

SUMMARY
Myotonic dystrophy type 2 (DM2) is a genetically defined disease caused by a toxic expanded repeat of r(CCUG) [r(CCUG)exp], harbored in intron 1 of CCHC-Type Zinc-Finger Nucleic-Acid Binding-Protein (CNBP) pre-mRNA. This r(CCUG)exp causes toxicity via a gain-of-function mechanism, resulting in three pathological hallmarks: aggregation into nuclear foci; sequestration of muscleblind-like-1 (MBNL1) protein, leading to splicing defects; and retention of CNBP intron 1. We studied two types of small molecules with different modes of action, ones that simply bind and ones that are templated by r(CCUG)exp in cells, i.e., the RNA synthesizes its own drug. Indeed, our studies completed in DM2 patient-derived fibroblasts showed that the compounds disrupt the r(CCUG)exp-MBNL1 complex, reduce intron retention, subjecting the liberated intronic r(CCUG)exp to native decay pathways, and rescue other DM2-associated cellular defects. Importantly, this study shows that small molecules can modulate RNA biology by shunting toxic transcripts towards native decay pathways.
Keywords: Chemical Biology, Drug Design, Medicinal Chemistry, Nucleic Acids, RNA, Click Chemistry, Myotonic Dystrophy, Repeat Expansion Disorder
Graphical Abstract
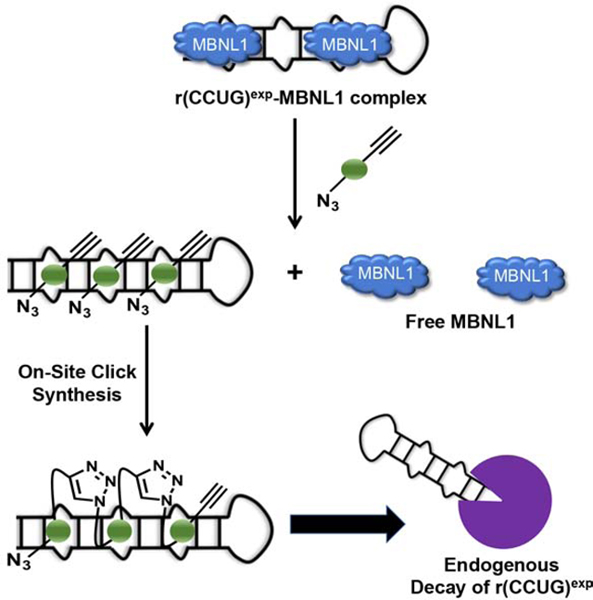
eTOC BLURB
RNA repeat expansions cause >30 diseases, and both oligonucleotides and small molecules have been developed to inhibit their dysfunction. Benhamou et al. show for the first time that small molecules targeting structured, disease-causing RNAs can shunt them towards native decay pathways by affecting their processing.
INTRODUCTION
A variety of human diseases are mediated by RNA structures (Bernat and Disney, 2015; Spitale et al., 2015; Wan et al., 2014), ideal targets for small molecules that could elucidate RNA biology or be exploited for therapeutic development (Connelly et al., 2016; Disney, 2019; Donlic and Hargrove, 2018). Estimates suggest that the number of potential human RNA drug targets are an order of magnitude greater than the number of protein targets (Connelly et al., 2016; Hangauer et al., 2013). Compounds that bind RNA have been identified by using screening (Chen et al., 2012; Gareiss et al., 2008; Lorenz et al., 2018), structure-based design (Davidson et al., 2009; Stelzer et al., 2011), or sequence-based design (Velagapudi et al., 2014). However, only a very limited number of these small molecules are bioactive and far fewer are known to target human RNAs and affect their function (Disney, 2019). As these tools are more fully developed and more broadly deployed, they will likely deliver many chemical probes to study human RNA biology.
RNA repeating transcripts are attractive targets for chemical probes, causing >30 genetically defined diseases with no known treatments (Ranum and Cooper, 2006). Although the RNA is the central toxin in each disease, their pathomechanisms and impact on biological processes are diverse. For example, repeats can cause transcriptional silencing by binding to promoters from which they are encoded, as has been observed in fragile X syndrome (FXS) (Colak et al., 2014). Toxic RNAs can be translated independently of a canonical start codon, in a process named repeat associated non-ATG (RAN) translation, as observed in C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia (c9ALS/FTD) (Zu et al., 2011). RNA repeats also cause toxicity by a gain-of-function mechanism in which they bind to and sequester proteins involved in RNA biogenesis, forming nuclear foci and causing defects in RNA processing, as observed in myotonic dystrophy types 1 and 2 (DM1 and DM2, respectively) (Liquori et al., 2001; Mankodi et al., 2000). More recently, various RNA repeats have been shown to affect the processing of the pre-mRNAs in which they reside, causing intron retention and providing potential biomarkers (Sznajder et al., 2018).
DM2 is caused by an expanded r(CCUG) repeat, or r(CCUG)exp, located in intron 1 of the CHC-Type Zinc Finger Nucleic Acid Binding Protein (CNBP) pre-mRNA. Like other RNA repeat expansions, the DM2 RNA is highly structured and forms a periodic array of 5’CCUG/3’GUCC 2×2 internal loops (Figure 1A). We previously designed two types of small molecules that recognize this repeating structure and improve DM2-associated defects (Childs-Disney et al., 2014; Lee et al., 2009b; Rzuczek et al., 2014). In particular, they rescue the aberrant splicing of bridging integrator 1 (BIN1) exon 11 and the formation of nuclear foci in a mouse myoblast cell line transfected with a plasmid encoding r(CCUG)300 (Childs-Disney et al., 2014; Rzuczek et al., 2014). The first compound, a dimer comprised of two RNA-binding modules, recognizes and binds two adjacent loops simultaneously, which affords avidity and selectivity (Childs-Disney et al., 2014). The second class of compounds are synthesized in cellulis and only in DM2-affected cells (Rzuczek et al., 2014); that is, r(CCUG)exp synthesizes its own inhibitor. In cell, on-site drug synthesis is enabled by appending the RNA-binding modules that target the 2×2 internal loops in r(CCUG)exp with azide and alkyne units that are unreactive in biological systems unless brought into high effective concentration or proximity by binding adjacent sites in r(CCUG)exp (Rzuczek et al., 2014).
