

Abstract
Sclerosing epithelioid fibrosarcoma (SEF) is a rare sarcoma subtype characterized by monomorphic epithelioid cells embedded in a densely sclerotic collagenous matrix. The overwhelming majority of tumors arise in soft tissues; however, rare cases have been documented to occur primarily in bone. The hallmarks of soft tissue SEF include MUC4 immunoreactivity and the presence of an EWSR1-CREB3L1 fusion. Rare cases with alternative fusions have also been reported such as EWSR1-CREB3L2 and FUS-CREB3L2 transcripts. The molecular alterations of skeletal SEF have not been well-defined, with only rare cases analyzed to date. In this study we investigated the clinicopathologic and molecular features of 7 patients presenting with primary osseous SEF. There were 3 males and 4 females, with a mean age at diagnosis of 38 years. All cases had microscopic features within the histologic spectrum of SEF and showed strong and diffuse MUC4 positivity, while lacking SATB2 expression. However, due to its unusual presentation within bone, 4 cases were initially misinterpreted as either osteosarcoma, Ewing sarcoma or chondroblastoma. Half of the patients with follow-up data developed metastasis. The cases were tested by targeted RNA sequencing, MSK-IMPACT, and/or FISH, showing EWSR1-CREB3L1 in 6 cases and EWSR1-CREB3L2 in one case. The fusion transcripts were composed of EWSR1 exon 11 to either exon 6 of CREB3L1 or CREB3L2. In summary, due to their rarity in the bone, skeletal SEF are often misdiagnosed, resulting in inadequate treatment modalities. Similar to their soft tissue counterpart, bone SEF follow an aggressive clinical behavior and show similar EWSR1-CREB3L1/CREB3L2 fusions.
Keywords: Sclerosing epithelioid fibrosarcoma, EWSR1, CREB3L1, CREB3L2, fusions
1. INTRODUCTION
Sclerosing epithelioid fibrosarcoma (SEF) is an aggressive sarcoma characterized by a distinctive epithelioid phenotype in a densely sclerotic collagenous stroma, that shows frequent MUC4 immunoreactivity and recurrent gene fusions, often involving the EWSR1 gene1. A pathogenetic link with low grade fibromyxoid sarcoma (LGFMS) has been suggested, due to cases with hybrid morphology as well as overlapping genetic signature2,3. Most cases occur in the deep soft tissues, but rare cases have been reported to arise in bone4–9,10. Only in a handful of these reports, the presence of EWSR1 or FUS gene rearrangements has been documented4,8,10. Triggered by 3 consecutive cases of primary skeletal SEF which were misdiagnosed as tumors that more often occur in the bone, ranging from ‘atypical chondroblastoma’, small cell osteosarcoma and Ewing sarcoma, we reviewed our files for cases that fit with this clinical presentation. This study investigates the clinicopathologic features and molecular abnormalities of the largest series to date of genetically confirmed osseous SEF. Moreover, we also compare our findings to the previously reported cases from the literature, as well as with the more common soft tissue SEF subset.
2. MATERIAL AND METHODS
2.1. Patients selection and data collection
The molecular files of two participating institutions (MSKCC, Mt Sinai Toronto), as well as the personal consultation files of the senior author (CRA) were searched. A total of 7 genetically confirmed SEF of bone cases were identified.
Hematoxylin and eosin-stained slides and immunohistochemical stains were re-reviewed. All cases were handled in accordance with the ethical rules of the respective institutions. The tumors were assessed for growth pattern, cytomorphology, cellular pleomorphism, nuclear features including nuclear contour, chromatin pattern and presence of nucleoli, mitotic activity, necrosis, and type of extracellular stroma. Immunohistochemical stains were performed or reviewed, including MUC4 and SATB2. However, in most cases a more exhaustive immunohistochemical work-up was performed.
Retrospective chart review was conducted to collect clinical information such as maximum tumor size, tumor location, stage at diagnosis (primary versus distant metastasis at diagnosis), modality of initial therapy, local recurrence or metastasis, vital status at last follow-up and survival time. One case with an EWSR1-CREB3L1 fusion has been previously reported10.
