

Abstract
Objective
Non-alcoholic steatohepatitis (NASH) is characterized by a robust pro-inflammatory component at both hepatic and systemic levels together with a disease-specific gut microbiome signature. Protein tyrosine phosphatase 1 B (PTP1B) plays distinct roles in non-immune and immune cells, in the latter inhibiting pro-inflammatory signaling cascades. In this study, we have explored the role of PTP1B in the composition of gut microbiota and gut barrier dynamics in methionine and choline-deficient (MCD) diet-induced NASH in mice.
Methods
Gut features and barrier permeability were characterized in wild-type (PTP1B WT) and PTP1B-deficient knockout (PTP1B KO) mice fed a chow or methionine/choline-deficient (MCD) diet for 4 weeks. The impact of inflammation was studied in intestinal epithelial and enteroendocrine cells. The secretion of GLP-1 was evaluated in primary colonic cultures and plasma of mice.
Results
We found that a shift in the gut microbiota shape, disruption of gut barrier function, higher levels of serum bile acids, and decreased circulating glucagon-like peptide (GLP)-1 are features during NASH. Surprisingly, despite the pro-inflammatory phenotype of global PTP1B-deficient mice, they were partly protected against the alterations in gut microbiota composition during NASH and presented better gut barrier integrity and less permeability under this pathological condition. These effects concurred with higher colonic mucosal inflammation, decreased serum bile acids, and protection against the decrease in circulating GLP-1 levels during NASH compared with their WT counterparts together with increased expression of GLP-2-sensitive genes in the gut. At the molecular level, stimulation of enteroendocrine STC-1 cells with a pro-inflammatory conditioned medium (CM) from lipopolysaccharide (LPS)-stimulated macrophages triggered pro-inflammatory signaling cascades that were further exacerbated by a PTP1B inhibitor. Likewise, the pro-inflammatory CM induced GLP-1 secretion in primary colonic cultures, an effect augmented by PTP1B inhibition.
Conclusion
Altogether our results have unraveled a potential role of PTP1B in the gut–liver axis during NASH, likely mediated by increased sensitivity to GLPs, with potential therapeutic value.
Keywords: NASH, PTP1B, Inflammation, Gut microbiota, GLP-1, GLP-2
Graphical abstract

Highlights
-
•
MCD diet induces a different shift of gut microbiota in PTP1BWT and PTP1B KO mice.
-
•
PTP1B KO mice present higher colonic mucosal inflammation under NASH conditions.
-
•
Gut barrier permeability was decreased in PTP1B KO mice with NASH.
-
•
PTP1B inhibition increases GLP-1 secretion in colonic cultures exposed to inflammatory stimulus.
Abbreviations
- A.U
arbitrary units
- AB
alcian blue
- CM
conditioned medium
- DMEM
Dulbecco modified Eagle medium
- DPP-IV
dipeptidyl peptidase-IV
- EECs
enteroendocrine cells
- FBS
fetal bovine serum
- FXR
farnesoid X receptor
- GLP
glucagon-like peptide
- H&E
hematoxylin and eosin
- IELs
intraepithelial lymphocytes
- IR
insulin receptor
- IRS1
insulin receptor substrate 1
- LPS
lipopolysaccharide
- MCD
methionine and choline-deficient
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- PAS
periodic acid-Schiff
- PICRUSt
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States
- PTP1B
protein tyrosine phosphatase 1 B
- STC-1
intestinal secretin tumor-1
- UHPLC
ultra-high performance liquid chromatography
- ZO-1
zonula occludens-1
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most frequent chronic hepatic disease in Western countries and is considered the hepatic manifestation of metabolic syndrome [1]. NAFLD is characterized by the accumulation of different lipid species within the hepatocyte that, together with pro-inflammatory cytokines, are the major triggers of disease progression. Moreover, 10%–40% of patients with NAFLD can progress to more advanced stages when adaptive mechanisms that protect hepatocytes from fatty acid-mediated lipotoxicity become overwhelmed, resulting in the development of non-alcoholic steatohepatitis (NASH), in which hepatic tissue presents a robust pro-inflammatory component. If unresolved, the disease can reach more severe stages such as fibrosis, cirrhosis (15%–25%), or even hepatocellular carcinoma (7%) [2]. NAFLD represents an important public health problem due the increased incidence of metabolic syndrome worldwide [3]. Therefore, elucidation of the molecular mechanisms and associated factors involved in the establishment and progression of NAFLD is crucially needed to identify new targets and implement efficient therapies.
Currently, the association between the gut–liver axis and gut microbiota is acquiring a major impact in the research on liver diseases as it is indispensable for metabolic regulation, gut hormone release, and immune responses [4]. A recent study of the metagenomic signature of hepatic steatosis in a cohort of non-diabetic women with morbid obesity has provided evidences on the association of hepatic steatosis with low microbial gene richness and dysregulation of aromatic and branched-chain amino acid metabolism [5]. The progression of NAFLD to more severe stages also correlates with alterations in the intestinal microbiota by affecting digestion, immune responses, and production of gut hormones [6]. Moreover, Loomba and co-workers [7] in a cohort of biopsy-proven patients with NAFLD with several degrees of fibrosis have described a panel of gut microbiome biomarkers that can be used as a non-invasive test to accurately diagnose advanced fibrosis in patients with NASH. In addition, interventional studies with probiotics such as VSL#3 and Lactobacillus casei Shirota and prebiotics such as fermentable dietary fructo-oligosaccharides suggest that their administration may improve NAFLD [[8], [9], [10]].
In the context of metabolic homeostasis, it is worth mentioning that the intestine has a complex network of specialized cells known as enteroendocrine cells (EECs) that secrete hormones to maintain energy balance by regulating food intake, digestion, absorption, satiation, storage, and disposal of digested nutrients [11]. Recent interest has emerged on the interaction of EECs with the immune system and, in this regard, plasma glucagon-like peptide (GLP)-1 levels rapidly increase on lipopolysaccharide (LPS) administration in mice via a toll-like receptor 4 (TLR4)-dependent mechanism [12,13]. Therefore, GLP-1 could be released to exert an anti-inflammatory effect in response to a pro-inflammatory situation such as an increase in endotoxin or cytokines [14].
Insulin resistance plays an important role in the development of NAFLD [15]. Protein tyrosine phosphatase 1 B (PTP1B) is a phosphatase ubiquitously expressed that acts as a negative regulator of insulin action since it dephosphorylates tyrosine residues, primarily insulin receptor (IR) and insulin receptor substrate 1 (IRS1), in critical nodes of the insulin signaling cascade [[16], [17], [18]]. Global PTP1B-deficient mice are resistant to diet-induced obesity [19] and do not show metabolic alterations during aging such as fat mass accretion, hepatic steatosis, and hyperinsulinemia [20]. Of relevance, PTP1B is also an immune modulator as it controls cytokine-mediated signaling by dephosphorylating JAK2 in the JAK-STAT pathway and the non-receptor tyrosine protein kinase 2 (TYK2), among others [[21], [22], [23]]. In this regard, a previous study in our laboratory has evidenced an exacerbated response of PTP1B-deficient versus wild-type (WT) macrophages to pro-inflammatory stimuli with increased expression of M1 polarization markers [24]. Furthermore, when challenged with a methionine and choline-deficient (MCD) diet, PTP1B knockout (KO) mice developed hepatic inflammation and NASH more rapidly than their WT counterparts. By contrast, when mice with established MCD-induced NASH were switched to a chow diet, those lacking PTP1B manifested an accelerated recovery due, at least in part, to hepatic oval cells proliferation [25]. Taking all these data into account, we hypothesized that in the context of the inflammatory environment associated with NASH, PTP1B could modulate specific cellular mechanisms in the gut. Based on that, in this work we have explored gut inflammation during NASH and, particularly, the role of PTP1B in the gut barrier function associated with this pro-inflammatory context. To achieve this goal, and considering that PTP1B-deficient mice are protected against obesity-induced NAFLD, an MCD dietary challenge was used as a preclinical model of non-obese NASH [26]. Although the MCD diet has been criticized because it causes body weight loss [27], it rapidly induces a NASH signature correlative to that found in humans and, importantly, it induces inflammation in the gut [28]. Our results have provided experimental evidences of a role of PTP1B in the gut during NASH, particularly with respect to gut microbiota, inflammation, and gut barrier integrity.
2. Materials and methods
2.1. Animals
In this study we used WT mice (C57BL/6 J) or Ptpn1 heterozygous mice of both sexes, maintained on a mixed genetic background (C57BL/6 J × 129 sv), that were intercrossed to yield the three genotypes of mice (WT, heterozygous, and KO), as described previously [20]. The mice were housed at the animal facilities of the Alberto Sols Biomedical Research Institute (CSIC-UAM, Madrid). To induce experimental NASH, 12- to 16-week-old PTP1B WT or Ptpn1 KO (PTP1B KO) male mice were fed a MCD diet (TD-90262 Harlan-Tecklad, Indianapolis, IN, USA) up to 4 weeks. The control animals were fed a chow diet during this period. Body weight was monitored weekly. Mice were maintained in 12-hour light/dark, temperature-controlled (22 °C), and humidity-controlled rooms. All animal experimentation was conducted in accordance with Spanish and European legislation and UK animals (Scientific Procedures) Act 1986 Amendment Regulations (SI 2012/3039), and approved by the CSIC and Comunidad de Madrid Animal Care and Use Committees and the University of Cambridge Animal Welfare and Ethical Review Body.
2.2. Microbiome analysis
Fecal content was freshly collected individually, weighed, and frozen at −80 °C until use. Genomic DNA was extracted using the FastDNA ® SPIN Kit for Soil (MP Biomedicals, CA, USA) following the manufacturer's instructions. The homogenization step was conducted in a FastPrep® Instrument (MP Biomedicals, USA) for 10 s at a speed setting of 5.5. DNA quantity and quality were measured using the NanoVue plus system (GE Healthcare Life Science, UK). All samples were diluted to a 100 ng/μL final concentration. Polymerase chain reaction (PCR) amplification of V3–V4 hypervariable regions of 16 S rRNA genes was performed using the following primers [29]: 16SV3V4-Fw: ACACTGACGACATGGTTCTACACCTACGGGNGGCWGCAG and 16SV3V4-Rv: TACGGTAGCAGAGACTTGGTCTGACTACHVGGGTATCTAATCC. Sequencing was performed in the Genomic Unit at Parque Cientifico de Madrid (Spain) using the Illumina-Myseq system (Illumina, San Diego, CA) following the manufacturer's guidelines. An average of 160,000 readings per sample was obtained. Paired reads were joined using the PandaSeq Assembler. Only sequences ranging from 300 to 460 bp were considered. Operational taxonomic units (OTUs) were determined using the GreenGenes 13_5 reference sequences database. The Miseq sequences derived from the 16 S profiling were deposited in the European Nucleotide Archive and are available under the following accession number: PRJEB13752. Community diversity (α-diversity) was calculated using Quantitative Insights into Microbial Ecology (QIIME) with the Shannon index [30]. QIIME was used to determine β-diversity by calculating the weighted and unweighted UniFrac distances [31]. EMPEROR was employed to visualize a UniFrac PCoA biplot. 16 S rDNA data were analyzed by the software Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) to predict metagenomics functional content by using OTUs picked against Greengenes 13_5 and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases [32].
2.3. FITC-dextran assay
A 60 mg per 100 g of body weight dose of fluorescein isothiocyanate (FITC)-Dextran (FITC-Dextran 4 kDa, Sigma Aldrich, USA) dissolved in sterile phosphate-buffered saline (PBS) was administered by oral gavage to PTP1B WT or PTP1B KO mice fed a MCD or chow diet. Blood samples were collected by submandibular bleeding after 4 h in special tubes for small volumes (SST™ Tubes BD Microtainer®). Serum was obtained after centrifugation at 3000 × g for 10 min at 4 °C, and fluorescence was measured at 485 nm (excitation) and 535 nm (emission) with the Fluorometer Biotek Synergy™ HT and analyzed with GEN 5™1.07 software. The FITC-Dextran concentrations were determined from a standard curve with serial dilutions of FITC-Dextran.
2.4. Measurement of circulating endotoxemia
Blood was collected from PTP1B WT and PTP1B KO mice fed a chow or MCD diet for 4 weeks in glass tubes (TU04-127-250, Labbox, Barcelona, Spain). Serum samples were obtained after centrifugation at 3000 × g for 10 min and stored in glass vials (SV02-A20-100, Labbox, Barcelona, Spain) at −80 °C. Endotoxemia was determined by enzyme-linked immunosorbent assay (ELISA) with the Mouse Endotoxin (ET) ELISA Kit (QAYEE-BIO, China) following the manufacturer's instructions.