Figure 1: The RNA that causes DM2, r(CCUG)exp, has multiple modes of toxicity.

(A) r(CCUG)exp in intron 1 of CNBP folds into a hairpin displaying a periodic array of internal loops that sequester MBNL1. Sequestration of MBNL1 by r(CCUG)exp causes (B) pre-mRNA splicing defects, for example increasing IR exon 11 exclusion and (C) aberrant retention of CNBP intron 1. (D) MBNL1 knock-down using siRNA and its effect on relative abundance of MBNL1, CNBP intron 1, and CNBP mature mRNA as measured by RT-qPCR. (E) MBNL1 knock-in using a transfected MBNL1 plasmid and its effect on relative abundance of MBNL1, CNBP intron 1, and CNBP mature mRNA as measured by RT-qPCR. Error bars represent SD. **P < 0.01, ***P < 0.001, ****P < 0.0001, as determined by a one-way ANOVA by comparison to untreated cells (“0”; n = 3). See also Figure S1.
Herein, we have evaluated these compounds in DM2 patient-derived fibroblasts for alleviating alternative pre-mRNA splicing defects, nuclear foci, and CNBP intron 1 retention. Intron retention caused by RNA repeat expansions was only recently discovered (Sznajder et al., 2018). Therefore, little is known about the mechanism of intron retention, whether this mechanism is unique to each repeat, or its relevance to disease. Importantly, the studies herein revealed that intron retention in DM2-affected cells is caused by MBNL1 and that small molecules can alleviate this defect by shunting the intron harboring the repeat expansion down endogenous decay pathways. Indeed, facilitating removal of the RNA toxin from the cell alleviated disease phenotypes as it both liberates MBNL1 to resume its normal function and prevents formation of nuclear foci. These studies are likely applicable to other repeat expansions harbored in introns but could provide a general strategy to direct malfunctioning RNAs down endogenous decay pathways.
RESULTS
Intron retention in DM2 patient-derived fibroblasts.
As aforementioned, r(CCUG)exp has at least three pathomechanisms: (i) formation of nuclear foci when it binds various RBPs; (ii) pre-mRNA splicing defects in MBNL1-regulated genes (Childs-Disney et al., 2007; Childs-Disney et al., 2014; Lee et al., 2009b; Llano-Sotelo et al., 2002); and, (iii) retention of intron 1 in CNBP mRNA, in which it is harbored (Sznajder et al., 2018) (Figure 1). The first two mechanisms were identified shortly after the discovery of the repeat expansion, facilitated by in-depth studies completed for the repeat that causes DM1. Intron 1 retention was recently discovered by the Swanson laboratory in which they used RT-PCR primers specific for the intron, which amplify both pre-mRNA species [i1-E2-i2-E3-i3-E4 and i1-E2-E3-i3-E4 where i indicates intron and E indicates exon] as well as mature mRNAs that have retained the intron [i1-E2-E3-E4] (Figure S1). In wild type (WT), healthy fibroblasts, the i1-E2-E3-E4 species comprises ~10% of all intron 1-containing CNBP transcripts. In DM2-affected fibroblasts, however, the intron retained species (i1-E2-E3-E4) is present at aberrant levels, ~40% of intron 1-containing transcripts. Since little is known about the mechanism of intron retention for any RNA repeat expansion, we sought to use our chemical probes to provide insight, hypothesizing that CNBP intron 1 retention is stimulated by MBNL1 binding to r(CCUG)exp (Figure 1C).
To study the role of MBNL1 in these events, a series of gain- and loss-of-function experiments were completed in DM2 patient-derived fibroblasts. When an siRNA directed at MBNL1 was delivered to DM2 fibroblasts, an ~65% decrease in the expression of MBNL1 was observed, leading to a significant decrease in intron 1 retention (Figures 1D & S1). In contrast, no effect was observed when cells were treated with a control, scrambled siRNA (Figure 1D).
To further support the role of MBNL1 in intron retention, a plasmid encoding MBNL1 was transfected into DM2 fibroblasts. An increase in the amount of intron retained product was observed upon overexpression of MBNL1 but not with a control plasmid (Figure 1E & S1). Collectively, both studies support a role for MBNL1 binding to r(CCUG)exp in the formation of intron retained products.
A designed dimeric compound (1) that targets the structure of r(CCUG)exp reduces nuclear foci and rescues alternative splicing.
Previously, a series of monomeric and dimeric ligands were designed that target the structure of r(CCUG)exp (Figure 2A) (Childs-Disney et al., 2014; Pushechnikov et al., 2009). These compounds are built on a kanamycin A scaffold in which the 6’ position is acylated. The consequence of this modification is that the compounds bind specifically to the internal loops in r(CCUG)exp and have no measurable affinity to the A-site in the bacterial ribosome (Childs-Disney et al., 2007; Childs-Disney et al., 2014; Lee et al., 2009b), a target of aminoglycosides (Llano-Sotelo et al., 2002). Based on these previous studies, a compound with two kanamycin modules, 1, was designed that binds two 5’CCUG/3’GUCC internal loops simultaneously (Figure 2B). The compound binds to r(CCUG)exp with low nanomolar affinity and selectively discriminates against a number of other RNA targets (Lee et al., 2009a; Lee et al., 2009b). Further, 1 rescued DM2-associated splicing defects and reduced the number of nuclear foci in a transfected model system, as DM2 patient-derived cells were not available (Childs-Disney et al., 2014). Studies on intron retention were not possible in this cellular model as only the repeats themselves were expressed, outside the context of the CNBP transcript. Notably, increasing the valency of 1 from two to three only modestly improved potency in cellulis despite significant gains in avidity and in vitro potency (Childs-Disney et al., 2014; Lee et al., 2009a). This effect can be traced to differences in their cellular permeability and subcellular localization as 1 localized to both the cytoplasm and nucleus (the latter of which is where the target is located) while the trimer is mainly present in the perinuclear region and cytoplasm; that is, the trimer co-localized with the RNA target to a lesser extent than the dimer (Lee et al., 2009a).
Figure 2: Design and biological impact of small molecules targeting r(CCUG)exp.

