

Abstract
Objective
Histaminergic neurons of the tuberomammillary nucleus (TMN) are wake-promoting and contribute to the regulation of energy homeostasis. Evidence indicates that melanocortin 4 receptors (MC4R) are expressed within the TMN. However, whether the melanocortin system influences the activity and function of TMN neurons expressing histidine decarboxylase (HDC), the enzyme required for histamine synthesis, remains undefined.
Methods
We utilized Hdc-Cre mice in combination with whole-cell patch-clamp electrophysiology and in vivo chemogenetic techniques to determine whether HDC neurons receive metabolically relevant information via the melanocortin system.
Results
We found that subsets of HDC-expressing neurons were excited by melanotan II (MTII), a non-selective melanocortin receptor agonist. Use of melanocortin receptor selective agonists (THIQ, [D-Trp8]-γ-MSH) and inhibitors of synaptic transmission (TTX, CNQX, AP5) indicated that the effect was mediated specifically by MC4Rs and involved a glutamatergic dependent presynaptic mechanism. MTII enhanced evoked excitatory post-synaptic currents (EPSCs) originating from electrical stimulation of the perifornical lateral hypothalamic area (PeFLH), supportive of melanocortin effects on the glutamatergic PeFLH projection to the TMN. Finally, in vivo chemogenetic inhibition of HDC neurons strikingly enhanced the anorexigenic effects of intracerebroventricular administration of MTII, suggesting that MC4R activation of histaminergic neurons may restrain the anorexigenic effects of melanocortin system activation.
Conclusions
These experiments identify a functional interaction between the melanocortin and histaminergic systems and suggest that HDC neurons act naturally to restrain the anorexigenic effect of melanocortin system activation. These findings may have implications for the control of arousal and metabolic homeostasis, especially in the context of obesity, in which both processes are subjected to alterations.
Keywords: Histaminergic neurons, Melanocortin, Lateral hypothalamus, Food intake, MC4R, Electrophysiology
Highlights
-
•
Histaminergic neurons of the tuberomammillary nucleus are excited by melanocortin receptor activation.
-
•
MC4Rs (but not MC3Rs) excite histaminergic neurons through a presynaptic glutamatergic mechanism.
-
•
MC4Rs in the perifornical lateral hypothalamus contribute toward melanocortin-induced activation of histaminergic neurons.
-
•
Chemogenetic inhibition of histaminergic neurons potentiates the anorexigenic effects of melanocortin agonism.
1. Introduction
Histaminergic neurons are confined to the tuberomammillary nucleus (TMN) in the posterior hypothalamus and provide the sole source of neuronal histamine to the brain [[1], [2], [3]]. These neurons are identified based on the expression of histidine decarboxylase (HDC), the enzyme required to convert l-histidine into histamine [4,5]. HDC neurons form a crucial part of the ascending arousal system and have well-established roles in regulating sleep and wakefulness [[6], [7], [8], [9], [10]]. In addition, HDC neurons innervate numerous hypothalamic nuclei and spinal regions known to regulate energy homeostasis [6,11,12]. Neuronal histamine release influences energy homeostasis through binding to histamine receptors located in these regions [[13], [14], [15], [16]]. Loss or inactivation of HDC neurons or histamine receptors is also associated with weight gain and obesity [[17], [18], [19]]. Despite the recognized role of the histaminergic system in the control of energy homeostasis, whether HDC neurons detect changes in metabolic status remains underexplored.
Assessing immediate early gene expression (c-fos) has shown that HDC neurons are activated by fasting [20] and that a small percentage of HDC neurons are sensitive to insulin-induced hypoglycemia [21]. Previous studies also implicate the histaminergic system in the leptin-induced suppression of food intake [18,22,23], although this effect appears to be indirect due to the absence of leptin receptor expression in the TMN [24]. Although these studies suggest that the histaminergic system may be sensitive to alterations in metabolic status, they do not provide any mechanisms by which HDC neurons detect such changes.
One potential way HDC neurons may detect changes in metabolic status is via the melanocortin system. The melanocortin system is comprised of anorexigenic pro-opiomelanocortin (POMC) neurons and their functionally antagonistic counterpart, the neuropeptide Y/agouti-related peptide (NPY/AgRP) neurons [25,26]. Both neuronal populations integrate multiple nutrient and hormonal factors indicative of energy status, including glucose, leptin, insulin, and fatty acids [27]. Once activated, POMC neurons convey this information to downstream targets through the release of the melanocortins, including α-MSH [28]. Melanocortins then bind to the cognate melanocortin receptors (MC3R and MC4R) located in different regions of the brain [29,30]. MC4Rs are expressed in the region of the TMN [30,31], and α-MSH fibers have been reported to form synaptic contacts with HDC neurons of the TMN [32]. Although the importance of MC4R in maintaining energy homeostasis has been described for over two decades [[33], [34], [35]], the role of the melanocortin system in the TMN remains surprisingly unexplored.
In the current study, we utilized Hdc-Cre mice in combination with whole-cell patch-clamp electrophysiology to examine the acute effects of melanocortin agonists on histaminergic neurons of the TMN. We also examined the role of these neurons in tandem with melanocortin signaling to regulate feeding using in vivo chemogenetic techniques. Together these data provide a novel mechanism by which HDC neurons detect and integrate changes in metabolic status and demonstrate an underappreciated role of the histaminergic system in the regulation of energy homeostasis.
2. Materials and methods
2.1. Animals
Mice were maintained on a 12-h light–dark cycle in a temperature-controlled conventional facility (ARC at UT Southwestern) with free access to food and water. Mice were fed a standard chow diet (Envigo Teklad Global 2016 Diet, 16% protein 4% fat). The Hdc-Cre mice were developed by Jeffrey Zigman and previously validated [36,37]. The Mc4r-GFP (green fluorescent protein) mice were obtained from Jeffrey Friedman (Rockefeller University) and previously validated [38]. tdTomato (Ai14, JAX stock # 007914) and R26-LSL-Gi-DREADD (JAX stock # 026219) were obtained from The Jackson Laboratory in Maine. All mice were maintained on a C57BL/6J background. All experiments adhered to the guidelines established by the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee (IACUC).
2.2. Electrophysiology
Whole-cell patch clamp recordings were obtained from tdTomato fluorescently-labelled HDC neurons in posterior hypothalamic brain slices from Hdc-cre::tdTomato mice. All animals were 8–12 weeks of age for electrophysiology studies. Mice were anaesthetized with chloral hydrate, decapitated, and the brain rapidly removed and cut in a modified, sucrose-based, ice-cold artificial cerebrospinal fluid (aCSF) containing (in mM): 213 sucrose; 2.8 KCl; 1.2 MgCl2; 2.5 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose. Posterior hypothalamic brain slices of 250 μm (coronal sections) containing the TMN were prepared on a Leica VT1000S vibratome. Slices were then incubated at 32οC in standard aCSF containing (in mM): 126 NaCl; 2.8 KCl; 2.5 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 1.2 MgSO4; and 10 d-glucose, and remained in this solution until slices were transferred to the recording chamber where they were continuously perfused with a reduced d-glucose concentration (5 mM) version of the standard heated aCSF.
Hdc-Cre::tdTomato neurons in the TMN were visually identified using an Axioskop FS2 (Zeiss) microscope fitted with DIC optics, infrared videomicroscopy and fluorescence. Patch pipettes were pulled from thin-walled borosilicate glass (TW150F-4, World Precision Instruments) with resistances between 4 and 7 MΩ when filled with an intracellular solution containing (in mM): 120 K-gluconate, 10 KCl, 1 NaCl, 1 MgCl2, 1 CaCl2, 5 EGTA, 10 HEPES, and 2 Mg2ATP), pH adjusted with KOH. Whole-cell patch-clamp recordings were made using a Multiclamp 700B amplifier (Molecular Devices LLC, Sunnyvale, CA, USA). Current clamp data was digitized and filtered at 4 kHz. Signals were stored on a personal computer for analysis with pClamp10 software (Molecular Devices). Resting membrane potential is reported as the read-out from the amplifier, uncorrected for the liquid junction potential offset (approx. -8 mV). Current voltage relations were performed to determine the input resistance (MΩ) of the cell and the reversal potential (Erev) of any effect. This was achieved by measuring the change in membrane potential (mV) in response to a series of current injections (nA) at baseline and following drug application. Voltage clamp data was filtered at 2 kHz. EPSCs were recorded using a holding potential of −60 mV to −70 mV and IPSCs −15 mV. PSC frequency and peak amplitude were measured using the Mini Analysis program (Synaptosoft, Inc.).