2.2. Fluorescence in situ hybridization (FISH)
FISH was conducted for EWSR1/FUS and CREB3L1/CREB3L2 in six cases (Case# 2- # 7). FISH for break-apart assay was applied on formalin-fixed and paraffin-embedded 4-micron sections as previously described11. Custom probes using bacterial artificial chromosomes (BACs) covering and flanking the EWSR1, FUS, CREB3L1 and CREB3L2 genes were utilized11. The BAC clones were selected according to the UCSC genome browser (http://genome.ucsc.edu) and obtained from the BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI)(Oakland, CA) (https://bacpacresources.org). DNA from individual BACs was isolated in line with manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI. Two hundred tumor nuclei were evaluated using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems, Newton, MA). A cut-off of >20% nuclei showing a break-apart signal was considered positive for rearrangement. Nuclei with incomplete set of signals were omitted from the score.
2.3. Targeted Sequencing
One case (Case# 1) was investigated with the MSK-IMPACT assay, a hybridization capture-based next-generation sequencing assay for targeted deep sequencing12,13. Two cases (Case# 3,4) were analyzed by targeted RNA sequencing using the TruSight RNA Fusion Panel (Illumina, San Diego, CA)14, as previously described. RNA was extracted from FFPE tissue using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA). Targeted RNA sequencing was performed on an Illumina MiSeq platform. Reads were independently aligned with STAR (version 2.3) against the human reference genome (hg19) and analyzed by STAR-Fusion. Transcript IDs used in this study were ENST00000397938.6 (EWSR1), ENST00000621158.4 (CREB3L1), and ENST00000330387.11 (CREB3L2).
3. RESULTS
3.1. Clinical presentation
The clinical features of SEF of bone are summarized in Table 1. There were 3 males and 4 females, with a mean age at diagnosis of 38 years (range, 8 to 55). The tumor location was femur in three patients, mandible in two, and one each in the skull and thoracic vertebrae. The tumor greatest dimension obtained from either imaging studies at presentation or gross descriptions had a mean of 5.0 cm (range, 3.0 to 6.0 cm).
Table 1.
Clinicopathologic, molecular and follow-up information of patients with SEF of bone from current study and published literature
| Case# | Age | Sex | Location | Size (cm) | Initial diagnosis | Fusion transcript | Mets Y/N | Metastatic site | Vital status | Follow-up (mo) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 23 | F | Femur | 5.7 | Atypical chondroblastoma | EWSR1 exon 11 /CREB3L1 exon6α | Y | Lung | AWD | 52 |
| 2 | 8 | M | Femur | NA | Ewing sarcoma, osteosarcoma | EWSR1 / CREB3L1β | Y | Skull | AWD | 24 |
| 310 | 55 | F | Mandible | 4.9 | NA | EWSR1 exon 11 / CREB3L1 exon 6β,γ | N | NA | NED | 36 |
| 4 | 47 | M | Thoracic vertebrae | NA | NA | EWSR1 exon 11 / CREB3L2 exon 6 β,γ | NA | NA | NA | NA |
| 5 | 52 | F | Skull | 6.0 | SEF | EWSR1 / CREB3L1β | Y | Rib, Spine, Skull | DOD | 58 |
| 6 | 28 | M | Mandible | 3.0 | Small round cell tumor | EWSR1 / CREB3L1β | N | NA | NED | 2 |
| 7 | 51 | F | Femur | 5.5 | Osteosarcoma | EWSR1 / CREB3L1β | N | NA | NED | 60 |
| 84 | 58 | M | Femur | 5.2 | NA | NA | Y | NA | NA | NA |
| 94 | 51 | F | Femur | 9.6 | NA | NA | Y | NA | NA | NA |
| 104 | 73 | F | Ulna | 4.0 | NA | NA | N | NA | NA | NA |
| 114 | 53 | M | Femur | 2.8 | NA | EWSR1 rearrangement | N | NA | NA | NA |
| 124 | 25 | M | Humerus | 11.5 | NA | EWSR1 rearrangement | Y | NA | NA | NA |
| 134 | 66 | M | Cervical spine | 4.2 | NA | FUS rearrangement | N | NA | NA | NA |
| 144 | 31 | F | Ulna | 3.0 | NA | NA | N | NA | NA | NA |
| 154 | 43 | M | 2nd rib | 8.0 | NA | NA | N | NA | NA | NA |
| 164 | 48 | M | Metacarpal | 4.4 | NA | NA | N | NA | NA | NA |
| 175 | 16 | F | Tibia | 4.0 | Ossifying fibroma | NA | N | NA | NED | 22 |
| 186 | 57 | F | Pelvis | NA | Metastatic cancer | NA | Y | Bone, Liver | DOD | 7 |
| 197 | 48 | F | Sacrum | 8.0 | SEF | NA | Y | Lung | DOD | 72 |
| 208 | 43 | M | Ilium | NA | Reactive change | EWSR1 / CREB3L1 | Y | Lung | AWD | 24 |
| 219 | 24 | M | Skull | 5.0 | SEF | NA | Y | Lung | DOD | 24 |
F, female; M, male; NA; not available, Cases confirmed by MSK-IMPACTα, FISHβ or targeted RNA sequencingγ; DOD, dead of disease; NED, no evidence of disease; AWD, alive with disease; mo, months.
