

Abstract
Myotonic dystrophy type 2 (DM2) is a genetically defined muscular dystrophy that is caused by an expanded repeat of r(CCUG) [r(CCUG)exp] in intron 1 of a CHC-type zinc finger nucleic acid binding protein (CNBP) pre-mRNA. Various mechanisms contribute to DM2 pathology including pre-mRNA splicing defects caused by sequestration of the RNA splicing regulator muscleblind-like-1 (MBNL1) by r(CCUG)exp. Herein, we study the biological impacts of the molecular recognition of r(CCUG)exp’s structure by a designer dimeric small molecule that directly cleaves the RNA in patient-derived cells. The compound is comprised of two RNA-binding modules conjugated to a derivative of the natural product bleomycin. Careful design of the chimera affords RNA-specific cleavage, as attachment of the bleomycin cleaving module was done in a manner that disables DNA cleavage. The chimeric cleaver is more potent than the parent binding compound for alleviating DM2-associated defects. Importantly, oligonucleotides targeting the r(CCUG)exp sequence for cleavage exacerbate DM2 defects due to recognition of a short r(CCUG) sequence that is embedded in CNBP, argonaute-1 (AGO1), and MBNL1, reducing their levels. The latter event causes a greater depletion of functional MBNL1 than the amount already sequestered by r(CCUG)exp. Thus, compounds targeting RNA structures can have functional advantages over oligonucleotides that target the sequence in some disease settings, particularly in DM2.
Graphical Abstract
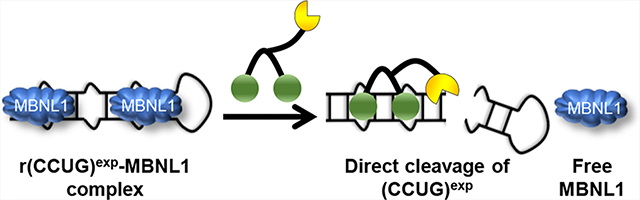
The most common way to target RNA for therapeutic benefit is to use antisense oligonucleotides (ASOs) that recognize the target’s sequence by Watson–Crick base pairing.1,2 The ASO–mRNA complex recruits endogenous ribonuclease (RNase) H to cleave the RNA strand in the hybrid duplex. This approach has proven broadly applicable for affecting RNA biology. The major advantages of this approach are ease of design and accessibility due to advances in their chemical synthesis.
An alternative and complementary way to target RNA is to use small molecules that target structured regions.3,4 Given that structured regions are often regions of function, it could be advantageous to target them. Various methods are available to identify RNA-binding compounds including screening, structure-based design, and sequence-based design.5–14 Simple binding compounds can elicit a biological response if they target a functional site, but they generally do not elicit a response if they target nonfunctional sites.15
One way to broaden the ability of RNA structure-binding small molecules to elicit a biological response is to imbue them with nuclease-like activities. Several approaches have been developed to engineer small molecule with cleaving ability in situ and in vivo, including ribonuclease targeting chimeras (RIBOTACs) that recruit an endogenous nuclease to enzymatically and selectively cleave an RNA target16,17 and appending small molecules with the natural product bleomycin A5 to directly and selectively cleave the target.18 This latter approach has been shown to cleave RNA targets selectively in myotonic dystrophy type 1 (DM1) patient-derived cells and in a DM1 mouse model. In particular, the compound specifically cleaved toxic expanded repeats of r(CUG) over short, nontoxic repeats; that is, limited off-target effects were observed.18 We recently reported a third approach, which is to shunt RNAs to decay pathways—as demonstrated in myotonic dystrophy type 2 (DM2) patient-derived fibroblasts. DM2 is caused by an expanded r(CCUG) repeat [r(CCUG)exp] harbored in an intron. Among other defects, r(CCUG)exp causes aberrant intron retention. The small molecule liberates the retained intron, which is subsequently decayed.19 Herein, we describe the development of a small molecule that directly cleaves r(CCUG)exp and rescues disease-causing defects. The cleavage approach allows us to assess the effects of structure-specific targeting versus sequence-specific targeting using an ASO. Importantly, we find that structure-specific targeting can have both functional and selectivity advantages over sequence-specific targeting in DM2.
RESULTS
Disease Features of DM2 Patient-Derived Fibroblasts
DM2-affected cells are characterized by a toxic mutation, an expanded repeat of r(CCUG) [r(CCUG)exp] located in intron 1 of CHC-type zinc finger nucleic acid binding protein (CNBP) pre-mRNA (Figure 1A). This genetic disorder causes identifiable features including: (i) formation of nuclear foci when r(CCUG)exp binds various RNA-binding proteins (RBPs) such as muscleblind-like 1 (MBNL1),20 (ii) pre-mRNA splicing defects in MBNL1-regulated genes,21–23 and (iii) retention of intron 1 in CNBP mRNA, in which it is harbored.24 The most common irregularity is pre-mRNA splicing defects due to MBNL1 sequestration, including defects in insulin receptor (IR) exon 11, which is excluded too frequently25 (Figure 1B).
Figure 1.

The RNA that causes DM2, r(CCUG)exp, has multiple modes of toxicity. (A) r(CCUG)exp in intron 1 of CNBP folds into a hairpin displaying a periodic array of internal loops that sequester MBNL1. Sequestration of MBNL1 by r(CCUG)exp causes (B) pre-mRNA splicing defects, for example increasing IR exon 11 exclusion.