The following drugs were used for electrophysiological experiments: MTII (Phoenix Pharmaceuticals), TTX, CNQX, AP5, THIQ, [D-Trp8]-ƴ-MSH, Orexin A (all from Tocris), and clozapine N-oxide (CNO, Sigma). All drugs were made as concentrated stocks in distilled water and stored at <4°C. Drugs were diluted to the required concentration in aCSF immediately prior to use. All drugs (≈20 mls) were bath applied to the slice by a peristaltic pump connected to the main perfusion line.
2.3. Electrical stimulation
Electrical stimulation of perifornical/lateral hypothalamic (PeFLH) afferents was performed using a concentric bipolar electrode (125 μm outer diameter, 12.5 μm inner diameter; FHC, Bowdoinham, ME, USA) placed in the PeFLH region directly superior to the fornix. The stimulus intensity required for minimum response in HDC neurons was determined, and then the intensity was increased until responses to two paired stimulation pulses 30 msec apart (33Hz) were consistently observed. Trains of paired-pulse stimuli were recorded before, during, and after administration of 100 nmol of MTII. The resulting evoked excitatory post-synaptic currents (eEPSCs) elicited by electrically stimulating the PeFLH were assessed for absolute amplitude and the paired pulse ratio (PPR), amplitude of the second response divided by the amplitude of the first response (R2/R1).
2.4. MC4R co-expression in HDC neurons
Mc4r-GFP mice were bred with Hdc-Cre::TdTomato mice. Cells were examined for expression of TdTomato and MC4R-tau Sapphire GFP using an Axioskop FS2 (Zeiss) microscope following patch-clamp preparation (methods described above), or immunohistochemistry. For immunohistochemistry preparation, mice were deeply anesthetized with an intraperitoneal injection of chloral hydrate (350 mg/kg) and perfused transcardially with diethyl pyrocarbonate (DEPC)-treated 0.9% saline followed by 10% neutral buffered formalin. Brains were removed, post-fixed in 10% neutral buffered formalin for 2 h on a shaker at room temperature, cryoprotected overnight at 4 °C in 20% sucrose made in DEPC-treated phosphate-buffered saline (PBS), pH 7.0, and cut coronally at 25 μM. Tissue was stored at 4 °C in PBS-azide until processed.
Immunohistochemistry detection of GFP was performed using a GFP polyclonal antibody made in rabbits (Invitrogen, Eugene, OR; Cat. no. A-6455, lot 743620). On an orbital shaker at room temperature, free-floating sections were rinsed 3 times for 10 min each in PBS at pH 7.4 and then incubated for 2 h in blocking solution made of 3% normal donkey serum (Equitech-Bio, Inc., Kerrville, TX) with 0.25% Triton X-100 in PBS (PBT) with 0.02% sodium azide. Sections were incubated overnight at room temperature with GFP antisera diluted to 1:10,000 in 3% normal donkey serum PBT with 0.02% sodium azide. After washing in PBS, tissue was incubated with Alexa Fluor 488 donkey anti-rabbit antibody (Life Technologies, Eugene, OR; Ref # A21206, lot 1275888) diluted to 1:200 in 3% donkey serum in PBT for 2 h at room temperature. The tissue was then rinsed in PBS and mounted on Fisherbrand Superfrost slides and coverslipped with VECTASHIELD Hardset Antifade mounting media (Vector Labs, Burlingame, CA; Cat. No. H-1400).
2.5. Generation of HDC Gi-DREADD mice
Hdc-Cre mice were bred with the R26-LSL-Gi-DREADD (hM4Di) to generate mice that express hM4Di in all histaminergic cells (referred to as Hdc-Cre::hM4Di). Additional R26-LSL-Gi-DREADD were bred with Hdc-Cre::tdTomato mice for patch-clamp electrophysiology.
2.6. Lateral ventricle cannulations
Eight-to 10-week-old male mice were stereotaxically implanted with a guide cannula (Plastics1, Roanoake, VA) into the right lateral ventricle (0.4 mm posterior, 1 mm lateral from bregma) under 1–3% (v/v) isoflurane in 1 L/min oxygen. Animals were allowed 6–7 days to recover before the ICV cannula placement was verified through administration of angiotensin II (Tocris, 20 ng in 2 uL) and observation of the dipsogenic response. For all in vivo and gene expression experiments, MTII (Phoenix Pharmaceuticals) was reconstituted in aCSF to a concentration of 500 μM (500 pM/μL), which allowed for delivery of 1 nmol of MTII in 2 μL, aCSF was used as the vehicle control.
2.7. Gene expression
Hdc gene expression was assessed in 10-week-old C57BL/6J male mice following recovery from stereotaxic surgery. Food was withdrawn 2.5 h before ICV injection (MTII 1 nmol vs aCSF) and brains were harvested 2 h later. Hypothalamic sections were used to isolate total mRNA using RNA STAT-60 reagent (Tel-Test, Inc.). The RNA concentrations were estimated from absorbance at 260 nm. cDNA synthesis was performed using the iScript Advanced cDNA Synthesis Kit (Bio-Rad, 172–5038). mRNA extraction and cDNA synthesis were performed following the manufacturer's instructions. cDNA was diluted in DNase-free water before quantification by real-time PCR. Relative quantification of Hdc gene expression was performed on diluted cDNA in duplicate samples using a CFX384 Touch™ real-time PCR (Bio-Rad). Fold differences in targeted mRNA expression were calculated using the ΔΔCt method and data were normalized to beta-microglobulin (B2m) expression. TaqMan® gene expression assays for B2m (Mm00437762_m1) and Hdc (Mm00456104_m1) were purchased from ThermoFisher Scientific.
2.8. Evaluation of food intake and locomotor activity
Following recovery from stereotaxic surgery, Hdc-Cre::hM4Di mice and littermate controls were acclimatized to experimental cages for 5 days before the beginning of data collection. After two full days of baseline recording, mice had their food withdrawn 1 h after the beginning of the light period, and underwent IP (CNO 1 mg/kg vs saline), and ICV injections (MTII 1 nmol vs aCSF), (1 h, and 30 min, respectively) prior to the beginning of the dark period when food was returned. Food intake and locomotor activity were measured using a combined TSE Labmaster monitoring system (TSE Systems GmbH, Bad Homburg, Germany). Locomotion was measured using a multi-dimensional infrared light beam system. Body weight measurements were obtained prior to IP and ICV injections, and again the following morning 1 h after the beginning of the light period. Administration of MTII ICV and CNO IP was performed in a randomized cross-over design so that all mice received all drug combinations. Mice were allowed a three-day wash-out period between experimental days.
2.9. Experimental design and statistical analysis
All electrophysiological data was analyzed using Clampfit 10 (MDS Analytical Technologies) or mini analysis. All data are presented as means ± SEM. Two-tailed paired t-tests were generated using GraphPad Prism 7 for comparisons between conditions. Chi-squared statistics were used to test differences in response type between groups. Parametric statistics were used when the data from both conditions adopted a Gaussian distribution, and nonparametric statistics were used when this assumption was not met. All in vivo and gene expression data were analyzed for comparisons between conditions. Significant differences were determined using two-way ANOVA and Tukey multiple comparison tests. All effects were considered significant when p values < 0.05 and were adjusted when multiple comparisons were performed.
3. Results
3.1. Melanocortin receptor agonism activates TMN histaminergic neurons
To determine if the melanocortin system is capable of influencing the activity of HDC neurons, we explored the effects of the non-selective melanocortin 3 and 4 receptor (MC3R/MC4R) agonist MTII on HDC-expressing neurons using patch-clamp electrophysiology. Whole-cell patch clamp recordings were obtained from tdTomato fluorescently-labeled HDC neurons in posterior hypothalamic brain slices from adult Hdc-cre::tdTomato mice. Bath application of MTII (100 nM) excited 37% of HDC neurons (15/41 cells) recorded from 7 male and 5 female mice. MTII-induced excitation of HDC neurons was associated with membrane depolarization from a resting membrane potential of −48.0 ± 1.1 mV to −44.2 ± 0.9 mV in MTII (n = 15, Wilcoxon matched-pairs signed-rank test, W = 120, p < 0.0001) and was associated with a significant increase in the spontaneous firing rate from 0.1 ± 0.1 Hz to 0.8 ± 0.1 Hz (n = 15, Wilcoxon matched-pairs signed-rank test, W = 105, p = 0.001) (Figure 1A–C). This effect was associated with a significant decrease in input resistance from 659 ± 88 MΩ to 614 ± 82 MΩ (n = 15, Wilcoxon matched-pairs signed-rank test, W = −110, p = 0.0005), with a mean reversal potential of −0.2 mV ± 7.4 mV (n = 12), suggesting activation of a non-selective cation conductance (Figure 1D–E). Supporting these observations, expression of Hdc mRNA was increased in the mediobasal-hypothalamus (where HDC neurons reside) following intracerebroventricular (icv) administration of MTII (1 nmol) in C57BL/6J mice (Mann–Whitney test, U = 5, p = 0.02) (Figure 1F).
Figure 1.