Follow-up data was available in 6 of our patients, summarized in Table 1, which also summarizes the clinical data from the cases previously reported in the literature. All our patients presented with localized disease at diagnosis. Case# 1 was diagnosed at the outside institution as an ‘atypical chondroblastoma’ arising in the femoral diaphysis, where it was resected. Two years later, local recurrence and pulmonary metastases developed. Case# 2 presented with a pathologic fracture in the femur and a diagnosis of Ewing sarcoma was rendered on the biopsy material due to a FISH positive result for EWSR1 gene rearrangement. The patient was then treated with neoadjuvant chemotherapy with vincristine/doxorubicin/cyclophosphamide (VDC) and ifosfamide/ etoposide (IE) per Ewing sarcoma regimen. Diagnosis was revised on the surgical resection specimen as a likely small cell osteosarcoma, showing 50% chemotherapy-related necrosis. Patient received adjuvant chemotherapy with high-dose methotrexate, doxorubicin, and cisplatin (MAP) with addition of ifosfamide/etoposide per osteosarcoma protocol. However, 12 months later, the patient developed skull metastasis. Similarly, case# 7, also presenting with a femur lesion, was diagnosed as a high grade osteosarcoma on the biopsy; patient receiving neoadjuvant chemotherapy per osteosarcoma regimen. The patient has no evidence of disease, 60 months follow-up from diagnosis. Case# 5 presented with a skull mass and underwent resection without neo/adjuvant chemotherapy. She developed multiple rib, spine and skull metastases, succumbing of disease after 58 months. Cases # 3 and 6, both with mandible tumors, underwent en-bloc resection without neo/adjuvant chemotherapy. Both patients are alive with no evidence of disease at last follow-up. The initial diagnosis for Case #3 was undifferentiated sarcoma and for Case #6 was a small blue round cell tumor.
In total, four cases were initially misinterpreted as either osteosarcoma, Ewing sarcoma or other small blue round cell tumors, and atypical chondroblastoma. Three of six (50%) patients with available clinical follow-up developed metastasis and one patient (17%) died of disease.
3.2. Radiologic findings
The imaging studies of primary sites were available in 4 cases. Case #1 presented with right thigh pain and X-ray showed lytic bone lesion within femoral diaphysis, associated with sclerotic cortex (Figure 1a). MRI showed an intraosseous mass surrounded by bone marrow edema (Figure 1b). Case #2 presented with progressive left thigh pain and inability to bear weight for 3 months. The patient developed sudden increase in pain and a pathologic fracture was subsequently diagnosed in the proximal femoral shaft, through a radiolucent lesion (Figure 1c). CT of Case #5 showed an osteolytic right skull mass associated with destruction of the cortex and soft tissue extension (Figure 1d). CT of Case# 6 also showed a left mandibular lytic lesion, with outer cortex destruction and soft tissue extension. Calcification/ossification within the tumor was suspected (Figure 1e).
Figure 1. Imaging studies demonstrating a primary bone lesion.
