

Abstract
Despite the recent advances in mass spectrometry (MS)-based methods for glycan structural analysis, characterization of glycomes remains a significant analytical challenge, in part due to the widespread presence of isomeric structures and the need to define the many structural variables for each glycan. Interpretation of the complex tandem mass spectra of glycans is often laborious and requires substantial expertise. Broad adoption of MS methods for glycomics, within and outside the glycoscience community, has been hindered by the shortage of bioinformatics tools for rapid and accurate glycan sequencing. Here, we developed an online porous graphitic carbon liquid chromatography (PGC-LC)-electronic excitation dissociation (EED) MS/MS method that takes advantage of the superior isomer resolving power of PGC and the structural details provided by EED MS/MS for characterization of glycan mixtures. We also made improvements to GlycoDeNovo, our de novo glycan sequencing algorithm, so that it can automatically and accurately identify glycan topologies from EED tandem mass spectra acquired online. The majority of linkages can also be determined de novo, although in some cases, biological insight may be needed to fully define the glycan structure. Application of this method to the analysis of N-glycans released from ribonuclease B not only revealed the presence of 18 high-mannose structures, including new isomers not previously reported, but also provided relative quantification for each isomeric structure. With fully automated data acquisition and topology analysis, the approach presented here holds great potential for automated and comprehensive glycan characterization.
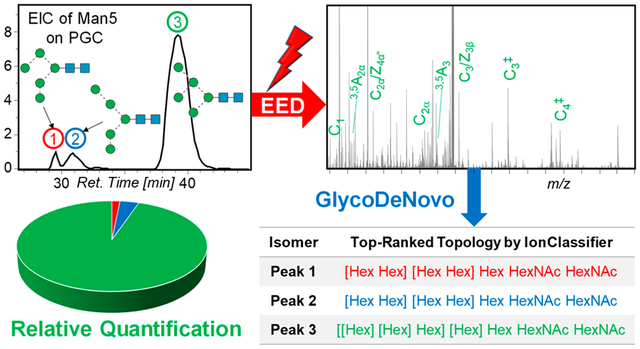
Graphical Abstract
Glycans are the most abundant biopolymers found in nature; they play crucial roles in a wide range of biological activities. Glycan biosynthesis does not follow genetic blueprints, and the resultant glycome is often a mixture of glycoforms with varying compositions, branching patterns, linkages, and stereochemical configurations. Subtle changes in the structure of a glycan can have a profound impact on its biological functions. For example, altered distribution between the α-2,3 and α-2,6 sialic acid linkages is associated with cancer, chronic inflammation, and infectious diseases.1,2 Changes in glycan expression are observed in many pathological processes, including tumorigenesis, viral infection, and autoimmune disorders.3–6 Gaining a better understanding of the disease mechanisms thus requires the development of analytical methods that can characterize and quantify all glycoforms present in a complex mixture.
Characterization of glycan structures remains a significant analytical challenge due to the need to define many structural variables, the often limited sample quantity available from biological sources, and the ubiquitous presence of structural isomers. While it is possible to detect each glycoform in a complex mixture by combining liquid chromatographic (LC) or capillary electrophoretic (CE) separation with UV or fluorescence detection of derivatized glycans, these optical detection methods do not provide structural information. Thus, each glycan structure must be determined by comparing its retention or electrophoretic migration time to those of the glycan standards, usually followed by examining the retention time or relative migration shift of the product(s) from its sequential exoglycosidase digestion.7 These approaches are limited by the availability of specific enzymes and the content of the LC and CE databases of glycan standards.
Mass spectrometry (MS) has become the method of choice for glycan structural characterization, as it offers high sensitivity and fast analysis, provides structural details through tandem MS (MS/MS), enables quantification with stable isotope labeling, and is compatible with online glycan separation.8–11 Permethylation is often performed before MS analysis to improve the ionization efficiency, stabilize the sialic acid linkages, prevent structural rearrangement, and facilitate spectral interpretation.12–15 Collision-induced dissociation (CID) is the most commonly used fragmentation method due to its broad availability on many commercial MS instruments. Because CID MS/MS generates only limited structural information, detailed structural elucidation often requires multistage tandem mass spectrometry (MSn),16 but the MSn approach lacks speed and sensitivity and is not suited for quantification. A major challenge in MSn is that the selection of product ions as precursors for further fragmentation often requires considerable expertise and time, since the most abundant fragment ion does not always produce the most structurally informative spectrum, and this makes MSn difficult to automate. Recent strategies for automating MSn analysis either rely on existing databases and are thus limited to branching pattern analysis,17 or require a significant amount of sample and multiple sample injections to fully define the structure of each glycan, even when it is as small as a trisaccharide.18,19 Finally, in MSn, specific glycoforms are identified by their observed fragmentation pathways, but it is not always possible to exhaustively investigate all relevant pathways, and some glycoforms may go undetected.
We recently showed that glycan cations can be extensively fragmented by irradiation with electrons that have energy exceeding the ionization potential of the target cations, in a process known as electronic excitation dissociation (EED).20,21 EED is capable of generating a complete series of glycosidic fragments for sequence determination and abundant cross-ring, secondary, and internal fragments for linkage elucidation.22–24 The ability of EED to produce structural details in a single stage of MS/MS analysis allows its efficient coupling to online glycan separation, including LC and ion mobility spectrometry (IMS).24,25 We showed that isomeric glycan standards in simple mixtures can be separated and identified by online reversed phase (RP)-LC-EED MS/MS analysis. However, RPLC has limited isomer resolving power, and we achieved only partial resolution for some linkage isomers.24 Porous graphitic carbon (PGC)-LC is one of the best chromatographic methods for glycan isomer separation and has been used in conjunction with CID or higher energy collisional dissociation (HCD) MS/MS for glycan mixture analysis, yet achieving definitive structural assignments still sometimes requires complementary information such as retention time shift upon exoglycosidase digestion.26–30 A recent study by Ashwood et al. showed some very promising results on differentiation of isomers in multicomponent glycan mixtures by online PGC-LC-negative CID (nCID) MS/MS, where nCID produced diagnostic fragments with different propensities for isomer discrimination.31 Differentiation was achieved at the substructure level, but complete structural determination of each isomer remained challenging, in part because of the insufficient structural details produced by nCID. The authors also acknowledged that small changes in the glycan structure, such as core fucosylation, could render some diagnostic fragments nonspecific and suggested that multiple diagnostic ions be used for better differentiation accuracy and for resolving isomers differing by more than one structural features.
Facile glycan analysis will demand automation in not only spectral acquisition but also spectral interpretation. Current bioinformatics tools for MS/MS-based glycan sequencing often achieve glycan identification by searching their tandem mass spectra against existing glycan databases.32–39 These approaches are limited to assignments of previously defined structures. Some software programs build their own theoretical glycan databases with predefined rules for glycan simulation,40,41 but these databases are incomplete, as restrictions must be placed to keep the database at a manageable size, and unusual structures not assembled via known biosynthetic pathways do exist in nature.42,43 To address this limitation, a number of de novo glycan sequencing algorithms have been developed, including STAT,44 StrOligo,45,46 GLYCH,47 Glyco-Master,48 glyfon,48 and others,49–52 but their performance is typically suboptimal for several reasons. First, they were generally applied to analyze CID tandem mass spectra, which may not contain the complete series of glycosidic cleavages needed for de novo glycan sequencing. Second, candidate structures are usually ranked by counting the number of their theoretical fragments present in the experimental tandem mass spectra, but this approach can be problematic, due to the common occurrences of isomeric structures and isobaric fragments that may yield ambiguous assignments, especially for low resolution/low-mass accuracy spectral data. Finally, CID MS/MS rarely produces a sufficient number of cross-ring or other linkage-diagnostic ions for reliable linkage determination.
We recently developed a novel algorithm, GlycoDeNovo, for de novo glycan sequencing from high-mass accuracy EED spectra of permethylated glycans.53 GlycoDeNovo builds an interpretation-graph from each glycan tandem mass spectrum by identifying all potential glycosidic fragments (sequence ions) that can be interpreted as a combination of a monosaccharide (root) and one or more previously interpreted peaks (branches) which lead to the assignment of the precursor peak. To reduce the peak assignment errors, we introduced an IonClassifier (IC) module that assigns a probability score to a peak assignment by considering its contextual features, such as the presence of its complementary and/or accompanying fragment ions. To establish the IonClassifier, we identified features from a set of tandem mass spectra of glycan standards by using a machine learning approach. We then ranked candidate topologies based on the cumulative IC scores of all supporting peaks. With IonClassifier, GlycoDeNovo was able to consistently identify the correct topologies as the top-ranked candidate structures for both permethylated glycans53 and native glycans with a reducing-end fixed charge label,23 based on their EED tandem mass spectra. To date, GlycoDeNovo has only been applied to analyze off-line EED spectra of glycan standards. Analysis of online EED spectra of glycans from biological sources is far more challenging, due to their likely lower spectral quality, especially for low-abundance glycoforms, potentially missed glycosidic cleavage(s), and spectral contamination from coeluted isomers. To address these challenges, we present here an integrated approach that combines high-resolution online nanoPGC-LC separation, EED MS/MS, and an improved GlycoDeNovo algorithm for rapid, de novo, comprehensive, and potentially quantitative characterization of complex mixtures of isomeric glycans.