MTII excites histaminergic neurons. A) Whole-cell current clamp recording showing MTII-induced depolarization and excitation of a HDC neuron. B) MTII induced a significant depolarization of the membrane potential. C) MTII induced a significant increase in firing rate. D) Current voltage relationships of a HDC neuron that was excited by MTII. E) Plot of data shown in D demonstrating a decrease in input resistance. Reversal potential averaged across all HDC neurons excited by MTII suggests activation of a non-selective cation conductance. F) Intracerebroventricular (ICV) MTII significantly increased Hdc mRNA expression in the mediobasal hypothalamus. Note: // denotes discontinuity in the recording (of 35 sec) where current voltage relations were performed. ∗p < 0.05, ∗∗∗p < 0.001.
In addition to the dominant excitatory effect of MTII on HDC neurons, a small proportion (4/41 cells) were inhibited by MTII, displaying membrane hyperpolarization and a decrease in firing rate (data not shown). The remaining 54% (22/41 cells) of HDC neurons were non-responsive to MTII, displaying no change in resting membrane potential (control −63.5 ± 1.4 mV vs MTII -64.3 ± 1.4 mV, t(19) = 0.99, p = 0.35, paired t-test) or firing rate in those cells that were active (control 2.2 ± 0.4 Hz vs MTII 2.1 ± 0.4 Hz, t(9) = 0.95, p = 0.37, paired t-test). Importantly, HDC neurons from male and female mice showed a similar proportion of cells responsive to MTII (12/25 and 7/16 cells responding respectively) (Fisher's exact test, p > 0.99). Male mice were utilized for all remaining in vitro and in vivo experiments.
3.2. Melanocortin receptors are presynaptic to TMN histaminergic neurons and their activation requires glutamate receptor activity
To determine whether MTII has a direct effect on HDC neurons, we administered MTII in the presence of tetrodotoxin (TTX, 500 nM). Under these conditions, 100% of HDC neurons (18/18 cells) failed to respond to MTII, with no change in membrane potential (control −56.5 ± 1.7 mV vs MTII -57.5 ± 1.7 mV, n = 18, Wilcoxon matched-pairs signed-rank test, W = −49, p = 0.30) (Figure 2A) or input resistance (control 683 ± 46 MΩ vs MTII 676 ± 48 MΩ, t(17) = 0.46, p = 0.65, paired t-test). These results indicate an indirect effect of MTII on HDC neurons that requires action potential-mediated synaptic transmission.
Figure 2.

Melanocortin receptors are presynaptic to, and their activation depolarizes histaminergic neurons via ionotropic glutamate receptors. A) Whole-cell current clamp recording showing that MTII has no effect on a HDC neuron when administered in the presence of TTX. B) Whole-cell voltage clamp recording showing MTII-induced increase in sEPSC amplitude as assessed by Kolmogorov-Smirnov test. ∗ indicates sEPSC enlarged below. C) Normalized increase in sEPSC amplitude or frequency, based off Kolmogorov-Smirnov test, following MTII administration. Numbers of cells are in parentheses. D) Whole-cell current clamp recording showing that MTII has no effect on a HDC neuron when administered in the presence of the ionotropic glutamate receptor antagonists CNQX and AP5. Note: // denotes discontinuity in the recording where current voltage relations were performed (35 sec and 45 sec, in A and D respectively).
Voltage-clamp experiments were performed to establish if pre-synaptic activation of melanocortin receptors by MTII modified excitatory or inhibitory inputs arriving onto histaminergic neurons. Spontaneous excitatory post-synaptic currents (sEPSCs) were assessed at a holding potential of −60 mV to −70 mV. MTII increased the amplitude of sEPSCs in three of 13 recorded HDC neurons (p < 0.05; Kolmogorov–Smirnov test) from 14.2 ± 1.6 pA to 17.5 ± 0.8 pA (Figure 2B) and increased the frequency of sEPSCs in three of the 13 HDC neurons (p < 0.05; Kolmogorov–Smirnov test) from 1.9 ± 0.6Hz to 2.4 ± 0.7Hz. This equates to an average 25.2% increase in amplitude and 36.7% increase in frequency in these cells compared to control conditions (Figure 2C). Only one of the HDC neurons was associated with a significant increase in both amplitude and frequency of sEPSCs. In contrast, when a separate population of HDC neurons were held at −15 mV to assess spontaneous inhibitory post-synaptic currents (sIPSCs), only one of 14 recorded HDC neurons had a significant decrease in the amplitude of sIPSCs. The remaining cells displayed no change in amplitude or frequency (p > 0.05; Kolmogorov–Smirnov test). These data suggest that MTII enhances overall excitatory synaptic input to excite HDC neurons.
The findings above suggested that glutamate may mediate the excitatory effects of MTII on HDC neurons. Therefore, CNQX 10 μM and AP-5 50 μM were used to block ionotropic glutamate receptors. In the presence of these antagonists, MTII failed to affect membrane potential (control −47.5 ± 1.4 mV vs MTII −47.5 ± 1.3 mV, t(14) = 0.04, p = 0.97, paired t-test) or firing rate (control 1.0 ± 0.3 Hz vs MTII 0.9 ± 0.3 Hz, n = 15, Wilcoxon matched-pairs signed-rank test, W = −24, p = 0.38) (Figure 2D). There was also no change in input resistance (control 849 ± 95 MΩ vs MTII 862 ± 99 MΩ, t(13) = 0.9, p = 0.38, paired t-test) in 100% (15/15) of HDC neurons. Together, these effects suggest that melanocortin receptors on presynaptic glutamatergic neurons mediate the excitatory effects of MTII on HDC neurons.
3.3. MC4R, not MC3R, mediates excitation of TMN histaminergic neurons
As MTII is a non-selective agonist for both the MC3R and MC4R, we next sought to determine which melanocortin receptor/s mediate the excitation of HDC neurons following MTII administration. To specifically activate the MC4R, we bath applied the MC4R selective agonist THIQ at 100 nM. THIQ excited 38% of HDC neurons (8/21 cells), inducing membrane depolarization from a resting membrane potential of −46.4 ± 2.4 mV in control aCSF to −43.9 ± 2.0 mV in THIQ (t(7) = 4.9, p = 0.0018, paired t-test) and an increase in firing frequency (control 0.6 ± 0.4 Hz vs THIQ 1.3 ± 0.4 Hz, n = 7, Wilcoxon matched-pairs signed-rank test, W = 28, p = 0.0156) (Figure 3A–C). This effect was also associated with a small but significant decrease in input resistance (control 768 ± 122 MΩ vs 704 ± 104 MΩ, n = 8, Wilcoxon matched-pairs signed-rank test, W = −36, p = 0.0078) with a mean reversal potential of −6.6 mV ± 15.4 mV (n = 8) (Figure 3D–E). The effects of THIQ on membrane potential, firing rate, and input resistance of HDC neurons recapitulated that of MTII, suggesting that the MC4R mediated the excitatory effects seen on HDC neurons.
Figure 3.

MC4R not MC3R mediates excitation of histaminergic neurons. A) Whole-cell current clamp recording showing THIQ (MC4R selective agonist) induced depolarization and excitation of a HDC neuron. B) THIQ induced a significant depolarization of the membrane potential. C) THIQ induced a significant increase in firing rate. D) Current voltage relationships of a HDC expressing neuron that was excited by THIQ. (THIQ IV was performed with -11.3pA of holding current). E) Plot of data in D demonstrating a decrease in input resistance. Reversal potential averaged across all HDC neurons excited by MTII suggests activation of a nonselective cation conductance. F) Whole-cell current clamp recording showing that [D-Trp8]-γ-MSH (MC3R selective agonist) had no effect on a HDC neuron. G) Summary of the responses to the non-selective and selective melanocortin receptor agonists and combination with various synaptic blockers. H) Immunocytochemistry of a posterior hypothalamic brain slice containing the TMN from a Mc4r-GFP::HDC-Cre::tdTomato mouse. The HDC expressing neurons (red) do not overlap with any of the MC4R (green) expressing neurons, demonstrating no co-expression of MC4R and HDC. Note: // denotes discontinuity in the recording where current voltage relations were performed (67 sec and 34 sec in A and F respectively). ∗p < 0.05, ∗∗p < 0.01.
To investigate whether MC4R activation alone was responsible for the excitation of histaminergic neurons, we bath applied the MC3R selective agonist [D-Trp8]-γ-MSH. 50 nM [D-Trp8]-γ-MSH had no effect on membrane potential (control −45.3 ± 1.4 mV vs [D-Trp8]-γ-MSH −45.8 ± 1.4 mV, t(12) = 1.8, p = 0.096, paired t-test) (Figure 3F) or input resistance (control 642 ± 53 MΩ vs [D-Trp8]-γ-MSH 626 ± 51 MΩ, t(12) = 1.8, p = 0.092, paired t-test) in 100% of recorded HDC neurons (13/13). However, a small but statistically significant increase in firing rate of HDC neurons was observed in those cells that were active (control 1.8 ± 0.3 Hz vs [D-Trp8]-γ-MSH 2.0 ± 0.3 Hz, t(7) = 3.09, p = 0.018, paired t-test). Taken together, the lack of effect of [D-Trp8]-γ-MSH on HDC neurons suggest that activation of MC4R, not MC3R, mediates melanocortin receptor driven excitation of HDC neurons.