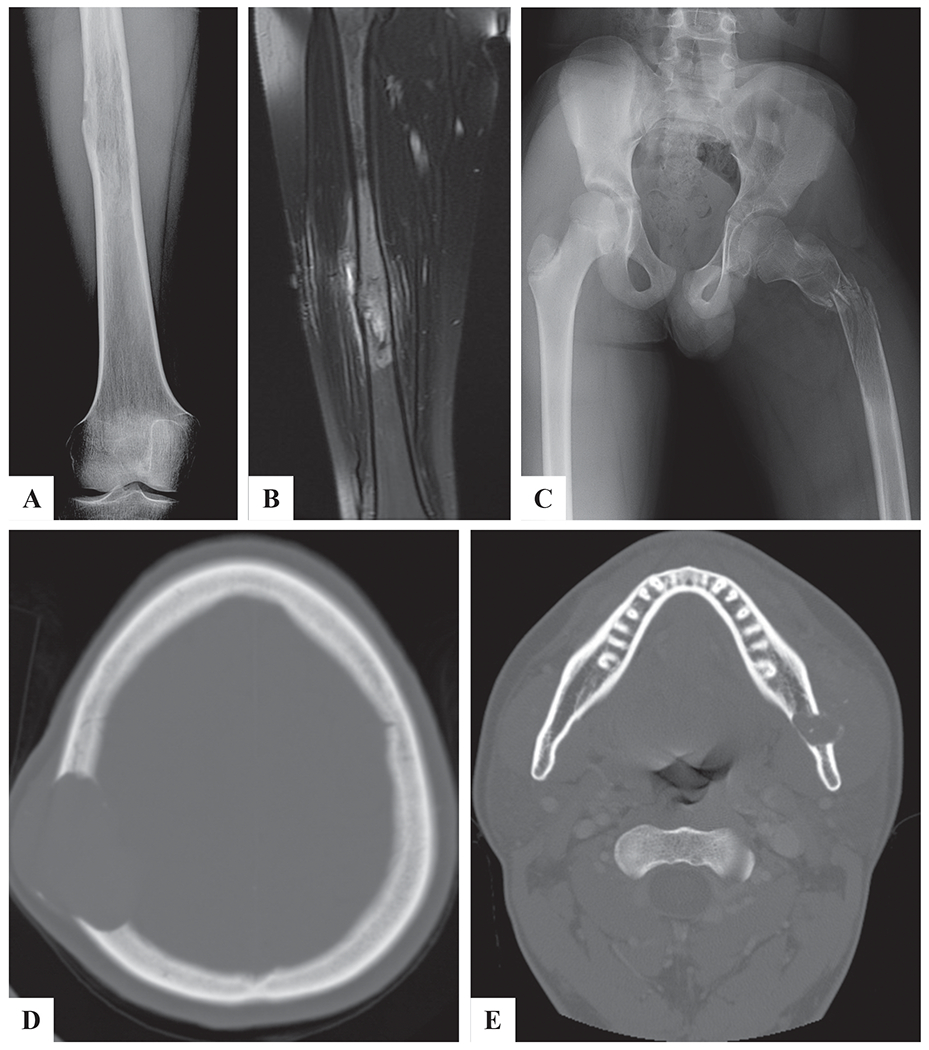
A) X-ray showing a destructive right femur lesion (Case# 1). B) MRI T2 weighted image showing a mass in the right femur with bone marrow edema (Case# 1). C) X-ray showing a left proximal femur lytic lesion associated with pathologic fracture (Case# 2). D) CT showing lytic skull mass with bone destruction and extra-osseous extension (Case# 5). E) CT showing a left mandibular destructive mass with extra-osseous extension (Case# 6).
3.3. Pathologic findings
The tumors were composed of solid sheets of monomorphic epithelioid cells with light eosinophilic to vacuolated cytoplasm and round nuclei with fine chromatin. A dense hyalinized stromal component was noted in all cases, although it varied in extent from case to case, or from area to area within the same case (Figure 2A). The tumor cells were divided in columns or nests by wiry collagen, in areas simulating lace-like osteoid. However, no mineralization or chondroid matrix was noted within the fibrous stroma. Occasionally the tumors showed hypocellular regions, with confluent areas of stromal hyalinization (Figure 2B–D). Some cases showed areas of increased cellularity, reminiscent of a round to undifferentiated neoplasm, in which the stroma was scant (Figure 2E). A few cases showed a more plasmacytoid phenotype or a nested pattern (Figure 2F). No areas of low grade fibromyxoid stroma were detected to suggest a hybrid morphology. However, Case #3 was found to have had a prior biopsy 20 years earlier that, on re-review, was morphology compatible with low grade fibromyxoid sarcoma, thereby raising the possibility of progression to SEF. All cases lacked SATB2 expression including in areas resembling osteoid matric (Figure 2 G,H) and showed strong and diffuse MUC4 positivity (Figure 2I).
Figure 2. Histologic features of skeletal SEF.