EXPERIMENTAL SECTION
Materials.
PNGase F was acquired from New England BioLabs (Ipswich, MA). Bovine ribonuclease B (RNase B) was purchased from Abnova (Taipei, Taiwan). HPLC grade water, acetonitrile, chloroform, and formic acid were obtained from Fisher Scientific (Pittsburgh, PA). Methyl iodide, dimethyl sulfoxide (DMSO), sodium hydroxide, sodium borodeuteride (NaBD4), and acetic acid were purchased from Sigma-Aldrich (St. Louis, MO).
Sample Preparation.
N-Linked glycans were released by PNGase F based on the protocol by New England Biolabs. Briefly, 20 μg of RNase B were dissolved in 10 μL of glycoprotein denaturing buffer (0.5% SDS, 40 mM DTT) and incubated at 60 °C for 45 min, followed by addition of 2 μL of NP-40 (10×), 2 μL of sodium phosphate buffer (50 mM), 6 μL of water, and 1 μL of PNGase F solution. The reaction mixture was incubated at 37 °C overnight. The released glycans were separated from proteins by passage of the product mixture through a C18 Sep-Pak cartridge (1 mL, Waters, Milford, MA). Deutero-reduction and permethylation were performed according to protocols described in detail else-where.24 Briefly, released glycans were dissolved in 200 μL of NaBD4 (250 mM) in NH4OH (100 mM) solution. After 2 h incubation at room temperature, the reaction was quenched by slowly adding 10% acetic acid until bubbling ceased. Reduced glycans were dissolved in a mixture of NaOH/DMSO/CH3I (40 mg/200 μL/50 μL); the tube containing the solution was vortexed for 3 h, with additions of 50 μL CH3I every hour. The permethylated glycans were desalted using a Pierce C18 microspin column (ThermoFisher Scientific, Waltham, MA), dried with a SpeedVac system (ThermoFisher Scientific, Waltham, MA), and stored at −80 °C.
Liquid Chromatography–Mass Spectrometry Analysis.
Online LC separation was performed on a nano-ACQUITY UPLC system (Waters, Milford, MA), equipped with a C18 trap column (5 μm Symmetry C18, 0.18 × 20 mm) and a PGC analytical nanocolumn (Hypercarb, 3 μm, 0.075 × 100 mm, Thermo Fisher Scientific). Sodium acetate (10 mM) was added to each glycan sample prior to injection to promote formation of sodium adducts.24 Mobile phase A contained 98.9% water, 1.0% acetonitrile, and 0.1% formic acid, and mobile phase B consisted of 1.0% water, 49.9% acetonitrile, 49.0% isopropanol, and 0.1% formic acid. Online desalting was done by trapping with 10% B for 2 min at a flow rate of 4 μL/min. For separation, the gradient used was 0–5 min, 10–35% B; 6–55 min, 35–60% B; 56–65 min, 60–95% B; 66–75 min, 95% B; 76–77 min, 95–10% B; and 78–90 min, 10% B. The flow rate was 0.5 μL/min, and the column temperature was kept at 60 °C for improved chromatographic resolution.54–57 About 5–10 pmol of released glycans were injected for each LC–MS/MS analysis.
The Waters nanoUPLC system was connected to a 12-T solariX hybrid Qh-Fourier-transform ion cyclotron resonance (FTICR) mass spectrometer (Bruker Daltonics, Bremen, Germany) via a Triversa nanoMate system (Advion, Ithaca, NY). The electrospray voltage was set to 1.7 kV. The external ion accumulation time was set to 0.1 and 2.5 s for MS and MS/MS scans, respectively. Both inclusion and exclusion lists were implemented for auto MS/MS, performed with one MS scan followed by two MS/MS scans. The cathode bias was set to 18 V with an electron irradiation time of 0.35 s. A 0.5 s transient was acquired for each mass spectrum.
Data Analysis.
All mass spectra were processed by DataAnalysis 4.4 (Bruker, Bremen, Germany). Fragments were annotated according to the Domon and Costello nomenclature (Scheme S1).58 For each LC–MS/MS run, a single MS/MS spectrum was chosen for internal calibration, using several fragments assigned with high confidence, typically B-, C-, Y-, Z-, or1,5 X ions. The same calibration constants were then automatically applied to all mass spectra acquired during the same LC run, resulting in a typical mass assignment accuracy of around 2 ppm. Peak picking was performed using the SNAP algorithm (Bruker Daltonics, Bremen, Germany) with the quality factor threshold set at 0.1, S/N cutoff at 5, maximum charge state at 3, and a relative ion abundance threshold at 0.01%. The precursor ion elemental composition was chosen to calculate the averagine formula.59 Automatic reconstruction of candidate topologies was achieved with GlycoDeNovo, and ranked by IonClassifier. Putative structures for all glycoforms, including their topologies and linkage configurations, were deduced through manual spectral interpretation.
RESULTS AND DISCUSSION
N-Linked, high-mannose glycans released from bovine RNase B are chosen as the model system for this study, as this set of glycoforms contains many structural isomers, including both topological and linkage isomers, and therefore serves as a rigorous test for the evaluation of the performance of the LC–MS/MS method and bioinformatics software developed here. Detailed structural characterization and relative quantification of high-mannose glycans are of significant biological relevance. For example, interaction between surfactant protein D and high-mannose structures on epidermal growth factor receptor (EGFR) leads to reduced EGF binding to EGFR and suppresses lung cancer progression.60 High-mannose glycans are also key targets in vaccine design, such as those against the human immunodeficiency virus.61–63 Analysis of hybrid and complex N-glycans from glycoproteins and biofluids will be presented in a follow-up report.
NanoPGC-LC–MS analysis of N-glycans released from RNase B.
Figure 1 shows the extracted ion chromatograms (EICs) of high-mannose glycans released from RNase B, separated on a nano-PGC column. With the lone exception of Man9, multiple peaks are observed for each glycan composition. The heterogeneity of high-mannose structures in RNase B has been the subject of several previous reports.27,56,64–67 One MSn study revealed the presence of 13 different isomers with the compositions of Man5, Man7, and Man8, although no quantitative information could be obtained, as glycoforms were identified on the basis of their unique MSn fragmentation pathways, with no prior isomer separation.64 Here, we are able to detect and characterize 18 high-mannose isomers by PGC-LC-EED MS/MS, with compositions ranging from Man4 to Man9, and putative structures shown in Figure 2. These high-mannose structures are annotated using the alphanumeric notation introduced by Reinhold et al.,64 where each of the antennal mannose residues is given a unique alphanumeric identifier. To facilitate discussion, we further assign the labels R0, R3, and R6 to the three core mannose residues, as depicted in Figure 2a. Since all high-mannose structures contain the three core mannose residues, we only need to specify antennal residues to define their structures. For example, a Man6 isomer containing residues A1, A2, and B1 would be annotated as 6A1,2B1. Note that some pauci-mannose structures may be missing the R3 or R6 residue, such as 4A3B3 and 4A1,2, but the missing core residue can be deduced from the antennal residues present in the structure.
Figure 1.