The proportion of HDC neurons that responded to the different melanocortin receptor agonists, including in the presence of antagonists, are summarized in Figure 3G. This demonstrates that not only was the magnitude of the response to the MC4R selective agonist similar to that induced by MTII, but the proportion of HDC neurons responding to MC4R selective agonism also recapitulated that of MTII.
We also examined MC4R expression in HDC neurons. We took advantage of the Mc4r-GFP transgenic mice and bred them to Hdc-Cre::tdTomato mice to examine GFP and TdTomato co-expression in posterior hypothalamic brain slices containing the TMN. Slices were prepared for patch-clamp electrophysiology and for immunocytochemistry using antibodies against GFP. Regardless of the preparation method, we failed to identify co-expression of GFP and tdTomato (Figure 3H). Together these data suggest that melanocortin receptors, specifically MC4R, are not expressed by HDC neurons.
When all HDC neurons exposed to MC3/4R or MC4R agonists (MTII and THIQ) were mapped across the rostral–caudal axis of the brain, no distinct anatomical distribution patterns were noted although the most caudal section of the TMN appeared to have more MTII non-responsive HDC neurons. Seventy-eight percent of cells for bregma −2.80 mm were non responsive to melanocortin receptor agonism compared to 55%, 54%, and 50% of cells for bregma −1.70 mm, −2.06 mm, and −2.46 mm, respectively (Figure 4); however, this difference was not statistically significant (chi-squared, X2 (3, n = 4) = 3.4, p = 0.3).
Figure 4.
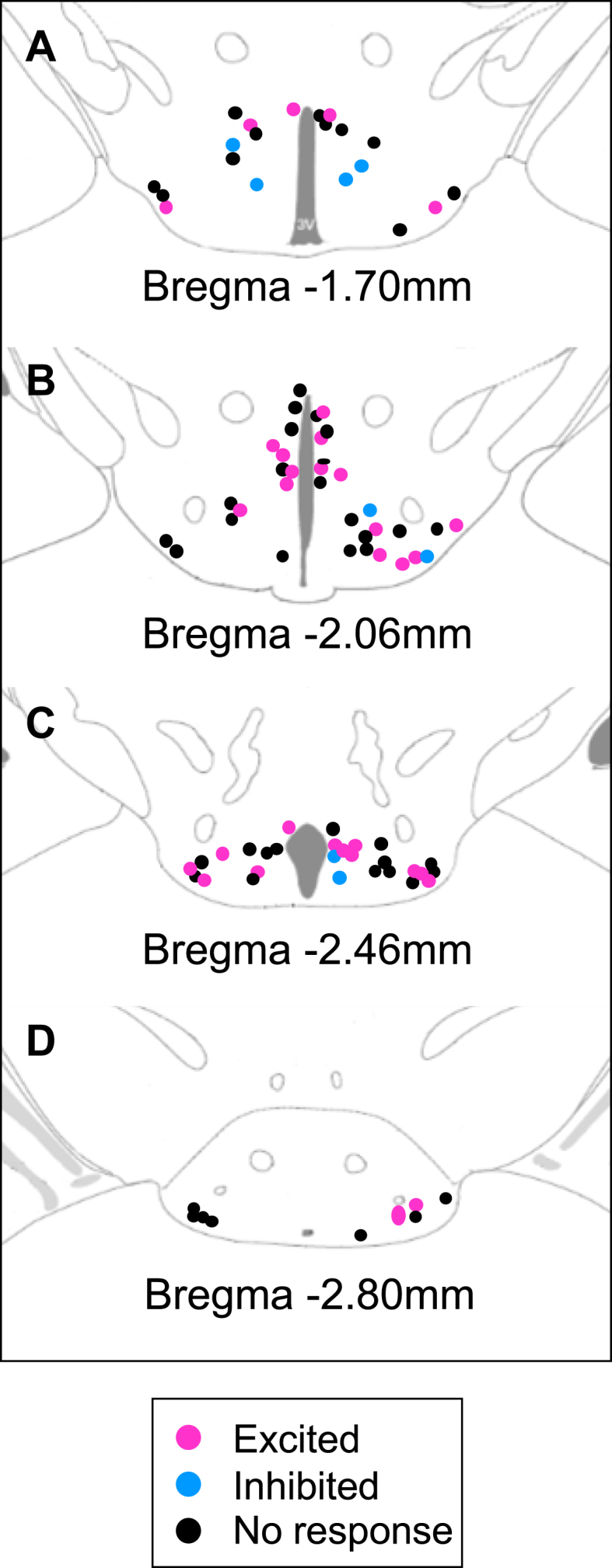
HDC neuron responsiveness to melanocortin receptor agonism did not differ across the TMN∗. HDC neurons that were exposed to selective and non-selective melanocortin receptor agonists (THIQ & MTII) were mapped across the TMN. Response to melanocortin receptor agonism did not reveal any distinct anatomical distribution. A) Bregma -1.70mm where 45% (9/20) of HDC neurons responded to melanocortin receptor agonism and 55% (11/20) were non responsive. B) Bregma -2.06mm where 46% (16/35) of HDC neurons responded to melanocortin receptor agonism and 54% (19/35) were non responsive. C) Bregma -2.46mm where 50% (14/28) of HDC neurons responded to melanocortin receptor agonism and 50% (14/28) were non responsive. D) Bregma -2.80mm where 2/9 (22%) of HDC neurons responded to melanocortin receptor agonism and 78% (7/9) were non-responsive. ∗Image modified from: Mouse Brain: In Stereotaxic Coordinates (3rd ed.). Franklin, K. B. J., & Paxinos, G. Copyright Elsevier (2007).
3.4. Perifornical lateral hypothalamic neurons innervate and contribute to the MTII-induced excitation of TMN histaminergic neurons
We found that the melanocortin receptor-induced activation of HDC neurons occurred through a presynaptic action potential mediated mechanism. This suggests that the MC4R-expressing neurons responsible for the effect were located within the same hypothalamic brain slices as the HDC cells. MC4R is highly expressed within the hypothalamus [30,31,38]; however, intra-hypothalamic nuclei projecting to the TMN are limited [39]. The lateral hypothalamus is one hypothalamic area that heavily innervates HDC neurons of the TMN [7,40] and contains cells expressing the MC4R [41,42]. Therefore, we investigated the lateral hypothalamus as a potential source of the MC4R-sensitive input to HDC neurons.
A concentric bipolar stimulating electrode was placed within the perifornical/lateral hypothalamus (PeFLH), superior to the fornix, an area known to contain orexin neurons [43,44]. A paired pulse protocol was initiated upon identification of HDC neurons that received evoked excitatory post-synaptic currents (eEPSCs) elicited by electrically stimulating the PeFLH. A minimum of 20 trains of paired pulses (323 ± 62 μA) were delivered to the PeFLH and the resultant postsynaptic currents (voltage clamp) measured at HDC neurons were averaged. Sixteen HDC neurons received short- and constant latency-eEPSCs (onset latency = 6.8 ± 0.4 ms) following simulation of the PeFLH, demonstrating that HDC neurons are synaptically connected to the PeFLH neurons. MTII (100 nM) enhanced the amplitude of constant-latency eEPSCs in 6 of 16 HDC neurons from 23.6 ± 3.3 pA in control to 31.1 ± 4.2 pA in MTII (129% increase from control; n = 6; Wilcoxon matched-pairs signed-rank test, W = 21, p = 0.03) suggesting that MTII had an excitatory effect on these synaptically-connected HDC-PeFLH cells (Figure 5A–B). The remaining 10 HDC neurons showed no change in the amplitude of constant-latency eEPSCs following MTII (32.4 ± 5.3 pA in control to 31.4 ± 4.9 pA in MTII, n = 10, Wilcoxon matched-pairs signed-rank test, W = −15, p = 0.492).
Figure 5.