Design of Compounds That Target r(CCUG)exp and Directly Cleave It
In our previous studies, we developed a potent dimer (1) that selectively targeted r(CCUG)exp and triggered native decay of the intron 1 species (Figure 2A).19,26 Dimer 1 rescued IR pre-mRNA splicing defects and, importantly, does not affect IR splicing in healthy cells (Figures S1 and S2). Additionally, no effect was observed on the inclusion of mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) exon 22a, a non-MBNL1 dependent splicing event (Figure S3). Although the designer binding compound liberated the retained intron, thereby subjecting it to decay, the percentage eliminated was only ~20%; notably, levels of CNBP mature mRNA were unaffected (Figure S4).19 Thus, to eliminate this transcript to a greater extent and more potently improve DM2 defects, we designed a version of the binding compound 1 that would directly subject the transcript to decay by appending it to bleomycin A5 (2, Figure 2A). Attachment of bleomycin A5 to an RNA-binding module using its free amine affords a compound that is capable of selective cleavage of an RNA target while ablating DNA cleavage at the active concentration,18,27 as the free amine forms key interactions that contribute to DNA affinity (Figure 2B).28 Such an approach has been used to cleave r(CUG)exp (causative agent of DM1) in a mouse model, robustly modulating disease phenotypes with limited off-targets.18 A control compound in which bleomycin A5 is conjugated to an assembly scaffold lacking RNA-binding modules was also synthesized (compound 3, Figure 2A).
Figure 2.

Design of small molecules targeting r(CCUG)exp. (A) Green spheres indicate the kanamycin RNA-binding modules. Compound 1 is a dimer composed of two kanamycin binding modules connected via a propylamine peptoid. Compound 2, the dimer linked to a bleomycin-cleaving module, where bleomycin is represented by the yellow partial circle. The control compound 3, a peptoid with the bleomycin cleavage module lacking kanamycin binding modules. (B) Schematic of 2’s mode of action, binding to r(CCUG)exp, cleaving the toxic repeat, and releasing MBNL1.
A series of in vitro studies was completed to assess the selectivity and cleaving capacity of 1, 2, and 3. First, affinity of the derivatives for RNA and DNA was measured in the absence of Fe(II) required for cleavage by microscale thermophoresis (MST).16,29 As expected, 1 and 2 bound only to r(CCUG)12 with similar affinities (Kd = 97 ± 15 nM and 106 ± 23 nM, respectively; Figures 3A,B and S5). No saturable binding of 3 to r(CCUG)12 was observed (Kd > 5 μM). Further, no binding of 1, 2, or 3 was observed to a base-paired control RNA (no internal loops) or to DNA (Kd > 5 μM).
Figure 3.

Binding affinities of compounds for various targets. (A) RNA and DNA sequences used in MST experiments. (B) Affinities of 1, 2, and 3 for Cy5-labeled (CCUG)12, Cy5-labeled RNA base paired control, or a Cy5-labeled AT-rich DNA hairpin, as determined by MST. (C) Studying the DNA cleaving activity of 2, 3, and bleomycin A5 in vitro. Error bars represent SD, n = 5.
In vitro cleavage of r(CCUG)10 was assessed using radioactively labeled RNA. Significant cleavage of r(CCUG)10 was observed upon incubation with 2, with ~50% of the RNA cleaved at 2.5 μM and nearly 100% cleavage at higher concentrations (Figure S6). No statistically significant cleavage of r(CCUG)10 was observed with 3, which does not have RNA-binding modules. The sites of cleavage by 2 were also mapped onto r(CCUG)10’s structure, revealing that the primary cleavage sites are between 5′-GC steps in base paired regions. Importantly, these sites of cleavage by 2 are different than the minimal cleavage sites induced by 3 without RNA binding modules that occur between 5′-UG steps (Figure S6). In contrast, the small molecule–bleomycin conjugate that targets r(CUG)exp cleaved between 5′-UG steps.30
The ability of these compounds to cleave DNA in vitro was also investigated. While robust DNA cleavage is observed with bleomycin A5, cleavage of DNA by 2 and 3 is ablated significantly (Figure 3C). This reduction in DNA cleavage is similar to previous results when bleomycin A5 is attached to an RNA binding compound to target r(CUG) repeats in DM1.18 Thus, 2 selectively cleaved r(CCUG) repeats over DNA in vitro, and the RNA-binding modules controlled the extent and sites of cleavage.
Biological Evaluation of 2, Which Binds and Cleaves r(CCUG)exp Directly
Given the favorable in vitro cleavage data, the ability of 2 to cleave r(CCUG)exp and more effectively reduce its abundance than 1 was studied in DM2 fibroblasts (Figure 4A). Application of 2 to DM2 fibroblasts reduced the amount of r(CCUG)exp intron retained product by an ~2.5-fold greater amount than the parent 1; 5 μM of 2 reduced levels of intron 1 by ~50% in comparison to ~20% reduction with a treatment of 5 μM of 1 (Figure 4A). Interestingly, we observed an ~10-fold difference between in vitro binding affinity and in cellulis activity. The reasons for this difference are diverse, such as cell permeability or subcellular localization. In previous studies of multivalent kanamycin compounds targeting RNA, a similar difference (~10-fold) between in vitro and in cellulis activity was observed.26,31 Moreover, a similar activity difference (~5 fold) was noticed with bleomycin-conjugated compounds in DM1 myotubes.18,30 Despite differences in affinity and cellular activity, cleavage of r(CCUG)exp occurs at similar concentrations in vitro and in cells. Levels of the CNBP mature mRNA were also reduced, by ~25%, supporting that 2 cleaves r(CCUG)exp-containing transcripts (Figure 4B). Interestingly, ~30% of CNBP transcripts contain intron 1 retention in DM2-affected cells. To evaluate the rate of cleavage in cells, DM2 fibroblasts were treated with 2, and intron 1 levels were evaluated at 24, 48, and 72 h (Figure S7). Cleavage of intron 1 increased from 24 to 48 h; however, cleavage does not further increase at 72 h. Therefore, for further cellular evaluation, compounds were treated for 48 h. No effect on the abundance of CNBP RNA was observed with control compound 3, as expected (Figure S8).