(A) Structures of a kanamycin derivative (green spheres) that avidly binds the internal loops found in r(CCUG)exp and a dimer composed of two kanamycin binding modules connected via a propylamine peptoid (1). (B) Schematic of 1’s mode of action, binding to r(CCUG)exp and releasing MBNL1. (C) Structures of compounds employed in an on-site drug synthesis approach. Compound 2, a modified kanamycin module, contains alkyne and azide moieties that react upon binding r(CCUG)exp, the catalyst. Compound 3 is a kanamycin module containing only the alkyne component and thus cannot oligomerize. (D) Schematic of on-site click synthesis of 2, catalyzed by binding to r(CCUG)exp, and release MBNL1 to relieve DM2-associated defects.
We therefore extended those studies to DM2 patient-derived cells, investigating 1’s ability to rescue formation of nuclear foci, pre-mRNA splicing defects, and importantly intron retention. Indeed, treatment with 1 reduced formation of nuclear foci, with a statistically significant effect observed at 10 μM (P <0.01; Figure 3A&B). Notably, both the presence of r(CCUG)exp and MBNL1 in foci were assessed in our analysis, indicating liberation of MBNL1 from r(CCUG)exp.
Figure 3: Biological activity of 1 in DM2 patient-derived fibroblasts.

(A) Representative microscopic images for the reduction of nuclear foci by 1, as completed by RNA-FISH and immunohistochemistry using an anti-MBNL1 antibody. (B) Quantification of r(CCUG)exp-MBNL1 foci/nucleus (n = 3 biological replicates, 40 nuclei counted per replicate). (C) Effect of 1 on IR pre-mRNA splicing regulated by MBNL1. (D) Effect of 1 on CNBP pre-mRNA splicing. (E) Effect of 1 on an MBNL1-regulated alternative splicing event, IR exon 11, as determined by RT-PCR; (F) Effect of 1 on aberrant CNBP pre-mRNA splicing caused by r(CCUG)exp, as determined by RT-PCR; that is, retention of intron 1; (G) Effect of 1 on total intron 1 levels and CNBP mature mRNA, as determined by RT-qPCR. Error bars represent SD. *P < 0.5, **P < 0.01, ***P < 0.001, ****P < 0.0001, as determined by a one-way ANOVA (n = 3). See also Figure S2.
The alternative splicing of insulin receptor (IR) exon 11 is controlled by MBNL1 and as such is aberrantly spliced in DM2-affected cells. In particular, exon 11 is excluded too frequently in DM2 fibroblasts (exon 11 is excluded in ~43% of transcripts vs. ~29% of transcripts from WT fibroblasts; Figure 1B) (Savkur et al., 2001). Thus, for example to rescue splicing by 50%, a compound would have to decrease exon 11 exclusion by ~7%. Compound 1 improved IR exon 11 alternative splicing defects dose-dependently while not affecting splicing in WT fibroblasts (Figures 3C, 3E, S2). Additionally, no effect was observed on the alternative splicing of mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) exon 22a, which is not MBNL1-dependent (Figure S2).
Designer compounds that target the structure of r(CCUG)exp decrease intron retention.
Two lines of experimental evidence suggest that 1 liberates a fraction of MBNL1 such that it can resume its normal function: (i) reduced nuclear foci containing both r(CCUG)exp and MBNL1 and (ii) improvement of a MBNL1-dependent splicing defect. We therefore hypothesized that 1 might also rescue MBNL1-dependent intron retention. Indeed, 1 reduced the amount of intron 1 retained in CNBP mRNA in DM2 fibroblasts dose-dependently as measured by RT-PCR (~20% reduction when cells were treated with 10 μM of 2, the highest concentration tested), indicating that the small molecule rescued aberrant CNBP pre-mRNA splicing (Figure 3D, 3F, and S2).
Intronic regions are subjected to decay when liberated from their pre-mRNAs (Hesselberth, 2013). Thus, we tested the possibility that 1 could stimulate endogenous decay of the freed intron containing r(CCUG)exp(Hesselberth, 2013). By using RT-qPCR primers for intron 1, we found that ~20% of the intron is eliminated from cells (Figure 3G). Importantly, 1 has no effect on CNBP mature mRNA levels (Figure 3G & S2) and did not affect the abundance of intron 1 in WT cells (Figure S2). Thus, binding of the ligand affects the processing of the pre-mRNA in a manner that then eliminates the toxic repeat. Collectively, these data further support the hypothesis that MBNL1 sequestration stimulates intron 1 retention of CNBP while simultaneously demonstrating a new activity for small molecules that bind RNA targets, subjecting the RNA to quality control pathways to facilitate its degradation.
The toxic r(CCUG)exp synthesizes its own inhibitor in patient-derived cells.
Although binding compound 1 rescued three hallmarks of DM2, its activity is not as robust as desired. We previously reported an approach to synthesize oligomeric compounds with enhanced potency (~100-fold more potent than the simple binding compound) and selectivity in a cell line transfected with r(CCUG)300 (Rzuczek et al., 2014). The approach, inspired by Sharpless to use click chemistry to assemble compounds on acetylcholinesterase in vitro (Lewis et al., 2002), was also used by the Dervan group to assemble polyamides on a DNA minor groove in vitro (Poulin-Kerstien and Dervan, 2003). We demonstrated that RNA repeats can template the synthesis of their own inhibitors using a click reaction in cellulis, including in DM1 patient-derived cells (Rzuczek et al., 2017; Rzuczek et al., 2014). Notably, the clickable compounds developed for DM1 and DM2 are the most potent RNA-targeting small molecules identified to date with pM and nM EC50’s for rescuing disease-associated defects in cells, respectively, and are 50,000-fold more potent than the corresponding repeat-targeting oligonucleotides (Costales et al., 2016; Rzuczek et al., 2017).