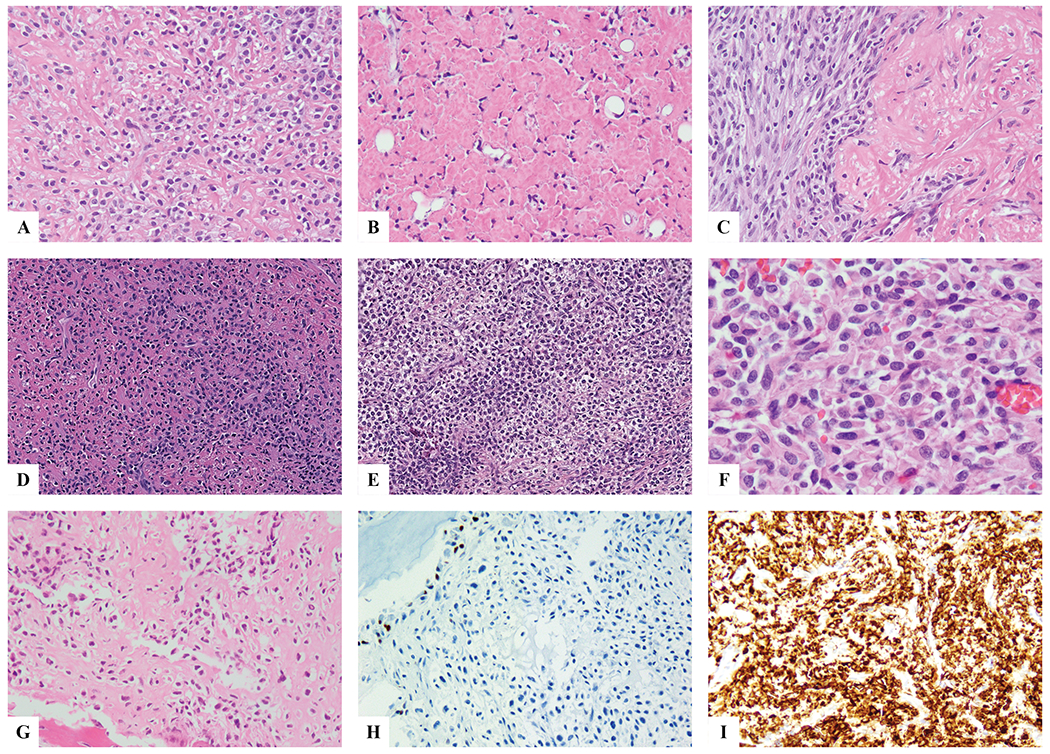
Classic morphologic appearance with uniform epithelioid cells separated by wiry collagen bundles in cord and columns (A); alternating markedly hyalinized hypocellular areas suggesting a benign/reactive process are noted (B). An abrupt transition between the more cellular, sometimes with a spindling phenotype, and hypocellular areas can be noted (C). Some tumors show highly cellular/primitive areas, almost completely devoid of fibrotic stroma, arranged in sheets of nests, reminiscent of an undifferentiated round cell sarcoma or Ewing sarcoma (D,E). In other tumors, the lesional cells have a more plasmacytoid or rhabdoid phenotype, with densely eosinophilic cytoplasm and eccentric round nuclei (F). This tumor showed a densely hyalinized matrix s/p neoadjuvant chemotherapy, highly suggestive of osteoid matrix deposition (G); however, the tumor cells were negative for SATB2 (with good internal positive control the adjacent osteoblasts), while being strongly positive for MUC4.
3.4. Molecular findings
FISH revealed EWSR1 gene rearrangements in all 7 cases tested. CREB3L1 gene break-apart was identified in 6 patients (Case # 2,3,5-7), while a CREB3L2 gene rearrangement was detected in the remaining patient (Case #4). Representative rearrangements are depicted in Supplem. Figure 1.
In one patient (Case# 1), MSK-IMPACT identified the presence of an EWSR1-CREB3L1 fusion, composed of EWSR1 exon 11 fused to exon 6 of CREB3L1. MSK-IMPACT also identified copy number amplification of CHECK2, and copy number losses of the NF2, RAC2, EP300, and CRKL genes.
TruSight RNA Fusion Panel detected two different in frame fusions, including one between EWSR1 exon 11 and CREB3L1 exon 6 (Case #3); and the other between EWSR1 exon 11 and CREB3L2 exon 6 (Case #4)(Figure 3).
Figure 3. Diagrammatic representation of the EWSR1-CREB3L1 and EWSR1-CREB3L2 fusions studied by targeted RNA sequencing.