2.5. Colon tissue collection
The colon was removed, and its length and weight were measured. Once the luminal content was removed with PBS, 5-mm pieces were cut and fixed with Methacarn (60% methanol, 30% chloroform, and 10% acetic acid) for 24 h at 4 °C. After fixation, pieces were washed with PBS and paraffin-embedded as follows: dehydration: 3 × 1 h in 96% ethanol, 3 × 1 h in 100% ethanol, 3 × 1 h in isoparaffin; paraffinization: 3 × 2 h in paraffin in a 60 °C oven. Next, 5-μm-thick sections were obtained using a standard microtome (Microm) and mounted on silanized glass slides for staining.
2.6. Evaluation of colon mucosal integrity and crypt length
Once the samples were dewaxed and rehydrated, they were stained using the hematoxylin and eosin (H&E) technique as follows. Sections were immersed in filtered Harris Hematoxylin for 10 s, washed with tap water, immersed in eosin for 30 s, washed with tap water, and dehydrated in ascending alcohol solutions. Colon sections stained with H&E were evaluated by a single blinded pathologist looking for inflammatory infiltrates, epithelial changes such as hyperplasia, and altered crypt architecture. Colon images were taken with the slide scanner Axio Scan. Z1 (Zeiss, Germany). Crypt lengths (μm) were quantified in periodic acid-Schiff (PAS)-stained sections using the Fiji version of Image J software (http://fiji.sc/Fiji). Measurements were taken from at least 27 full-length crypts obtained from five different longitudinal sections (per mouse).
2.7. Analysis of colonic neutral and acid mucins
Colonic mucins were determined following the PAS histochemical technique [33] and alcian blue (AB) stains. Briefly, 5-μm sections were stained with AB for 15 min and washed with distilled water. Then, samples were treated with periodic acid (1%) for 5 min, washed with distilled water, treated with the Schiff reagent for 5 min, and washed again. Finally, nuclei were counterstained using Harris Hematoxylin for 2 min. The neutral mucins were stained in magenta, whereas the acid mucins were stained in blue. The Fiji version of ImageJ software was used to obtain neutral and acid mucin scores. Results, expressed in arbitrary units (A.U.), correspond to the optical intensity × surface of magenta and blue staining after spectral separation of both colors. Only intact crypts were quantified.
2.8. Measurement of colon edema
The colon was sectioned in 1-cm pieces after its extraction. Sections were homogenously distributed over a filter and placed into the moisture analyzer MB120 (Ohaus, Switzerland). Moisture was determined from the weight loss of the sample dried by heating. The temperature profile selected was a standard drying at 140 °C. The drying was considered finished when no changes at the level of milligrams were produced for at least 180 s.
2.9. ZO-1 immunofluorescence
Five-micron colonic paraffin sections were dewaxed and rehydrated. Antigen retrieval was performed with 10 mM sodium citrate and 0.05% Tween-20 (pH 7.4) for 10 min at 100 °C. H2O2 (3%): methanol (1:1) was used to block endogenous peroxidase for 30 min. Blocking was performed with 5% bovine serum albumin (BSA) and 4% goat serum for 1 h at room temperature. Samples were incubated with 1:50 anti-zonula occludens-1 (ZO-1) antibody (ZO-1-1A12, Invitrogen) overnight at 4 °C. Incubation with secondary antibody Rhodamine Red-X (Jackson ImmunoResearch) and 4,6-diamino-2-phenylindole (DAPI; Sigma) was performed for 1 h at room temperature. Images were taken using the confocal microscope LSM710 (Zeiss, Oberkochen, Germany).
2.10. RNA isolation and quantitative real-time PCR analysis
The colon was immediately frozen in liquid nitrogen after its collection. Colon homogenization was performed using the Tissue Lyser LT (Qiagen, Hilden, Germany) for 4 min, 30 s in TRIzol® Reagent (Invitrogen, USA) followed by a chloroform and isopropanol extraction. We used the Turbo DNA-free™ kit (Invitrogen by ThermoFisher Scientific, USA) for DNA digestion and the iScript™ cDNA Synthesis Kit (BioRad, USA) to conduct the cDNA synthesis following the manufacturer's instructions. SYBER Green-based quantitative PCR was performed with Kapa SYBR® FAST (Kapa Biosystems, Roche, Switzerland). Quantification was done through delta Ct analysis.
2.11. GLP-1 measurement in plasma
Overnight fasted mice were administered a dose of 6 g/kg glucose by oral gavage. After 10 min, blood samples were collected in cold ethylenediamine tetraacetic acid (EDTA) tubes (BD Vacutainer® spray-coated K2EDTA Tubes, BD, USA) containing dipeptidyl peptidase-IV (DPP-IV) inhibitor (DPP4, EMD Millipore, USA) at a final concentration of 25 μM. Blood samples were centrifuged at 3000 × g for 10 min at 4 °C. After collection of plasma samples, DPP-IV inhibitor was added up to a final concentration of 50 μM, and samples were then immediately frozen and stored at −80 °C. GLP-1 was determined using a total GLP-1 assay kit (MesoScale Discovery, USA).
2.12. Acute pro-inflammatory systemic model in mice
C57BL/6 J male mice were treated with a single intraperitoneal injection of LPS (2 mg/kg body weight). For GLP-1 measurement, plasma samples were collected 16 h after the LPS challenge following 4 h of fasting and administration of 6 g/kg glucose by oral gavage as previously described.
2.13. Plasma bile acids profiling
After protein precipitation with 3 volumes of cold (−20 °C) acetonitrile, samples were centrifuged. The separated supernatant solvent was evaporated, and the residue was resuspended in methanol:water (same volume as plasma). The plasma bile acids content was determined by ultra-high performance liquid chromatography (UHPLC) using a 1290 Infinity II LC System connected to a 6460 Triple Quadrupole MS as the instrument detector, both from Agilent Technologies (Santa Clara, CA, USA). A Zorbax Eclipse Plus C8 2.1 × 150-mm 1.8-μm column (Agilent Technologies, Spain) and an optimized gradient with acetonitrile, methanol, and ammonium acetate (pH 4) were used for bile acid separation. The analysis was performed at the Center for Metabolomics and Bioanalysis (CEMBIO), Universidad CEU-San Pablo, Madrid, Spain, based on previously described methodology [34] with slight modifications. Commercial standards from Sigma (St. Louis, MO, USA) were used for all the bile acid measurements. D4-deoxycholic acid was used as the internal standard (IS). Calibration curves were prepared with seven concentrations of each standard.
2.14. Conditioned media production in macrophages
Raw 246.7 murine macrophages cell line was used to obtain a pro-inflammatory macrophage-conditioned medium (mCM) for performing in vitro experiments. Macrophages were cultured in T75 flasks (BD Falcon™, USA) in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (1% P/S; 100 U/mL penicillin, 100 μg/mL streptomycin) at 37 °C in a humidified atmosphere with 5% CO2. Medium was collected after the treatment of confluent macrophages with 100 ng/mL LPS (InvivoGen, California, USA) with or without 20 μM of PTP1B inhibitor (Ref. 539,741, Sigma–Aldrich) in Dulbecco modified Eagle medium (DMEM) supplemented with 2% FBS and 1% P/S for 16 h [referred to as mCM-lipopolysaccharide (mCM-LPS)]. As a control, mCM was collected from unstimulated macrophages maintained for the same time period in DMEM-2% FBS-1% P/S (referred to as mCM-C).
Peritoneal macrophages were plated at 10-12 × 106 cells per dish and maintained in RPMI medium supplemented with 10% FBS and 1% P/S for 2 h. Then, the medium was changed to DMEM supplemented with 2% FBS and 1% P/S for 24 h. To obtain the CM, macrophages were treated or not with 150 ng/mL LPS for 18 h in DMEM-2% FBS-1% P/S (referred as pCM-LPS or pCM-C, respectively).
2.15. Culture of STC-1 enteroencocrine cells
Confluent STC-1 (intestinal secretin tumor-1) cells were maintained in DMEM supplemented with 10% FBS and 1% P/S. For experiments, STC-1 cells were treated with mCM-LPS or mCM-C for several time-periods to analyze pro-inflammatory signaling cascades or for 16 h prior to stimulation with 1 nM insulin (Sigma–Aldrich, Missouri, USA) for 10 min to analyze insulin signaling. To inhibit PTP1B enzymatic activity, STC-1 cells were pretreated with the PTP1B inhibitor (20 μM) for 2 h prior to the treatment of the cells with either mCM. We also supplemented the mCM with the same concentration of the inhibitor thorough the experiment. Then, the cells were washed twice with PBS and collected for further analysis.
2.16. Analysis of protein expression by western blot
STC-1 cells were homogenized in lysis buffer containing 10 mM Tris–HCl, 1 mM MgCl2, 1 mM Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 10% glycerol, 0.5% 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS), 1 mM β-mercaptoethanol, 0.1 mM phenymethylsulfonyl fluoride (PMSF), and 5 mg/mL of a protease and phosphatase inhibitor cocktail (Sigma–Aldrich, USA), pH 7.5. The extracts were centrifuged for 20 min at 13,000 × g at 4 °C, and supernatants were stored at −20 °C. Protein levels were determined using the Bradford dye method (Bio-Rad, USA). Protein extracts were boiled at 95 °C for 5 min in loading buffer [250 mM Tris–HCl, pH 6.8, 2% sodium dodecyl sulfate (SDS), 10% glycerol, and 2% β-mercaptoethanol] and subjected to SDS polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred onto nylon membranes (Immobilon P, Millipore, USA). Blots were incubated for 2 h at room temperature or overnight at 4 °C with primary antibodies against phospho-AKT Ser308, phospho-AKT Ser473, phospho-IR, phospho-JNK and phospho-p65 NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) (Cell Signaling Technology, USA), AKT, (IκBα) inhibitor of kappa B, insulin receptor (IR), Vinculin, and JNK (Santa Cruz Biotechnology, USA). Finally, the blots were developed with Clarity ECL Western Blot (BioRad). Values of scanning densitometry were determined using the Quantity One software (Bio-Rad, UK).
2.17. Lentiviral silencing of Ptpn1 in Caco-2 cells
A stable cell line of Caco2 silenced for Ptpn1 gene, that encodes PTP1B protein, was generated by transduction with a battery of lentiviral particles TRCN0000218522, TRCN0000226109, TRCN0000226108, TRCN0000226107, and TRCN0000226106 (Mission shRNA Transduction Particles, Sigma Aldrich, USA) following the manufacturer's protocol. Lentiviral particles with Scramble shRNA were used as a control. Transduced cells were selected with puromycin (2.5 μg/mL) for 2 weeks. Transduction was confirmed by measuring Ptpn1 expression by RT-qPCR. Stable cell-lines were obtained and referred as Caco-2 Scramble (Caco-2 shScrb) and Caco-2 shPtpn1.
2.18. Determination of TransEpithelial Electrical Resistance (TEER)
For the analysis of TEER, Caco-2 cells (epithelial cells from human colon adenocarcinoma) were grown in (EMEM) Minimum Essential Medium - Eagle with Earle's BSS (Lonza, Switzerland) supplemented with 20% FBS and 1% P/S on porous filter membranes (0.4 μm-diameter pore) (Transwell® Permeable Support 0.4 μM Polyester Membrane Ref. 3460, Costar, Strasbourg, France). TEER was measured with a Millicell-ERS system (Millipore, USA) every 3 days until cells formed a monolayer. A monolayer was considered to be formed when three consecutive stable TEER measurements were obtained. Caco-2 cells were treated with mCM-C or mCM-LPS, and TEER was measured at the indicated time points.