Figure 4.

Biological activity of 2 in DM2 fibroblasts. (A) Effect of 2 on r(CCUG)exp-containing CNBP levels measured by RT-qPCR. (B) Effect of 2 on CNBP mature mRNA levels measured by RT-qPCR. (C) Effect of competitive assay between 1 and 2 on CNBP mature mRNA levels measured by RT-qPCR. (D) Ability of 2 to rescue the IR exon 11 splicing defect. (E) Representative microscopic images for the reduction of nuclear foci by 2, as completed by RNA-FISH and immunohistochemistry (IHC) using an anti-MBNL1 antibody.32 (F) Quantification of r(CCUG)exp-MBNL1 foci/nucleus (n = 3 biological replicates, 40 nuclei counted per replicate). Error bars represent SD. *P < 0.5, **P < 0.01, ***P < 0.001, ****P < 0.0001 as determined by a one-way ANOVA (n = 3).
The selective cleavage of r(CCUG)exp by 2 was assessed by studying DNA damage in cells. In particular, we investigated the presence of the phosphorylated form of H2A histone family member X (γ-H2AX) foci that form due to DNA double strand breaks. When cancer cells are exposed to bleomycin A5, γ-H2AX foci are observed.33 Indeed, DM2 fibroblasts treated with 5 μM of bleomycin A5 have an ~6-fold increase in γ-H2AX foci compared to untreated cells, as expected (Figure S9). In contrast, treatment with the same amount of 2 showed no significant increase in γ-H2AX foci (Figure S9). Thus, both in vitro and in cellulis data support the hypothesis that the appendage of an RNA-binding module onto bleomycin A5 using its free amine affords specific cleavage of the RNA target while ablating DNA cleavage.
Competitive Cleavage of CNBP Mature mRNA to Assess Target Engagement in DM2 Fibroblasts
To validate the target of 2, the levels of CNBP mature mRNA were evaluated in DM2 fibroblasts treated with a constant concentration of 2 and increasing concentrations of 1. Interestingly, using 0.1-, 1-, and 10-fold excess concentrations of 1 relative to 2 reduced the cleavage of mature mRNA by 2 dose-dependently and restored levels to that observed in untreated cells (Figure 4C), as 1 only affects levels of the intron-retained product, not mature CNBP mRNA levels (Figure S4). Thus, this competitive cleavage experiment supports the fact that 1 and 2 share the same target and that 2 directly cleaves r(CCUG)exp and does not, for example, affect transcription. Previously, compounds have been developed that decreased levels of RNA repeat expansions by inhibiting transcription,34,35 and this direct cleavage provides a route to cleave RNA directly and has been demonstrated in multiple settings such as DM1 and now DM2.
Cleaving Compound 2 Improves Downstream DM2 Defects
Application of 5 μM of 2 improved the IR pre-mRNA splicing defect, regulated by MBNL1, by ~50% (Figure 4D). No effect was observed with compound 3 (Figure S8). Moreover, the cleaving compound 2 is more effective than 1, which only rescued the IR splicing defect by ~20% at a 2-fold higher concentration (10 μM). These results are in agreement with 1’s and 2’s effect on r(CCUG)exp abundance. Additionally, there was no effect of 2 on the alternative splicing of MAP4K4, as it is not regulated by MBNL1 (Figure S10). Thus, the rescue of IR splicing is due to 2 increasing levels of free MBNL1.
The toxic r(CCUG)exp RNA binds and sequesters MBNL1 (and other RBPs) in nuclear foci. Thus, compound 2 and control compound 3 were tested for reducing foci. Compound 2 at 5 μM reducedthe number of r(CCUG)exp-containing foci by ~50%, which correlates with the reduction in abundance of r(CCUG)exp (Figure 4E,F). As expected, 3 had no effect on the number of nuclear foci present (Figure S11).
Biological Evaluation of 2 in Healthy Fibroblasts
To further evaluate selectivity, 2 was tested in healthy fibroblasts. Since the target [r(CCUG)exp] is absent in healthy fibroblasts, an effect on CNBP levels or IR alternative splicing is not expected. Indeed, this was the case at concentrations ranging from 0.005 to 5 μM (Figure S12). Thus, 2 does not cleave the wild-type allele that contains a short, nonpathogenic r(CCUG) repeat and thereby recognizes the structure adopted by r(CCUG)exp.
An ASO Targeting r(CCUG)exp Cleaves r(CCUG)exp and Exacerbates pre-mRNA Splicing Defects
The most common way to cleave RNAs is via RNase H recruitment by oligonucleotides.1,36 Thus, to serve as a positive control for cellular experiments, we designed an antisense oligonucleotide (ASO) that would target r(CCUG)exp via base pairing and target it for destruction. Indeed, application of a r(CCUG)exptargeting ASO, but not a control oligonucleotide, resulted in reduction of CNBP intron 1 levels in DM2 patient-derived fibroblasts (Figures 5 and S13). In particular, transfection of 10 nM of ASO caused an ~50% reduction of the intron, similar to the reduction observed with 5 μM of 2 (Figure 5A).
Figure 5.

Comparison of the selectivity of an ASO and 2. (A) Precise recognition of r(CCUG)exp by 2 compared to ASO-treated cells. Effect on RNAs containing r(CCUG) repeats as determined by RTqPCR; WT CNBP is that derived from a non-DM2 or healthy fibroblast that does not express r(CCUG)exp. (B) ASO RNA-targeting strategy. (C) Small molecule RNA-targeting strategy. Error bars represent SD. **P < 0.01, ***P < 0.001, ****P < 0.0001 as determined by a one-way ANOVA (n = 3).