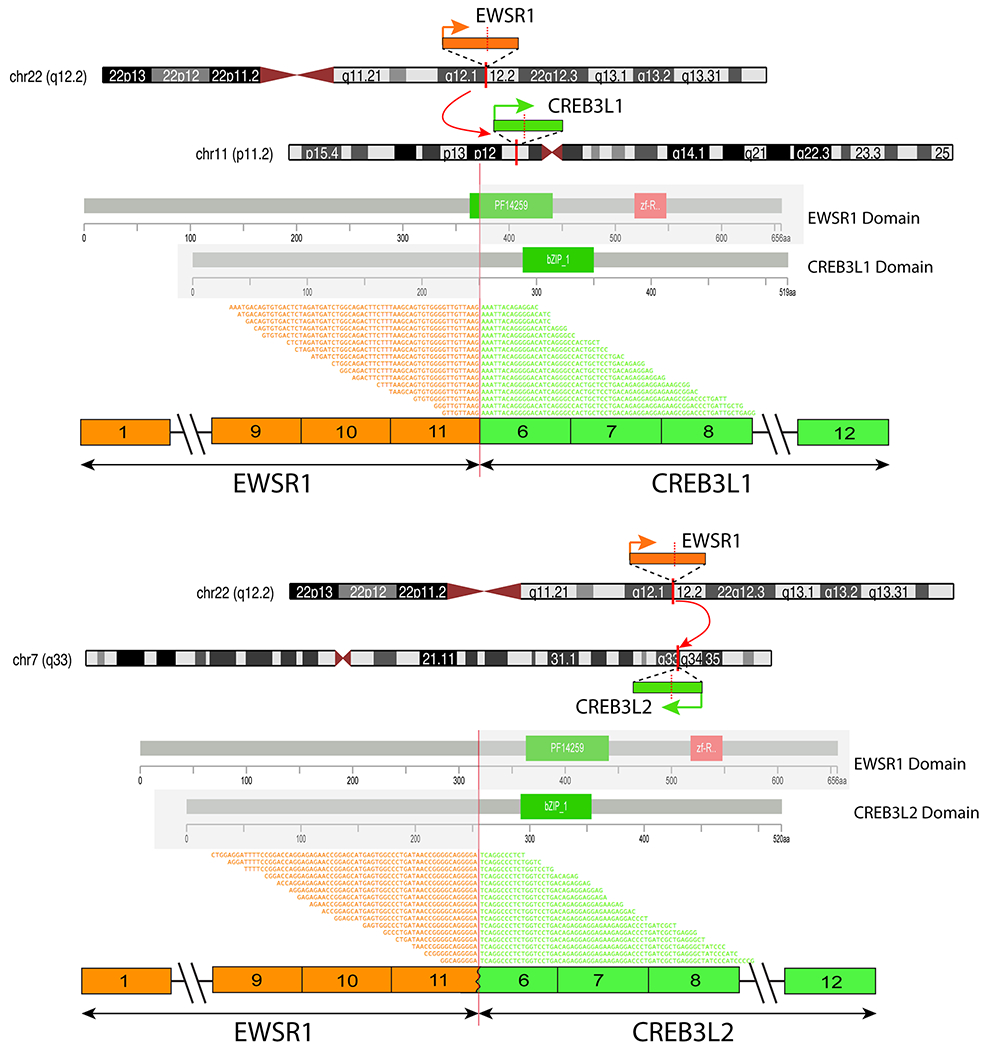
Upper and middle panel show chromosomal localization of EWSR1 on 22q12.2, CREB3L1 on 11p11.2 and CREB3L2 on 7q33. Red vertical lines depict the genomic breakpoints. Orange arrows and bars show the direction of transcription of each gene and the exonic breaks, respectively. The protein domains of each gene are shown below. Fusion variants are depicted at the bottom of each panel showing EWSR1 exon 11 fused to CREB3L1 exon 6 and EWSR1 exon 11 to CREB3L2 exon 6.
3.5. Literature review
In addition to our current series, there are 14 patients with primary SEF of bone previously reported, 8 males and 6 females, with a mean age of 45 years (range, 16 to 73 years)(Table 1)4–9. Eight tumors (57%) were located in the extremity; with the most frequent primary site being the femur (21%). Three cases were initially misdiagnosed as ossifying fibroma, metastatic cancer, or reactive change. Half of the patients with available clinical follow-up developed metastasis and 60% (3 of 5) patients died of their diseases.
Of the 11 patients with available histologic evaluation, all except 2 (82%) were composed of pure SEF, with small nests and linear arrays of epithelioid cells embedded in a dense collagenous matrix. The remaining 2 cases contained areas of bland spindle cells embedded in a fibromyxoid stroma, in keeping with a LGFMS component (hybrid SEF-LGFMS type). Of 9 cases with available data, 7 tumors (78%) were positive for MUC4 immunostain. Three cases showed EWSR1 rearrangement and one case showed FUS rearrangement. None were tested to determine their gene partner or fusion breakpoints.