Perifornical lateral hypothalamic neurons mediate MTII-induced excitation of HDC neurons. A) Averaged voltage-clamp traces showing that MTII (100nM) enhanced the amplitude of the first constant-latency eEPSC evoked after PeFLH paired-pulse stimulation. B) Absolute increase in the evoked EPSC amplitude of the first response following PeFLH stimulation. C) MTII induced a significant decrease in the paired-pulse ratio (PPR) of eEPSCs recorded in HDC neurons following PeFLH stimulation. D) Whole-cell current clamp recording demonstrating a HDC neuron that was excited by the MC4R agonist THIQ, was also excited by orexin A (200nM). E) The majority of HDC neurons that were responsive to MC4R agonism (THIQ) were also excited by orexin A, whereas only about 50% of HDC neurons that were non-responsive (NR) to MC4R agonism where also excited by orexin A. Numbers of cells are in parentheses. Note: // denotes discontinuity in the recording where current voltage relations were performed (83 sec for THIQ and 45 sec for orexin A). ∗p < 0.05, ∗∗p < 0.01, ns = non significant.
The paired pulse ratio (PPR) was examined to assess whether MTII acted presynaptically in the PeFLH to enhance constant-latency eEPSCs. Stimuli paired 30 ms apart (33Hz) revealed either a paired-pulse depression or facilitation in eEPSC amplitude, where the amplitude of the second eEPSC was either smaller or larger than the first. This is consistent with previous studies demonstrating both paired pulse depression and paired pulse facilitation in HDC neurons following optogenetic stimulation of orexin neurons [40]. Bath application of MTII enhanced the amplitude of the first eEPSC to a larger degree than the second in all but one affected cell, overall revealing a decrease in the paired-pulse ratio (R2/R1) from 1.1 ± 0.1 in control to 0.8 ± 0.1 in MTII (n = 5; Friedman test, p = 0.0085; Dunn's multiple comparison test p = 0.0089 control vs MTII, and p = 0.69 control vs wash) (Figure 5C). In all cases, MTII enhanced the amplitude of one evoked response more than the other, consistent with a presynaptic mechanism of action of MTII. These data indicate that activation of melanocortin receptors in the PeFLH area enhances the excitatory input to HDC neurons. Moreover, these data strongly suggest that MC4Rs located in the PeFLH mediate the MC4R agonist-induced excitation of HDC neurons.
In the lateral hypothalamus, MC4R is expressed in neurons with different peptidergic profiles, including the orexin/hypocretin neurons [41,45]. Orexin/hypocretin neurons are a major input to the histaminergic neurons of the TMN and also co-express glutamate [40,46], the fast-excitatory neurotransmitter shown in the present study to mediate the MC4R-induced excitation of HDC neurons. Therefore, to determine if the HDC neurons excited by melanocortin agonism were also potentially innervated by orexin neurons, we bath exposed MC4R-activated HDC neurons to orexin A (200 nM). Of 11 recorded HDC neurons that were excited by MC4R agonism (THIQ 100 nM), 90.1% (10/11) of these neurons were also activated by orexin A. This is in contrast to only 52.6% (10/19) of HDC neurons that were non-responsive to MC4R agonism (Figure 5D–E). These results suggest that HDC neurons that respond to MC4R agonism are also sensitive to orexin.
3.5. Chemogenetic inhibition of TMN histaminergic neurons potentiates the anorexigenic effects of MTII
We next sought to determine the potential physiological relevance of our findings. Previous studies have utilized a suicide inhibitor of HDC to eliminate histaminergic neuron's contribution to drug and hormone-induced food suppression [23,47]. We inhibited HDC neurons in vivo, using a chemogenetic approach to determine the normal contribution of HDC neurons to MTII-induced anorexia. Briefly, Hdc-Cre mice were bred with R26-LSL-Gi-DREADD to generate Hdc-Cre::hM4Di mice in which HDC neurons can be inhibited by the DREADD ligand clozapine-N-Oxide (CNO). We first validated the efficiency of this approach using whole-cell patch-clamp recordings. As shown in Figure 6A, CNO induced membrane hyperpolarization (from −52.2 ± 2.4 mV to −56.3 ± 2.4 mV (t(8) = 7.7, p < 0.0001, paired t-test) in 90% (9/10) of recorded HDC neurons.
Figure 6.

Histaminergic neurons modulate the anorexigenic effects of MTII. A) Whole-cell current clamp recording showing CNO-induced hyperpolarization and inhibition of a HDC expressing cell from a Hdc-Cre::hM4Di mice mouse. B) Left: Cumulative food intake across the night was significantly different between the groups studied. Right: Hourly analysis of cumulate food intake (up to 6hrs post food return) showed MTII significantly reduced food intake in control animals and this effect was significantly enhanced when HDC neurons were inhibited. C) Left: Cumulative locomotor activity across the night was significantly different between the groups studied. Right: Hourly analysis of cumulative locomotor activity (up to 6hrs post food return) showed MTII had no effect on locomotor activity in control animals but was significantly reduced with MTII by 5hrs when HDC neurons were inhibited. Note: // denotes discontinuity (of 40sec) in the recording where current voltage relations were performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 for comparisons between aCSF and MTII treated animals of the same genotype and #p < 0.05, ##p < 0.01 for comparisons between MTII treated hM4Di and Hdc-Cre::hM4Di animals.
Having confirmed the inhibitory effects of CNO on brain slices (Figure 6A), we ICV injected Hdc-Cre::hM4Di mice and littermate controls with MTII (1 nmol) or aCSF 30 min prior to the dark phase when they received food. All animals were also IP injected with CNO (1 mg/kg) 60 min prior to the dark phase, thereby controlling for any potential effects of CNO itself. Two-way analysis of variance (ANOVA) revealed differences in cumulative food intake between groups (two-way ANOVA, F(3, 23) = 13, p < 0.0001) (Figure 6B). Consistent with previous reports [48,49], MTII suppressed cumulative food intake compared to aCSF in control animals, an effect that was evident 1 h after food was returned (Tukey's multiple comparison, p = 0.0084). However, when histaminergic neurons were inhibited, the MTII-induced food suppression was dramatically enhanced. Hourly analysis indicated that cumulative food intake was significantly less in MTII-treated Hdc-Cre::hM4Di mice compared to MTII-treated littermate control animals as early as 1 h after food was returned (Tukey's multiple comparison, p = 0.0331). CNO alone had no effect on cumulative food intake between littermate control or Hdc-Cre::hM4Di mice (Tukey's multiple comparison, p = 0.7070) (Figure 6B). The enhanced suppression of food intake that occurred with MTII when HDC neurons were inhibited was also associated with significant weight loss through the night (aCSF 1.25 ± 0.88 g vs MTII -2.23 ± 0.6 g) that was not seen in the control animals (aCSF 0.75 ± 1.18 g vs MTII 0.50 ± 1.08 g) (F(1, 10) = 10.64, two-way ANOVA interaction p = 0.0085) (Sidak's multiple comparisons test HDC-Gi p = 0.0031, control p = 0.8916). These data suggest that inhibiting HDC neurons enhances the anorexigenic effects of melanocortin receptor agonism.
Histaminergic neurons play an important role in regulating sleep/wake states. Therefore, we also assessed locomotor activity to evaluate whether inactivity could explain the drastic reduction in food intake observed in MTII-treated Hdc-Cre::hM4Di mice. Two-way analysis of variance (ANOVA) revealed differences in cumulative locomotor activity between groups (F(3, 23) = 3.439, p = = 0.0336, two-way ANOVA). Multiple comparison analysis over the 6-h period following food being returned showed that inhibition of histaminergic neurons alone had no effect on locomotor activity (Tukey's multiple comparison, p > 0.05 at all time points); however, the combination of MTII and inhibition of HDC neurons led to a reduction in cumulative locomotor activity (Figure 6C). When assessed hourly, it was evident that cumulative locomotor activity was significantly reduced 5 h after food was returned in MTII-treated Hdc-Cre::hM4Di mice, when compared to both MTII-treated control and aCSF-treated Hdc-Cre::hM4Di mice (Tukey's multiple comparison, p = 0.0088 and p = 0.0055, respectively) (Figure 6C). The food intake effects were observed in the first hour after food was returned, and the decrease in locomotor activity did not occur until the fifth hour. These data suggest that inhibiting histaminergic neurons enhances the anorexigenic effects of MTII via mechanisms other than induction of sleep or decreased arousal. The reduction in locomotor activity may, however, explain some of the potentiation of anorexigenic effects later in the night.
We proposed a model to summarize the mechanism by which MC4R agonism excites histaminergic neurons of the TMN to regulate food intake (Figure 7).
Figure 7.

Proposed model by which the melanocortin system excites HDC neurons to regulate food intake. 1) Endogenous melanocortin ligand (α-MSH) binds to and activates 2) MC4Rs on glutamatergic neurons in the PeFLH. 3) Binding of glutamate on postsynaptic receptors located on HDC neurons contributes to 4) increased electrical excitability of HDC neurons. 5) Melanocortin-induced activation of HDC neurons restrains the anorexigenic effects of melanocortin system activation.
