M. 2003. Complement component 3 is required for optimal expansion of CD8 T cells during a systemic viral infection. J Immunol 170:788–794. doi: 10.4049/jimmunol.170.2.788. [DOI] [PubMed] [Google Scholar]
- 50.Gunn BM, Jones JE, Shabman RS, Whitmore AC, Sarkar S, Blevins LK, Morrison TE, Heise MT. 2018. Ross River virus envelope glycans contribute to disease through activation of the host complement system. Virology 515:250–260. doi: 10.1016/j.virol.2017.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson JB, Lyles DS, Alexander-Miller MA, Parks GD. 2012. Virion-associated complement regulator CD55 is more potent than CD46 in mediating resistance of mumps virus and vesicular stomatitis virus to neutralization. J Virol 86:9929–9940. doi: 10.1128/JVI.01154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson JB, Aguilar HC, Lee B, Parks GD. 2011. Interactions of human complement with virus particles containing the Nipah virus glycoproteins. J Virol 85:5940–5948. doi: 10.1128/JVI.00193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hicks JT, Klutch MJ, Albrecht P, Frank MM. 1976. Analysis of complement-dependent antibody-mediated lysis of target cells acutely infected with measles. J Immunol 117:208–215. [PubMed] [Google Scholar]
- 54.McSharry JJ, Pickering RJ, Caliguiri LA. 1981. Activation of the alternative complement pathway by enveloped viruses containing limited amounts of sialic acid. Virology 114:507–515. doi: 10.1016/0042-6822(81)90230-0. [DOI] [PubMed] [Google Scholar]
- 55.Ehrnst A. 1978. Separate pathways of C activation by measles virus cytotoxic antibodies: subclass analysis and capacity of F(ab) molecules to activate C via the alternative pathway. J Immunol 121:1206–1212. [PubMed] [Google Scholar]
- 56.Sreekumar E, Issac A, Nair S, Hariharan R, Janki MB, Arathy DS, Regu R, Mathew T, Anoop M, Niyas KP, Pillai MR. 2010. Genetic characterization of 2006–2008 isolates of Chikungunya virus from Kerala, South India, by whole genome sequence analysis. Virus Genes 40:14–27. doi: 10.1007/s11262-009-0411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leddy JP, Simons RL, Douglas RG. 1977. Effect of selective complement deficiency on the rate of neutralization of enveloped viruses by human sera. J Immunol 118:28–34. [PubMed] [Google Scholar]
- 58.Mills BJ, Beebe DP, Cooper NR. 1979. Antibody-independent neutralization of vesicular stomatitis virus by human complement. II. Formation of VSV-lipoprotein complexes in human serum and complement-dependent viral lysis. J Immunol 123:2518–2524. [PubMed] [Google Scholar]
- 59.Johnson JB, Grant K, Parks GD. 2009. The paramyxoviruses simian virus 5 and mumps virus recruit host cell CD46 to evade complement-mediated neutralization. J Virol 83:7602–7611. doi: 10.1128/JVI.00713-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sahu A, Isaacs SN, Soulika AM, Lambris JD. 1998. Interaction of vaccinia virus complement control protein with human complement proteins: factor I-mediated degradation of C3b to iC3b1 inactivates the alternative complement pathway. J Immunol 160:5596–5604. [PubMed] [Google Scholar]
- 61.Lim CK. 2016. Virus isolation and preparation of sucrose-banded Chikungunya virus samples for transmission electron microscopy. Methods Mol Biol 1426:153–162. doi: 10.1007/978-1-4939-3618-2_14. [DOI] [PubMed] [Google Scholar]
- 62.Lachmann PJ. 2010. Preparing serum for functional complement assays. J Immunol Methods 352:195–197. doi: 10.1016/j.jim.2009.11.003. [DOI] [PubMed] [Google Scholar]