2.19. Two-dimensional (2D) organoid culture and analysis of GLP-1 secretion
Transgenic mice (GLU-Epac2camps) bearing glucagon promoter-driven YFP expression that marks functional enteroendocrine (EEC) L cells were used. Colonic intestinal organoid lines were established using the procedure described previously [35] and underwent a differentiation protocol, which involved withdrawal of Wnt3A 5–7 days before plating in a 2D culture. For plating 2D organoid cultures, differentiated organoids were collected in ice cold advanced DMEM:F12 (ADF) medium (Life Technologies, USA) and centrifuged at 200 × g for 5 min. The pellet was broken up enzymatically with TrypLE (Gibco, Thermofisher, USA) for 2 min at 37 °C and neutralized with ADF supplemented with 10% FBS and 10 μM ROCK inhibitor y27632 (Tocris, USA). Resulting single cells and clusters were centrifuged at 300 × g for 5 min, resuspended in organoid medium, and seeded onto 2% Matrigel-coated 48-well plates for GLP-1 secretion measurement [36]. After overnight incubation (37 °C, 5% CO2), 2D cultures were washed three times in warm standard saline solution 138 buffer [consisting of (in mM) NaCl (138), KCl (4.5), HEPES (10), NaHCO3 (4.2), NaH2PO4 (1.2), d-glucose (1), CaCl2 (2.6), MgCl2 (1.2); pH 7.4 with NaOH] and 0.1% fatty acid-free BSA. Cells were incubated with pCM-C or pCM-LPS at 37 °C for 6 h. Supernatants were removed from the organoids and centrifuged at 2000 × g for 5 min at 4 °C, transferred to a fresh tube, and snap frozen on dry ice. Meanwhile, cells were lysed in 150 μL of lysis buffer on ice, and frozen overnight before lysates were scraped, collected, and centrifuged at 10,000 × g for 10 min at 4 °C. Samples were stored at −80 °C until measurement by the Core Biochemistry Assay Lab facility (Cambridge, UK). GLP-1 levels were measured using the total GLP-1 ELISA kit (MesoScale Diagnostics, USA) following the manufacturer's instructions [36].
2.20. Colonic epithelial cultures and analysis of GLP-1 secretion
Colonic crypts were isolated and cultured from 3- to 6-month-old C57BL/6 J mice of either sex as previously described [37]. Mice were euthanized, and the colon was excised. Luminal contents were flushed with cold PBS, and the outer muscle layer was removed. Tissues were minced and digested with collagenase type XI (0.35 mg/mL). Crypt-containing fractions were centrifuged at 100 × g for 3 min and resuspended with DMEM supplemented with 10% FBS, 1% P/S, and 2 mM l-glutamine (Gibco, Life Technologies) before filtering through a 70-μm cell strainer. Colonic crypts were plated onto Matrigel (BD Bioscience) coated 48-well plates and placed overnight in a 5% CO2 incubator at 37 °C. For analysis of GLP-1 secretion, primary colonic epithelial cultures were seeded into 48-well plates as already described. After 1 day in culture, the culture medium was exchanged for fresh warmed medium with or without the PTP1B inhibitor (20 μM) or vehicle (DMSO) as a control. After incubation at 37 °C for 2 h, the culture medium was replaced by pCM-C or pCM-LPS for a further 6 h. An additional control group of culture medium was included (RPMI-2% FBS not exposed to macrophage cultures). The PTP1B inhibitor was kept during all the treatments. Supernatant and lysate samples were collected and processed for GLP-1 as previously indicated.
2.21. Statistical analysis
Statistical analysis was performed using GraphPad Prism v5. Sample outliers were found using the Grubbs test. The Shapiro–Wilk test was run to determine whether the samples showed a parametric or non-parametric distribution. Unpaired two-tailed Student's t-tests were used to do pairwise comparisons in parametric distributions, and the Mann–Whitney U test was used for non-parametric distributions. Two-way analysis of variance (ANOVA) with post-hoc Tukey test was employed to compare two different categorical independent variables (i.e., diet and genotype). When another statistical analysis different from the one described was used, it is indicated in the description of the corresponding result. The data are presented as the mean ± standard error of the mean (SEM). Differences were considered statistically significant when p < 0.05.
3. Results
3.1. Characterization of gut microbiota in PTP1B WT and PTP1B KO mice during NASH
PTP1B WT and PTP1B KO mice were fed a chow or MCD diet for 4 weeks, and 16s rRNA microbiome analysis of feces was performed at the end of the treatment. α-diversity, determined by the Shannon index [38], showed a trend to a decrease in mice from both genotypes fed a MCD diet; this effect was statistically significant only for PTP1B KO mice (Figure 1A). The analysis of β-diversity showed a marked difference between fecal microbiota of mice fed chow or MCD diets with a clear separation of both clusters of samples along the experimental protocol (Figure 1B). Furthermore, a separation in PTP1B WT and PTP1B KO clusters was appreciated particularly during NASH. Fecal profiling analyzed by unweighted UniFrac algorithm evidenced significant differences in microbiota composition between groups at different taxonomic levels (Table 1). As expected, the phyla Firmicutes and Bacteroidetes represent more than 80% of all bacterial species in mice fed a chow diet, and no differences between genotypes were found in the Firmicutes/Bacteroidetes (F/B) ratio (Figure 1C). However, in PTP1B WT mice under a MCD diet, the abundance of Firmicutes increased, whereas a marked decrease was observed in Bacteroidetes, leading to a higher F/B ratio. Of note, no changes in F/B ratio were found in MCD diet-fed PTP1B KO mice compared with their genotype counterparts fed a chow diet. Phylum TM7 abundance also exhibited a decrease in animals fed a MCD diet; these changes were statistically significant only for PTP1B KO mice (Table 1). At the family level, we found that S24-7, Lactobacillaceae, and Mogibacteriaceae families decreased similarly in both genotypes during NASH, whereas Lachnospiraceae and Dehalobacteriaceae decreased only in PTP1B WT mice. Moreover, families Turicibacteriaceae and Verrucomicrobiaceae appear to be decreased in PTP1B KO mice after 4 weeks on the MCD diet. Conversely, families Porphyromonadaceae, Rikenellaceae, and Erysipelotrichaceae were increased in both genotypes during NASH, whereas Turicibacteriaceae increased only in MCD diet-fed PTP1B WT mice. Finally, at the genus level, Adlercreutzia, Lactobacillus, Roseburia, and Desulfovibrio showed a decrease with NASH in both genotypes, whereas an increase in Parabacteroides and Allobaculum relative levels were observed. In this regard, only MCD diet-fed PTP1B KO mice exhibited an increase in Prevotella and a decrease in Turicibacter, whereas PTP1B WT mice showed an increase in Turicibacter genera during NASH (Table 1).
Figure 1.

Gut microbiota composition and functional changes. A) Alpha-diversity analysis for PTP1B WT and PTP1B KO mice fed a chow or MCD diet for 4 weeks. Shannon index is expressed as mean ± SEM. ###p < 0.001 PTP1B KO chow vs PTP1B KO MCD. B) Phylogenetic β-diversity of fecal microbiota. Graph represents principal coordinate analysis (PCoA) plot based on 16 S rRNA gene sequencing of stool samples from PTP1B WT (blue symbols) and PTP1B KO (red symbols) mice fed a chow or MCD diet for 4 weeks. Differences between genotypes in this case are indicated with different clouds. Pink cloud corresponds to animals fed chow diet and green cloud surrounds animals fed a MCD diet. C) Ratio Firmicutes/Bacteroidetes (F/B) is expressed as mean ± SEM. ∗p < 0.05 PTP1B WT chow vs PTP1B WT MCD. D-E) Predicted functional changes in gut microbiota analyzed by PICRUSt tool. Bar graphs show significant differences in KEGG pathways. MCD_1 M refers to animals fed a MCD diet for 4 weeks; Chow_ refers to animals fed a chow diet. Significant differences are shown as p-value at the right panel. n = 5–7 mice per experimental group.
Table 1.
Changes in gut microbiota composition at different taxonomic levels during NASH in PTP1B WT and PTP1B KO mice.
 |
1Comparison between the different conditions. Two-way ANOVA with a Tukey post-hoc test was performed for statistical analysis. aChanges in gut microbiota due to the administered diet. ↑ expresses higher abundance and ↓ indicates lower abundance after 4 weeks of MCD diet relative to chow diet. bChanges in gut microbiota due to the genotype. ↑ indicates higher and ↓ indicates lower abundance in PTP1B KO mice vs PTP1B WT mice. Taxa with differences due to diet and genotype are shown highlighted in purple. w refers to significant differences found only for PTP1B WT animals; k refers to significant differences found only for PTP1B KO animals. m refers to significant differences found only in MCD-fed animals. ∗p<0.05; ∗∗p<0,01; ∗∗∗p<0.001. n=5-7 animals per condition.
Biochemical function of the microbial community was determined using PICRUSt software, which allows prediction of the functional characteristics of the gut microbiota under the different experimental conditions. PICRUSt analysis showed that the genes related to metabolism of cofactors/vitamins and energy balance were significantly underrepresented in PTP1B WT mice fed a MCD diet compared with their corresponding controls fed a chow diet (Figure 1D), and these changes were not found in PTP1B KO mice. Moreover when we compared both genotypes in MCD diet-fed animals, we found that microbiota from PTP1B WT mice were significantly enriched in genes for secondary bile acid biosynthesis compared with PTP1B KO mice, whereas microbiota from PTP1B KO mice were enriched in genes related to propanoate metabolism compared PTP1B WT mice during NASH (Figure 1E).
3.2. Effect of PTP1B deficiency on serum bile acids during NASH
Bile acids are mediators in the cross-talk between the liver and gut and can modulate gut microbiota and also be modulated by the host gut microbiota [39]. We quantified bile acids in serum to verify if their levels were altered in the context of PTP1B deficiency and/or MCD diet-induced NASH. Cholic (CA), chenodeoxycholic (CDCA), and ursodeoxycholic (UDCA) primary bile acids in mice were significantly augmented in serum of PTP1B WT mice receiving the MCD diet, but this increase did not reach significance in PTP1B KO mice as depicted in Figure 2A–C. Moreover, taurocholic (TCA) and taurochenodeoxycholic (TCDCA) acids, also primary bile acids resulting from the conjugation with taurine of CA and CDCA, respectively, exhibited higher levels in serum in the NASH condition, but no differences were found between genotypes (Figure 2D–E). We also found changes in CDCA-derived tauro α-muricholic acid (TαMCA), a primary bile acid in mice [40]. As shown in Figure 2F, its serum levels were more elevated in PTP1B KO mice. In addition, the secondary bile acid taurodeoxycholic (TDCA) increased in MCD diet-induced NASH exclusively in PTP1B WT mice (Figure 2G). The analysis of Nr1h4 mRNA, encoding the nuclear bile acid receptor farnesoid X receptor (FXR), in the gut showed higher levels in PTP1B KO mice under NASH (Figure 2H).
Figure 2.

Bile acid levels in serum samples from PTP1B WT and PTP1B KO mice fed chow or MCD diet. A-G) Mean ± SEM of bile acids concentrations in serum samples are expressed as part per billion (ppb). n = 10–15 mice per experimental group. H) Nr1h4 mRNA levels in the gut (encoding FXR protein). White dots represent mice fed a chow diet and black dots represent mice fed a MCD diet. n = 10–15 mice per experimental group. ∗p< 0.05; ∗∗p < 0.01; PTP1B WT chow vs PTP1B WT MCD; #p < 0.05; ##p < 0.01 PTP1B KO chow vs PTP1B KO MCD; +p < 0.05; ++p < 0.01 PTP1B WT MCD vs PTP1B KO MCD.
3.3. Differential alterations in gut barrier permeability in PTP1B WT and PTP1B KO mice during NASH
We measured endotoxin levels in serum samples as an indicator of microbial product translocation, and higher circulating endotoxin levels were found in mice from both genotypes fed a MCD diet during 4 weeks compared with animals receiving a chow diet (Figure 3A). However, endotoxin levels were lower in MCD diet fed-PTP1B KO mice. Intestinal permeability was also analyzed by an in vivo FITC-dextran assay. Figure 3B shows that, in agreement with endotoxin levels, PTP1B WT mice showed a more pronounced increase in serum FITC-dextran levels after NASH induction, reflecting increased intestinal permeability compared with PTP1B KO mice. Better preservation of the gut barrier under NASH conditions in PTP1B KO mice was also confirmed by ZO-1 immunostaining (Figure 3C).
Figure 3.

Alterations in the gut barrier permeability in PTP1B WT and PTP1B KO mice during NASH. A) LPS levels in serum after 4 weeks of feeding mice a chow or MCD diet. n = 6–9 mice per group. B) FITC-dextran assay in PTP1B WT and PTP1B KO mice fed a chow or MCD diet for 4 weeks. Data are expressed as percentage of FITC-dextran levels absorbed in serum samples. n = 9–16 animals per experimental group. ∗∗∗p < 0.001 PTP1B WT chow vs PTP1B WT MCD; ###p < 0.001 PTP1B KO chow vs PTP1B KO MCD; ++p < 0.01 PTP1B WT MCD diet vs PTP1B KO MCD diet. C) Representative images of ZO-1 immunostaining in colon samples of PTP1B WT and PTP KO mice. Scale bar 50 μm. D) Effect of the pro-inflammatory environment on TEER values in Caco-2 shScrb and Caco-2 shPtpn1 monolayers. TEER values from Caco-2 shScrb or shPtpn1 cultured in Transwell plates treated with mCM-C or mCM-LPS are represented as percentage of baseline. Data are expressed as mean ± SEM of three independent experiments performed in triplicate. ∗∗∗p < 0.001 Caco-2 shScb mCM-LPS vs mCM-C; ###p < 0.001 Caco-2 shPtpn1 mCM-LPS vs mCM-C.