4. Discussion
The histaminergic system is involved in the regulation of energy homeostasis. However, little is known about the ability of HDC neurons to detect changes in metabolic status. Using complementary electrophysiological, molecular genetics, and behavioral/physiological approaches, we explored the prediction that HDC neurons in the TMN receive metabolically relevant information via the melanocortin system. We found that subpopulations of HDC neurons are excited by MC4R activation via a glutamatergic-dependent presynaptic mechanism. Importantly, we revealed that this input originates from the perifornical lateral hypothalamus. In vivo chemogenetic silencing of HDC neurons enhanced the anorexigenic effects of the melanocortin receptor agonist MTII, suggesting that physiologic melanocortin-induced activation of HDC neurons may constrain the metabolic effects of the melanocortin system. Our findings provide the first evidence that activation of MC4R has excitatory effects within the TMN and demonstrate a functional interaction between the melanocortin and histaminergic systems. Collectively, our results support a model predicting that MC4R agonists excite histaminergic neurons of the TMN to regulate food intake (Figure 7). Our results further suggest that MC4R activation of histaminergic neurons may restrain the anorexigenic effects of melanocortin system activation.
4.1. Presynaptic MC4Rs mediate excitatory inputs to TMN histaminergic neurons
Using inhibitors of synaptic transmission and dual-labeling studies, we demonstrated that the MC4R mediates excitatory effects on HDC neurons via increased glutamatergic signaling. These findings establish that the MC4Rs expressed within the region of the TMN [30,31] are not expressed on HDC neurons themselves. While the increased excitability of HDC neurons following MTII treatment were presynaptic in nature, it is currently unclear how MTII enhanced sEPSC amplitude independent of frequency in some HDC neurons. These findings may indicate a more complex mechanism of regulation of neuronal activity. In particular, activation of the MC4R is associated with an increase in intracellular calcium concentrations [[50], [51], [52]] and may therefore have the potential to alter the readily releasable pool or probability of release of neurotransmitters in the presynaptic neurons [53]. We also cannot rule out the possibility of astrocyte-mediated glutamate release [54], possibly triggered by synaptically-released glutamate [55] or by other neurotransmitters, including histamine [56,57]. These multiple possible mechanisms warrant further investigation.
4.2. Perifornical/lateral hypothalamic (PeFLH) neurons expressing MC4Rs innervate TMN histaminergic neurons
HDC neurons receive heavy innervation from the LH, particularly from the orexin/hypocretin expressing neuronal population [7,40,58]. The perifornical region of the lateral hypothalamus (PeFLH) is heavily enriched in orexin/hypocretin neurons [43]. A proportion of the orexin neurons express the MC4R [41], and orexin neurons mediate TTX- and CNQX-sensitive glutamatergic currents arriving at HDC neurons [58]. Accordingly, we observed eEPSCs in HDC neurons following electrical stimulation of the PeFLH. Importantly, we found that MTII enhanced this excitatory input. These data suggest that MC4Rs located in the PeFLH are a source of the presynaptic MC4R-expressing neurons mediating the MC4R agonist-induced excitation of HDC neurons, as summarized in our model (Figure 7). In addition, TMN neurons express orexin receptor 2 [59]. Notably, the majority of HDC neurons activated by MC4R agonism were also excited by orexin. Together, this raises the possibility that the MC4R-expressing PeFLH neurons mediating the melanocortin-induced excitation of HDC neurons may also express orexin. One technical limitation to consider is that electrical stimulation protocols may lead to stimulation of fibers of passage. However, it should be noted that the identified inputs to the TMN are relatively limited [39].
4.3. Mechanisms by which TMN histaminergic neurons may regulate food intake
Given the potential role of the histaminergic system in regulating food intake [14,60], we predicted that the melanocortin-induced excitation of HDC neurons would reduce food intake. We used a chemogenetic approach to inhibit HDC neurons and minimize any potential contribution of these neurons to the physiological effects of MTII. We found that chemogenetic inhibition of HDC neurons significantly enhanced the anorexigenic actions of MTII, effects opposite to our expectations. Decreased histamine levels and prevention of histamine binding at histamine H1 receptors (H1Rs) have been shown to increase food intake [47,61,62]. Our results, however, suggest that a potential physiological role of activation of HDC neurons by MTII is to restrain the food intake suppression, and therefore implicates the involvement of a neurotransmitter and/or mechanism other than histamine binding at H1Rs.
Activation of HDC neurons by MTII also has the potential to influence multiple other neurotransmitter systems via the histamine 3 receptor (H3R). Although the H3R was originally described as an auto-receptor that could suppress the release of histamine itself [63], recent studies suggest that the H3R also functions as a heteroreceptor [14]. H3R agonism suppresses serotonin, dopamine, noradrenaline, and acetylcholine release [64,65], neurotransmitters all capable of altering food intake. The H3R also has the ability to modify GABA-mediated synaptic transmission [66,67]. In addition, HDC neurons co-express GABA and project to multiple hypothalamic nuclei involved in the regulation of energy homeostasis [14,68]. Removing a GABAergic inhibitory tone in these areas may allow melanocortin receptor agonism an unrestrained opportunity to suppress food intake. While these theories are intriguing, further investigation is required to determine any potential for these mechanisms in contributing to the enhanced anorexigenic effects of MTII following chemogenetic inhibition of HDC neurons.
4.4. The role of TMN histaminergic neurons in regulating homeostatic processes
Recent studies have questioned the role of HDC neurons in the regulation of arousal. Acute activation or inhibition of HDC neurons was shown to have minimal effect on sleep or wakefulness under basal conditions [69]. However, HDC neuron activity is thought to be physiologically relevant in the context of arousal when facing additional behavioral challenge [8,68,69]. Similarly, in the present study, chemogenetic inhibition of HDC neurons had no effect on food intake or activity alone. It was only following additional (metabolic) stimulation (with MTII) that dramatically enhanced reductions in food intake were observed. Together, these studies suggest that HDC neurons may be important for integrating or regulating responses to homeostatic challenges. This points to a role for HDC neurons in fine tuning ‘normal’ responses to metabolic and behavioral challenges, rather than basal control of these processes.
The melanocortin system is widely recognized as a key regulator of energy homeostasis, with the MC4R itself being crucial for the maintenance of normal body weight [25,33,70]. Notably, pharmacological therapeutics targeting the melanocortin system for the treatment of obesity are uncommon, except in the case of rare monogenic forms of obesity [71]. The present results demonstrate that the HDC neurons act to restrain MC4R-mediated effects on food intake and may limit the effectiveness of MC4R agonists in reducing food intake and body weight. Therefore, pharmacological approaches aimed at inhibiting HDC neurons in combination with activating MC4R may represent a novel strategy to effectively reduce appetite. Further studies are required to assess the long-term effectiveness of such an approach and any potential for undesirable outcomes associated with decreased activity levels.
In conclusion, subpopulations of HDC neurons are activated by MC4R agonism via an indirect glutamatergic mechanism. Electrical stimulation showed that melanocortin receptors located in the PeFLH contribute to these excitatory effects. In vivo studies using chemogenetic inhibition of HDC neuron activity revealed that the normal activation of HDC neurons by MTII could represent an important metabolic response to melanocortin system activation. These findings demonstrate, for the first time, a striking interaction between the melanocortin and histaminergic systems, and suggest that the histaminergic system may act to restrain the anorexigenic effects of melanocortin (MC4R) activation. Together, the ability of this circuitry to integrate both arousal and metabolic information may provide an important facet for understanding links between sleep and metabolic dysfunction.
Author contributions
Conceptualization, NJM, AC, and JKE; Methodology, NJM, AC, CEL, CMC, and KWW; Formal Analysis, NJM and AC; Investigation, NJM, AC, and CEL; Writing - Original Draft, NJM; Writing - Review & Editing, NJM, AC, KWW, SL, and JKE; Funding Acquisition, JKE; Supervision, JKE and KWW; Resources, JKE, KWW, and JMZ. All authors contributed to editing the manuscript and approved the manuscript for publication.
Acknowledgments
We thank the NIH for its support (R01 DK008423 and R01 DK118725 to JKE, R01 DK119169, R01 DK100699 and P01 DK119130 to KWW, K99 DK120894 to AC, K01 DK111644 to CMC), as well as a gift from the David and Teresa Disiere Foundation to JMZ. AC is a Canadian Institutes of Health Research (CIHR) Banting fellow. The authors would also like to thank the staff of the UT Southwestern Metabolic Phenotyping Core.
Contributor Information
Kevin W. Williams, Email: Kevin.williams@utsouthwestern.edu.
Joel K. Elmquist, Email: Joel.elmquist@utsouthwestern.edu.
Conflict of interest
None declared.