Interestingly, sequence analysis of MBNL1 revealed that it contains eight regions with two r(CCUG) repeats each and thus has partial complementarity to the ASO (Table S1). Therefore, we measured the effect of the ASO on MBNL1 expression. In DM2 fibroblasts, the ASO reduced MBNL1 mRNA abundance to a similar extent to that for the reduction of CNBP intron 1 levels at all concentrations tested (Figures 5A and S13). Thus, an ASO that targets r(CCUG)exp has an important off-target, the mRNA that encodes the protein that it is meant to free (Figure 5B). Moreover, the ASO was also tested in healthy fibroblasts, and levels of CNBP mature mRNA, which also contain short r(CCUG) repeats, were drastically reduced (75% decrease at 10 nM), further indicating the ASO’s lack of selectivity (Figures 5A and S14).
A search for other targets in the human transcriptome that contain stretches of sequences that are complementary to the ASO revealed many potential off-targets (Table S1). Interestingly, Argonaute RISC Component 1 (AGO1), a key component in the RNA-induced transcriptional silencing complex (RISC) that controls a wide variety of RNA regulatory processes, contains eight consecutive r(CCUG) repeats. As expected, in the DM2 patient-derived cells, the ASO reduced the abundance of the mRNA encoding AGO1. Importantly, the small molecule 2 that targets the r(CCUG)exp structure, not sequence, did not affect the abundance of MBNL1 or AGO1 targets (Figure 5A,C).
To provide insight into these molecular recognition events, the structures of the repeating units and surrounding regions in wild-type CNBP, mutant CNBP, AGO1, and MBNL1 were modeled, and thermodynamic stability was predicted as previously described.18 These calculations show that r(CCUG)exp in mutant CNBP forms a robust and highly stable structure (−1995.6 kcal mol−1, Table S3). In contrast, a notably less stable structure was predicted in regions containing r(CCUG) repeats in wild-type CNBP (−22.8 kcal mol−1), while in AGO1 or MBNL1 the r(CCUG) repeats do not fold into a structure containing the 5′CCUG/3′GUCC motif (Figure S15). Furthermore, the thermodynamic stability of the complex formed between the ASO and each mRNA was predicted (Table S4). The similarities in ΔG values further support that the ASO targets all of these mRNAs and cannot discriminate between binding long and short r(CCUG) repeats. An additional factor that can drive the selectivity difference between the ASO and the cleaving compound 2 is that the ASO is an efficient cleaver of RNA targets it engages that contain r(CCUG) sequences, and thus selectivity between long and short r(CCUG) repeats containing RNAs would be modest. If 2 is not efficient in cleaving of RNAs that have short stretches of the r(CCUG)exp-like structures, then there would be a desirable bias in cleaving the r(CCUG)exp RNA over RNAs with shorter repeats of this motif.
DISCUSSION
The most common way to target RNA is to bind to unstructured regions using oligonucleotides. Given the importance of RNA structure in biology, there is increasing interest in the development of organic ligands that target structured RNAs and affect their function. A wide variety of functional outputs can occur when ligands target structured RNAs, including affecting pre-mRNA splicing outcomes, protein binding, and translation.3,37,38 This study demonstrates that compounds targeting RNA can facilitate their elimination by disrupting a key RNA–protein interaction that promotes intron retention. Compound activity can be further enhanced by conjugating compounds with cleaving modules to enable direct cleavage.16,17 This study suggests that there may be many ways to influence RNA biology, including stimulating cleavage programmably.
The use of oligonucleotides to target r(CCUG)exp for cleavage should perhaps be pursued with caution. Given that the toxic r(CCUG)exp is in an intronic region, there is limited sequence space by which to target this RNA with ASOs outside of the repeat. Due to the presence of r(CCUG) repeats in other RNAs such as MBNL1, wild-type CNBP, and AGO1, these off-targets must be carefully considered during lead optimization. Further, the cellular model used for evaluation must be carefully selected. For example, an oligonucleotide targeting r(CCUG) repeats improved splicing of a BIN1 exon 11 mini-gene in a transfected mouse myoblast cell line where the desired target is over-expressed.31 Notably, mouse MBNL1 only contains two regions of two r(CCUG) repeats each, as compared to eight regions of two repeats in human MBNL1. Thus, mouse MBNL1 may not be down-regulated by the ASO to the same extent in the transfected cellular model, obscuring off-target effects observed in human patient-derived cells. It is clear that there is no one approach to target RNA and affect biology for functional studies or for therapeutic benefit. In some cases, oligonucleotide-based approaches could be beneficial, while in others, small molecules that interact with structured RNA regions will be preferable for eliciting a favorable response.
Interestingly, the small molecule cleaver 2 is more selective than the ASO for targeting RNAs with long and toxic r(CCUG) repeats versus RNAs with shorter repeats. A variety of factors could help explain this observation, including the following: (i) The cleaving capacity of 2 is not as efficient as the ASO, giving a larger bias for cleaving a target with many binding sites in r(CCUG)exp over other targets with shorter repeats. (ii) RNA targets that are not structured are most tightly bound by complementary oligonucleotides compared to structured RNAs as sequence-based recognition requires unfolding of the targeted structure and the targeting oligonucleotide,39,40 and (iii) there are key differences in molecular recognition of oligonucleotides and compounds targeting structure in the context of solvent accessible sites. Perhaps, not all structures that have multiple copies of the 5′CCUG/3′GUCC motif are accessible by ligands that target structure.
Studies that elucidate druggable, structured regions within RNAs and deploying the corresponding compounds that bind them will allow for more in-depth studies on the functional consequences of binding. The downstream biological effects of binding to functional sites will likely be predictable; binding to nonfunctional sites could have unanticipated effects in some cases. Such studies will be critical in order to leverage information on the composite of RNAs, especially folded RNAs, to develop targeted chemical probes and therapeutics. That is, RNA is starting its journey from its perception as undruggable to a tried-and-true human small molecule drug target.