We used human Caco-2 colon cells infected with lentiviral particles encoding scrambled shRNA (shScrb) or Ptpn1 shRNA (shPtpn1) to measure TEER, a common method for in vitro analysis of the barrier properties in epithelial intestinal cells [41,42]. A comparison between the TEER values of Caco-2 shScrb and Caco-2 shPtpn1 cell lines treated with mCM collected from Raw 264.7 macrophages without (mCM-C) or with (mCM-LPS) LPS stimulation as described in the Materials and Methods is shown in Figure 3D, and the raw data of TEER values are included in Supplementary Table 1. After 24 and 48 h of treatment of Caco-2 shScrb or shPtpn1 cell lines with mCM-LPS, TEER measurements decreased significantly to approximately half the levels detected in cells treated with mCM-C regardless of PTP1B expression. These results indicate that PTP1B deficiency does not directly impact on the barrier properties in the monolayer system of Caco-2 cells.
3.4. Colonic changes in PTP1B WT and PTP1B KO mice during NASH
Interestingly, under basal conditions (chow diet), colonic crypts were 51.6% ± 22.46% deeper in mice with deletion of the Ptpn1 gene compared with PTP1B WT control mice (p < 0.05). Colitis is usually associated with the presence of edema due to alterations in lymphatic vessel function triggered by the inflammatory mediators [[43], [44], [45]]. Because MCD diet-induced NASH generates a pro-inflammatory environment [46], we tested the presence of edema through the analysis of colon moisture content. Figure 4A shows an increase in colon water content of 3% and 4% in MCD diet-fed PTP1B WT and PTP1B KO animals, respectively, which reached significance only in PTP1B KO mice. However, when we determined the colon weight/length-to-body weight ratio, a parameter used to determine colon edema [47,48], no differences were found between the four groups of animals (Supplementary Fig. 1A), an effect probably due to the decrease of body mass associated with MCD administration. Therefore, it seems that this parameter does not determine colon edema in a pathological model such as MCD diet-induced NASH. Furthermore, we analyzed the bowel content searching for diarrhea, a symptom of advanced colitis. Surprisingly, MCD diet-fed animals exhibited lower number of fecal pellets and lower water content in colon feces (Supplementary Figs. 1B–C).
Figure 4.

Analysis of anatomical and histological parameters in colon samples from PTP1B WT and PTP1B KO mice fed a chow or MCD diet for 4 weeks. A) Colon moisture content is expressed as percentage of water regarding total colon weight. n = 7–8 mice per experimental group. B) Colonic epithelial granuloma areas expressed as percentage of total epithelium area. n = 4 mice per experimental group. C) Representative images of H&E colonic stained sections from PTP1B WT and PTP1B KO mice. Images were taken from 5 μm colon paraffin-embedded sections stained with H&E with an Axio Scan. Z1 (Zeiss). Granuloma areas are marked with a white symbol. D) Mucin layer analysis in colonic samples from mice under NASH conditions. Images show AB (left) (n = 8–16 crypts per experimental group) and PAS (right) stained representative crypts from colonic sections that stain acidic and neutral mucins, respectively (n = 17–24 crypts per experimental group). E) Muc2 (mucin) mRNA levels are shown as fold change referred to the control PTP1B WT fed a chow diet. Data are expressed as mean ± SEM. n = 5–8 mice per experimental group. ∗p < 0.05; ∗∗p < 0.01 PTP1B WT chow vs PTP1B WT MCD; #p < 0.05; ###p < 0.001 PTP1B KO chow vs PTP1B KO MCD.
The histopathological analysis of colon samples did not show substantial changes in tissue epithelium such as hyperplasia or altered mucosa architecture. However, colonic mucosa from PTP1B KO mice contained unspecific granulomas not only after the challenge with MCD diet, but also before the induction of NASH (Figure 4B–C). Furthermore, PAS and AB staining was performed in colonic sections to evaluate colon mucosal integrity. Figure 4D shows decreased mucin levels in both genotypes of mice during NASH. Results for Muc2 (the gene encoding for Mucin-2) mRNA levels in colon corroborated this decrease (Figure 4E).
Beyond epithelial and endocrine cells [49], the intestine comprises a complex network of immune cells. Among them, a specialized gut immune cell population known as intraepithelial lymphocytes (IELs) modulates innate immunity and controls gut barrier function, epithelial turnover, and the host response to enteric pathogens [13]. Based on that, we analyzed the expression of T-cell surface glycoprotein CD3γ (Cd3g) as the IEL marker in colon samples. As depicted in Figure 5A, PTP1B KO mice exhibited a greater than two-fold increase in Cd3g mRNA levels compared with PTP1B WT mice on a chow diet. Similar results were obtained when we evaluated the natural killer and cytotoxic T lymphocyte marker granzyme B (Gzmb) (Figure 5B). Likewise, the analysis of interferon gamma (Ifng) expression levels, a natural killer and CD4 Th1/CD8 cytotoxic T lymphocyte marker, showed an increase in PTP1B KO mice fed a MCD diet compared with PTP1B WT animals under the same conditions (Figure 5C). In light of these results, the expression levels of the inducible nitric oxide synthase (Nos2) were higher in PTP1B KO mice compared with PTP1B WT animals after NASH induction (Figure 5D). Taken together, these results along with the presence of edema (Figure 4A) and the mucosal inflammation noted in PTP1B KO mice, particularly during NASH (Figure 4B–C), shed light on an effect of PTP1B deficiency on constitutive colonic mucosal inflammation further exacerbated during MCD diet-induced NASH. Interestingly, Ptpn1 levels were decreased in colon samples from PTP1B WT mice, reflecting an inverse relationship between this phosphatase and inflammation (Figure 5E). Conversely, the evaluation of mRNA levels of genes encoding pro-inflammatory cytokines [interleukin-1 beta (IL-1β), tumor necrosis factor alpha (TNFα), and transforming growth factor beta (TFG-β)] and fibroblast growth factor 7 (FGF7) in colon samples did not show significant differences among the experimental groups (data not shown).
Figure 5.

mRNA expression levels of pro-inflammatory markers in colon samples of PTP1B WT and PTP1B KO mice during NASH. A-E) Cd3g (T-cell surface glycoprotein CD3 γ), GzmB (granzyme B), Ifng (interferon gamma), Nos2 (inducible nitric oxide synthase), and Ptp1n (PTP1B) mRNA levels are shown as fold change referred to the control PTP1B WT fed a chow diet. White dots correspond to mice fed a chow diet and black dots to mice fed a MCD diet during 4 weeks. Data are expressed as mean ± SEM. n = 5–10 mice per experimental group. ∗∗∗p < 0.001 PTP1B WT chow vs PTP1B WT MCD; ##p < 0.01 PTP1B KO chow vs PTP1B KO MCD; +p < 0.05; ++ p< 0.01; +++p < 0.001 PTP1B WT MCD vs PTP1B KO MCD.
3.5. Impact of PTP1B deficiency on GLP-1 secretion, EECs pro-inflammatory-mediated signaling, and expression of GLP-2 target genes
It has been demonstrated that glucagon-like peptide-1 (GLP-1) binds to GLP-1R in IELs and suppresses the pro-inflammatory cytokine program, thereby protecting against gut inflammation [13]. To assess the ability of a pro-inflammatory stimulus to induce GLP-1 secretion in vivo, we injected C57BL/6 J mice with a single dose of LPS (2 mg/kg), and plasma GLP-1 levels in response to an oral glucose challenge were analyzed 16 h later. As shown in Figure 6A, a three-fold increase in circulating GLP-1 was found in LPS-injected mice in agreement with previous studies [12,13]. Since GLP-1 is secreted by EECs of the gut, we evaluated the impact of a pro-inflammatory environment in GLP-1 secretion in colonic organoid cultures generated from GLU-Epac2camps mice (see Materials and Methods). Organoids were stimulated with either regular medium (DMEM-2% FBS) or with CM collected from peritoneal macrophages stimulated with LPS (pCM-LPS) or left untreated (pCM-C) for 6 h. A significant increase in GLP-1 released to the medium was found in organoids cultured in pCM-LPS compared with those cultured in regular medium, indicating that a pro-inflammatory stimulus is able to trigger GLP-1 secretion both in vivo and ex vivo (Figure 6B).
Figure 6.

Effect of PTP1B deficiency on GLP-1 release by the gut under pro-inflammatory stimuli. A) GLP-1 serum levels from C57BL/6 J mice injected with LPS (2 mg/kg body weight) as described in Materials and Methods. Black dots represent control animals and gray dots correspond to animals under an LPS acute pro-inflammatory stimulus. n = 4–7 mice per experimental group. ∗p<0.05 PTP1B WT vs PTP1B WT LPS. B) Upper panel represents GLP-1 release from colonic organoids stimulated with pCM-LPS compared with organoids treated with pCM-C and DMEM-2%FBS. n = 2 independent experiments performed in triplicate. ∗∗ p< 0.01 DMEM vs CM-LPS. Lower panel shows colonic organoids images used in this experiment. Upper panel shows a colonic organoid with budding crypt-like domains. Bottom panel shows an organoid with mature EEC L cells. Both images correspond to the same organoid, left image under light microscopy, right image under fluorescence microscopy. L cells (yellow fluorescence YFP) are shown in the right image. C) GLP-1 levels measured in the culture media from colonic crypts cultured under pro-inflammatory conditions. Data are expressed as fold change referred to control medium (DMEM alone). n = 3 independent experiments performed in quadruplicate. ∗∗p < 0.01; ∗∗∗p < 0.001 data referred to basal conditions; ###p < 0.001 referred to pCM-LPS. D) GLP-1 serum levels measured in animals under chow or MCD diet during 4 weeks. n = 5–7 mice per experimental group. ∗∗∗p < 0.001 PTP1B WT chow vs PTP1B WT MCD; ++p < 0.01 PTP1B WT chow vs PTP1B KO chow. E) Gcg mRNA levels (encoding proglucagon) are shown as fold difference referred to the control PTP1B WT fed a chow diet. White dots correspond to mice fed a chow diet and black dots to mice fed a MCD diet during 4 weeks. All data are expressed as mean ± SEM. n = 5–8 animals per experimental group. ∗∗∗p < 0.001 PTP1B WT chow vs PTP1B WT MCD ; ###p < 0.001 PTP1B KO chow vs PTP1B KO MCD.
Next, we analyzed the impact of chemical inhibition of PTP1B in GLP-1 secretion in primary cultures of colonic crypts under the aforementioned pro-inflammatory conditions. Treatment of primary colonic cultures with pCM-LPS increased GLP-1 secretion, an effect further enhanced by the presence of a PTP1B inhibitor (Figure 6C). Of note, GLP-1 secretion was also augmented when the PTP1B inhibitor was directly added to the primary cultures of colonic crypts. We extended these observations by evaluating GLP-1 levels in serum of PTP1B WT and PTP1B KO mice after a 4-hour fast and an oral glucose challenge as described in Materials and Methods and, as shown in Figure 6D, they were decreased in PTP1B WT mice fed a MCD diet compared with the levels found under a chow diet. Unexpectedly, PTP1B KO animals exhibited lower GLP-1 levels in serum under a chow diet, but these levels were preserved under NASH conditions. Of note, mRNA levels of Gcg, the precursor of GLP-1, were similarly downregulated in MCD diet-fed mice from both genotypes (Figure 6E), and no differences between genotypes were found in Dpp4 expression in the colon (data not shown).
Since LPS or IL-6 has been shown to induce GLP-1 secretion by EECs [12,50], we studied the impact of the mCM-LPS medium in the activation of pro-inflammatory signaling cascades in STC-1 cells. To achieve this, STC-1 cells were stimulated with mCM-LPS for short time periods. As depicted in Figure 7A, phosphorylation of JNK and p65 NFκB was markedly increased in STC-1 cells stimulated with mCM-LPS. Likewise, under these conditions, a more rapid degradation of IκBα was observed, likely reflecting an enhancement of NFκB pathway activation. Of note, these pro-inflammatory cascades in STC-1 cells were further enhanced by the presence of the PTP1B inhibitor in the mCM-LPS.