4. DISCUSSION
In this study we investigated the clinicopathologic and molecular features of 7 cases of primary SEF of bone. Our study showed the presence of an EWSR1-CREB3L1 fusion in 6 cases and EWSR1-CREB3L2 fusion in one case. Our results also reinforce that SEF of bone is a sarcoma with a highly aggressive clinical course and that the differential diagnosis is much more challenging than in the soft tissues due to the rarity of this disease in bone and its resemblance to other more common bone sarcomas, particularly with osteosarcoma and Ewing sarcoma. As a result of these misdiagnoses in the initial biopsies or subsequent resections, 3 patients were subjected to neoadjuvant or adjuvant therapies for osteosarcoma or Ewing sarcoma, with mostly no benefit.
In this series, SEF of bone exhibited an aggressive behavior with a high frequency of metastasis, seen in half of the patients with available follow-up. A literature review of previously reported cases showed a similar incidence of metastatic disease, occurring in 50% of the patients 4–9. Similar to a previous report5, one of our cases showed a poor response to Ewing sarcoma chemotherapy regimen. The aggressive clinical features and chemoresistance noted in the skeletal SEF recapitulate the experience seen with the more common soft-tissue SEF, showing high rates of metastasis (86 to 92%) and death of disease (57% to 77%)3,15.
In contrast to soft tissue SEF where the diagnosis appears straightforward, the differential diagnosis of primary SEF of bone appears much more challenging5,6,8. One of the reasons being the rarity of SEF as a primary bone lesion. Additionally, the dense extracellular collagenous stroma can be misinterpreted as osteoid matrix, and thus misdiagnosed as osteosarcoma. Immunostaining for SATB2 might be critical in confirming the osteoblastic nature of the tumor cells, staining which was consistently negative in our cases and a previous study4. Furthermore, close radiographic correlation might refute the presence of mineralized matrix on images. Differentiating the diagnosis from osteosarcoma is clinically important, as skeletal SEF have been resistant to osteosarcoma regimen chemotherapy5,15. Moreover, due to its round and monomorphic appearance, SEF can also be misdiagnosed as an Ewing sarcoma family of round cell tumors or even small cell osteosarcoma. Based on the reported literature, skeletal SEF can also be mistaken as a benign neoplasm, due to its bland histologic appearance. One case in our series was also initially misdiagnosed as an ‘atypical chondroblastoma’.
Morphologically SEF of bone shared similar features to the more common soft tissue SEF, including a monomorphic epithelioid phenotype, arranged in cords and nests, separated by a dense collagenous stroma. Also similar to the soft tissue counterpart, MUC4 was diffusely and strongly positive which can be used to confirm the diagnosis.
At extraskeletal locations, an EWSR1-CREB3L1 gene fusion is the most common abnormality in pure SEF, while FUS-CREB3L2 transcript is commonly present in the hybrid SEF/LGFMS16,17. In keeping with these findings, our study composed of pure SEF morphology demonstrated the presence of an EWSR1-CREB3L1 fusion in all except one case. The only case harboring a EWSR1-CREB3L2 fusion occurred from the thoracic vertebrae. The predicted structure of the fusion protein is similar to other EWSR1-related fusions, including the N-terminal serine-tyrosine-glutamine-glycine transactivation domain of EWSR1. CREB3L1 and CREB3L2 belong to the CREB3 family of transcription factors, which encode proteins localized to the endoplasmic reticulum and act as transcription factors activated by cyclic AMP stimulation18. Further studies are warranted to define the oncologic role of fusion protein in SEF.
In conclusion, this is the largest study to date investigating the clinical and molecular abnormalities of primary SEF of bone using a combined approach of targeted RNA/DNA sequencing and/or FISH. Our results stress the diagnostic challenges encountered in establishing a correct diagnosis outside the molecular testing, mainly due to its resemblance to other more common bone sarcomas. As seen with the soft tissue tumors, skeletal SEF emerges as a highly aggressive sarcoma type, with a high rate of distant metastases and resistance to both Ewing sarcoma and osteosarcoma regimens. Adjunct immunohistochemical markers (MUC4, SATB2) and FISH/NGS molecular methods are critical in reaching the correct diagnosis.
Supplementary Material
Supplementary Figure 1. FISH assay using custom break-apart probes showing split apart signals of EWSR1 and CREB3L2 (A,B, case 4) and EWSR1 and CREB3L1 (C,D, case 7); (red, centromeric; green, telomeric).