Briefly, for r(CCUG)exp-catalyzed on-site, in cell drug synthesis, a kanamycin moiety was appended with biorthogonal azide and alkyne moieties, affording 2 (Figure 2C). When 2 binds adjacent 2×2 nucleotide internal loops in r(CCUG)exp, the azide displayed by one 2 molecule is brought into close proximity with the alkyne of another 2 molecule, reacting to form stable triazole units and hence multimeric compounds (Figure 2D). This reaction is catalyzed by the RNA and does not require copper(I). We therefore studied if on-site drug synthesis using 2 is also operational in DM2 fibroblasts and occurs selectively; that is, on-site synthesis should not occur in fibroblasts lacking r(CCUG)exp (i.e., WT). For these studies, DM2 and WT fibroblasts were co-treated with 2 and 3, a kanamycin derivative that only contains an alkyne moiety. Compound 3 limits the extent of polymerization, thereby allowing mass spectrometry to be used to assess reactivity in cells. Indeed, mass spectral analysis of lysates from treated DM2 fibroblasts indicated formation of both trimer and tetramer products, in agreement with our previously reported studies in transfected cells (Figure S3) (Rzuczek et al., 2014). Importantly, oligomerization was not observed in WT fibroblasts treated with 2 and 3, since no r(CCUG)exp is present to catalyze the click reaction (Figure S3).
In cell synthesized compounds reduce foci and rescue splicing defects more potently than 1.
To compare the potency of 1 to compounds synthesized on-site derived from 2, we evaluated the ability of 2 to reduce the number of r(CCUG)exp-MBNL1 foci in cells. On average, DM2 fibroblasts contain 10.6 ± 0.9 foci per cell. Upon treatment with 1 μM of 2, the number of foci was reduced by ~45% (5.7± 0.9) (Figure 4A&B), a significant improvement over 1, which reduced the number of foci by only ~20% at 10 μM concentration (Figure 3A&B). As a control, DM2 fibroblasts were treated with 3, which cannot oligomerize. No significant decrease in foci was observed (Figure S3).
Figure 4: Biological activity of 2 in DM2 patient-derived fibroxblasts.

(A) Representative microscopic images for the reduction of nuclear foci by 2, as completed by RNA-FISH and immunohistochemistry using an anti-MBNL1 antibody. (B) Quantification of r(CCUG)exp-MBNL1 foci/nucleus (n = 3 biological replicates, 40 nuclei counted per replicate). (C) Effect of 2 on IR pre-mRNA splicing regulated by MBNL1. (D) Effect of 2 on CNBP pre-mRNA splicing. (E) Effect of 2 on an MBNL1- regulated alternative splicing event, IR exon 11, as determined by RT-PCR; (F) Effect of 2 on aberrant CNBP pre-mRNA splicing caused by r(CCUG)exp, as determined by RT-PCR; that is, retention of intron 1; (G) Effect of 2 on total intron 1 levels and CNBP mature mRNA, as determined by RT-qPCR. Error bars represent SD. *P < 0.5, **P < 0.01, ***P < 0.001, as determined by a one-way ANOVA (n = 3). See also Figure S3.
Given these encouraging results, we tested the ability of 2 to correct the IR alternative pre-mRNA splicing defect (Figures 4C & 4E). Notably,~45% rescue of IR exon 11 splicing was observed at 1 μM of 2 (Figure 4E & S3), compared to only ~20% rescue with the dimer 1 at 10 μM (Figure 3C). Control compound 3 showed no significant rescue of the IR splicing defects in DM2 fibroblasts, as expected (Figure S3). Additionally, 2 does not affect IR alternative splicing in WT fibroblasts (Figure S3) nor splicing of MAP4K4 exon 22a in DM2 fibroblasts (Figure S3).
In cell synthesized oligomers derived from 2 decrease intron retention more potently than 1.
To further confirm that compounds derived from the oligomerization of 2 liberate MBNL1 from r(CCUG)exp to improve DM2 pathology, we studied its ability to rescue the aberrant splicing of CNBP pre-mRNA and to activate the cellular decay of the liberated intron containing r(CCUG)exp, as observed for 1. Clickable compound 2 reduced the amount of intron 1 retained in CNBP mRNA dose dependently, with an ~30% reduction observed upon treatment with 1 μM (Figure 4D & 4F), an ~10-fold improvement over 1 (Figure 3F & S3). Likewise, using RT-qPCR primers for intron 1 itself, 2 reduced overall intron 1 levels dose-dependently with an ~40% reduction at 1 μM (Figure 4G). Importantly, this reduction can be traced directly to rescue of aberrant CNBP splicing as levels of mature CNBP mRNA were not affected (Figure 4G). Further, 2 did not affect the abundance of intron 1 in WT cells (Figure S3). For comparison, 1 only reduced intron 1 levels by ~20% when DM2 fibroblasts were treated with 10 μM compound.
Factors contributing to the enhanced potency of molecules synthesized on-site.
Our mass spectral data indicated that at least trimers and tetramers were formed in DM2 fibroblasts, and that they are the source of the improved potency of 2 vs. 1 for rescue of all three defects studied. However, the dimer derived from 2 (4; Figure S4) could also contribute to this enhancement, as its linker is different than that in 1. We therefore synthesized and tested 4 for its ability to affect intron retention in cells. Both dimers decreased intron retention to a similar extent, by ~20% at 10 μM dose (Figure S4), indicating that the on-site synthesis of only a dimer would not significantly improve potency. Further, differences in cellular permeability could account for 2 inhibiting DM2-associated molecular pathology. However, our previous studies have shown that both 3 and 6”-azido kanamycin A (5) are ~7-fold less permeable than 1 (Lee et al., 2011), and both 3 and 5 are inactive in DM2 fibroblasts (Figure S4). Collectively, these data demonstrate that on-site drug synthesis converts an inactive compound into a potent modulator of function due to oligomerization in cells expressing r(CCUG)exp.
DISCUSSION
Two significant challenge have precluded exploiting RNA as a drug target: (i) the lack of approaches and rules that govern their molecular recognition with non-oligonucleotide-based modalities; and (ii) a companion set of data that defines which RNA-small molecule binding events elicit a biological response (Disney et al., 2018). Various tools have emerged to study molecular recognition events and to validate RNA targets in vitro and in cellulis, which have begun to provide these much needed data (Disney, 2019). Such tools include small molecules with novel activities in situ and in vivo that directly and selectively cleave the RNA target (Angelbello et al., 2019) or recruit cellular nucleases to effect their degradation (Costales et al., 2018).