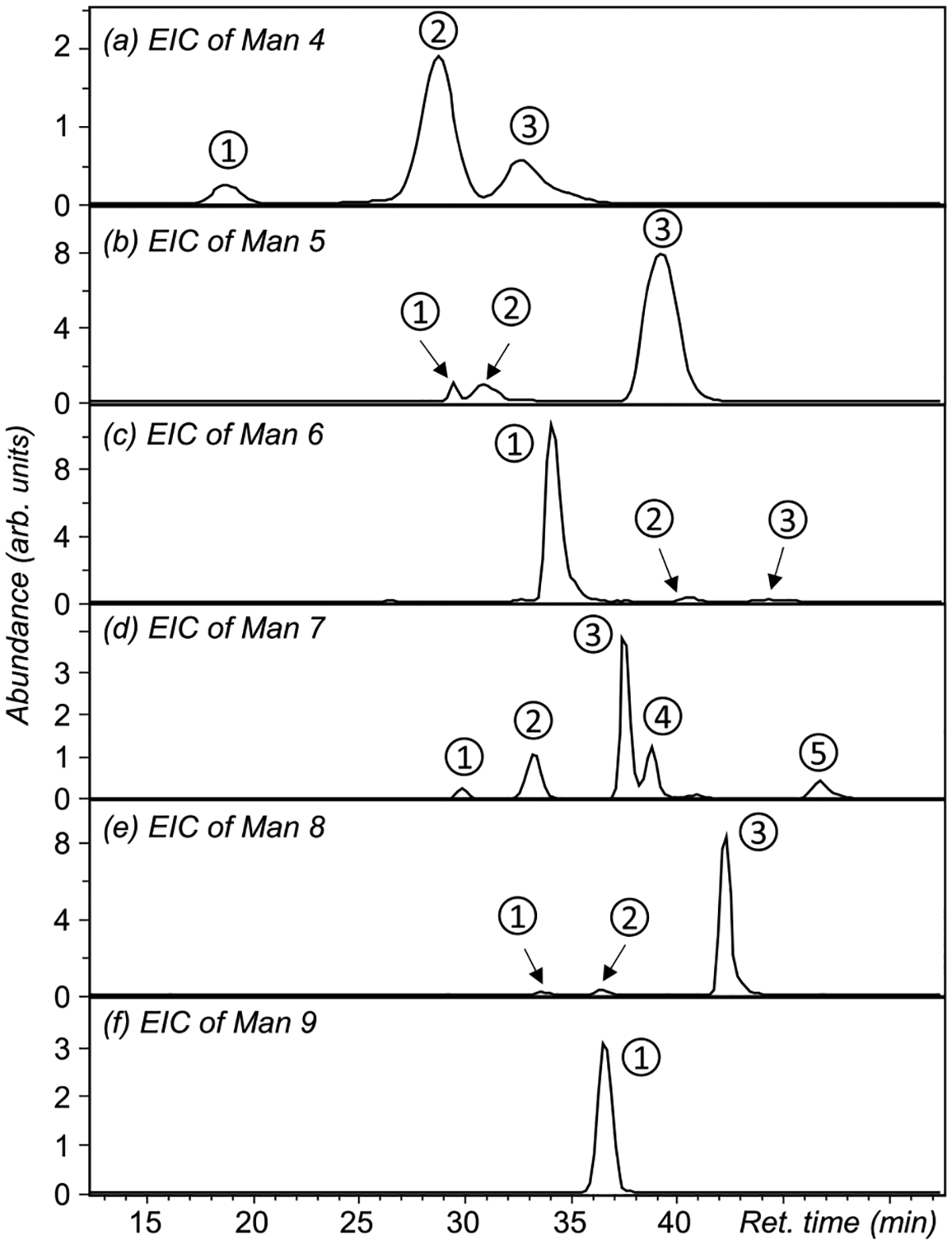
Extracted ion chromatograms (EICs) of high-mannose compounds released from RNase B and separated on a nano-PGC column.
Figure 2.
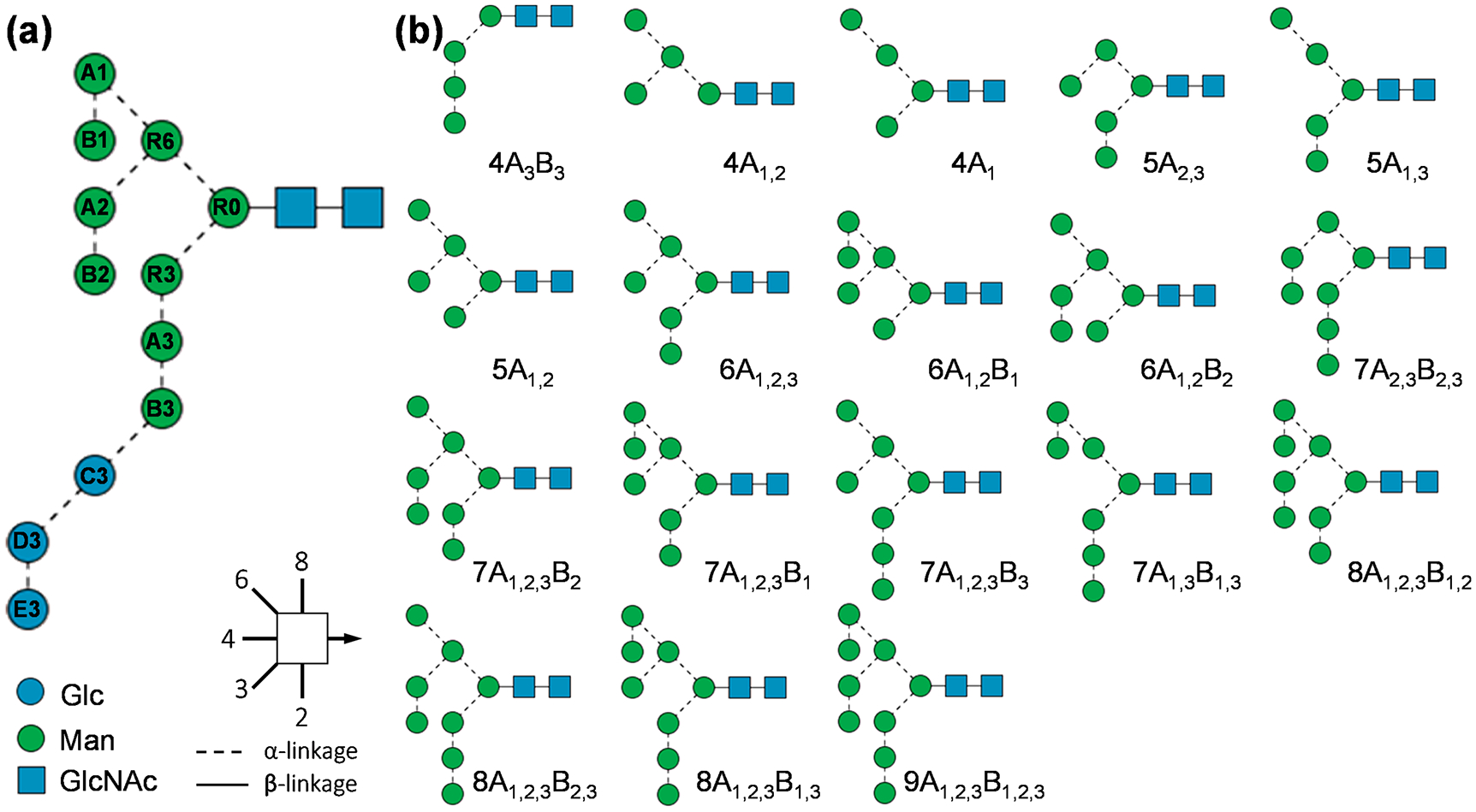
SNFG representation and alphanumeric notation of the N-glycan precursor (a) and the 18 identified high-mannose structures from RNase B (b).
Manual Determination of Putative Structures.
For manual structural determination, we assume that all high-mannose structures derive from the tetradecameric N-glycan structure, shown in Figure 2a. This structure corresponds to the glycan derived from the lipid-linked Glc3Man9GlcNAc2 that is cotranslationally transferred to the nascent protein and then trimmed by the action of glucosidases 1 and 2 that trim the α1,2-linked terminal glucose and then the internal α1,3-linked residues, respectively. With this constraint, topology deduction may be made based on the observation of 3,5A and 0,4A ions, as they can only be produced at 1 → 6-linked mannose residues, thus providing key structural information on the 6-antenna. Specifically, if two sets of 3,5A and 0,4A ions are observed, the m/z values of the lighter set are indicative of the 6-branch on the 6-antenna, whereas the mass difference between these two sets is reflective of the 3-branch on the 6-antenna. All unaccounted hexose residues must have formed a linear sequence on the 3-antenna. Similarly, if only one set of 3,5A and 0,4A ions is observed, their m/z values may be used to determine the C3 substitution on the R6 residue, and the rest of hexose residues may be assigned to the 3-antenna. If no 3,5A or 0,4A ions are observed, all hexose residues will be assigned to a linear sequence attached to the 3-antenna. Here, we focus our discussion on Man5 and Man7 isomers and leave discussions on other high-mannose structures to the Supporting Information.
Three chromatographic peaks are observed for Man5 isomers (Figure 1b), with their EED spectra shown in Figure 3, and assigned fragments listed in Tables S2–S4. Among them, isomer 3 is the most abundant, accounting for nearly 95% of the Man5 population. The presence of 0,4A2α, 3,5A2α, 0,4A3, and 3,5A3 ions establishes a branched trimannose 6-antenna and a monomannose 3-antenna. Thus, this major Man5 isomer may be assigned as 5A1,2, consistent with the canonical Man5 structure reported in the literature.68 For the two lower-abundance Man5 isomers, isomers 1 and 2 in Figure 1b, EED produced 0,4A3 and 3,5A3 ions at m/z 505.226 and 533.257, suggesting a branched topology with two dimannose antennae. The Man-Man linkage within the 6-antenna may be determined as 1 → 6 for isomer 2, based on the presence of 0,4A2α and 3,5A2α ions at m/z 301.126 and 329.157, and as 1 → 3 for isomer 1, whose EED spectrum does not include these two cross-ring fragments. Thus, isomers 1 and 2 can be assigned as 5A2,3 and 5A1,3, respectively.
Figure 3.