METHODS
Antisense Oligonucleotides
Antisense oligonucleotide (ASO) and scrambled control LNA oligonucleotide were purchased from QIAGEN. The sequence of the CCAG Gapmer Custom LNA Oligonucleotide is +C+A+GG*C*A*G*G*C*A*G*G*C*+A+G+G where + indicates LNA modifications and * indicates phosphorothioate modifications. The sequence of the scrambled LNA control is G*C*+T*A*T*+A*C*C*+A*G*C*+G*T*C*+G*T*C*+A*T.
Cell Lines
Compounds were tested in two cell lines: (i) DM2 patient-derived fibroblasts (DM11; University of Florida, Center for NeuroGenetics) and (ii) fibroblasts from a healthy donor (wild type; WT GM07492; Coriell Institute).
Microscale Thermophoresis (MST)27
MST measurements were performed on a Monolith NT.115 system (NanoTemperTechnologies) with Cy5-labeled AT-rich DNA hairpin (5′-Cy5-CGCGAATTCGCGTTTTCGCGAATTCGCG; IDT), Cy5-labeled (CCUG)12 (5′-Cy5-GCG(CCUG)12CGC; Dharmacon), and Cy5-labeled base pair control (BP; 5′-Cy5-GCG(CCUG)7(GCAG)5CGC; Dharmacon), which were deprotected according to the manufacturer’s protocol. RNA or DNA (10 nM) was prepared in 1 × MST Buffer (8 mM Na2HPO4, 185 mM NaCl, 1 mM EDTA, and 0.05% (v/v) Tween-20) and folded by heating at 60 °C for 5 min and slowly cooling to RT. Compound 1, 2, or 3 was then added to a final concentration of 10 μM, followed by 1:1 dilutions in 10 μL of 1 × MST Buffer. Nucleic acid and the compound were then mixed 1:1 (v/v). Samples were incubated for 30 min at room temperature and then loaded into standard capillaries (NanoTemper Technologies). The following parameters were used: 5–20% LED, 80% MST power, Laser-On time = 30 s, Laser-Off time = 5 s. Fluorescence was detected using excitation wavelengths of 605–645 nm and emission wavelengths of 680–685 nm. The resulting data were analyzed by thermophoresis analysis and fitted using a quadratic binding equation in the MST analysis software (NanoTemper Technologies). The dissociation constant was then determined using a single-site model to fit the curve. The reported Kd values are an average of two independent sets of experiments.
DNA Cleavage in Vitro27
Compounds 2 and 3 and bleomycin A5 (156, 312, 625, 1250, 2500, 5000 nM) were preactivated by the addition of 1 equiv of freshly prepared (NH4)2Fe(SO4)2·6H2O. A plasmid encoding GFP (40 ng/μL) was dissolved in 5 mM Na2HPO4, and 12 μL was added to 2 μL of preactivated compound. An additional equivalent of (NH4)2Fe(SO4)2·6H2O was added 30 min and then 60 min later, and the reaction mixture was incubated at 37 °C overnight. The reaction mixture was mixed with with 6 × Gel Loading Dye [Purple; New England BioLabs (NEB)], and the cleavage fragments were separated on 0.8% agarose gel stained with ethidium bromide (110 V, 1 h). Gels were imaged using a Molecular Dynamics Typhoon 9410 variable mode imager, and percent cleavage was quantified using QuantityOne (BioRad). Values are an average of two independent experiments.
Radiolabeling of RNA
The r(CCUG)10 oligonucleotide was purchased from Dharmacon and deprotected according to the manufacturer’s standard protocol. The RNA (500 pmoles) was then 5′-end labeled. The RNA was then purified on a denaturing 15% polyacrylamide gel, excised from the gel, and extracted as previously described.30 The RNA was then dissolved in 50 μL of nanopure water.
RNA Cleavage in Vitro18
A 4 μL aliquot of 5′−32P-r(CCUG)10 was diluted in 150 μL of 5 mM NaH2PO4 (pH 7.4) and folded by heating at 95 °C for 30 s. The solution was cooled to room temperature, and 2 was added at varying concentrations (10, 5, 2.5, 1.25, and 0.6 μM). An equimolar amount of freshly prepared (NH4)2Fe(SO4)2·6H2O in 5 mM NaH2PO4, pH 7.4, was then added. The solutions were incubated at 37 °C and supplemented with additional equimolar aliquots of (NH4)2Fe(SO4)2·6H2O in 5 mM NaH2PO4, pH 7.4, after both 30 min and 1 h. The RNA was treated with the compound for a total of 24 h at 37 °C.
A T1 ladder was prepared by mixing 1 μL of radiolabeled RNA with 30 μL of T1 Buffer (20 mM sodium citrate, 1 mM EDTA, 7 M urea) and heating to 95 °C for 30 s. After cooling to room temperature, RNase T1 (3 units/μL final concentration) was added. The sample was incubated at room temperature for 20 min and stopped by adding an equal volume of Loading Buffer (95% formamide, 20 mM EDTA, pH 8.0). A hydrolysis ladder was prepared by mixing 1 μL of radiolabeled RNA with 10 μL of 1 × Alkaline Hydrolysis Buffer (50 mM NaHCO3, pH 9.2, and 1 mM EDTA) and heating at 95 °C for 5 min.
All reactions were stopped by adding an equal volume of Loading Buffer, and the fragments were separated on a denaturing 15% polyacrylamide gel (70 W for 3 h in 1 × TBE Buffer). Gels were imaged using a Molecular Dynamics Typhoon 9410 variable mode imager. Percent cleavage was quantified using QuantityOne (BioRad).