Of particular interest, this study has shown that a toxic RNA can template the synthesis of its own inhibitor in patient-derived cells to rescue multiple disease mechanisms. The observation that 2 can be transformed into a bioactive oligomeric species upon binding to adjacent sites in r(CCUG)exp suggests that disease-affected cells can be provoked to manufacture a cellular active inhibitor from an inactive monomer (3 in this case) at the needed site of action. Healthy cells that have the same genetic makeup but do not express the disease-causing RNA target were therefore unexposed to the active compounds, as oligomerization did not occur. Many microsatellite disorders, such as c9ALS/FTD, are caused by RNAs that are expressed in the brain. An on-site synthesis approach could therefore be advantageous, providing low molecular weight, tissue and cell penetrant compounds that can engage the disease-causing targets and be assembled into potent modulators of RNA function in situ.
A second aspect of special interest is the observation that RNA repeat expansions harbored in introns may be especially sensitive to small molecule intervention, the activity of which is then interfaced with endogenous RNA quality control decay mechanisms. A variety of other RNA repeating transcripts are present in introns and form intron retained products. A previous study showed that GC-rich repeats harbored in introns [such as r(CCUG)exp] cause the greatest extent of retention (as compared to AU-rich repeats), indicating a potential role for protein loading in intron retention observed in each disease (Sznajder et al., 2018). These GC-rich repeat expansions, which cause c9ALS/FTD [r(G4C2)exp] repeat expansions, Fuchs endothelial corneal dystrophy [FECD; r(CUG)exp] and others, would be high priority to complete similar intron-retention studies, as chemical probes have already been developed (Rzuczek et al., 2017; Rzuczek et al., 2015; Su et al., 2014; Wang et al., 2019). Clearly, such studies should be completed in patient-derived cell lines whenever possible.
Collectively, these studies show that there are a great many ways to affect RNA function that can be leveraged for therapeutic development. Further, systems that model native processing will be required to understand and capture the full potential of compounds to target RNA and rescue disease biology. The development of chemical probes and lead medicines targeting structured RNA, particularly human RNAs, are in their infancy. It will be interesting to see the types of chemical structures, their features, and the functional responses that emerge.
Significance.
RNA is the central toxin in many diseases, particularly microsatellite disorders caused by RNA repeat expansions. Repeat expansions can be located in open reading frames as observed in Huntington’s disease and spinocerebellar ataxias, untranslated regions as observed in DM1 and fragile X-associated tremor ataxia syndrome, and introns disorders such as DM2 and amyotrophic lateral sclerosis and frontotemporal dementia (c9ALS/FTD). Microsatellite disorders can share disease mechanisms or have unique mechanisms, depending the sequence, structure, and location of the RNA repeat expansion within a transcript. Recently, it was discovered that expanded RNA repeats harbored introns, particularly those that are GC rich, cause aberrant splicing of the transcript in which they are embedded (Sznajder et al., 2018). Here, we have uncovered for the first time a cause of intron retention that operates DM2-affected cells, binding of MBNL1. It is likely that a similar mechanism causes intron retention in other diseases, and our studies provide a framework for such investigations. Further, we have shown that small molecule lead medicines can bind an aberrant RNA structure, which templates the synthesis of a potent inhibitor on-site in patient-derived fibroblasts. This molecular recognition event shunts the toxic RNA down natural decay pathways to eliminate it from disease-affected cells. Such an approach could be effective for other RNA repeat expansions in particular and disease-causing RNAs in general.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
All requests for additional information and reagents should be directed to the Lead Contact, Matthew D. Disney (disney@scripps.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines:
DM2 patient-derived fibroblasts were obtained from the University of Florida, Center for NeuroGenetics (DM11, male, 55 years old at sampling ). Fibroblasts from a healthy donor (wild type; WT) were obtained from the Coriell Institute (GM07492; male, 17 years old at sampling).
Authentication of cell lines:
Fibroblasts obtained from the Coriell Institute were used without further authentication. DM2 patient-derived fibroblasts were confirmed to have r(CCUG)exp by Southern blotting and were compared to Southern blot analysis of lymphoblastoid cells obtained from the same patient. Both types of fibroblasts were confirmed to be free of mycoplasma contamination.
Cell culture maintenance:
All cells were maintained at 37 °C with 5% CO2. DM2 fibroblasts were cultured in 1× DMEM, High Glucose medium (HyClone) supplemented with 20% fetal bovine serum (FBS; Sigma), and 1% antibiotic-antimycotic solution (Corning). WT fibroblasts were cultured in MEM (Corning) supplemented with 10% FBS, 1% Glutagro (Corning), and 1% antibiotic-antimycotic solution.
Animals, human subjects, microbe strains, primary cell cultures: N/A
METHOD DETAILS
Treatment of fibroblasts with siRNAs or compounds.
Cells were transfected with EGFP (empty vector control) and MBNL1 plasmids (to overexpress MBNL1) in 6-well plates with Lipofectamine 3000 per the manufacturer’s protocol. After 5 h, the medium was replaced with growth medium, and the cells were grown for an additional 24 h. Transfection of fibroblasts with siRNAs, wheter siRNA-MBNL1 (Sense:5’-CACUGGAAGUAUGUAGAGAdTdT-3’; Antisense: 5’-UCUCUACAUACUUCCAGUGdTdT-3’; both purchased from Dharmacon) or a control siRNA (purchased from Santa S-39 Cruz Biotechnology; control siRNA catalog #: sc-37007) was completed using Lipofectamine RNAiMAX Reagent per the manufacturer’s protocol for 48 h.
Cells were treated with compounds 1 – 5 at the indicated concentration in growth medium for 48 h at 37 °C with 5% CO2.
Analysis of abundance of CCUG-containing transcripts:
Cells were grown in 6-well plates and treated or transfected as described in “Cell Culture”. After 48 h, the cells were lysed, and total RNA was harvested using a Zymo Quick RNA miniprep kit. Approximately 1 μg of total RNA was reverse transcribed using a qScript cDNA synthesis kit (20 μL total reaction volume, Quanta BioSciences). A 2 μL aliquot of the RT reaction was used for each primer pair for qPCR, which was completed with SYBR Green Master Mix on an Applied Biosystems 7900HT Fast Real-Time PCR System. Relative abundance of each transcript was calculated by normalizing to GAPDH.