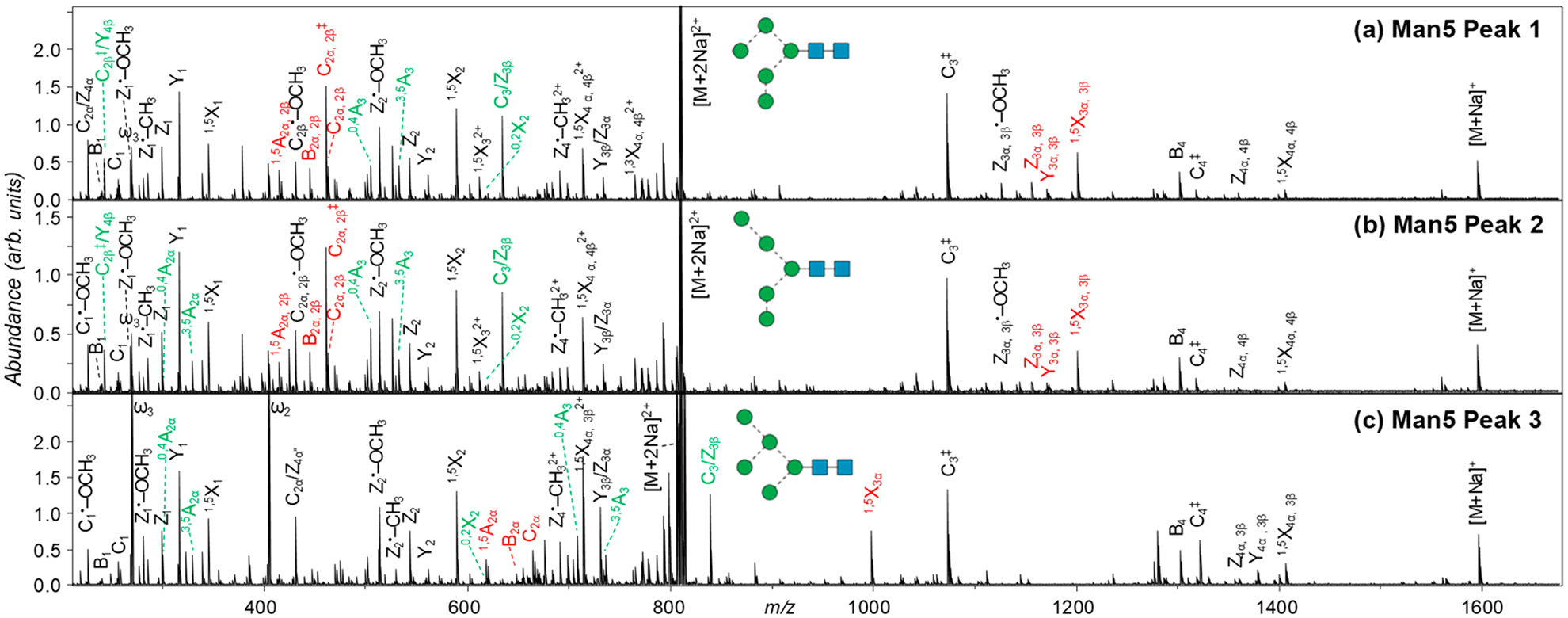
EED spectra of the three Man5 isomers, acquired at retention times marked in Figure 1b. Glycosidic and 1,5-cross ring fragments unique to each topology are labeled in red; linkage-diagnostic cross-ring and secondary fragments are labeled in green.
Five chromatographic peaks are observed for Man7 isomers (Figure 1d), with their EED spectra shown in Figure 4, and assigned fragments in Tables S5–S9. For isomers 2 and 3, the presence of 0,4A4 and 3,5A4 ions at m/z 913.425 and 941.456 suggests a tetra-mannose 6-antenna and a dimannose 3-antenna. For isomer 2, the observation of 0,4A2α and 3,5A2α ions at m/z 301.126 and 329.157 suggests a single mannose 6-branch on the 6-antenna, whereas for isomer 3, the observation of 0,4A3α and 3,5A3α ions at m/z 505.226 and 533.257 suggests a dimannose 6-branch on the 6-antenna. Thus, isomers 2 and 3 of Man7 can be assigned as 7A1,2,3B2 and 7A1,2,3B1, respectively. For isomers 1, 4, and 5, the observation of 0,4A and 3,5A ions at m/z 709.325 and 737.357 suggests the presence of a trimannose 6-antenna and a trimannose 3-antenna. For isomer 5, the observation of 0,4A3α and 3,5A3α ions at m/z 505.226 and 533.257 suggests a dimannose motif attached to the C6-position of the R6 residue, forming a linear trimannose 6-antenna; for isomer 4, the observation of 0,4A2α and 3,5A2α ions at m/z 301.126 and 329.157 suggests a branched trimannose structure as its 6-antenna; for isomer 1, the absence of lighter 0,4A or 3,5A ions suggests a dimannose 3-branch on the 6-antenna. Taken altogether, isomers 1, 4, and 5 may be assigned as 7A2,3B2,3, 7A1,2,3B3, and 7A1,3B1,3, respectively.
Figure 4.
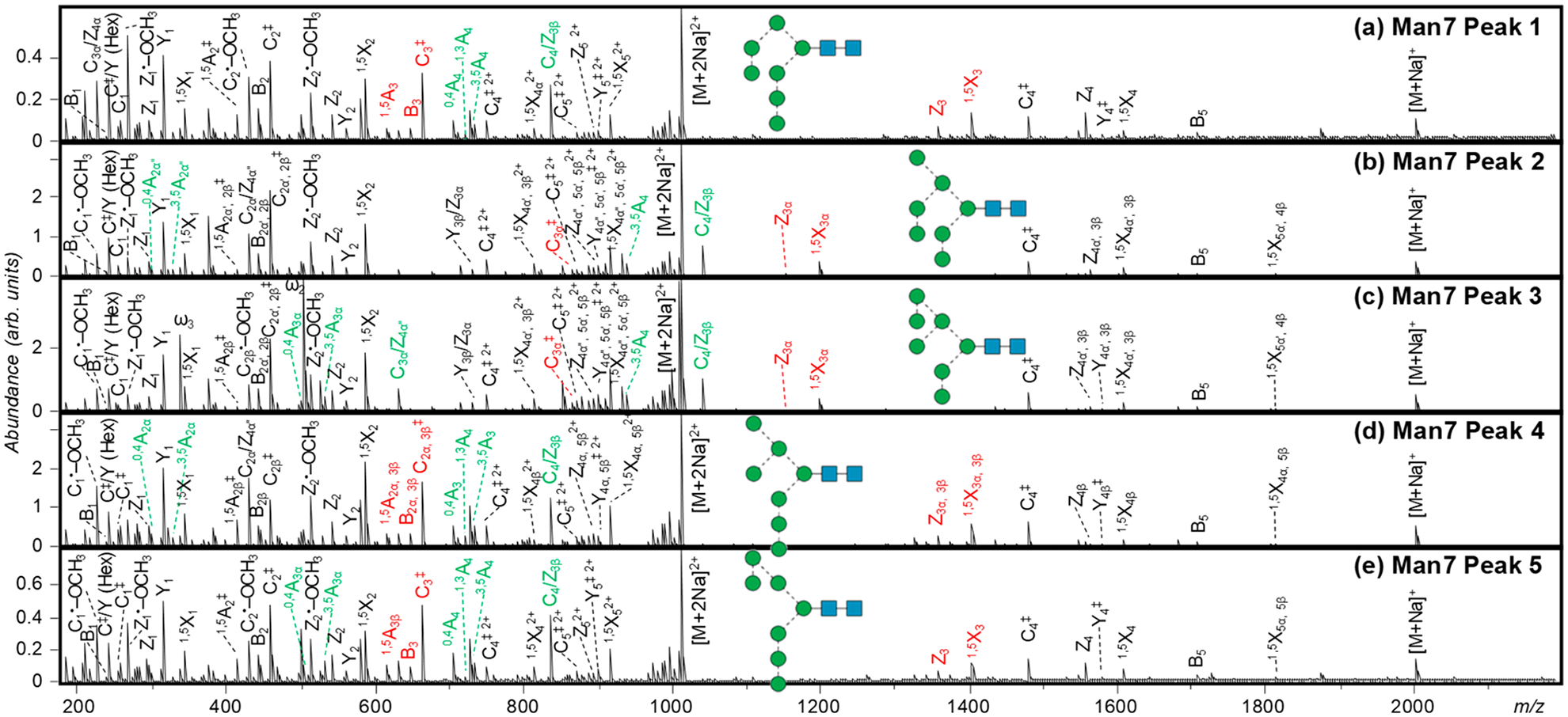
EED spectra of the five Man7 isomers, acquired at retention times marked in Figure 1d. Glycosidic and 1,5-cross ring fragments unique to each topology are labeled in red; linkage-diagnostic cross-ring and secondary fragments are labeled in green.