Cell Culture
All cells were maintained at 37 °C with 5% CO2. DM2 fibroblasts were cultured in DMEM/high glucose (HyClone) supplemented with 20% FBS (Sigma) and 1% Antibiotic–Antimycotic solution (Corning). WT fibroblasts were cultured in MEM (Corning) supplemented with 10% FBS, 1% Glutagro (Corning), and 1% Antibiotic–Antimycotic solution. LNA Gapmer (QIAGEN Inc.) was transfected into cells using Lipofectamine RNAiMAX Reagent per the manufacturer’s protocol for 48 h. Cells were treated with compounds 1, 2, and 3 at concentrations ranging from 10 μM to 0.1 μM, 5 μM to 0.005 μM, and 5 μM to 0.005 μM, respectively, in growth medium for 48 h at 37 °C with 5% CO2.
Analysis of Abundance of CCUG-Containing Transcripts
Cells were grown in 6-well plates and treated or transfected as described in the Cell Culture section. After 48 h, the cells were lysed, and total RNA was harvested using a Zymo Quick RNA miniprep kit per the manufacturer’s protocol. Approximately 1 μg of total RNA was reverse transcribed using a qScript cDNA synthesis kit (20 μL total reaction volume, Quanta BioSciences); 2 μL of the reverse transcription (RT) reaction was used for each primer pair for quantitative (q) PCR with SYBR Green Master Mix, performed on a 7900HT Fast Real-Time PCR System. The relative abundance of each transcript was determined by normalizing to GAPDH.
Evaluation of Nuclear Foci Using Fluorescence in Situ Hybridization (FISH)
FISH was used to determine the small molecules’ effects on the number of nuclear foci as previously described.41 Briefly, DM2 fibroblasts were grown to ~40% confluence in a Mat-Tek 96-well glass bottom plate in growth medium. Then, cells were treated with 2 or 3 (5 μM) for 48 h. To fix the cells, the growth medium was removed and washed with 1 × DPBS followed by the addition of 100 μL of 4% formaldehyde in 1 × DPBS. The cells were incubated at 37 °C for 10 min and then washed five times with 1 × DPBS at 37 °C for 2 min each. Fibroblasts were permeabilized with 100 μL of 1 × DPBS containing 0.1% TritonX-100 for 5 min at 37 °C, followed by incubation with 100 μL of 30% formamide in 2 × SSC for 10 min at room temperature. Then, 100 μL of the FISH probe Cy3-(CAGG)10 (purchased from Integrated DNA Technologies) was added to each well (140 nM of Cy3-(CAGG)10 in 30% formamide in 2 × SSC, 2 μg/μL BSA, 1 μg/μL yeast tRNA, and 2 mM vanadyl complex) and incubated at 37 °C overnight. The cells were then washed with 100 μL of 30% formamide in 2 × SSC buffer at 37 °C for 30 min and 100 μL of 2 × SSC buffer at 37 °C for 30 min. MBNL1 was imaged by adding 20 μL of 1:5 anti-MBNL132 in 2 × SSC and incubated at 37 °C for 1 h. The cells were washed three times with 100 μL of 0.1% Triton X-100 in 1 × DPBS for 5 min at 37 °C and then incubated with 1:200 dilution antimouse IgG Dylight 488 in 2 × SSC at 37 °C for 1 h. After washing three times with 100 μL of 1 × DPBS containing 0.1% Triton X-100 at 37 °C for 5 min, the cells were washed with 1 × DPBS for 5 min at 37 °C. Nuclei were stained by incubating with 100 μL of 1 μg/mL DAPI for 5 min at 37 °C. The cells were washed with 1 × DPBS twice and imaged in 100 μL of 1 × DPBS using an Olympus Fluoview 1000 confocal microscope at 100× magnification.
Evaluation of pre-mRNA Splicing by RT-PCR
Cells were grown in 6-well plates and were treated as described in the Cell Culture section. After 48 h, the cells were lysed, and total RNA was harvested using a Zymo Quick RNA miniprep kit. Approximately 1 μg of total RNA was reverse transcribed using a qScript cDNA synthesis kit (20 μL of total reaction volume, Quanta BioSciences); 2 μL of the RT reaction was used for PCR using GoTaq DNA polymerase (Promega). RT-PCR products were observed after 35 cycles of 95 °C for 30 s, 58 °C for 30s, 72 °C for 1 min, and a final extension at 72 °C for 5 min. Products were separated on a 2% agarose gel run at 110 V for 1 h in 1 × TBE buffer, visualized by staining with ethidium bromide, and imaged using a Typhoon 9410 variable mode imager. Gels were quantified using ImageJ. Percent rescue was calculated by dividing the difference between treated and untreated DM2 samples by the difference between untreated DM2 and WT samples (eq 1).
| (1) |
Evaluation of γ-H2AX Foci42
DNA damage induced by small molecules was assessed by imaging γ-H2AX, associated with DNA double strand breaks. DM2 fibroblasts were grown to ~40% confluence in Mat-Tek 96-well glass bottom plates in growth medium and treated with 2 [5 μM] or bleomycin A5 [5 μM] for 48 h. Cells were washed with 1 × DPBS three times and then fixed with 100 μL of 4% paraformaldehyde for 10 min at 37 °C. Cells were washed with 1 × DPBS three times and then with 1 × DPBS containing 0.1% Triton X-100 three times for 5 min at 37 °C. Cells were then incubated with a 1:500 dilution of anti-γH2AX (Abcam) at 37 °C for 1 h and washed three times with 1 × DPBS containing 0.1% Triton X-100 for 5 min at 37 °C. A 1:200 dilution of goat anti-mouse IgG-DyLight 488 conjugate (Thermo Scientific) was added, and the cells were incubated at 37 °C for 1 h. After washing the cells with 1 × DPBS containing 0.1% Triton X-100 twice with 1 × DPBS for 5 min at 37 °C, nuclei were stained with DAPI (100 μL of 1 μg/mL), and cells were imaged as described above.