Western blotting:
Western blotting was used to determine the expression of MBNL1 protein as previously described (Li and Disney, 2018). Briefly, DM2 cells, grown in 6-well plates (~50% confluency), were transfected with siRNAs directed against MBNL1 or a scrambled control siRNA as described above. Approximately 24 h later, total protein was extracted using M-PER Mammalian Protein Extraction Reagent (Pierce Biotechnology) following the manufacturer’s protocol and quantified using a Micro BCA Protein Assay Kit (Pierce Biotechnology). Approximately 50 μg of total protein was separated on a 10% SDS-polyacrylamide gel, and then transferred to a PVDF membrane. The membrane was washed with 1× Tris-buffered saline (TBS) and then blocked in 1× TBST (1× TBS containing 0.1% Tween-20) containing 5% (w/v) milk for 1 h at room temperature. After washing with 1× TBST, the membrane was incubated with a 1:2000 dilution of anti-MBNL1 antibody (Clone 4A8; catalog number: MABE70; EMD Millipore) in 1× TBST containing 5% milk overnight at 4 °C. The membrane was then washed with 1× TBST and incubated with 1:3000 anti-mouse IgG horseradish-peroxidase secondary antibody conjugate (catalog number: 7076; Cell Signaling Technology) in 1×TBS for 2 h at room temperature. The membrane was again washed with 1× TBST, and protein expression was quantified using SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology) per the manufacturer’s protocol. To quantify Vinculin expression, used for normalization, the membrane was stripped using 1× Stripping Buffer (200 mM glycine, pH 2.2 and 0.1% SDS) followed by washing in 1× TBST. The membrane was blocked and probed for Vinculin as described above for MBNL1 using a 1:5000 dilution of anti-Vinculin antibody (E1E9V; catalog number: 13901S; Cell Signaling Technology) at room temperature for 2 h and 1:5000 anti-rabbit IgG horseradish-peroxidase secondary antibody conjugate (catalog number: 7074; Cell Signaling Technology) in 1× TBS for 1 h at room temperature. The fold change of MBNL1 expression was calculated by normalizing MBNL1 band intensity to Vinculin band intensity using ImageJ.
Evaluation of nuclear foci using fluorescence in situ hybridization (FISH):
FISH was used to determine the small molecules’ effects on formation and disruption of nuclear foci as previously described (Rzuczek et al., 2017). Briefly, DM2 fibroblasts were grown to ~40% confluence in a Mat-Tek 96-well glass bottom plate in growth medium. Cells were then treated with 1, 2 or 3 for 48 h. To fix the cells, the compound-containing growth medium was removed, the cells were washed with 1× DPBS, and 100 μL of 4% formaldehyde in 1× DPBS was added. After incubating at 37°C for 10 min, the cells were washed five times with 1× DPBS at 37°C for 2 min and then twice with 100 μL of 0.1% Triton X-100 in 1× DPBS for 5 min at 37°C. Next, 100 μL of 30% formamide in 2× SSC buffer was added, and the cells were incubated for 10 min at room temperature. The FISH probe, 5’-Cy3-(CAGG)10 (140 nM; purchased from Integrated DNA Technologies, Inc.) (Margolis et al. 2006) was added to each well in 2× SSC containing 30% formamide, 2 μg/μL BSA, 1 μg/μL yeast tRNA, and 2 mM vanadyl complex, and the cells were incubated at 37 °C overnight. The cells were then washed with 100 μL of 2× SSC containing 30% formamide at 37 °C for 30 min and then 100 μL of 2× SSC buffer for 30 min at 37°C. For MBNL1 immunostaining, 20 μL of 1:5 anti-MBNL1 in 2× SSC was added, and the cells were incubated at 37 °C for 1 h. The cells were washed three times with 100 μL of 1× DPBS containing 0.1% Triton X-100 for 5 min at 37 °C and then incubated with 1:200 dilution of anti-mouse IgG Dylight 488 (catalog number: PI35502; Thermo Fisher/Pierce) in 2× SSC at 37°C for 1 h. The cells were then washed three times with 100 μL of 1× DPBS containing 0.1% Triton X-100 for 5 min at 37 °C and then with 1× DPBS for 5 min at 37°C. Finally, 100 μL of 1 μg/mL DAPI was added, and cells were incubated for 5 min at 37 °C. After washing with 1× DPBS twice, the cells were placed in 100 μL of 1× DPBS and imaged using an Olympus Fluoview 1000 confocal microscope at 100× magnification.
Evaluation of pre-mRNA splicing:
Cells were grown in 6-well plates and treated as described in “Cell Culture”. After 48 h, the cells were lysed, and total RNA was harvested using a Zymo Quick RNA miniprep kit. Approximately 1 μg of total RNA was reverse transcribed using a qScript cDNA synthesis kit (20 μL total reaction volume, Quanta BioSciences); 2 μL of the RT reaction was used for PCR amplification using GoTaq DNA polymerase (Promega). RT-PCR products were observed after 30 cycles of: 95 °C for 30 s; 58°C for 30 s; and 72°C for 1 min followed by an additional extension at 72 °C for 5 min. Products were separated on a 2% agarose gel (110 V for 1 h in 1× TBE buffer), visualized by staining with ethidium bromide, and imaged using a Typhoon 9410 variable mode imager. Gels were quantified using ImageJ. Percent rescue was calculated by dividing the difference between treated and untreated DM2 samples by the difference between untreated DM2 and WT samples (Equations 1 & 2):
| (Eq. 1) |
| (Eq. 2) |
Identification of in cellulis clicked products by mass spectrometry.
DM2 fibroblasts were grown in 100 mm dishes in growth medium. Cells were treated with 25 μM of 2 and/or 25 μM of 3. Compound 3 was added to limit the molecular weight of the oligomeric products in order to enable detection by LC-MS. After 48 h, the cells were scraped from the plate and lysed by freezing and thawing in 10% water in acetonitrile. The thawed lysate was concentrated and re-suspended in 1 mL of 10% water in acetonitrile. Insoluble cellular debris was pelleted, and the supernatant was used for mass spectral analysis. Mass spectrometry was performed with an Applied Biosystems MALDI ToF/ToF Analyzer 4800 Plus using an α-cyano-4-hydroxycinnamic acid matrix.
Compounds synthesis
Compounds 1 – 5 were synthesized as previously described and dissolved in Nanopure water (Lee et al., 2009b; Rzuczek et al., 2014). A description for the synthesis of each compound is provided below.