Not surprisingly, the Man7 canonical structures, 7A1,2,3B1 (isomer 3), 7A1,2,3B2 (isomer 2), and 7A1,2,3B3 (isomer 4), are the most abundant Man7 isomers observed in this study. An earlier MSn study identified two additional Man7 isomers in released glycans from RNase B: 7A1,3B1,3 and 7A1,3B3C3.64 Our current study confirmed the presence of 7A1,3B1,3 (isomer 5) but did not find evidence for the presence of 7A1,3B3C3. Instead, we detected the isomer 7A2,3B2,3 (isomer 1), as described above. A careful examination of the MSn results in the previous report revealed the possible reason for the discrepancy between the results of the earlier study and those we report here. The following MSn pathways were used to confirm the presence of 7A1,3B1,3: precursor → B5 → B5/Y5β → B5/Y4β → B5/Y3β → 3,5A4 → C3α → B2α + C2α + C/Y (Hex), but these pathways are common to both 7A1,3B1,3 and 7A2,3B2,3. The 6-antenna trisaccharide was assigned as Man(1–2)Man(1–6)Man, based on the dissimilarity between its fragmentation pattern and that produced by Man(1–2)Man(1–3)Man, the C3α ion from the 5A2B2 structure (Figures 6e and 4c, ref 47). However, this assignment seems questionable, since the MS7 fragment at m/z 667.3 from Man7 was at least a mixture of C3α from 7A1,3B1,3, or Man(1–2)Man(1–6)Man, and C2α from 7A1,2,3B3, or [Man(1–3)][Man(1–6)]Man, whose fragmentation pattern would have been different from that of Man(1–2)Man(1–3)Man from 5A2B2, regardless of whether the latter species, likely originating from 7A2,3B2,3, was also present in the mixture. More importantly, the tandem mass spectrum of the 6-antenna trisaccharide C-ion from Man7 contained a prominent peak at m/z 563.4 that could not have been produced by either 7A1,2,3B3 or 7A1,3B1,3 and should therefore be assigned as a 1,4A3α ion, likely derived from 7A2,3B2,3. This example underlines a major limitation of the MSn approach, in that coisolation of isomeric or isobaric structures can sometimes lead to erroneous interpretation, especially when chromatographic separation is omitted. In contrast, our PGC-LC–MS analysis clearly identified the presence of five Man7 isomers, whose structures can be assigned with high confidence based on structurally informative fragments generated by EED MS/MS.
Automatic Topology Deduction by GlycoDeNovo.
The glycan EED spectra are generally very complex due to the occurrence of many fragmentation pathways. Without any structural constraint, a given fragment may be assigned to multiple structures, making manual spectral interpretation an extremely challenging task. In the previous section, putative structures of high-mannose isomers were deduced from their EED tandem mass spectra based on the structure of the N-glycan precursor. This knowledge significantly reduced the number of topologies needing to be considered and greatly simplified spectral interpretation by allowing one to specifically look for just a handful of diagnostic cross-ring fragments for structural assignment. However, current knowledge of glycan biosynthesis is incomplete, and discovery of new glycan structures requires a de novo approach. In this section, we will demonstrate de novo glycan topology reconstruction with minimal reference to the biological knowledge. Instead, topologies are reconstructed based on observed glycosidic fragments and ranked by the cumulative IonClassifier scores of all supporting fragments.
For automatic topology deduction, the SNAP-identified lists of fragments with their m/z, charge states, and relative ion abundances subjected to GlycoDeNovo analysis, and the results are shown in Table 1. GlycoDeNovo was able to reconstruct topologies in 13 out of 18 cases. The five spectra from which GlycoDeNovo failed to generate topology candidates were all recorded for low abundance glycoforms and hence have lower spectral quality, i.e., they do not contain complete series of glycosidic cleavages. For example, isomer 3 of Man6 accounts for less than 4% of the Man6 population, and no glycosidic fragment resulting from cleavage between the R0 and R6 residues was automatically identified by SNAP. Manual inspection did reveal the presence of two peaks at m/z 869.398 and 871.412, corresponding to C3α‡(‡ indicates 2 hydrogen losses) and C3α ions, respectively (Figure S2, inset), but the A+1 isotope peaks of these two ions were too low in abundance to be accepted for assignment by SNAP.
Table 1.
GlycoDeNovo Analysis of EED Spectra of 18 High-Mannose Isomersa
| glycans | number of isomers | Peak no. | number of supporting fragments | rank by the IonClassifier | reconstructed topology |
|---|---|---|---|---|---|
| Man 4 | 3 | peak 1* | 11 | 1 | Hex Hex Hex Hex HexNAc HexNAc |
| peak 2 | 9 | 1 | [Hex] [Hex] Hex Hex HexNAc HexNAc | ||
| peak 3 | 9 | 1 | [Hex Hex] [Hex] Hex HexNAc HexNAc | ||
| Man 5 | 3 | peak 1 | 9 | 1 | [Hex Hex] [Hex Hex] Hex HexNAc HexNAc |
| peak 2 | 9 | 1 | [Hex Hex] [Hex Hex] Hex HexNAc HexNAc | ||
| peak 3 | 10 | 1 | [[Hex] [Hex] Hex] [Hex] Hex HexNAc HexNAc | ||
| Man 6 | 3 | peak 1 | 12 | 1(2) | [Hex Hex Hex] [Hex Hex] Hex HexNAc HexNAc |
| [[Hex] [Hex] Hex] [Hex Hex] Hex HexNAc HexNAc | |||||
| peak 2* | 9 | 1 | [[Hex Hex] [Hex] Hex] [Hex] Hex HexNAc HexNAc | ||
| peak 3* | 10 | 1 | [[Hex Hex] [Hex] Hex] [Hex] Hex HexNAc HexNAc | ||
| Man 7 | 5 | peak 1 | 11 | 1(2) | [Hex Hex Hex] [Hex Hex Hex] Hex HexNAc HexNAc |
| [[Hex] [Hex] Hex] [Hex Hex Hex] Hex HexNAc HexNAc | |||||
| peak 2 | 11 | 1 | [[Hex Hex] [Hex] Hex] [Hex Hex] Hex HexNAc HexNAc | ||
| peak 3 | 11 | 1 | [[Hex Hex] [Hex] Hex] [Hex Hex] Hex HexNAc HexNAc | ||
| peak 4 | 13 | 1(2) | [Hex Hex Hex] [Hex Hex Hex] Hex HexNAc HexNAc | ||
| [[Hex] [Hex] Hex] [Hex Hex Hex] Hex HexNAc HexNAc | |||||
| peak 5 | 12 | 1(2) | [Hex Hex Hex] [Hex Hex Hex] Hex HexNAc HexNAc | ||
| [[Hex] [Hex] Hex] [Hex Hex Hex] Hex HexNAc HexNAc | |||||
| Man 8 | 3 | peak 1* | 8 | 1 | [[Hex Hex] [Hex Hex] Hex] [Hex Hex] Hex HexNAc HexNAc |
| peak 2* | 12 | 1(5) | [Hex Hex Hex Hex] [Hex Hex Hex] Hex HexNAc HexNAc | ||
| [[Hex Hex] [Hex] Hex] [Hex Hex Hex] Hex HexNAc HexNAc | |||||
| [[Hex] [Hex] Hex Hex] [Hex Hex Hex] Hex HexNAc HexNAc | |||||
| [[Hex] [Hex] Hex] [Hex Hex Hex Hex] Hex HexNAc HexNAc | |||||
| [[Hex] [Hex] Hex] [[Hex Hex] [Hex] Hex] Hex HexNAc HexNAc | |||||
| peak 3 | 12 | 1(5) | [Hex Hex Hex Hex] [Hex Hex Hex] Hex HexNAc HexNAc | ||
| [[Hex Hex] [Hex] Hex] [Hex Hex Hex] Hex HexNAc HexNAc | |||||
| [[Hex] [Hex] Hex Hex] [Hex Hex Hex] Hex HexNAc HexNAc | |||||
| [[Hex] [Hex] Hex] [Hex Hex Hex Hex] Hex HexNAc HexNAc | |||||
| [[Hex] [Hex] Hex] [[Hex Hex] [Hex] Hex] Hex HexNAc HexNAc | |||||
| Man 9 | 1 | peak 1 | 12 | 1(5) | [[Hex Hex Hex] [Hex] Hex] [Hex Hex Hex] Hex HexNAc HexNAc |
| [[Hex Hex] [Hex Hex] Hex] [Hex Hex Hex] Hex HexNAc HexNAc | |||||
| [[Hex] [Hex] Hex] [[Hex Hex Hex] [Hex] Hex] Hex HexNAc HexNAc | |||||
| [[Hex] [Hex] Hex] [[Hex Hex] [Hex Hex] Hex] Hex HexNAc HexNAc | |||||
| [[[Hex] [Hex] Hex] [Hex] Hex] [Hex Hex Hex] Hex HexNAc HexNAc |
Putative topologies are listed in bold. Asterisks denote peaks where topologies can only be reconstructed by invoking the gap option (see text). Reconstructed topologies are listed using a modified condensed linear IUPAC nomenclature, with branching indicated by square brackets.