Prediction of RNA Secondary Structures and Thermodynamic Stability
To analyze how r(CCUG) repeats may fold in CNBP, AGO1, and MBNL1, the program Tandem Repeats Finder43 was used to predict tandem repeats in each gene sequence. To model the secondary structures of CCUG repeats, the identified repeats in each gene, plus 100 nt of gene sequence at both ends, was analyzed using the ScanFold-Scan web server.44 This program uses a 100 nt sliding analysis window and a single nt step size to predict potential secondary structures across each gene region. Resulting secondary structures for windows centered (approximately) on each repeat region are shown in Figure S14.
ΔG Calculations for ASO-mRNA Complex
RNA sequences were folded using the RNAstructure web server. To calculate the hybrid free energy between the RNA sequence and the ASO, the Duplexfold web server was used (http://rna.urmc.rochester.edu/RNAstructureWeb/Servers/DuplexFold/DuplexFold.html).45 The Duplexfold server allows only intermolecular interaction and no intramolecular to determine the free energy.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Childs-Disney for help writing this manuscript and the agencies that funded this work including the National Institutes of Health (DP1-NS096898 to M.D.D., R00GM112877 to W.N.M., and F31-NS110269 to A.J.A.), the Muscular Dystrophy Association (grant 380467 to M.D.D.), and a Fullbright Fellowship (to R.I.B.). We thank the Roy J. Carver Charitable Trust (startup funds for W.N.M.). We also thank the University of Florida, Center for NeuroGenetics for their generous gift of the DM2 fibroblast cell line used in this study.
Footnotes
The authors declare the following competing financial interest(s): M.D.D is a founder of Expansion Therapeutics. M.D.D. and E.T.W are consultants for Expansion Therapeutics.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.9b00958.
Seventeen figures, four tables, and general synthetic procedures (PDF)
Contributor Information
Raphael I. Benhamou, The Scripps Research Institute, Jupiter, Florida
Alicia J. Angelbello, The Scripps Research Institute, Jupiter, Florida
Ryan J. Andrews, Iowa State University, Ames, Iowa.
Eric T. Wang, University of Florida, Gainesville, Florida
Walter N. Moss, Iowa State University, Ames, Iowa
Matthew D. Disney, The Scripps Research Institute, Jupiter, Florida.
REFERENCES
- (1).Stojic L, Lun ATL, Mangei J, Mascalchi P, Quarantotti V, Barr AR, Bakal C, Marioni JC, Gergely F, and Odom DT (2018) Specificity of RNAi, LNA and CRISPRi as loss-of-function methods in transcriptional analysis. Nucleic Acids Res. 46, 5950–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Stein CA, and Castanotto D (2017) FDA-approved oligonucleotide therapies in 2017. Mol. Ther. 25, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Disney MD (2019) Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc. 141, 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Disney MD, Dwyer BG, and Childs-Disney JL (2018) Drugging the RNA world. Cold Spring Harbor Perspect. Biol. 10 (pii), No. a034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Velagapudi SP, Gallo SM, and Disney MD (2014) Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol. 10, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Disney MD, Winkelsas AM, Velagapudi SP, Southern M, Fallahi M, and Childs-Disney JL (2016) Inforna 2.0: A Platform for the Sequence-Based Design of Small Molecules Targeting Structured RNAs. ACS Chem. Biol. 11, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Morgan BS, Forte JE, and Hargrove AE (2018) Insights into the development of chemical probes for RNA. Nucleic Acids Res. 46, 8025–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, and Miller BL (2008) Dynamic combinatorial selection of molecules capable of inhibiting the (CUG) repeat RNAMBNL1 interaction in vitro: Discovery of lead compounds targeting myotonic dystrophy (DM1). J. Am. Chem. Soc. 130, 16254–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Davidson A, Leeper TC, Athanassiou Z, Patora-Komisarska K, Karn J, Robinson JA, and Varani G (2009) Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl. Acad. Sci. U. S. A. 106, 11931–11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, and Al-Hashimi HM (2011) Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 7, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Garcia-Lopez A, Tessaro F, Jonker HRA, Wacker A, Richter C, Comte A, Berntenis N, Schmucki R, Hatje K, Petermann O, Chiriano G, Perozzo R, Sciarra D, Konieczny P, Faustino I, Fournet G, Orozco M, Artero R, Metzger F, Ebeling M, Goekjian P, Joseph B, Schwalbe H, and Scapozza L (2018) Targeting RNA structure in SMN2 reverses spinal muscular atrophy molecular phenotypes. Nat. Commun. 9, 2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sztuba-Solinska J, Chavez-Calvillo G, and Cline SE (2019) Unveiling the druggable RNA targets and small molecule therapeutics. Bioorg. Med. Chem. 27, 2149–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vo DD, Becquart C, Tran TPA, Di Giorgio A, Darfeuille F, Staedel C, and Duca M (2018) Building of neomycin−nucleobase−amino acid conjugates for the inhibition of oncogenic miRNAs biogenesis. Org. Biomol. Chem. 16, 6262–6274. [DOI] [PubMed] [Google Scholar]