Figure 7.

Inflammatory and insulin signaling in enteroendocrine STC-1 cells stimulated with pro-inflammatory conditioned media. A) STC-1 cells were pre-incubated or not for 2 h with the PTP1B inhibitor (20 μM) before the treatment with mCM-LPS for 5, 15, or 30 min. The dishes pretreated with the PTP1B inhibitor were used throughout the experiment. At the end of the culture time, cells were lysed and protein extracts were analyzed by western blot with the corresponding antibodies. Vinculin was used as a loading control. Right panel shows representative western blots. In the left panel, graphs show quantification of three independent experiments performed in duplicate. ∗∗p < 0.01 mCM-LPS plus PTP1B inhibitor vs mCM-LPS. B) PTP1B inhibition protected against the drop of insulin signaling in EECs stimulated with pro-inflammatory conditioned medium. STC-1 cells were pre-incubated with the PTP1B inhibitor (20 μM) before the treatment with mCM-LPS or mCM-C for 16 h. Then, insulin was added for an additional 15 min. Protein extracts were prepared, and the phosphorylation of the IR and AKT was analyzed by western blot with the antibodies against phospho-IR, phospho-AKT Ser473, phospho-AKT Thr308, total IR, and total AKT. An anti-p85-PI3K antibody was used as a loading control. Right panel shows representative western blots. In the left panel, graphs show quantification of three independent experiments. The different experimental conditions are indicated under the blots or graphs. Data are shown as mean ± SEM. + p < 0.05 mCM-LPS plus PTP1B inhibitor vs mCM-LPS.
It has been reported that insulin via IR-mediated signaling stimulates proglucagon gene expression and, subsequently, the secretion of GLP-1 [51,52]. Since PTP1B dephosphorylates and switches off IR tyrosine kinase activity, we analyzed the impact of PTP1B inhibition in insulin signaling in STC-1 cells. Supplmentary Fig. 2A shows that the pre-treatment with a selective PTP1B inhibitor for 2 h before insulin stimulation further enhanced IR and AKT (Ser473 and Thr308 residues) phosphorylation. A step further, insulin signaling was decreased by the presence of the pro-inflammatory environment of the mCM-LPS (Supplementary Fig. 2B) and, importantly, PTP1B inhibition increased insulin signaling even in these pro-inflammatory conditions (Figure 7B). Altogether, these results suggest that PTP1B plays a role in GLP-1 secreting EECs in the context of inflammation and IR-mediated signaling.
In addition to GLP-1, GLP-2 has also a beneficial effect on gut mucosal integrity as an intestinotrophic factor mainly by stimulating crypt cell proliferation, an effect mediated by the intestinal IGF-1-IGF-1R system [[53], [54], [55]]. Because PTP1B deficiency enhances IGF-1R-mediated effects in a cell autonomous manner [56], we hypothesized that constitutive enhancement of GLP-2 effects would contribute to the better preservation of the gut barrier integrity in PTP1B KO mice. As depicted in Figure 8A–C, constitutive expression levels of Lyz (encoding lysozyme) and Kgf (encoding keratinocyte growth factor), two GLP-2 responsive genes [57], were significantly elevated in colon samples from PTP1B KO mice under basal conditions (chow diet) together with a trend toward elevation of Vip (encoding vasoactive intestinal peptide) mRNA compared with the expression levels of PTP1B WT mice.
Figure 8.
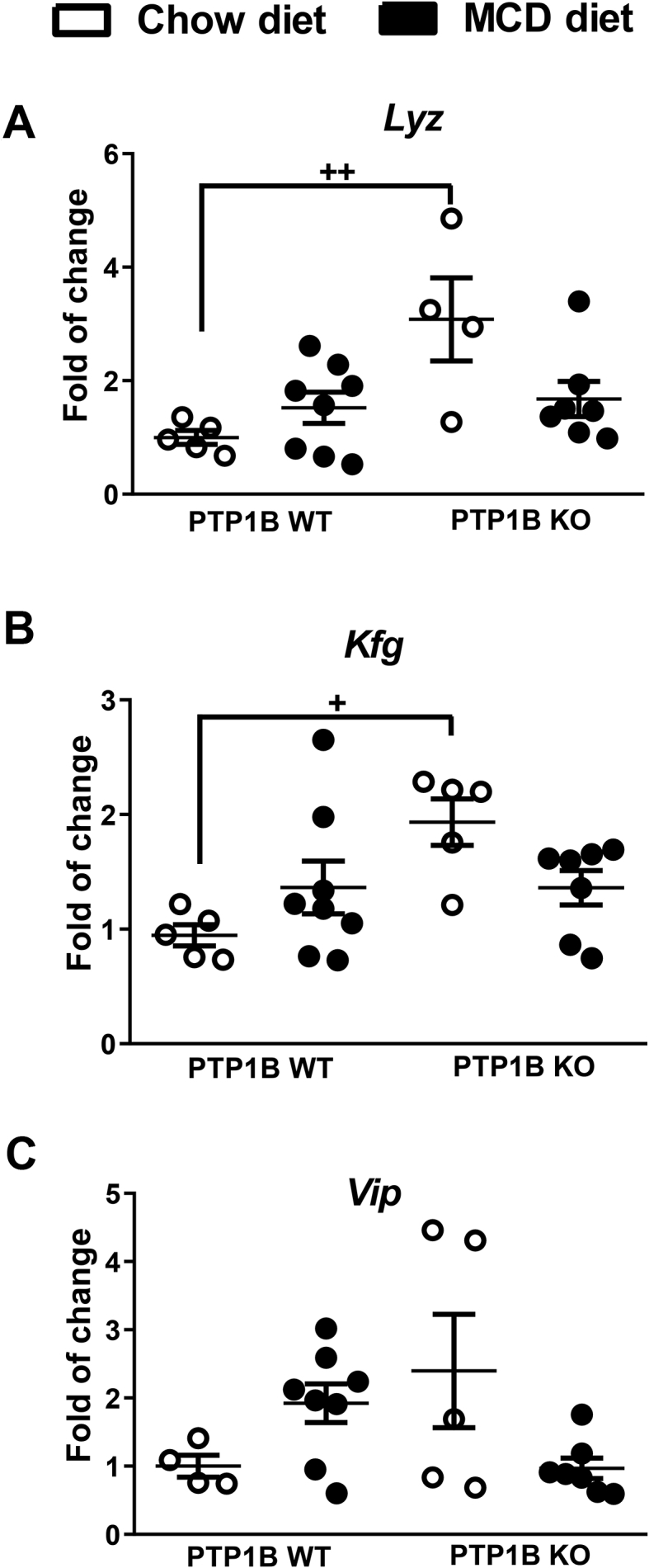
mRNA expression levels of GLP-2 responsive genes in colon samples of PTP1B WT and PTP1B KO mice under NASH conditions. A-C) Lyz (lysozyme), Kfg (keratinocyte growth factor), and Vip (vasointestinal peptide) mRNA levels are shown as fold change referred to the control PTP1B WT fed a chow diet. White dots correspond to mice fed a chow diet, and black dots correspond to mice fed a MCD diet. Data are expressed as mean ± SEM. n = 4–8 animals per experimental group. +p< 0.05; ++p < 0.01 PTP1B WT chow diet vs PTP1B KO chow diet.
4. Discussion
This work has been focused on the study of gut barrier alterations during NASH, a chronic liver disease characterized by a robust pro-inflammatory component, as well as in deciphering a previously unknown role of PTP1B in this process. Since PTP1B displays cellular-specific dual effects, on the one hand, promoting insulin resistance and susceptibility to cell death in non-immune cells such as hepatocytes [20,58] and, on the other, inhibiting the pro-inflammatory responses in immune cells [[21], [22], [23], [24],59], the research on the duality of actions of PTP1B in gut cells (immune and non-immune) during NASH-mediated inflammation is relevant. Overall, our results have showed that both diversity and gut microbial composition are different between mice with a healthy liver and mice with NASH. In addition, some NASH-mediated alterations in gut microbiota are different in PTP1B KO and PTP1B WT mice, as will be discussed following.
It is well known that microbiota diversity is essential to maintain the stability and efficiency of this dynamic micro ecosystem [60]. Our results show that the α-diversity, measured by Shannon index, is lower during MCD diet-induced NASH. Furthermore, the unweighted UniFrac PCoA plot analysis reflects a clustering of the microbial populations from animals fed a MCD diet far away from that of the controls fed a chow diet, confirming that MCD diet-induced NASH alters intestinal microbial populations. Moreover, taxonomy analysis showed that the main changes between the four experimental groups (PTP1B WT chow, PTP1B WT MCD, PTP1B KO chow, and PTP1B KO MCD) are due to NASH as a disease.
In PTP1B WT mice under a MCD diet, the abundance of Firmicutes increased, whereas a marked decrease was observed in Bacteroidetes leading to a higher F/B ratio. By contrast, we did not find changes in MCD diet-fed PTP1B KO mice, either in F/B ratio or in Bacteroidetes and Firmicutes abundance. In this regard, it has been previously described that patients with NASH have a significantly lower proportion of Bacteroidetes compared with healthy controls or those with NAFLD [61]; whereas, decreased F/B ratio was found in patients with inflammatory bowel disease [62]. Moreover, patients with NASH under a probiotic-supplemented diet exhibit a higher level of Bacteroidetes in parallel to a decrease in intrahepatic triglyceride content [8].
Our data also show an increase in the families Porphyromonadaceae (and the corresponding genus Parabacteroides), Erysipelotrichaceae (and the corresponding genus Allobaculum), and Rikenellaceae in mice receiving a MCD diet for 4 weeks. In addition, an increase in the abundance of family Turicibacteraceae (and the corresponding genus Turicibacter) was also observed, but only in PTP1B WT animals. These variations agree with observations from other studies in mice under a MCD diet or in patients with NASH [28,[63], [64], [65]]. Families Rikenellaceae and Turicibacteraceae are known to be associated with inflammatory states [28] and might contribute to gut inflammation in mice receiving a MCD diet. Conversely, a significant decrease of families S24-7, Dehalobacteriaceae, Mogibacteriaceae, Lactobacillae, Lachnospiraceae, and genera Adlercreutzia, Lactobacillus, Roseburia, and Desulfovibrio was found in mice with NASH. In particular, a low presence of Dehalobacteriaceae was detected only in PTP1B WT mice, and the same was observed for Lachnospiraceae; but, its genus Roseburia decreased in mice from both phenotypes. Of special interest is the decrease observed in MCD diet-fed mice of Lactobacillus and Roseburia, two beneficial organisms producing lactate and butyrate, respectively. Lactate-producing bacteria promote lactate-converting organisms, such as Roseburia, that produce butyrate favoring the abundance of Lactobacilli, thereby contributing to the maintenance of the anti-inflammatory effects of these microorganisms [28,66,67]. The observed decrease in genus Desulfovibrio with the MCD diet should cause a lower production of H2S in the gut, a compound that has been related to regulation of epithelial mucus production and decrease of systemic inflammation [65,68]. Likewise, a lower abundance of Adlercreutzia and Dehalobacteriaceae might be related to altered lipid and glucose metabolism. In this regard, Adlercreutzia is characterized by its ability to produce equol, which binds to the G-protein-coupled estrogen receptor (GPER) and improves glucose tolerance [69]. Conversely, Dehalobateriaceae has been reported to decrease in situations of hepatic lipid accumulation induced by environmental chemicals [70]. Notably, two marked alterations are observed only in PTP1B KO mice. First, at the phylum level, TM7 relative levels decreased in these mice during NASH. Phylum TM7 has been described as a potential immunomodulatory due to its ability to inhibit TNFα production in macrophages [71,72] and, consequently, its decrease could be associated with the exacerbation of inflammation in NAFLD in PTP1B KO mice [25]. Because PTP1B KO mice present a constitutive pro-inflammatory phenotype, it is possible to speculate that the lower presence of phylum TM7 might play a role in their immune responses during NASH progression. Second, the LPS-producing genus Prevotella increases in PTP1B KO mice under a MCD diet. This finding is somehow unexpected, since PTP1B KO mice under a MCD diet show a lower level of circulating LPS than WT animals. It is worth noting that some changes in microbiota described in the MCD diet-induced NASH, such as decreased level of Lactobacillus and Roseburia and increased level of Prevotella, are similar to those reported in individuals with lean NASH [73], many of them with malnutrition (choline deficiency) or genetic impairment of phosphatidylcholine synthesis [74].