References
- 1.Panula P., Yang H.Y., Costa E. Histamine-containing neurons in the rat hypothalamus. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(8):2572–2576. doi: 10.1073/pnas.81.8.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watanabe T., Taguchi Y., Shiosaka S., Tanaka J., Kubota H., Terano Y. Distribution of the histaminergic neuron system in the central nervous system of rats; a fluorescent immunohistochemical analysis with histidine decarboxylase as a marker. Brain Research. 1984;295(1):13–25. doi: 10.1016/0006-8993(84)90811-4. [DOI] [PubMed] [Google Scholar]
- 3.Takeda N., Inagaki S., Taguchi Y., Tohyama M., Watanabe T., Wada H. Origins of histamine-containing fibers in the cerebral cortex of rats studied by immunohistochemistry with histidine decarboxylase as a marker and transection. Brain Research. 1984;323(1):55–63. doi: 10.1016/0006-8993(84)90264-6. [DOI] [PubMed] [Google Scholar]
- 4.Taylor K.M., Snyder S.H. Isotopic microassay of histamine, histidine, histidine decarboxylase and histamine methyltransferase in brain tissue. Journal of Neurochemistry. 1972;19(5):1343–1358. doi: 10.1111/j.1471-4159.1972.tb01459.x. [DOI] [PubMed] [Google Scholar]
- 5.Green J.P., Prell G.D., Khandelwal J.K., Blandina P. Aspects of histamine metabolism. Agents & Actions. 1987;22(1–2):1–15. doi: 10.1007/BF01968810. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz J.C., Arrang J.M., Garbarg M., Pollard H., Ruat M. Histaminergic transmission in the mammalian brain. Physiological Reviews. 1991;71(1):1–51. doi: 10.1152/physrev.1991.71.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Saper C.B., Scammell T.E., Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437(7063):1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 8.Parmentier R., Ohtsu H., Djebbara-Hannas Z., Valatx J.L., Watanabe T., Lin J.S. Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. Journal of Neuroscience. 2002;22(17):7695–7711. doi: 10.1523/JNEUROSCI.22-17-07695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin J.S., Sakai K., Jouvet M. Evidence for histaminergic arousal mechanisms in the hypothalamus of cat. Neuropharmacology. 1988;27(2):111–122. doi: 10.1016/0028-3908(88)90159-1. [DOI] [PubMed] [Google Scholar]
- 10.Monti J.M., Pellejero T., Jantos H. Effects of H1- and H2-histamine receptor agonists and antagonists on sleep and wakefulness in the rat. Journal of Neural Transmission. 1986;66(1):1–11. doi: 10.1007/BF01262953. [DOI] [PubMed] [Google Scholar]
- 11.Ericson H., Watanabe T., Kohler C. Morphological analysis of the tuberomammillary nucleus in the rat brain: delineation of subgroups with antibody against L-histidine decarboxylase as a marker. Journal of Comparative Neurology. 1987;263(1):1–24. doi: 10.1002/cne.902630102. [DOI] [PubMed] [Google Scholar]
- 12.Inagaki N., Yamatodani A., Ando-Yamamoto M., Tohyama M., Watanabe T., Wada H. Organization of histaminergic fibers in the rat brain. Journal of Comparative Neurology. 1988;273(3):283–300. doi: 10.1002/cne.902730302. [DOI] [PubMed] [Google Scholar]
- 13.Haas H., Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nature Reviews Neuroscience. 2003;4(2):121–130. doi: 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- 14.Haas H.L., Sergeeva O.A., Selbach O. Histamine in the nervous system. Physiological Reviews. 2008;88(3):1183–1241. doi: 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K., Suwa H., Ishikawa T., Kotani H. Targeted disruption of H3 receptors results in changes in brain histamine tone leading to an obese phenotype. Journal of Clinical Investigation. 2002;110(12):1791–1799. doi: 10.1172/JCI200215784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasuda T., Masaki T., Sakata T., Yoshimatsu H. Hypothalamic neuronal histamine regulates sympathetic nerve activity and expression of uncoupling protein 1 mRNA in brown adipose tissue in rats. Neuroscience. 2004;125(3):535–540. doi: 10.1016/j.neuroscience.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 17.Tabarean I.V. Histamine receptor signaling in energy homeostasis. Neuropharmacology. 2016;106:13–19. doi: 10.1016/j.neuropharm.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masaki T., Yoshimatsu H., Chiba S., Watanabe T., Sakata T. Targeted disruption of histamine H1-receptor attenuates regulatory effects of leptin on feeding, adiposity, and UCP family in mice. Diabetes. 2001;50(2):385–391. doi: 10.2337/diabetes.50.2.385. [DOI] [PubMed] [Google Scholar]
- 19.Jorgensen E.A., Vogelsang T.W., Knigge U., Watanabe T., Warberg J., Kjaer A. Increased susceptibility to diet-induced obesity in histamine-deficient mice. Neuroendocrinology. 2006;83(5–6):289–294. doi: 10.1159/000095339. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y.Q., Li R., Wu X., Zhu F., Takata Y., Zhang Z. Fasting activated histaminergic neurons and enhanced arousal effect of caffeine in mice. Pharmacology Biochemistry and Behavior. 2015;133:164–173. doi: 10.1016/j.pbb.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Miklos I.H., Kovacs K.J. Functional heterogeneity of the responses of histaminergic neuron subpopulations to various stress challenges. European Journal of Neuroscience. 2003;18(11):3069–3079. doi: 10.1111/j.1460-9568.2003.03033.x. [DOI] [PubMed] [Google Scholar]
- 22.Morimoto T., Yamamoto Y., Mobarakeh J.I., Yanai K., Watanabe T., Watanabe T. Involvement of the histaminergic system in leptin-induced suppression of food intake. Physiology & Behavior. 1999;67(5):679–683. doi: 10.1016/s0031-9384(99)00123-7. [DOI] [PubMed] [Google Scholar]
- 23.Toftegaard C.L., Knigge U., Kjaer A., Warberg J. The role of hypothalamic histamine in leptin-induced suppression of short-term food intake in fasted rats. Regulatory Peptides. 2003;111(1–3):83–90. doi: 10.1016/s0167-0115(02)00260-4. [DOI] [PubMed] [Google Scholar]
- 24.Elmquist J.K., Bjorbaek C., Ahima R.S., Flier J.S., Saper C.B. Distributions of leptin receptor mRNA isoforms in the rat brain. Journal of Comparative Neurology. 1998;395(4):535–547. [PubMed] [Google Scholar]
- 25.Cone R.D. Anatomy and regulation of the central melanocortin system. Nature Neuroscience. 2005;8(5):571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz M.W., Woods S.C., Porte D., Seeley R.J., Baskin D.G. Central nervous system control of food intake. Nature. 2000;404(6778):661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 27.Belgardt B.F., Okamura T., Bruning J.C. Hormone and glucose signalling in POMC and AgRP neurons. Journal of Physiology. 2009;587(Pt 22):5305–5314. doi: 10.1113/jphysiol.2009.179192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mountjoy K.G. Pro-opiomelanocortin (POMC) neurones, POMC-derived peptides, melanocortin receptors and obesity: how understanding of this system has changed over the last decade. Journal of Neuroendocrinology. 2015;27(6):406–418. doi: 10.1111/jne.12285. [DOI] [PubMed] [Google Scholar]
- 29.Roselli-Rehfuss L., Mountjoy K.G., Robbins L.S., Mortrud M.T., Low M.J., Tatro J.B. Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(19):8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mountjoy K.G., Mortrud M.T., Low M.J., Simerly R.B., Cone R.D. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Molecular Endocrinology. 1994;8(10):1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 31.Kishi T., Aschkenasi C.J., Lee C.E., Mountjoy K.G., Saper C.B., Elmquist J.K. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. Journal of Comparative Neurology. 2003;457(3):213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- 32.Fekete C., Liposits Z. Histamine-immunoreactive neurons of the tuberomammillary nucleus are innervated by alpha-melanocyte stimulating hormone-containing axons. Generation of a new histamine antiserum for ultrastructural studies. Brain Research. 2003;969(1–2):70–77. doi: 10.1016/s0006-8993(03)02279-0. [DOI] [PubMed] [Google Scholar]