Compound 1:
Compound 1 was synthesized as previously described (Lee et al., 2009b). Briefly, Kanamycin-6’-N-hexynoate (1.5 mg, 2.6 μmol, 2 eq) was clicked to a peptoid linker with two azide moieties (1 mg, 1.3 μmol, 1 eq) using copper (I) catalyst (0.7 mg, 1.3 μmol, 1 eq), N,N-diisopropylethylamine (0.2 μL, 1.3 μmol, 1 eq) in 200 μL of DMF. The reaction was stirred at 60 °C overnight and purified via HPLC, performed with a linear gradient from 0% to 100% B [methanol + 0.1% trifluoroacetic acid (TFA)] in A (water + 0.1% TFA) over 60 min and a flow rate of 5 mL/min. tR = 31 min.
Compound 2:
Compound 2 was synthesized as previously described (Rzuczek et al., 2014). Briefly, 6”-azido-Kanamycin (10 mg, 19.6 μmol, 1.1 eq) was coupled to a N-(6-hexynoyloxy)-5-norbornene-2,3-dicarboximide (4.7 mg, 17.2 μmol, 1 eq) by stirring in 2:1 acetonitrile : water for 24 h at room temperature. The solvent was evaporated in vacuo and purified via HPLC, performed as described for 1. tR = 23 min.
Compound 3:
Compound 3 was synthesized as previously described (Lee et al., 2009b). Briefly, the 6’ primary amine of Kanamycin sulfate (500 mg, 0.8 mmol, 1.1 eq) was coupled to a N-(6-hexynoyloxy)-5-norbornene-2,3-dicarboximide (212 mg, 0.78 mmol, 1 eq), and stirred in 2:1 acetonitrile : water with potassium carbonate (215 mg, 1.5 mmol, 2 eq) for 24 h at room temperature. The solvent was evaporated in vacuo, and the crude material was purified by column chromatography on SiO2, using 10% NH4OH in MeOH/DCM 10:90 as the eluent to afford 3.
Compound 4:
Compound 4 was synthesized as previously described (Rzuczek et al., 2014). Briefly, 6”-azido-Kanamycin (1 mg, 1.9 μmol, 1.1 eq) was clicked to a Kanamycin-6’-N-hexynoate (0.9 mg,1.7 μmol, 1 eq) using copper (I) catalyst (0.9 mg, 1.7 μmol, 1 eq) and N,N-Diisopropylethylamine (0.3 μL, 1.7 μmol, 1 eq) in 200 μL of DMF. The reaction was stirred at 60 °C, overnight and purified via HPLC as described for 1. tR = 37 min
Compound 5:
Compound 5 was synthesized as previously described (Rzuczek et al., 2014). Briefly, to NH-Boc-6’’-O-trisyl-Kanamycin (10 mg, 8.6 μmol, 1 eq) in 1mL DMF was added sodium azide (2.7 mg, 43 μmol, 5 eq), and the reaction was stirred under argon at 70 °C overnight. Upon completion, the solvent was evaporated in vacuo, and the crude material was purified by column chromatography on SiO2, using MeOH/DCM 6:94 as eluent to afford the NH-Boc-protected compound. Deprotection was carried out using 1mL TFA for 5 min, which was then removed under reduced pressure.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are reported as means with error bars representing S.D., unless noted otherwise. Experiments were completed with at least n = 3. Data were plotted and statistical significance (one-way ANOVA) were calculated using GraphPad Prism 7 software. Details are provided in the figure legends.
Methods to determine whether data met assumptions of the statistical approach: N/A
DATA AND CODE AVAILABILITY
This study did not generate datasets or codes.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-MBNL1 | EMD Millipore Corporation | CAT# MABE70 |
| anti-mouse IgG Dylight 488 | Thermo Scientific Pierce | CAT# PI35502 |
| anti-mouse IgG horseradish-peroxidase | Cell Signaling Technology | CAT# 7076 |
| anti-rabbit IgG Vinculin (E1E9V) | Cell Signaling Technology | CAT# 13901 |
| anti-rabbit IgG horseradish-peroxidase | Cell Signaling Technology | CAT# 7074 |
| Critical Commercial Assays | ||
| Quick-RNA Miniprep Kit | Zymo | CAT# R1055 |
| qScript cDNA synthesis | Quanta BioSciences | CAT# 101414–100 |
| SYBR Green Master Mix | Life Technologies | CAT# 4368708 |
| GoTaq DNA polymerase | Promega | CAT# M8295 |
| Oligonucleotides | ||
| Control siRNA-A | Santa Cruz Biotechnology | CAT# sc-37007 |
| siRNA-MBNL1 | Dharmacon | N/A |
| Primers used for RT-qPCR; see Table S1 | Integrated DNA Technologies/ Eurofins MWG Operon | N/A |
| 5’-Cy3-(CAGG)10 | Margolis et al., 2006; Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | N/A |
| Olympus Fluoview software version 3.0 | Olympus | N/A |
| GraphPad Prism 7 | GraphPad Prism Software, Inc. | N/A |
HIGHLIGHTS.
RNA repeat expansions can cause Intron retention by binding proteins
Small molecules that bind RNA repeats and inhibit protein binding trigger decay
A toxic RNA repeat can catalyze the synthesis of its own inhibitor on-site
On-site drug synthesis most potently affects repeat expansion disease biology
Acknowledgements.
We dedicate this work to our inspirational colleague Prof. K. Barry Sharpless on the occasion of him being awarded the Priestly Medal. We thank Jessica Childs-Disney for help writing this manuscript and the agencies that funded this work including the National Institutes of Health (DP1-NS096898 to MDD and F31-NS110269 to AJA), the Muscular Dystrophy Association (Grant #380467 to MDD), and a Fullbright Fellowship (to RIB). We also thank the University of Florida’s Center for Neurogenetics for the DM2 fibroblast cell line used in this study.