A step forward, the PICRUSt functional approach showed that a MCD diet shifts metabolic pathways of the microbial community toward a reduction in genes involved in metabolism of cofactors and vitamins and energy balance in PTP1B WT, but not in PTP1B KO mice, pointing to an impact of PTP1B deletion in the changes of bacterial metabolism under the NASH condition. Of relevance, when we compared both genotypes, gut microbiota from PTP1B WT mice fed a MCD diet overexpressed genes involved in the synthesis of secondary bile acids, whereas PTP1B KO mice exhibited an increase in genes linked to propionate metabolism. Since propionate (a short-chain fatty acid) is considered to be a health promoting microbial metabolite that possesses biological activity at the intestine and intestinal epithelium level [75], these differences likely associate PTP1B WT mice with a more harmful microbiota compared with their counterparts lacking PTP1B.
There is a growing interest in revealing the function of bile acids during NASH and their possible role in the onset and progression of this disease. In agreement with the work of Puri and co-workers [76], we found a marked increase in primary bile acids in plasma in mice with NASH. It is known that primary bile acid synthesis is modulated by gut microbiota through the regulation of key enzymes of this metabolic pathway such as CYP7A1. Related to this, ileal FXR activation by its agonist CDCA leads to CYP7A1 inhibition [77] and, therefore, decreases bile acid synthesis. During NASH, CA levels are much higher than CDCA levels and, as a result, FXR activation by CDCA is reduced, thereby increasing bile acid synthesis. Thus, the lower levels of CA found in serum from PTP1B KO mice during NASH point to a lower bile acid synthesis, which was manifested by decreased amounts of CA, CDCA, and UDCA. These data are reinforced by increased expression of the Nr1h4 gene (coding FXR protein) observed in the gut of MCD diet-fed PTP1B KO mice compared with their PTP1B WT counterparts. In addition, as we remarked previously, PICRUSt predicted that gut microbiome from PTP1B WT mice fed a MCD diet is enriched in genes for secondary bile acid biosynthesis compared with PTP1B KO mice, and this could be the reason of the lower plasma levels of TDCA during NASH found in mice lacking PTP1B.
Although a number of studies have addressed the molecular mechanisms involved in the progression of NAFLD toward NASH, little is known about how the intestine is affected by this inflammatory disease. A recent study [28] has reported the beneficial effect of butyrate on MCD diet-induced gut barrier dysfunction and inflammation in the liver. A previous clinical study [78] demonstrated that both intestinal permeability and alterations in gut microbiota were increased in patients with NAFLD. In the present study, we found that colonic edema and, thus, colonic inflammation, was significantly enhanced only in PTP1B KO mice with NASH, suggesting exacerbated pro-inflammatory responses in this genotype as we previously reported [24]. Moreover, the presence of inflammatory infiltrates in the colonic mucosa together with a marked increase in the expression levels of inflammatory markers such as Ifng and Nos2 were found in PTP1B KO animals fed a MCD diet compared with PTP1B WT mice. Of note, PTP1B KO mice fed a chow diet showed increased expression levels of Cd3g and GzmB compared with PTP1B WT mice, reinforcing the fact that PTP1B KO mice exhibit an intrinsic pro-inflammatory susceptibility, which is exacerbated after NASH induction. Taking into account that PTP1B is a negative modulator of key immune signaling cascades [24], our results suggesting that PTP1B deficiency likely facilitates the activation of macrophages (through increasing Nos2) and T lymphocytes (through Ifng, Cd3g and GzmB) in the gut are in line with data previously reported by our laboratory and others in different inflammatory contexts [24,79]. Because NASH concurs with a systemic pro-inflammatory environment [80], the enhancement of gut inflammation in PTP1B KO mice is somehow expected due to the dynamic cross-talk in the gut-to-liver axis.
To obtain further insights on differential gut barrier disruption in PTP1B WT and PTP1B KO mice with NASH, we measured their intestinal permeability by a FITC-dextran assay, which showed major flux of this fluorescent compound and, thus, major leakage in PTP1B WT mice. Similar results were obtained by the analysis of serum endotoxin content, confirming a disruption in the gut barrier function by the MCD diet and, importantly, a partial protection in PTP1B KO mice. We also explored gut barrier permeability in a cellular autonomous manner and found decreased TEER values in Caco-2 cells and, thus, disrupted barrier integrity under pro-inflammatory culture conditions in agreement with the work of Moran and co-workers showing decreased TEER in Caco-2 cells cultured with TNFα [81]. However, no differences were observed between Caco-2 cells with silenced Ptpn1 and the controls (shScrb). Taking into account that TEER measurement allows the study of barrier integrity in an epithelial monolayer depending only on the unions between cells [42], these results suggest that specific PTP1B deficiency in the epithelial cells of the gut might not affect permeability in a relevant way. By contrast, the analysis of the tight junction protein ZO-1 in colon sections showed a better preservation of the gut barrier features in PTP1B KO mice fed a MCD diet. Thus, the results of the in vivo NASH model in which PTP1B deficiency confers protection against gut barrier disruption triggered by pro-inflammatory stimuli strongly suggest that environmental factors such as the aforementioned gut microbiota diversity rather than the gut epithelial cells may have a major contribution in maintaining the gut barrier integrity under this pathologic condition.
Recent findings have expanded the classical concept of enteroendocrine L cell biology toward sensors of inflammatory stimuli and compromised mucosal integrity, thereby linking GLP-1 secretion to gut inflammation. Yusta and co-workers showed that binding of GLP-1 to its receptor in IELs switches the pattern of the inflammatory response [13]. In a recent study, Lebrun and co-workers [12] reported increases in GLP-1 levels in circulation by LPS administration as well as in a dextran sulfate sodium- and ischemia/reperfusion-induced colitis model in mice, this effect being dependent on TLR4. A step further, the same study showed elevations in GLP-1 in human healthy volunteers receiving a single injection of LPS and patients subjected to ischemia/reperfusion in a jejunal segment during a pancreaticoduodenectomy. Moreover, Keller and co-workers [82] showed increased release of GLP-1 in patients with inflammatory bowel disease. Despite the effects of acute pro-inflammatory stimuli or severe gut inflammation in triggering GLP-1 release to the circulation, our results showed that under a mild chronic pro-inflammatory environment (i.e. MCD diet), PTP1B WT animals had reduced circulating GLP-1 levels in response to an oral glucose challenge. A possible explanation for such effect could be supported by the existence of a cross-talk between EECs and IELs aimed to counteract elevated inflammation, although MCD diet-fed PTP1B WT mice might not have reached enough inflammatory thresholds to activate this compensatory mechanism through time. In line with these data, a human study reported reduced peak (15 min) GLP-1 concentrations in a cohort of 52 non-diabetic patients with NAFLD (including simple steatosis and NASH) compared with controls, as well as total GLP-1 secretion in an oral glucose tolerance test [83]. Interestingly, PTP1B KO mice that already showed lower glucose-triggered GLP-1 responses on a chow diet were protected against the decrease of this incretin under a MCD diet. We speculate that the reduced GLP-1 responses found in PTP1B KO animals on a chow diet would prevent excessive postprandial insulin excursions in these systemic hypoinsulinemic and insulin-hypersensitive animals [19,84] to avoid excessive hypoglycemia. Future research could unravel differences in the amount of IELs between PTP1B WT and PTP1B KO mice.
To further investigate the role of PTP1B in GLP-1 release by the gut under pro-inflammatory conditions as previously reported [12,13], we performed in vitro studies in EECs, colonic organoids, and primary cultures of colonic crypts. Our results showed for the first time on the one hand, an effect of PTP1B inhibition by enhancing the pro-inflammatory signaling cascades triggered by the mCM-LPS (JNK, NFκB) in STC-1 cells, reinforcing our experimental evidence on the impact of this phosphatase in the interplay between immune (likely IELs) and EECs in the gut and, on the other, an unknown role of this phosphatase as a negative modulator of insulin signaling (IR, Akt) in EECs cells under basal or pro-inflammatory conditions. A step further, in a more physiological model using primary cultures of colonic crypts, we found a significant increase in GLP-1 release on treatment with the pro-inflammatory media and, more importantly, this effect was further increased in the presence of the PTP1B inhibitor that, as previously mentioned, increased the pro-inflammatory signaling cascades in EECs. Altogether, these results led us to suggest that PTP1B deficiency might affect both IELs and EECs by enhancing pro-inflammatory and insulin signaling, respectively, thereby boosting GLP-1 secretion. However, a limitation of the present study is the use of global PTP1B-deficient mice and, in this regard, future research will be needed to characterize the outcome of deficiency of this phosphatase in different gut cell types.
The results of the present study are apparently contradictory with our previous data [25] showing accelerated NASH in PTP1B KO mice on a MCD diet due to enhanced immune responses in the liver. The protective mechanism in the intestine reported here seems to be tissue-specific and could be explained by the maintenance of GLP-1 levels in MCD diet-fed PTP1B KO mice that elicit an anti-inflammatory protective effect targeting IELs (as proposed by Yusta and co-workers [13]) and probably other gut cell types, thereby preserving gut barrier permeability. In contrast, the severe hepatic phenotype of PTP1B KO mice on a MCD diet reported in our previous manuscript [25] likely reflects a profound inflammation within the liver beyond endotoxemia that results from the enhancement of pro-inflammatory signaling cascades in immune cells of the liver, either resident or recruited, on NASH insult.
GLP-1 and GLP-2 are simultaneously produced via posttranslational processing of proglucagon by prohormone convertase 1/3 and co-secreted from intestinal L cells [85]. In the context of gut inflammation, Lebrun and co-workers showed increases in plasma GLP-2 in mice receiving an acute injection of LPS or subjected to intestinal ischemia/reperfusion [12]. In this regard, we found higher constitutive expression of GLP-2 sensitive genes in colon samples from PTP1B KO mice. These data are relevant since gut epithelial IGF-1R signaling has been shown to mediate the effects of GLP-2 in the proliferation of the gut crypts [55], and it is plausible that PTP1B-deficiency might confer increased GLP-2 secretion together with IGF-1R-mediated sensitivity in the gut as occurs in pancreatic beta cells [56,86], as demonstrated by the greater depth of the colonic crypts in PTP1B KO mice, thereby favoring gut mucosal integrity. Another possible explanation for the better preservation of the gut barrier in PTP1B KO mice is based on the requirement of insulin for the maintenance of normal rates of protein synthesis in the mucosa of the small intestine, an essential component of the remodeling process during damage [87]. Thus, the decrease in the synthesis rate of small intestine mucosa under hypoinsulinemia in MCD-induced NASH might be attenuated in PTP1B KO mice due to their hypersensitivity to insulin.
5. Conclusions
Taking all the results together, this study has identified a protective role of PTP1B deficiency against the shift in gut microbiome during NASH that was accompanied with less gut barrier permeability, preserved GLP-1 levels under a pro-inflammatory environment, and a potential sensitivity to GLP-2. Further investigation is required to identify the accurate mechanisms responsible for the ameliorated phenotype of PTP1B KO mice in the gut during this liver pathology with a possible therapeutic value.
Funding
This work was funded by grants SAF2015-65267-R (MINECO/FEDER, UE), RTI2018-094052-B-100 (MCI/AEI/FEDER, UE), S2010/BMD-2423, S2017/BMD-3684 (Comunidad de Madrid, Spain), CIBERdem, CIBERhed and PI 16/02096 (ISCIII, Spain). We also acknowledge grants H2020 Marie Sklodowska-Curie ITN-TREATMENT (Grant Agreement 721236, European Commission), Spanish Ministry of Education-MECD (FPU13/05802 grant to C.R) and Fundación Ramón Areces (to A.M.V.). CBMSO is recipient of institutional aid from Fundación Ramón Areces. VBL, FMG and FR received funding from a Wellcome Trust joint investigator award (106262/Z/14/Z and 106263/Z/14/Z) and a joint MRC programme within the Metabolic Diseases Unit (MRC_MC_UU_12012/3), United Kingdom. VBL received a project support grant from the Society for Endocrinology, United Kingdom. The MRL Histology and Biochemistry Assay Lab Core facilities received funding from the MRC Metabolic Diseases Unit [MRC_MC_UU_12012/5] and the Imaging Core through a Wellcome Trust Strategic Award [100574/Z/12/Z], United Kingdom.
Author contributions
C.R.; participated in study design, acquisition, analysis, interpretation of data and writing of the manuscript; M.P and J.J G-R performed analysis of gut permeability; V.B.L performed the organoid studies; I. G-M and R.A performed the signaling studies; P.S–S and M.V. T-L contributed to gut characterization; J.S, J.R and C.B contributed to the analysis of bile acids; L.M; F.M.G; F.R; L.G.G contributed to supervision of the work and scientific discussion; A.M.V and J.M.C participted in design and coordination of the study and wrote the manuscript. All authors revised and approved the final manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2020.01.018.