It is possible to reconstruct topologies from an MS/MS spectrum with no more than one consecutively missed cleavage by introducing a gap option. The original gap option allows artificial addition of a glycosidic fragment to the interpretation-graph as a linear elongation of a previously identified substructure by one monosaccharide residue. However, turning on the linear gap option did not result in the recovery of the putative Man6 #3 topology here, since the absence of a C3α ion left a four-saccharide gap between C2α′ and C4 ions. It is thus necessary to implement a branched gap option, allowing insertion of a branched substructure that consists of two or more previously identified substructures connected to one monosaccharide residue, to complete the pathway leading to the interpretation of the precursor. Turning on the gap option inevitably results in a significant increase in the number of reconstructed candidate topologies. Four topologies, [Hex Hex Hex] [Hex Hex] Hex HexNAc HexNAc, [[Hex] [Hex] Hex] [Hex Hex] Hex HexNAc HexNAc, [[Hex Hex] [Hex] Hex] [Hex] Hex HexNAc HexNAc, and [Hex Hex] [Hex Hex] Hex Hex HexNAc HexNAc, could be reconstructed based on the Man6 isomer 3 spectrum with the gap option on. These topologies share the same four B/C-type substructures found in the EED spectrum, Hex, Hex2, Hex6, and Hex6HexNAc, and thus cannot be differentiated by the number of supporting peaks. Since each topology was deduced with one artificial supporting peak, it should be possible to rank them based on the IC score of the added peak. The assumption is that when a glycosidic fragment is not identified by the peak picking algorithm due to its lower signal-to-noise ratio or interference from nearby isotopic clusters, some of its contextual features may still exist in the spectrum. In the case of Man6 isomer 3, the [[Hex Hex] [Hex] Hex] [Hex] Hex HexNAc HexNAc topology receives the highest IC score because the presence of 1,5X3α and 3,5A4 ions supports the addition of a Hex4 fragment, whereas contextual features supporting the addition of Hex3 or Hex5 fragment that might have enabled the reconstruction of the other three topologies are not present in the EED spectrum. With the gap option turned on, GlycoDeNovo was able to reconstruct topologies from the remaining five tandem mass spectra (marked with an asterisk in Table 1). More importantly, manually deduced putative structures, listed in bold font in Table 1, were ranked by the IonClassifier as the top candidates in all 18 cases. This marks the first demonstration that GlycoDeNovo, with improved gap option, can correctly determine glycan topologies from tandem mass spectra with missing glycosidic cleavage(s). This capability is especially important for characterization of low-abundance glycoforms when relatively sparse data may be acquired during online LC–MS/MS.
In some cases, more than one of the candidate topologies received the top IC score because their supporting peaks are identical. Current biological knowledge enables the analyst to recognize easily the correct topology from among the candidates. For example, five different topologies received the same IC score based on the EED spectra of Man9 (Figure S5b), but only structure 2 may be derived from the tetradecameric N-glycan precursor. Thus, we showed that correct topologies of all isomeric structures present in a complex glycan mixture can be deduced automatically by GlycoDeNovo from LC-EED MS/MS data.
De Novo Linkage Analysis.
For high-mannose glycans, once the topology is identified, the majority of linkages are also defined based on the accepted structure of the N-glycan precursor. Occasionally, more than one linkage configuration may exist for a given topology, but these can generally be differentiated based on a few diagnostic cross-ring fragments, as shown earlier in the case of isomers 1, 4, and 5 of Man7 glycans. Without taking into account established biological knowledge, linkage analysis would be very difficult. We showed earlier that structural constraints may also be generated by GlycoDeNovo with the IonClassifier. Identification of the true topology among a small number of top-ranked structures is an important first step that facilitates subsequent linkage analysis. While it remains possible that extraordinary structures may sometimes be encountered, especially during the study of unique branches of the evolutionary tree, ranking of candidates based on the established structures will usually be the reasonable course of action.
For linkage elucidation, we use a combination of linkage-diagnostic cross-ring and secondary fragments, as listed in Table 2. Glycosidic linkages are usually determined based on the observation of characteristic cross-ring fragments. However, some linkages cannot be defined with certainty, even if a complete set of cross-ring fragments is observed. This is especially true for a hexose residue in its pyranose form, which has an identical group (OH for native or OCH3 for permethylated glycans) attached to its C2, C3, and C4 positions. Consequently, some cross-ring fragments, e.g., 1,3A for 1 → 2- or 1 → 3-linked and 2,4A ions for 1 → 3- or 1 → 4-linked hexose residues, are isomeric. It is thus difficult to unambiguously assign 1 → 2 and 1 → 3 linkages at a hexose residue, since a 1 → 2- or 1 → 3-linked hexose residue does not produce unique cross-ring fragments that cannot originate from a 1 → 4-linked hexose residue. Fortunately, EED also generates abundant secondary fragments, some of which can facilitate linkage determination. For example, C•-OCH3, C‡/Y, and C/Z ions are formed via alpha cleavages from C1–C2 diradical intermediates (Scheme S2), thus their presence may be used to deduce the composition of substituents at the C2 and C3 positions.
Table 2.
Characteristic Cross-Ring and Secondary Fragments Used for Determination of the Linkage Position at a Hexose Residuea
| 0,2X | 0,4A | 3,5A | 1,3A, 2,4A | C•-OCH3 | C/Z | C‡/Y | |
|---|---|---|---|---|---|---|---|
| 1→2 | − | − | − | + | + | − | + |
| 1→3 | + | − | − | + | − | + | − |
| 1→4 | + | − | + | + | + | − | − |
| 1→6 | + | + | + | − | + | − | − |
Observed fragments are marked with “+”, and missing fragments are indicated by “−”.
The topology of Man5 isomer 3 was identified by GlycoDeNovo as [[Hex] [Hex] Hex] [Hex] Hex HexNAc HexNAc, based on observed C ions with compositions of Hex, Hex3, and Hex5. The presence of 0,4A3 and 3,5A3 ions establishes a 1 → 6-linked trimannose antenna and places the monomannose branch, or the β-branch, to either the C2 or C3 position of the R0 residue. The presence of 0,2X2 and C3/Z3β ions and the absence of a C‡/Y (Hex) fragment then specify that the β-branch is at the C3-position. The two terminal mannose residues on the 6-antenna can be localized to the C3- and C6-positions, respectively, based on the presence of 0,4A2α and 3,5A2α ions and the absence of a C‡/Y (Hex) fragment. Thus, the linkage configuration of this Man5 isomer can be assigned from first-principles, without reference to biological knowledge.
For the two low-abundance Man5 isomers, the presence of C ion series with compositions of Hex, Hex2, and Hex5 is supportive of a branched topology with two dimannose antennae, as determined by GlycoDeNovo. Both isomers produced 0,4A3 and 3,5A3 ions with two mannose residues attached, suggesting that one dimannose branch is connected to the C6 position of the R0 residue. The location of the second dimannose antenna can be assigned at the C3 position, based on the presence of 0,2X2 and C3/Z3β ions. For both isomers, the presence of C2β/Y4β and C2•-OCH3 ions indicates a 1 → 2-linkage between the two mannose residues on one antenna, but the MS2 data are insufficient to determine whether this branch is located at the C3 or C6 position of the core mannose. Similarly, the Man-Man linkage on the other antenna may be deduced as 1 → 6 for isomer 2 and 1 → 3 for isomer 1, based on the presence of 0,4A2α and absence of 3,5A2α ions, but its location on the core mannose residue cannot be defined. This is an inherent limitation for MS2-based linkage analysis: when there exist multiple branches with the same sequence but different linkage configurations, the exact location of each branch cannot be determined by MS/MS alone. One potential solution is to perform a targeted MSn analysis. In this case, MS3 of the 3,5A3 ion at m/z 533.257 may be sufficient to define the linkage on the 6-antenna. Such a MS2-guided MSn approach is advantageous over the conventional MSn method, as the latter is difficult to automate and requires investigation of many MSn pathways, oftentimes involving a high number of MS/MS stages. With the majority of the features of each glycan structure already elucidated by EED MS/MS, the remaining structural parameters may be determined by even a single MS3 experiment, thus paving the way for automated analysis on the chromatographic time scale.
Relative Quantification of Isomeric Structures.
Assuming similar ionization efficiencies for isomeric glycans, insignificant matrix effects (e.g., minimal ionization suppression by coeluted species), and no column saturation or collision cell overfill, the chromatographic peak areas of the precursor ions in their respective EICs during the MS scan may be used for relative quantification. Figure 5 shows the averaged percentage distribution of each isomer from three technical replicates of LC–MS analysis. For clarity, error bars are only shown for each composition. The LC–MS data used to generate this plot may be found in Table S1.
Figure 5.