- (14).Staedel C, Tran TPA, Giraud J, Darfeuille F, Di Giorgio A, Tourasse NJ, Salin F, Uriac P, and Duca M (2018) Modulation of oncogenic miRNA biogenesis using functionalized polyamines. Sci. Rep. 8, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Costales MG, Haga CL, Velagapudi SP, Childs-Disney JL, Phinney DG, and Disney MD (2017) Small Molecule Inhibition of microRNA-210 Reprograms an Oncogenic Hypoxic Circuit. J. Am. Chem. Soc. 139, 3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Costales MG, Matsumoto Y, Velagapudi SP, and Disney MD (2018) Small Molecule Targeted Recruitment of a Nuclease to RNA. J. Am. Chem. Soc. 140, 6741–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Costales MG, Suresh B, Vishnu K, and Disney MD (2019) Targeted Degradation of a Hypoxia-Associated Non-coding RNA Enhances the Selectivity of a Small Molecule Interacting with RNA. Cell Chem. Biol. 26, 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Angelbello AJ, Rzuczek SG, McKee KK, Chen JL, Olafson H, Cameron MD, Moss WN, Wang ET, and Disney MD (2019) Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U. S. A. 116, 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Benhamou RI, Angelbello AJ, Wang E, and Disney MD (2019) A toxic RNA catalyzes the cellular synthesis of its own inhibitor, shunting it to endogenous decay pathways. bioRxiv, 741926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Perdoni F, Malatesta M, Cardani R, Giagnacovo M, Mancinelli E, Meola G, and Pellicciari C (2009) RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type 2: an immunocytochemical study. Eur. J. Histochem. 53, No. 18. [DOI] [PubMed] [Google Scholar]
- (21).Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, Tran T, Sarkar P, Schatz GC, and Disney MD (2014) Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem. Biol. 9, 538–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, and Disney MD (2007) A small molecule microarray platform to select RNA internal loop-ligand interactions. ACS Chem. Biol. 2, 745–754. [DOI] [PubMed] [Google Scholar]
- (23).Llano-Sotelo B, Azucena EF Jr., Kotra LP, Mobashery S, and Chow CS (2002) Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 9, 455–463. [DOI] [PubMed] [Google Scholar]
- (24).Sznajder LJ, Thomas JD, Carrell EM, Reid T, McFarland KN, Cleary JD, Oliveira R, Nutter CA, Bhatt K, Sobczak K, Ashizawa T, Thornton CA, Ranum LPW, and Swanson MS (2018) Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. U. S. A. 115, 4234–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Savkur RS, Philips AV, and Cooper TA (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 29, 40–47. [DOI] [PubMed] [Google Scholar]
- (26).Lee MM, Pushechnikov A, and Disney MD (2009) Rational and modular design of potent ligands targeting the RNA that causes myotonic dystrophy 2. ACS Chem. Biol. 4, 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Li Y, and Disney MD (2018) Precise small molecule degradation of a noncoding RNA identifies cellular binding sites and modulates an oncogenic phenotype. ACS Chem. Biol. 13, 3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Boger DL, and Cai H (1999) Bleomycin: synthetic and mechanistic studies. Angew. Chem., Int. Ed. 38, 448–476. [DOI] [PubMed] [Google Scholar]
- (29).Moon MH, Hilimire TA, Sanders AM, and Schneekloth JS (2018) Measuring RNA−ligand interactions with microscale thermophoresis. Biochemistry 57, 4638–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R, and Disney MD (2017) Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 13, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Costales MG, Rzuczek SG, and Disney MD (2016) Comparison of small molecules and oligonucleotides that target a toxic, non-coding RNA. Bioorg. Med. Chem. Lett. 26, 2605–2609. [DOI] [PubMed] [Google Scholar]
- (32).Holt I, Mittal S, Furling D, Butler-Browne GS, David Brook J, and Morris GE (2007) Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells 12, 1035–1048. [DOI] [PubMed] [Google Scholar]
- (33).Burma S, Chen BP, Murphy M, Kurimasa A, and Chen DJ (2001) ATM phosphorylates histone H2AX in response to DNA double-strandb Breaks. J. Biol. Chem. 276, 42462–42467. [DOI] [PubMed] [Google Scholar]
- (34).Lee J, Bai Y, Chembazhi UV, Peng S, Yum K, Luu LM, Hagler LD, Serrano JF, Chan HYE, Kalsotra A, and Zimmerman SC (2019) Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc. Natl. Acad. Sci. U. S. A. 116, 8709–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Siboni RB, Nakamori M, Wagner SD, Struck AJ, Coonrod LA, Harriott SA, Cass DM, Tanner MK, and Berglund JA (2015) Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep. 13, 2386–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Childs-Disney JL, and Disney MD (2016) Approaches to validate and manipulate RNA targets with small molecules in cells. Annu. Rev. Pharmacol. Toxicol. 56, 123–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Connelly CM, Moon MH, and Schneekloth JS Jr. (2016) The emerging role of RNA as a therapeutic target for small molecules. Cell Chem. Biol. 23, 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Donlic A, and Hargrove AE (2018) Targeting RNA in mammalian systems with small molecules. Wiley Interdiscip. Rev. RNA 9, No. e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lima WF, Monia BP, Ecker DJ, and Freier SM (1992) Implication of RNA structure on antisense oligonucleotide hybridization kinetics. Biochemistry 31, 12055–12061. [DOI] [PubMed] [Google Scholar]
- (40).Mathews DH, Burkard ME, Freier SM, Wyatt JR, and Turner DH (1999) Predicting oligonucleotide affinity to nucleic acid targets. RNA 5, 1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Pinto BS, Saxena T, Oliveira R, Méndez-Gómez HR, Cleary JD, Denes LT, McConnell O, Arboleda J, Xia G, Swanson MS, and Wang ET (2017) Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell 68, 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rzuczek SG, Park H, and Disney MD (2014) A toxic RNA catalyzes the in cellulo synthesis of its own inhibitor. Angew. Chem., Int. Ed. 53, 10956–10959. [DOI] [PubMed] [Google Scholar]
- (43).Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Andrews RJ, and Moss WN (2019) Computational approaches for the discovery of splicing regulatory RNA structures. Biochim. Biophys. Acta, Gene Regul. Mech 1862, 194380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Reuter JS, and Mathews DH (2010) RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.