Contributor Information
Jose M. Carrascosa, Email: josemaria.carrascosa@uam.es.
Ángela M. Valverde, Email: avalverde@iib.uam.es.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary Figure 1.

Gut characterization in PTP1B WT and PTP1B KO mice under NASH conditions. A) Graph represents the ratio colon weight/length for body weight. White and black bars represent animals fed a chow or MCD diet, respectively. n= 5-7 mice per experimental group. B) Upper panel shows quantification of number of feces in the colon from the four experimental animal groups. Lower panel shows representative images of colon samples. n= 4-8 mice per experimental group. C) Feces humidity was measured with the moisture analyzer MB120 (Ohaus). Percentage of humidity was calculated from the difference between final dried feces and starting fresh feces. Data are expressed as mean ± SEM. n=6-11 mice per experimental group. ∗p<0.05; ∗∗p<0.01 PTP1B WT chow vs PTP1B WT MCD diet; ##p<0.01; ###p<0. 001 PTP1B KO chow vs PTP1B KO MCD.
Supplementary Figure 2.

Effect of PTP1B inhibition in insulin signaling in EECs under proinflammatory stimulus. A) STC-1 cells were pretreated or not for 2 h with the PTP1B inhibitor (20 μM) and then stimulated with insulin for 15 min. Protein extracts were prepared, and the phosphorylation of the IR and AKT was analyzed by western blot with the antibodies against phosho-IR, phospho-AKT Ser473, phospho-AKT Thr308, total IR, and total AKT. An anti-p85-PI3K antibody was used as a loading control. Left panel shows representative western blots. In the right panel, graphs show quantification of three independent experiments. Data are shown as mean ± SEM. n=3 independent experiments. ∗p<0.05; ∗∗∗p<0.001 DMEM alone vs insulin; ++p<0.01; +++p<0.001 insulin plus PTP1B inhibitor vs insulin. B) STC-1 cells were pre-incubated with the PTP1B inhibitor (20 μM) before the treatment with mCM-LPS or mCM-C for 16 h. Then, insulin was added for a further 15 min. Protein extracts were prepared and the phosphorylation of the IR and AKT was analyzed by western blot with the antibodies against phosho-IR, phospho-AKT Ser473, phospho-AKT Thr308, total IR, and total AKT. An anti-p85-PI3K antibody was used as a loading control. Left panel shows representative western blots. In the right panel, graphs show quantification of three independent experiments. Data are shown as mean ± SEM. +++p<0.001 mCM-LPS vs mCM-C.
Supplementary Table 1.

TEER values of Caco-2 shScrb and Caco-2 shPtp1n cells under mCM-C or mCM-LPS treatment. Two stable cell lines, Caco-2 scramble (Caco-2 shScb) and Caco-2 siPtpn1 (Caco-2 silenced for Ptpn1 gene, encoding for PTP1B protein), were cultured with mCM-C or mCM-LPS. TEER values were measured after 24 and 48 h. Data are expressed as mean values ± SEM. n=3 independent experiments performed in triplicate.
References
- 1.Byrne C.D., Targher G. NAFLD: a multisystem disease. Journal of Hepatology. 2015;62(1 Suppl):S47–S64. doi: 10.1016/j.jhep.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Wree A., Broderick L., Canbay A., Hoffman H.M., Feldstein A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nature Reviews Gastroenterology & Hepatology. 2013;10(11):627–636. doi: 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
- 3.Chalasani N., Younossi Z., Lavine J.E., Diehl A.M., Brunt E.M., Cusi K. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142(7):1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Henao-Mejia J., Elinav E., Thaiss C.A., Flavell R.A. The intestinal microbiota in chronic liver disease. Advances in Immunology. 2013;117:73–97. doi: 10.1016/B978-0-12-410524-9.00003-7. [DOI] [PubMed] [Google Scholar]
- 5.Hoyles L., Fernandez-Real J.M., Federici M., Serino M., Abbott J., Charpentier J. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nature Medicine. 2018;24(7):1070–1080. doi: 10.1038/s41591-018-0061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betrapally N.S., Gillevet P.M., Bajaj J.S. Changes in the intestinal microbiome and alcoholic and nonalcoholic liver diseases: causes or effects? Gastroenterology. 2016;150(8):1745–1755 e3. doi: 10.1053/j.gastro.2016.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loomba R., Seguritan V., Li W., Long T., Klitgord N., Bhatt A. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metabolism. 2017;25(5):1054–1062 e5. doi: 10.1016/j.cmet.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pachikian B.D., Essaghir A., Demoulin J.B., Catry E., Neyrinck A.M., Dewulf E.M. Prebiotic approach alleviates hepatic steatosis: implication of fatty acid oxidative and cholesterol synthesis pathways. Molecular Nutrition & Food Research. 2013;57(2):347–359. doi: 10.1002/mnfr.201200364. [DOI] [PubMed] [Google Scholar]
- 9.Velayudham A., Dolganiuc A., Ellis M., Petrasek J., Kodys K., Mandrekar P. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology. 2009;49(3):989–997. doi: 10.1002/hep.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagnerberger S., Spruss A., Kanuri G., Stahl C., Schroder M., Vetter W. Lactobacillus casei Shirota protects from fructose-induced liver steatosis: a mouse model. The Journal of Nutritional Biochemistry. 2013;24(3):531–538. doi: 10.1016/j.jnutbio.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Sternini C., Anselmi L., Rozengurt E. Enteroendocrine cells: a site of 'taste' in gastrointestinal chemosensing. Current Opinion in Endocrinology Diabetes and Obesity. 2008;15(1):73–78. doi: 10.1097/MED.0b013e3282f43a73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebrun L.J., Lenaerts K., Kiers D., Pais de Barros J.P., Le Guern N., Plesnik J. Enteroendocrine L cells sense LPS after gut barrier injury to enhance GLP-1 secretion. Cell Reports. 2017;21(5):1160–1168. doi: 10.1016/j.celrep.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Yusta B., Baggio L.L., Koehler J., Holland D., Cao X., Pinnell L.J. GLP-1R Agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes. 2015;64(7):2537–2549. doi: 10.2337/db14-1577. [DOI] [PubMed] [Google Scholar]
- 14.Insuela D.B.R., Carvalho V.F. Glucagon and glucagon-like peptide-1 as novel anti-inflammatory and immunomodulatory compounds. European Journal of Pharmacology. 2017;812:64–72. doi: 10.1016/j.ejphar.2017.07.015. [DOI] [PubMed] [Google Scholar]
- 15.Bugianesi E., Moscatiello S., Ciaravella M.F., Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Current Pharmaceutical Design. 2010;16(17):1941–1951. doi: 10.2174/138161210791208875. [DOI] [PubMed] [Google Scholar]
- 16.Salmeen A., Andersen J.N., Myers M.P., Tonks N.K., Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Molecular Cell. 2000;6(6):1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 17.Seely B.L., Staubs P.A., Reichart D.R., Berhanu P., Milarski K.L., Saltiel A.R. Protein tyrosine phosphatase 1B interacts with the activated insulin receptor. Diabetes. 1996;45(10):1379–1385. doi: 10.2337/diab.45.10.1379. [DOI] [PubMed] [Google Scholar]
- 18.Zabolotny J.M., Bence-Hanulec K.K., Stricker-Krongrad A., Haj F., Wang Y., Minokoshi Y. PTP1B regulates leptin signal transduction in vivo. Developmental Cell. 2002;2(4):489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 19.Elchebly M., Payette P., Michaliszyn E., Cromlish W., Collins S., Loy A.L. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283(5407):1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Rodriguez A., Mas-Gutierrez J.A., Mirasierra M., Fernandez-Perez A., Lee Y.J., Ko H.J. Essential role of protein tyrosine phosphatase 1B in obesity-induced inflammation and peripheral insulin resistance during aging. Aging Cell. 2012;11(2):284–296. doi: 10.1111/j.1474-9726.2011.00786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aoki N., Matsuda T. A cytosolic protein-tyrosine phosphatase PTP1B specifically dephosphorylates and deactivates prolactin-activated STAT5a and STAT5b. Journal of Biological Chemistry. 2000;275(50):39718–39726. doi: 10.1074/jbc.M005615200. [DOI] [PubMed] [Google Scholar]
- 22.Lu X., Malumbres R., Shields B., Jiang X., Sarosiek K.A., Natkunam Y. PTP1B is a negative regulator of interleukin 4-induced STAT6 signaling. Blood. 2008;112(10):4098–4108. doi: 10.1182/blood-2008-03-148726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myers M.P., Andersen J.N., Cheng A., Tremblay M.L., Horvath C.M., Parisien J.P. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. Journal of Biological Chemistry. 2001;276(51):47771–47774. doi: 10.1074/jbc.C100583200. [DOI] [PubMed] [Google Scholar]
- 24.Traves P.G., Pardo V., Pimentel-Santillana M., Gonzalez-Rodriguez A., Mojena M., Rico D. Pivotal role of protein tyrosine phosphatase 1B (PTP1B) in the macrophage response to pro-inflammatory and anti-inflammatory challenge. Cell Death & Disease. 2014;5:e1125. doi: 10.1038/cddis.2014.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Rodriguez A., Valdecantos M.P., Rada P., Addante A., Barahona I., Rey E. Dual role of protein tyrosine phosphatase 1B in the progression and reversion of non-alcoholic steatohepatitis. Molecular Metabolism. 2018;7:132–146. doi: 10.1016/j.molmet.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haczeyni F., Yeh M.M., Ioannou G.N., Leclercq I.A., Goldin R., Dan Y.Y. Mouse models of non-alcoholic steatohepatitis: a reflection on recent literature. Journal of Gastroenterology and Hepatology. 2018;33(7):1312–1320. doi: 10.1111/jgh.14122. [DOI] [PubMed] [Google Scholar]
- 27.Oz H.S., Chen T.S., Neuman M. Methionine deficiency and hepatic injury in a dietary steatohepatitis model. Digestive Diseases and Sciences. 2008;53(3):767–776. doi: 10.1007/s10620-007-9900-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye J., Lv L., Wu W., Li Y., Shi D., Fang D. Butyrate protects mice against methionine-choline-deficient diet-induced non-alcoholic steatohepatitis by improving gut barrier function, attenuating inflammation and reducing endotoxin levels. Frontiers in Microbiology. 2018;9:1967. doi: 10.3389/fmicb.2018.01967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research. 2013;41(1):e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lozupone C., Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology. 2005;71(12):8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langille M.G., Zaneveld J., Caporaso J.G., McDonald D., Knights D., Reyes J.A. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology. 2013;31(9):814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roman I.D., Cano-Martinez D., Lobo M.V., Fernandez-Moreno M.D., Hernandez-Breijo B., Sacristan S. Infliximab therapy reverses the increase of allograft inflammatory factor-1 in serum and colonic mucosa of rats with inflammatory bowel disease. Biomarkers. 2017;22(2):133–144. doi: 10.1080/1354750X.2016.1252950. [DOI] [PubMed] [Google Scholar]
- 34.Alnouti Y., Csanaky I.L., Klaassen C.D. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. Journal of Chromatography B Analytical Technologies in the Biomedical and Life Sciences. 2008;873(2):209–217. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato T., Vries R.G., Snippert H.J., van de Wetering M., Barker N., Stange D.E. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 36.Goldspink D.A., Lu V.B., Billing L.J., Larraufie P., Tolhurst G., Gribble F.M. Mechanistic insights into the detection of free fatty and bile acids by ileal glucagon-like peptide-1 secreting cells. Molecular Metabolism. 2018;7:90–101. doi: 10.1016/j.molmet.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reimann F., Habib A.M., Tolhurst G., Parker H.E., Rogers G.J., Gribble F.M. Glucose sensing in L cells: a primary cell study. Cell Metabolism. 2008;8(6):532–539. doi: 10.1016/j.cmet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim B.R., Shin J., Guevarra R., Lee J.H., Kim D.W., Seol K.H. Deciphering diversity indices for a better understanding of microbial communities. Journal of Microbiology and Biotechnology. 2017;27(12):2089–2093. doi: 10.4014/jmb.1709.09027. [DOI] [PubMed] [Google Scholar]