- 33.Huszar D., Lynch C.A., Fairchild-Huntress V., Dunmore J.H., Fang Q., Berkemeier L.R. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 34.Yeo G.S., Farooqi I.S., Aminian S., Halsall D.J., Stanhope R.G., O'Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nature Genetics. 1998;20(2):111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 35.Vaisse C., Clement K., Guy-Grand B., Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nature Genetics. 1998;20(2):113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 36.Walker A.K., Park W.M., Chuang J.C., Perello M., Sakata I., Osborne-Lawrence S. Characterization of gastric and neuronal histaminergic populations using a transgenic mouse model. PloS One. 2013;8(3) doi: 10.1371/journal.pone.0060276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yanovsky Y., Zigman J.M., Kernder A., Bein A., Sakata I., Osborne-Lawrence S. Proton- and ammonium-sensing by histaminergic neurons controlling wakefulness. Frontiers in Systems Neuroscience. 2012;6:23. doi: 10.3389/fnsys.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu H., Kishi T., Roseberry A.G., Cai X., Lee C.E., Montez J.M. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. Journal of Neuroscience. 2003;23(18):7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sherin J.E., Elmquist J.K., Torrealba F., Saper C.B. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. Journal of Neuroscience. 1998;18(12):4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schone C., Cao Z.F., Apergis-Schoute J., Adamantidis A., Sakurai T., Burdakov D. Optogenetic probing of fast glutamatergic transmission from hypocretin/orexin to histamine neurons in situ. Journal of Neuroscience. 2012;32(36):12437–12443. doi: 10.1523/JNEUROSCI.0706-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mickelsen L.E., Kolling F.W.t., Chimileski B.R., Fujita A., Norris C., Chen K. Neurochemical heterogeneity among lateral hypothalamic hypocretin/orexin and melanin-concentrating hormone neurons identified through single-cell gene expression analysis. eNeuro. 2017;4(5) doi: 10.1523/ENEURO.0013-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui H., Sohn J.W., Gautron L., Funahashi H., Williams K.W., Elmquist J.K. Neuroanatomy of melanocortin-4 receptor pathway in the lateral hypothalamic area. Journal of Comparative Neurology. 2012;520(18):4168–4183. doi: 10.1002/cne.23145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peyron C., Tighe D.K., van den Pol A.N., de Lecea L., Heller H.C., Sutcliffe J.G. Neurons containing hypocretin (orexin) project to multiple neuronal systems. Journal of Neuroscience. 1998;18(23):9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chou T.C., Lee C.E., Lu J., Elmquist J.K., Hara J., Willie J.T. Orexin (hypocretin) neurons contain dynorphin. Journal of Neuroscience. 2001;21(19):RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harthoorn L.F., Sane A., Nethe M., Van Heerikhuize J.J. Multi-transcriptional profiling of melanin-concentrating hormone and orexin-containing neurons. Cellular and Molecular Neurobiology. 2005;25(8):1209–1223. doi: 10.1007/s10571-005-8184-8. [DOI] [PubMed] [Google Scholar]
- 46.Chemelli R.M., Willie J.T., Sinton C.M., Elmquist J.K., Scammell T., Lee C. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98(4):437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 47.Ookuma K., Yoshimatsu H., Sakata T., Fujimoto K., Fukagawa F. Hypothalamic sites of neuronal histamine action on food intake by rats. Brain Research. 1989;490(2):268–275. doi: 10.1016/0006-8993(89)90244-8. [DOI] [PubMed] [Google Scholar]
- 48.Fan W., Boston B.A., Kesterson R.A., Hruby V.J., Cone R.D. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385(6612):165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 49.Marsh D.J., Hollopeter G., Huszar D., Laufer R., Yagaloff K.A., Fisher S.L. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nature Genetics. 1999;21(1):119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 50.Mountjoy K.G., Kong P.L., Taylor J.A., Willard D.H., Wilkison W.O. Melanocortin receptor-mediated mobilization of intracellular free calcium in HEK293 cells. Physiological Genomics. 2001;5(1):11–19. doi: 10.1152/physiolgenomics.2001.5.1.11. [DOI] [PubMed] [Google Scholar]
- 51.Nickolls S.A., Fleck B., Hoare S.R., Maki R.A. Functional selectivity of melanocortin 4 receptor peptide and nonpeptide agonists: evidence for ligand-specific conformational states. Journal of Pharmacology and Experimental Therapeutics. 2005;313(3):1281–1288. doi: 10.1124/jpet.105.083337. [DOI] [PubMed] [Google Scholar]
- 52.Newman E.A., Chai B.X., Zhang W., Li J.Y., Ammori J.B., Mulholland M.W. Activation of the melanocortin-4 receptor mobilizes intracellular free calcium in immortalized hypothalamic neurons. Journal of Surgical Research. 2006;132(2):201–207. doi: 10.1016/j.jss.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 53.Thanawala M.S., Regehr W.G. Presynaptic calcium influx controls neurotransmitter release in part by regulating the effective size of the readily releasable pool. Journal of Neuroscience. 2013;33(11):4625–4633. doi: 10.1523/JNEUROSCI.4031-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parpura V., Basarsky T.A., Liu F., Jeftinija K., Jeftinija S., Haydon P.G. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369(6483):744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 55.Dani J.W., Chernjavsky A., Smith S.J. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8(3):429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 56.Inagaki N., Wada H. Histamine and prostanoid receptors on glial cells. Glia. 1994;11(2):102–109. doi: 10.1002/glia.440110205. [DOI] [PubMed] [Google Scholar]
- 57.Shelton M.K., McCarthy K.D. Hippocampal astrocytes exhibit Ca2+-elevating muscarinic cholinergic and histaminergic receptors in situ. Journal of Neurochemistry. 2000;74(2):555–563. doi: 10.1046/j.1471-4159.2000.740555.x. [DOI] [PubMed] [Google Scholar]
- 58.Schone C., Apergis-Schoute J., Sakurai T., Adamantidis A., Burdakov D. Coreleased orexin and glutamate evoke nonredundant spike outputs and computations in histamine neurons. Cell Reports. 2014;7(3):697–704. doi: 10.1016/j.celrep.2014.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marcus J.N., Aschkenasi C.J., Lee C.E., Chemelli R.M., Saper C.B., Yanagisawa M. Differential expression of orexin receptors 1 and 2 in the rat brain. Journal of Comparative Neurology. 2001;435(1):6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- 60.Sakata T., Yoshimatsu H., Kurokawa M. Hypothalamic neuronal histamine: implications of its homeostatic control of energy metabolism. Nutrition. 1997;13(5):403–411. doi: 10.1016/s0899-9007(97)91277-6. [DOI] [PubMed] [Google Scholar]
- 61.Morimoto T., Yamamoto Y., Yamatodani A. Brain histamine and feeding behavior. Behavioural Brain Research. 2001;124(2):145–150. doi: 10.1016/s0166-4328(01)00225-x. [DOI] [PubMed] [Google Scholar]
- 62.Ookuma K., Sakata T., Fukagawa K., Yoshimatsu H., Kurokawa M., Machidori H. Neuronal histamine in the hypothalamus suppresses food intake in rats. Brain Research. 1993;628(1–2):235–242. doi: 10.1016/0006-8993(93)90960-u. [DOI] [PubMed] [Google Scholar]
- 63.Arrang J.M., Garbarg M., Schwartz J.C. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302(5911):832–837. doi: 10.1038/302832a0. [DOI] [PubMed] [Google Scholar]
- 64.Schlicker E., Malinowska B., Kathmann M., Gothert M. Modulation of neurotransmitter release via histamine H3 heteroreceptors. Fundamental & clinical Pharmacology. 1994;8(2):128–137. doi: 10.1111/j.1472-8206.1994.tb00789.x. [DOI] [PubMed] [Google Scholar]
- 65.Deng C., Weston-Green K., Huang X.F. The role of histaminergic H1 and H3 receptors in food intake: a mechanism for atypical antipsychotic-induced weight gain? Progress In Neuro-Psychopharmacology & Biological Psychiatry. 2010;34(1):1–4. doi: 10.1016/j.pnpbp.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 66.Garcia M., Floran B., Arias-Montano J.A., Young J.M., Aceves J. Histamine H3 receptor activation selectively inhibits dopamine D1 receptor-dependent [3H]GABA release from depolarization-stimulated slices of rat substantia nigra pars reticulata. 1997;80(1):241–249. doi: 10.1016/s0306-4522(97)00100-0. [DOI] [PubMed] [Google Scholar]
- 67.Barrett P., van den Top M., Wilson D., Mercer J.G., Song C.K., Bartness T.J. Short photoperiod-induced decrease of histamine H3 receptors facilitates activation of hypothalamic neurons in the Siberian hamster. Endocrinology. 2009;150(8):3655–3663. doi: 10.1210/en.2008-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu X., Ye Z., Houston C.M., Zecharia A.Y., Ma Y., Zhang Z. Wakefulness is governed by GABA and histamine cotransmission. Neuron. 2015;87(1):164–178. doi: 10.1016/j.neuron.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Venner A., Mochizuki T., De Luca R., Anaclet C., Scammell T.E., Saper C.B. Reassessing the role of histaminergic tuberomammillary neurons in arousal control. Journal of Neuroscience. 2019;39(45):8929–8939. doi: 10.1523/JNEUROSCI.1032-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farooqi I.S., Keogh J.M., Yeo G.S., Lank E.J., Cheetham T., O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. New England Journal of Medicine. 2003;348(12):1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 71.Collet T.H., Dubern B., Mokrosinski J., Connors H., Keogh J.M., Mendes de Oliveira E. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Molecular Genetics and Metabolism. 2017;6(10):1321–1329. doi: 10.1016/j.molmet.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]