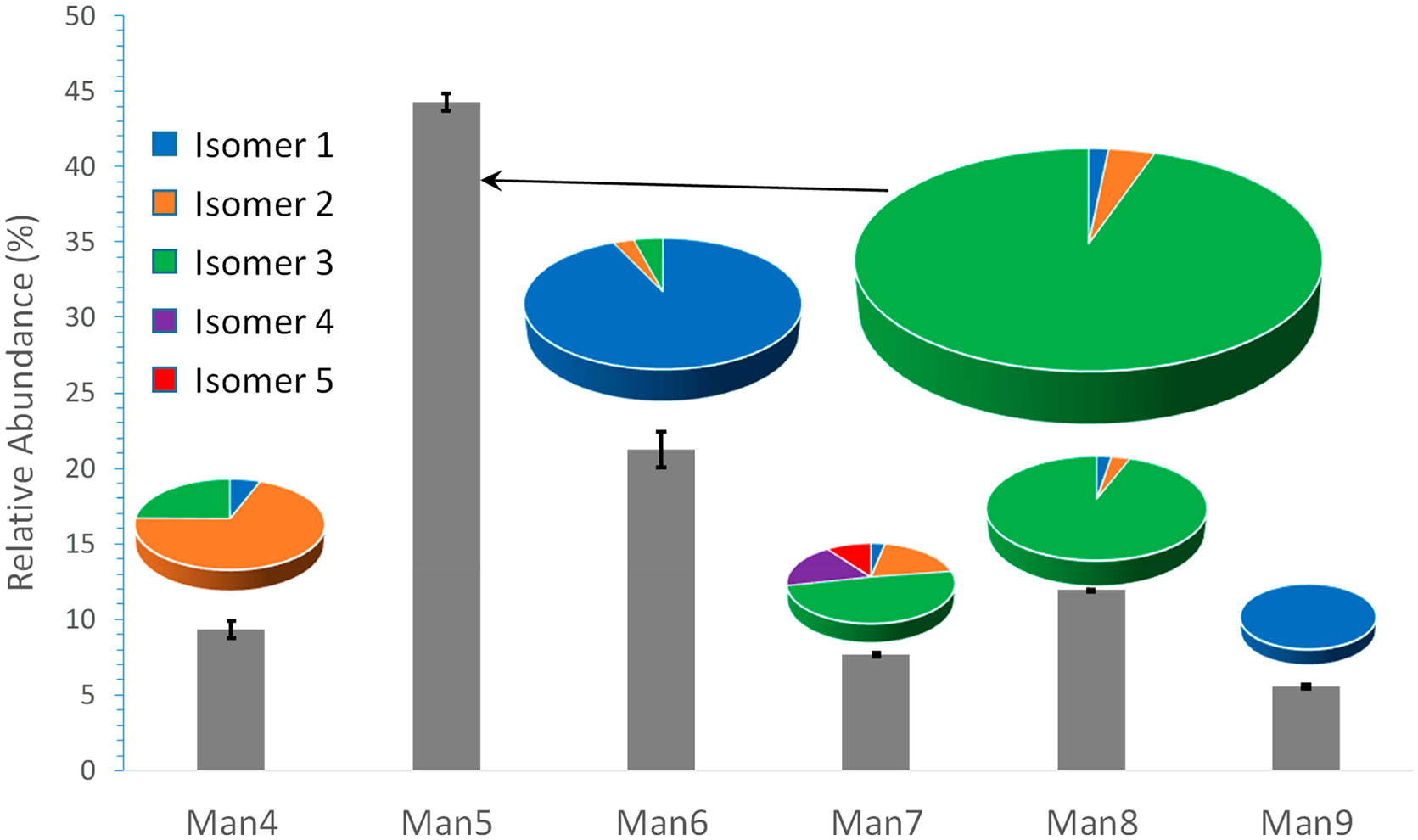
Percentage abundances of 18 high-mannose structures released from RNase B, measured by PGC-LC–MS. The size of the pie chart is reflective of the relative abundance of each composition.
Quantification of N-glycans from RNase B was the subject of an interlaboratory study,69 where glycan profiling was achieved using either normal-phase HPLC or MALDI-TOF MS, but only at the compositional level, as neither method was capable of resolving isomeric structures. Our quantitative results is comparable with the findings of that study. We found that Man5 and Man6 are the most abundant components, with relative abundances of 44.3 ± 0.6% and 21.24 ± 1.2%, respectively, right in line with the reported values of 39.7 ± 10.3% and 24.7 ± 2.6%. A key advantage of our approach is that it also allows relative quantification at the structural level, achieving identification and quantification of isomeric structures in a single LC–MS/MS experiment. This capability can be beneficial for many applications, since changes in the relative quantities of isomeric glycans may impact their biological functions, including their binding affinity and immunogenicity, among others, and could serve as markers for various diseases. Additionally, the method developed here provides a powerful tool for evaluating similarity among biopharmaceutical drugs, at the structural level in glycosylation.
CONCLUSIONS
We demonstrate here an integrated analytical and bioinformatics workflow, based on PGC-LC-EED MS/MS analysis, for separation, characterization, and potential relative quantification of isomeric glycans. The superior chromatographic resolving power provided by PGC allowed separation of both topological and linkage isomers. In all, 18 high-mannose structures were detected in N-linked glycans released from RNase B, including structures that have not been previously reported. Online EED MS/MS analysis generated structural details for all isomers, even for low-abundance glycoforms that account for less than a quarter percent of the total glycan population, while consuming only picomoles of glycans per LC run. The topologies of all structures were accurately and automatically determined from their online EED tandem mass spectra, without utilizing prior biological knowledge, by an improved de novo glycan sequencing algorithm, GlycoDeNovo. Ranking by the IonClassifier significantly reduced the number of candidate topologies needing to be considered for manual spectral interpretation, allowing elucidation of linkages based on characteristic cross-ring and secondary fragments. Finally, the presented approach enabled relative quantification of isomeric structures with good reproducibility. In conclusion, combining PGC-LC-EED MS/MS with GlycoDeNovo analysis provides a facile and sensitive method for characterization of glycan mixtures. Although additional development, possibly including an MS2-guided MSn method, is needed to achieve complete de novo structural determination, the results presented here represent an important step toward automatic and comprehensive glycome characterization.
Supplementary Material
ACKNOWLEDGMENTS
This research is supported by the NIH grants P41 GM104603, R21 GM122635, U01CA221234, R01 GM132675, and S10 RR025082 and a grant from the National Natural Science Foundation of China (21728501). The contents are solely the responsibility of the authors and do not represent the official views of the awarding offices.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b03183.
Manual determination of Man4, Man6, Man8, and Man9 putative structures; Figure S-1, EED spectra of Man4 isomers; Figure S-2, EED spectra of Man6 isomers; Figure S-3, EED spectra of Man8 isomers; Figure S-4, the EED spectrum of Man9; Figure S-5, top-ranked Man9 topologies; Scheme S-1, the Domon-Costello nomenclature for glycan fragmentation; Scheme S-2, proposed EED mechanisms for linkage-diagnostic fragments; Table S-1, LC–MS data used for relative quantitation; Tables S-2 to S-19, lists of assigned EED fragments (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Varki A Trends Mol. Med 2008, 14, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Shinya K; Ebina M; Yamada S; Ono M; Kasai N; Kawaoka Y Nature 2006, 440, 435. [DOI] [PubMed] [Google Scholar]
- (3).Fuster MM; Esko JD Nat. Rev. Cancer 2005, 5, 526. [DOI] [PubMed] [Google Scholar]
- (4).Dube DH; Bertozzi CR Nat. Rev. Drug Discovery 2005, 4, 477. [DOI] [PubMed] [Google Scholar]
- (5).Vigerust DJ; Shepherd VL Trends Microbiol. 2007, 15, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chui D; Sellakumar G; Green RS; Sutton-Smith M; McQuistan T; Marek KW; Morris HR; Dell A; Marth JD Proc. Natl. Acad. Sci. U. S. A 2001, 98, 1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mariño K; Bones J; Kattla JJ; Rudd PM Nat. Chem. Biol 2010, 6, 713–723. [DOI] [PubMed] [Google Scholar]
- (8).Zaia J Mass Spectrom. Rev 2004, 23, 161–227. [DOI] [PubMed] [Google Scholar]
- (9).Kailemia MJ; Ruhaak LR; Lebrilla CB; Amster IJ Anal. Chem 2014, 86, 196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mechref Y; Hu Y; Desantos-Garcia JL; Hussein A; Tang H Mol. Cell. Proteomics 2013, 12, 874–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ruhaak LR; Xu G; Li Q; Goonatilleke E; Lebrilla CB Chem. Rev 2018, 118, 7886–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ciucanu I; Kerek F Carbohydr. Res 1984, 131, 209–217. [Google Scholar]
- (13).Ciucanu I; Costello CE J. Am. Chem. Soc 2003, 125, 16213–16219. [DOI] [PubMed] [Google Scholar]
- (14).Morelle W; Michalski J-C Nat. Protoc 2007, 2, 1585–1602. [DOI] [PubMed] [Google Scholar]
- (15).Zhou S; Veillon L; Dong X; Huang Y; Mechref Y Analyst 2017, 142, 4446–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ashline D; Singh S; Hanneman A; Reinhold V Anal. Chem 2005, 77, 6250–6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sun S; Huang C; Wang Y; Liu Y; Zhang J; Zhou J; Gao F; Yang F; Chen R; Mulloy B; Chai W; Li Y; Bu D Anal. Chem 2018, 90, 14412–14422. [DOI] [PubMed] [Google Scholar]
- (18).Hsu HC; Liew CY; Huang S-P; Tsai S-T; Ni C-KJ Am. Soc. Mass Spectrom 2018, 29, 470–480. [DOI] [PubMed] [Google Scholar]
- (19).Ni C-K; Tsai S-T; Liew CY; Hsu HC; Huang S-P; Weng W-C; Kuo Y-H ChemBioChem 2019, 20, 2351–2359. [DOI] [PubMed] [Google Scholar]