- 39.Wahlstrom A., Sayin S.I., Marschall H.U., Backhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metabolism. 2016;24(1):41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 40.Chiang J.Y.L., Ferrell J.M. Bile acid metabolism in liver pathobiology. Gene Expression. 2018;18(2):71–87. doi: 10.3727/105221618X15156018385515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Artursson P., Ungell A.L., Lofroth J.E. Selective paracellular permeability in two models of intestinal absorption: cultured monolayers of human intestinal epithelial cells and rat intestinal segments. Pharmaceutical Research. 1993;10(8):1123–1129. doi: 10.1023/a:1018903931777. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez-Feo J.A., Puerto M., Fernandez-Mena C., Verdejo C., Lara J.M., Diaz-Sanchez M. A new role for reticulon-4B/NOGO-B in the intestinal epithelial barrier function and inflammatory bowel disease. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2015;308(12):G981–G993. doi: 10.1152/ajpgi.00309.2014. [DOI] [PubMed] [Google Scholar]
- 43.Cibor D., Domagala-Rodacka R., Rodacki T., Jurczyszyn A., Mach T., Owczarek D. Endothelial dysfunction in inflammatory bowel diseases: pathogenesis, assessment and implications. World Journal of Gastroenterology. 2016;22(3):1067–1077. doi: 10.3748/wjg.v22.i3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitajima S., Takuma S., Morimoto M. Histological analysis of murine colitis induced by dextran sulfate sodium of different molecular weights. Experimental Animals. 2000;49(1):9–15. doi: 10.1538/expanim.49.9. [DOI] [PubMed] [Google Scholar]
- 45.Ordas I., Eckmann L., Talamini M., Baumgart D.C., Sandborn W.J. Ulcerative colitis. Lancet. 2012;380(9853):1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 46.Machado M.V., Michelotti G.A., Xie G., Almeida Pereira T., Boursier J., Bohnic B. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PloS One. 2015;10(5) doi: 10.1371/journal.pone.0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montbarbon M., Pichavant M., Langlois A., Erdual E., Maggiotto F., Neut C. Colonic inflammation in mice is improved by cigarette smoke through iNKT cells recruitment. PloS One. 2013;8(4) doi: 10.1371/journal.pone.0062208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motavallian-Naeini A., Andalib S., Rabbani M., Mahzouni P., Afsharipour M., Minaiyan M. Validation and optimization of experimental colitis induction in rats using 2, 4, 6-trinitrobenzene sulfonic acid. Research in Pharmaceutical Sciences. 2012;7(3):159–169. [PMC free article] [PubMed] [Google Scholar]
- 49.Peterson L.W., Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nature Reviews Immunology. 2014;14(3):141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 50.Ellingsgaard H., Hauselmann I., Schuler B., Habib A.M., Baggio L.L., Meier D.T. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nature Medicine. 2011;17(11):1481–1489. doi: 10.1038/nm.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lim G.E., Huang G.J., Flora N., LeRoith D., Rhodes C.J., Brubaker P.L. Insulin regulates glucagon-like peptide-1 secretion from the enteroendocrine L cell. Endocrinology. 2009;150(2):580–591. doi: 10.1210/en.2008-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi F., Sun J., Lim G.E., Fantus I.G., Brubaker P.L., Jin T. Cross talk between the insulin and Wnt signaling pathways: evidence from intestinal endocrine L cells. Endocrinology. 2008;149(5):2341–2351. doi: 10.1210/en.2007-1142. [DOI] [PubMed] [Google Scholar]
- 53.Benjamin M.A., McKay D.M., Yang P.C., Cameron H., Perdue M.H. Glucagon-like peptide-2 enhances intestinal epithelial barrier function of both transcellular and paracellular pathways in the mouse. Gut. 2000;47(1):112–119. doi: 10.1136/gut.47.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cameron H.L., Perdue M.H. Stress impairs murine intestinal barrier function: improvement by glucagon-like peptide-2. Journal of Pharmacology and Experimental Therapeutics. 2005;314(1):214–220. doi: 10.1124/jpet.105.085373. [DOI] [PubMed] [Google Scholar]
- 55.Dong C.X., Zhao W., Solomon C., Rowland K.J., Ackerley C., Robine S. The intestinal epithelial insulin-like growth factor-1 receptor links glucagon-like peptide-2 action to gut barrier function. Endocrinology. 2014;155(2):370–379. doi: 10.1210/en.2013-1871. [DOI] [PubMed] [Google Scholar]
- 56.Fernandez-Ruiz R., Vieira E., Garcia-Roves P.M., Gomis R. Protein tyrosine phosphatase-1B modulates pancreatic beta-cell mass. PloS One. 2014;9(2) doi: 10.1371/journal.pone.0090344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yusta B., Holland D., Waschek J.A., Drucker D.J. Intestinotrophic glucagon-like peptide-2 (GLP-2) activates intestinal gene expression and growth factor-dependent pathways independent of the vasoactive intestinal peptide gene in mice. Endocrinology. 2012;153(6):2623–2632. doi: 10.1210/en.2012-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sangwan V., Paliouras G.N., Cheng A., Dube N., Tremblay M.L., Park M. Protein-tyrosine phosphatase 1B deficiency protects against Fas-induced hepatic failure. Journal of Biological Chemistry. 2006;281(1):221–228. doi: 10.1074/jbc.M507858200. [DOI] [PubMed] [Google Scholar]
- 59.Heinonen K.M., Dube N., Bourdeau A., Lapp W.S., Tremblay M.L. Protein tyrosine phosphatase 1B negatively regulates macrophage development through CSF-1 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2776–2781. doi: 10.1073/pnas.0508563103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Almada C.N., Nunes de Almada C., Martinez R.C., Sant'Ana Ade S. Characterization of the intestinal microbiota and its interaction with probiotics and health impacts. Applied Microbiology and Biotechnology. 2015;99(10):4175–4199. doi: 10.1007/s00253-015-6582-5. [DOI] [PubMed] [Google Scholar]
- 61.Mouzaki M., Comelli E.M., Arendt B.M., Bonengel J., Fung S.K., Fischer S.E. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58(1):120–127. doi: 10.1002/hep.26319. [DOI] [PubMed] [Google Scholar]
- 62.Sokol H., Seksik P., Furet J.P., Firmesse O., Nion-Larmurier I., Beaugerie L. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflammatory Bowel Diseases. 2009;15(8):1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 63.Henao-Mejia J., Elinav E., Jin C., Hao L., Mehal W.Z., Strowig T. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wong V.W., Tse C.H., Lam T.T., Wong G.L., Chim A.M., Chu W.C. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis-a longitudinal study. PloS One. 2013;8(4) doi: 10.1371/journal.pone.0062885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ye J.Z., Li Y.T., Wu W.R., Shi D., Fang D.Q., Yang L.Y. Dynamic alterations in the gut microbiota and metabolome during the development of methionine-choline-deficient diet-induced nonalcoholic steatohepatitis. World Journal of Gastroenterology. 2018;24(23):2468–2481. doi: 10.3748/wjg.v24.i23.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tamanai-Shacoori Z., Smida I., Bousarghin L., Loreal O., Meuric V., Fong S.B. Roseburia spp.: a marker of health? Future Microbiology. 2017;12:157–170. doi: 10.2217/fmb-2016-0130. [DOI] [PubMed] [Google Scholar]
- 67.Wang Y., Gu Y., Fang K., Mao K., Dou J., Fan H. Lactobacillus acidophilus and Clostridium butyricum ameliorate colitis in murine by strengthening the gut barrier function and decreasing inflammatory factors. Beneficial Microbes. 2018:1–14. doi: 10.3920/BM2017.0035. [DOI] [PubMed] [Google Scholar]
- 68.Wallace J.L., Motta J.P., Buret A.G. Hydrogen sulfide: an agent of stability at the microbiome-mucosa interface. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2018;314(2):G143–G149. doi: 10.1152/ajpgi.00249.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharma G., Hu C., Brigman J.L., Zhu G., Hathaway H.J., Prossnitz E.R. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology. 2013;154(11):4136–4145. doi: 10.1210/en.2013-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lai K.P., Ng A.H., Wan H.T., Wong A.Y., Leung C.C., Li R. Dietary exposure to the environmental chemical, PFOS on the diversity of gut microbiota, associated with the development of metabolic syndrome. Frontiers in Microbiology. 2018;9:2552. doi: 10.3389/fmicb.2018.02552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He X., McLean J.S., Edlund A., Yooseph S., Hall A.P., Liu S.Y. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(1):244–249. doi: 10.1073/pnas.1419038112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang Y., Liang S., Zhang Y., Deng Y., He Y., Chen Y. Oral administration of compound probiotics ameliorates HFD-induced gut microbe dysbiosis and chronic metabolic inflammation via the G protein-coupled receptor 43 in non-alcoholic fatty liver disease rats. Probiotics and Antimicrobial Proteins. 2019;11(1):175–185. doi: 10.1007/s12602-017-9378-3. [DOI] [PubMed] [Google Scholar]
- 73.Duarte S.M.B., Stefano J.T., Miele L., Ponziani F.R., Souza-Basqueira M., Okada L. Gut microbiome composition in lean patients with NASH is associated with liver damage independent of caloric intake: a prospective pilot study. Nutrition, Metabolism, and Cardiovascular Diseases. 2018;28(4):369–384. doi: 10.1016/j.numecd.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 74.Younes R., Bugianesi E. NASH in lean individuals. Seminars in Liver Disease. 2019;39(1):86–95. doi: 10.1055/s-0038-1677517. [DOI] [PubMed] [Google Scholar]
- 75.Hosseini E., Grootaert C., Verstraete W., Van de Wiele T. Propionate as a health-promoting microbial metabolite in the human gut. Nutrition Reviews. 2011;69(5):245–258. doi: 10.1111/j.1753-4887.2011.00388.x. [DOI] [PubMed] [Google Scholar]
- 76.Puri P., Daita K., Joyce A., Mirshahi F., Santhekadur P.K., Cazanave S. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology. 2018;67(2):534–548. doi: 10.1002/hep.29359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sayin S.I., Wahlstrom A., Felin J., Jantti S., Marschall H.U., Bamberg K. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metabolism. 2013;17(2):225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 78.Miele L., Valenza V., La Torre G., Montalto M., Cammarota G., Ricci R. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49(6):1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 79.Medgyesi D., Hobeika E., Biesen R., Kollert F., Taddeo A., Voll R.E. The protein tyrosine phosphatase PTP1B is a negative regulator of CD40 and BAFF-R signaling and controls B cell autoimmunity. Journal of Experimental Medicine. 2014;211(3):427–440. doi: 10.1084/jem.20131196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farrell G.C., van Rooyen D., Gan L., Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut and Liver. 2012;6(2):149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moran G.W., O'Neill C., McLaughlin J.T. GLP-2 enhances barrier formation and attenuates TNFalpha-induced changes in a Caco-2 cell model of the intestinal barrier. Regulatory Peptides. 2012;178(1–3):95–101. doi: 10.1016/j.regpep.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 82.Keller J., Binnewies U., Rosch M., Juul Holst J., Beglinger C., Andresen V. Gastric emptying and disease activity in inflammatory bowel disease. European Journal of Clinical Investigation. 2015;45(12):1234–1242. doi: 10.1111/eci.12542. [DOI] [PubMed] [Google Scholar]
- 83.Bernsmeier C., Meyer-Gerspach A.C., Blaser L.S., Jeker L., Steinert R.E., Heim M.H. Glucose-induced glucagon-like Peptide 1 secretion is deficient in patients with non-alcoholic fatty liver disease. PloS One. 2014;9(1) doi: 10.1371/journal.pone.0087488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klaman L.D., Boss O., Peroni O.D., Kim J.K., Martino J.L., Zabolotny J.M. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Molecular and Cellular Biology. 2000;20(15):5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park M.K. Glucagon-like peptide-2. In: Takei Y., Ando H., Tsutsui K., editors. Handbook of hormones. Academic Press; San Diego: 2016. Sub[chapter 17]D. p. 138-e17D-5. [Google Scholar]
- 86.Kushner J.A., Haj F.G., Klaman L.D., Dow M.A., Kahn B.B., Neel B.G. Islet-sparing effects of protein tyrosine phosphatase-1b deficiency delays onset of diabetes in IRS2 knockout mice. Diabetes. 2004;53(1):61–66. doi: 10.2337/diabetes.53.1.61. [DOI] [PubMed] [Google Scholar]
- 87.Charlton M., Ahlman B., Nair K.S. The effect of insulin on human small intestinal mucosal protein synthesis. Gastroenterology. 2000;118(2):299–306. doi: 10.1016/s0016-5085(00)70212-5. [DOI] [PubMed] [Google Scholar]