Acknowledgments
Disclosures: Supported in part by: P50 CA 140146-01 (CRA), P50 CA217694 (CRA), P30 CA008748, Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA)
REFERENCES
- 1.Fletcher C, Bridge JA, Hogendoorn PC, et al. WHO Classification of Tumours of Soft Tissue and Bone. 4th Edition: IARC: Lyon; 2013. [Google Scholar]
- 2.Guillou L, Benhattar J, Gengler C, et al. Translocation-positive low-grade fibromyxoid sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: a study from the French Sarcoma Group. Am J Surg Pathol. 2007;31:1387–1402. [DOI] [PubMed] [Google Scholar]
- 3.Antonescu CR, Rosenblum MK, Pereira P, et al. Sclerosing epithelioid fibrosarcoma: a study of 16 cases and confirmation of a clinicopathologically distinct tumor. Am J Surg Pathol. 2001;25:699–709. [DOI] [PubMed] [Google Scholar]
- 4.Wojcik JB, Bellizzi AM, Dal Cin P, et al. Primary sclerosing epithelioid fibrosarcoma of bone: analysis of a series. Am J Surg Pathol. 2014;38:1538–1544. [DOI] [PubMed] [Google Scholar]
- 5.Grunewald TG, von Luettichau I, Weirich G, et al. Sclerosing epithelioid fibrosarcoma of the bone: a case report of high resistance to chemotherapy and a survey of the literature. Sarcoma. 2010;2010:431627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G, Eyden B. A primary sclerosing epithelioid fibrosarcoma of the pubic bone, with evidence of divergent epithelial differentiation. Ultrastructural Pathology. 2010;34:99–104. [DOI] [PubMed] [Google Scholar]
- 7.Chow LT, Lui YH, Kumta SM, et al. Primary sclerosing epithelioid fibrosarcoma of the sacrum: a case report and review of the literature. Journal of Clinical Pathology. 2004;57:90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdulkader I, Cameselle-Teijeiro J, Fraga M, et al. Sclerosing epithelioid fibrosarcoma primary of the bone. International Journal of Surgical Pathology. 2002;10:227–230. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Wang J, Zhang M, et al. Skull sclerosing epithelioid fibrosarcoma: A case report and review of the literature. Oncology Letters. 2016;11:3417–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laliberte C, Leong IT, Holmes H, et al. Sclerosing Epithelioid Fibrosarcoma of the Jaw: Late Recurrence from a Low Grade Fibromyxoid Sarcoma. Head and Neck Pathology. 2018;12:619–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prieto-Granada C, Zhang L, Chen HW, et al. A genetic dichotomy between pure sclerosing epithelioid fibrosarcoma (SEF) and hybrid SEF/low-grade fibromyxoid sarcoma: a pathologic and molecular study of 18 cases. Genes Chromosomes Cancer. 2015;54:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of Molecular Diagnostics : JMD. 2015;17:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature Medicine. 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickson BC, Sung YS, Rosenblum MK, et al. NUTM1 Gene Fusions Characterize a Subset of Undifferentiated Soft Tissue and Visceral Tumors. Am J Surg Pathol. 2018;42:636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chew W, Benson C, Thway K, et al. Clinical Characteristics and efficacy of chemotherapy in sclerosing epithelioid fibrosarcoma. Med Oncol. 2018;35:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arbajian E, Puls F, Antonescu CR, et al. In-depth Genetic Analysis of Sclerosing Epithelioid Fibrosarcoma Reveals Recurrent Genomic Alterations and Potential Treatment Targets. Clin Cancer Res. 2017;23:7426–7434. [DOI] [PubMed] [Google Scholar]
- 17.Arbajian E, Puls F, Magnusson L, et al. Recurrent EWSR1-CREB3L1 gene fusions in sclerosing epithelioid fibrosarcoma. Am J Surg Pathol. 2014;38:801–808. [DOI] [PubMed] [Google Scholar]
- 18.Dewaele B, Libbrecht L, Levy G, et al. A novel EWS-CREB3L3 gene fusion in a mesenteric sclerosing epithelioid fibrosarcoma. Genes Chromosomes Cancer. 2017;56:695–699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. FISH assay using custom break-apart probes showing split apart signals of EWSR1 and CREB3L2 (A,B, case 4) and EWSR1 and CREB3L1 (C,D, case 7); (red, centromeric; green, telomeric).