- (20).Yu X; Huang Y; Lin C; Costello CE Anal. Chem 2012, 84, 7487–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Huang Y; Pu Y; Yu X; Costello CE; Lin CJ Am. Soc. Mass Spectrom 2016, 27, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yu X; Jiang Y; Chen Y; Huang Y; Costello CE; Lin C Anal. Chem 2013, 85, 10017–10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tang Y; Pu Y; Gao J; Hong P; Costello CE; Lin C Anal. Chem 2018, 90, 3793–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tang Y; Wei J; Costello CE; Lin CJ Am. Soc. Mass Spectrom 2018, 29, 1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pu Y; Ridgeway ME; Glaskin RS; Park MA; Costello CE; Lin C Anal. Chem 2016, 88, 3440–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Itoh S; Kawasaki N; Ohta M; Hyuga M; Hyuga S; Hayakawa TJ Chromatogr. A 2002, 968, 89–100. [DOI] [PubMed] [Google Scholar]
- (27).Costell CE; Contado-Miller JM; Cipollo JF J. Am. Soc. Mass Spectrom 2007, 18, 1799–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ruhaak LR; Deelder AM; Wuhrer M Anal. Bioanal. Chem 2009, 394, 163–174. [DOI] [PubMed] [Google Scholar]
- (29).Hua S; An HJ; Ozcan S; Ro GS; Soares S; DeVere-White R; Lebrilla CB Analyst 2011, 136, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhou S; Dong X; Veillon L; Huang Y; Mechref Y Anal. Bioanal. Chem 2017, 409, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ashwood C; Lin C-H; Thaysen-Andersen M; Packer NH J. Am. Soc. Mass Spectrom 2018, 29, 1194–1209. [DOI] [PubMed] [Google Scholar]
- (32).Lohmann KK; von der Lieth C-W Nucleic Acids Res. 2004, 32, W261–W266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Joshi HJ; Harrison MJ; Schulz BL; Cooper CA; Packer NH; Karlsson NG Proteomics 2004, 4, 1650–1664. [DOI] [PubMed] [Google Scholar]
- (34).Goldberg D; Bern M; Li B; Lebrilla CB J. Proteome Res 2006, 5, 1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Apte A; Meitei NS In Functional Glycomics; Springer, 2010; pp 269–281. [Google Scholar]
- (36).Morimoto K; Nishikaze T; Yoshizawa AC; Kajihara S; Aoshima K; Oda Y; Tanaka K Bioinformatics 2015, 31, 2217–2219. [DOI] [PubMed] [Google Scholar]
- (37).Lisacek F; Mariethoz J; Alocci D; Rudd PM; Abrahams JL; Campbell MP; Packer NH; Ståhle J; Widmalm G; Mullen E In High-Throughput Glycomics and Glycoproteomics; Springer, 2017; pp 235–264. [DOI] [PubMed] [Google Scholar]
- (38).Klein J; Zaia JJ Proteome Res. 2019, 18, 3532–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Rojas-Macias MA; Mariethoz J; Andersson P; Jin C; Venkatakrishnan V; Aoki NP; Shinmachi D; Ashwood C; Madunic K; Zhang T Nat. Commun 2019, 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kronewitter SR; An HJ; De Leoz ML; Lebrilla CB; Miyamoto S; Leiserowitz GS Proteomics 2009, 9, 2986–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Xiao K; Wang Y; Shen Y; Han Y; Tian Z Rapid Commun. Mass Spectrom 2018, 32, 142–148. [DOI] [PubMed] [Google Scholar]
- (42).GUERARDEL Y; BALANZINO L; MAES E; LEROY Y; CODDEVILLE B; ORIOL R; STRECKER G Biochem. J 2001, 357, 167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).De Castro C; Molinaro A; Piacente F; Gurnon JR; Sturiale L; Palmigiano A; Lanzetta R; Parrilli M; Garozzo D; Tonetti MG; Van Etten JL Proc. Natl. Acad. Sci. U. S. A 2013, 110, 13956–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gaucher SP; Morrow J; Leary JA Anal. Chem 2000, 72, 2331–2336. [DOI] [PubMed] [Google Scholar]
- (45).Ethier M; Saba JA; Ens W; Standing KG; Perreault H Rapid Commun. Mass Spectrom 2002, 16, 1743–1754. [DOI] [PubMed] [Google Scholar]
- (46).Ethier M; Saba JA; Spearman M; Krokhin O; Butler M; Ens W; Standing KG; Perreault H Rapid Commun. Mass Spectrom 2003, 17, 2713–2720. [DOI] [PubMed] [Google Scholar]
- (47).Tang H; Mechref Y; Novotny MV Bioinformatics 2005, 21, i431–i439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Shan B; Ma B; Zhang K; Lajoie GJ Bioinf. Comput. Biol 2008, 06, 77–91. [DOI] [PubMed] [Google Scholar]
- (49).Mizuno Y; Sasagawa T; Dohmae N; Takio K Anal. Chem 1999, 71, 4764–4771. [DOI] [PubMed] [Google Scholar]
- (50).Bocker S; Kehr B; Rasche F IEEE/ACM Trans. Comput. Biol. Bioinf 2011, 8, 976–986. [DOI] [PubMed] [Google Scholar]
- (51).Liang D; Bing S; Guangdong T; YanBo L; Bing W; MengChu Z IEEE/ACM Trans. Comput. Biol. Bioinf 2015, 12, 568–578. [Google Scholar]
- (52).Sun W; Lajoie GA; Ma B; Zhang K In International Symposium on Bioinformatics Research and Applications; Springer, 2015; pp 320–330. [Google Scholar]
- (53).Hong P; Sun H; Sha L; Pu Y; Khatri K; Yu X; Tang Y; Lin CJ Am. Soc. Mass Spectrom 2017, 28, 2288–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Ritamo I; Räbinä J; Natunen S; Valmu L Anal. Bioanal. Chem 2013, 405, 2469–2480. [DOI] [PubMed] [Google Scholar]
- (55).Zhou S; Hu Y; Mechref Y Electrophoresis 2016, 37, 1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zhou S; Huang Y; Dong X; Peng W; Veillon L; Kitagawa DA; Aquino AJ; Mechref Y Anal. Chem 2017, 89, 6590–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Hinneburg H; Chatterjee S; Schirmeister F; Nguyen-Khuong T; Packer NH; Rapp E; Thaysen-Andersen M Anal. Chem 2019, 91, 4559–4567. [DOI] [PubMed] [Google Scholar]
- (58).Domon B; Costello CE Glycoconjugate J. 1988, 5, 397–409. [Google Scholar]
- (59).Senko MW; Beu SC; McLaffertycor FW J. Am. Soc. Mass Spectrom 1995, 6, 229–233. [DOI] [PubMed] [Google Scholar]
- (60).Hasegawa Y; Takahashi M; Ariki S; Asakawa D; Tajiri M; Wada Y; Yamaguchi Y; Nishitani C; Takamiya R; Saito A; et al. Oncogene 2015, 34, 838–845. [DOI] [PubMed] [Google Scholar]
- (61).Scanlan CN; Offer J; Zitzmann N; Dwek RA Nature 2007, 446, 1038–1045. [DOI] [PubMed] [Google Scholar]
- (62).Crispin M; Doores KJ Curr. Opin. Virol 2015, 11, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Nguyen DN; Xu B; Stanfield RL; Bailey JK; Horiya S; Temme JS; Leon DR; LaBranche CC; Montefiori DC; Costello CE; et al. ACS Cent. Sci 2019, 5, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Prien JM; Ashline DJ; Lapadula AJ; Zhang H; Reinhold VN J. Am. Soc. Mass Spectrom 2009, 20, 539–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Pabst M; Grass J; Toegel S; Liebminger E; Strasser R; Altmann F Glycobiology 2012, 22, 389–399. [DOI] [PubMed] [Google Scholar]
- (66).Zhu F; Lee S; Valentine SJ; Reilly JP; Clemmer DE J. Am. Soc. Mass Spectrom 2012, 23, 2158–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Abrahams JL; Campbell MP; Packer NH Glycoconjugate J. 2018, 35, 15–29. [DOI] [PubMed] [Google Scholar]
- (68).Fu D; Chen L; O’Neill RA Carbohydr. Res 1994, 261, 173–186. [DOI] [PubMed] [Google Scholar]
- (69).Thobhani S; Yuen C-T; Bailey MJ; Jones C Glycobiology 2008, 19, 201–211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.