Footnotes
Supplemental Information
Supplemental Information includes four figures and one table and can be found with this article online at: [to be filled in]
DECLARATION OF INTERESTS
M.D.D. is a founder of Expansion Therapeutics and M.D.D. and E.T.W. are scientific consultants for Expansion Therapeutics
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Angelbello AJ, Rzuczek SG, McKee KK, Chen JL, Olafson H, Cameron MD, Moss WN, Wang ET, and Disney MD (2019). Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U. S. A 116, 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernat V, and Disney MD (2015). RNA structures as mediators of neurological diseases and as drug targets. Neuron 87, 28–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CZ, Sobczak K, Hoskins J, Southall N, Marugan JJ, Zheng W, Thornton CA, and Austin CP (2012). Two high-throughput screening assays for aberrant RNA-protein interactions in myotonic dystrophy type 1. Anal. Bioanal. Chem 402, 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, and Disney MD (2007). A small molecule microarray platform to select RNA internal loop-ligand interactions. ACS Chem. Biol 2, 745–754. [DOI] [PubMed] [Google Scholar]
- Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, Tran T, Sarkar P, Schatz GC, and Disney MD (2014). Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem. Biol 9, 538–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, and Jaffrey SR (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly CM, Moon MH, and Schneekloth JS Jr. (2016). The emerging role of RNA as a therapeutic target for small molecules. Cell Chem. Biol 23, 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costales MG, Matsumoto Y, Velagapudi SP, and Disney MD (2018). Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc 140, 6741–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costales MG, Rzuczek SG, and Disney MD (2016). Comparison of small molecules and oligonucleotides that target a toxic, non-coding RNA. Bioorg. Med. Chem. Lett 26, 2605–2609. [DOI] [PubMed] [Google Scholar]
- Davidson A, Leeper TC, Athanassiou Z, Patora-Komisarska K, Karn J, Robinson JA, and Varani G (2009). Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl. Acad. Sci. U. S. A 106, 11931–11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney MD (2019). Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc 141, 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney MD, Dwyer BG, and Childs-Disney JL (2018). Drugging the RNA World. Cold Spring Harb. Perspect. Biol. 10, pii: a034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlic A, and Hargrove AE (2018). Targeting RNA in mammalian systems with small molecules. Wiley Interdiscip. Rev. RNA 9, e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, and Miller BL (2008). Dynamic combinatorial selection of molecules capable of inhibiting the (CUG) repeat RNA-MBNL1 interaction in vitro: Discovery of lead compounds targeting myotonic dystrophy (DM1). J. Am. Chem. Soc 130, 16254–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer MJ, Vaughn IW, and McManus MT (2013). Pervasive Transcription of the Human Genome Produces Thousands of Previously Unidentified Long Intergenic Noncoding RNAs. PLOS Genet. 9, e1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR (2013). Lives that introns lead after splicing. Wiley Interdiscip. Rev. RNA 4, 677–691. [DOI] [PubMed] [Google Scholar]
- Lee MM, Childs-Disney JL, Pushechnikov A, French JM, Sobczak K, Thornton CA, and Disney MD (2009a). Controlling the specificity of modularly assembled small molecules for RNA via ligand module spacing: targeting the RNAs that cause myotonic muscular dystrophy. J. Am. Chem. Soc 131, 17464–17472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MM, French JM, and Disney MD (2011). Influencing uptake and localization of aminoglycoside-functionalized peptoids. Mol. Biosyst 7, 2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MM, Pushechnikov A, and Disney MD (2009b). Rational and modular design of potent ligands targeting the RNA that causes myotonic dystrophy 2. ACS Chem. Biol 4, 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, and Sharpless KB (2002). Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew. Chem. Int. Ed. Engl 41, 1053–1057. [DOI] [PubMed] [Google Scholar]
- Li Y, and Disney MD (2018). Precise small molecule degradation of a noncoding RNA identifies cellular binding sites and modulates an oncogenic phenotype. ACS Chem. Biol 13, 3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, and Ranum LP (2001). Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867. [DOI] [PubMed] [Google Scholar]
- Llano-Sotelo B, Azucena EF Jr., Kotra LP, Mobashery S, and Chow CS (2002). Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol 9, 455–463. [DOI] [PubMed] [Google Scholar]
- Lorenz DA, Kaur T, Kerk SA, Gallagher EE, Sandoval J, and Garner AL (2018). Expansion of cat-ELCCA for the discovery of small molecule inhibitors of the pre-let-7–Lin28 RNA–protein interaction. ACS Med. Chem. Lett 9, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, and Thornton CA (2000). Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1773. [DOI] [PubMed] [Google Scholar]
- Margolis JM, Schoser BG, Moseley ML, Day JW, Ranum LPW (2006). DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum. Mol. Genet 15, 1808–1815. [DOI] [PubMed] [Google Scholar]
- Poulin-Kerstien AT, and Dervan PB (2003). DNA-templated dimerization of hairpin polyamides. J. Am. Chem. Soc 125, 15811–15821. [DOI] [PubMed] [Google Scholar]
- Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, and Disney MD (2009). Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J. Am. Chem. Soc 131, 9767–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranum LP, and Cooper TA (2006). RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci 29, 259–277. [DOI] [PubMed] [Google Scholar]
- Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R, and Disney MD (2017). Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol 13, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzuczek SG, Park H, and Disney MD (2014). A toxic RNA catalyzes the in cellulo synthesis of its own inhibitor. Angew. Chem. Int. Ed. Engl 53, 10956–10959. [DOI] [PubMed] [Google Scholar]
- Rzuczek SG, Southern MR, and Disney MD (2015). Studying a drug-like, RNA-focused small molecule library identifies compounds that inhibit RNA toxicity in myotonic dystrophy. ACS Chem. Biol 10, 2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur RS, Philips AV, and Cooper TA (2001). Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet 29, 40–47. [DOI] [PubMed] [Google Scholar]
- Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, et al. (2015). Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, and Al-Hashimi HM (2011). Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol 7, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, et al. (2014). Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 84, 239. [DOI] [PubMed] [Google Scholar]
- Sznajder ŁJ, Thomas JD, Carrell EM, Reid T, McFarland KN, Cleary JD, Oliveira R, Nutter CA, Bhatt K, Sobczak K, et al. (2018). Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. U. S. A 115, 4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velagapudi SP, Gallo SM, and Disney MD (2014). Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol 10, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, et al. (2014). Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505, 706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZF, Ursu A, Childs-Disney JL, Guertler R, Yang WY, Bernat V, Rzuczek SG, Fuerst R, Zhang YJ, Gendron TF, et al. (2019). The hairpin form of r(G4C2)exp in c9ALS/FTD is repeat-associated non-ATG translated and a target for bioactive small molecules. Cell Chem. Biol 26, 179–190.e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, et al. (2011). Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U. S. A 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or codes.