

Abstract
Metabolic pathways must be adapted to support cell processes required for transformation and cancer progression. Amino acid metabolism is deregulated in many cancers, with changes in branched chain amino acid metabolism specifically affecting cancer cell state as well as systemic metabolism in individuals with malignancy. This review highlights key concepts surrounding the current understanding of branched chain amino acid metabolism and its role in cancer.
Introduction
Metabolism is altered in disease, including cancer (Vander Heiden and DeBerardinis, 2017; Zhu and Thompson, 2019). Metabolic reprogramming can be important to sustain aberrant proliferation, particularly in contexts where nutrients are limiting (Dang, 2012; Vander Heiden et al., 2009; Ward and Thompson, 2012). Metabolites can also propagate signals that coordinate processes such as gene expression and nutrient utilization. Cancer cells in particular must adapt their metabolism to support biomass production, ATP generation, and maintain redox state, and disrupting these processes can interfere with both cell proliferation and tumor growth. Thus, understanding how cells use and sense nutrients provides an opportunity to exploit metabolic changes found in cancer and improve patient care.
A barrier to targeting metabolism for cancer therapy is identifying which specific cancers are dependent on a particular pathway. While dysregulated glucose metabolism is widely appreciated to be prevalent in many cancer types, an elevated demand for amino acids must also be met to support cell proliferation and cancer progression. Amino acid metabolism varies across tumors (Commisso et al., 2013; Mayers et al., 2016), suggesting that a better understanding of amino acid metabolism could be leveraged to effectively target metabolism.
Some amino acids can be synthesized by mammalian cells, while other essential amino acids, must be derived from the diet (Reeds, 2000). However, many cancers also rely on access to non-essential amino acids from their environment and can obtain both essential and non-essential amino acids from many sources including scavenged protein (Finicle et al., 2018; Zhang and Commisso, 2019). Differences in how specific cancers obtain amino acids have been reported, and this choice can depend on both oncogenic signaling and where a tumor originates in the organism (Birsoy et al., 2014; Mayers et al., 2016; Muir et al., 2017; Palm et al., 2015; Yuneva et al., 2012). Nevertheless, additional work is needed to better understand and exploit these metabolic differences for therapeutic development.
Branched chain amino acids (BCAAs) are one class of amino acids whose metabolism has been associated with specific cancer phenotypes. Altered BCAA metabolism can impact both cell intrinsic cancer properties and reflect systemic changes in metabolism associated with certain cancers. Thus, BCAA metabolism can both influence multiple cancer phenotypes and serve as a marker of disease pathology. Here we provide an overview of BCCA synthesis and breakdown, discuss both cellular and systemic regulation of BCAA metabolism, and review data for how BCAA metabolism is involved in cancer.
Overview of BCAA metabolism
The three BCAAs are leucine, isoleucine, and valine. As essential amino acids, BCAAs are not synthesized in humans, however, they are catabolized by highly reversible enzymes (Figure 1A). Many of the enzymes that act on these amino acids can use all three BCAAs as substrates and thus similarly affect levels of all three BCAAs. Consequently, changes in levels of one BCAA are accompanied by changes in levels of the other two BCAAs with the same directionality and magnitude, reflecting the similar chemical properties and metabolism of these amino acids.
Figure 1: Schematic depicting BCAA metabolism and signaling.
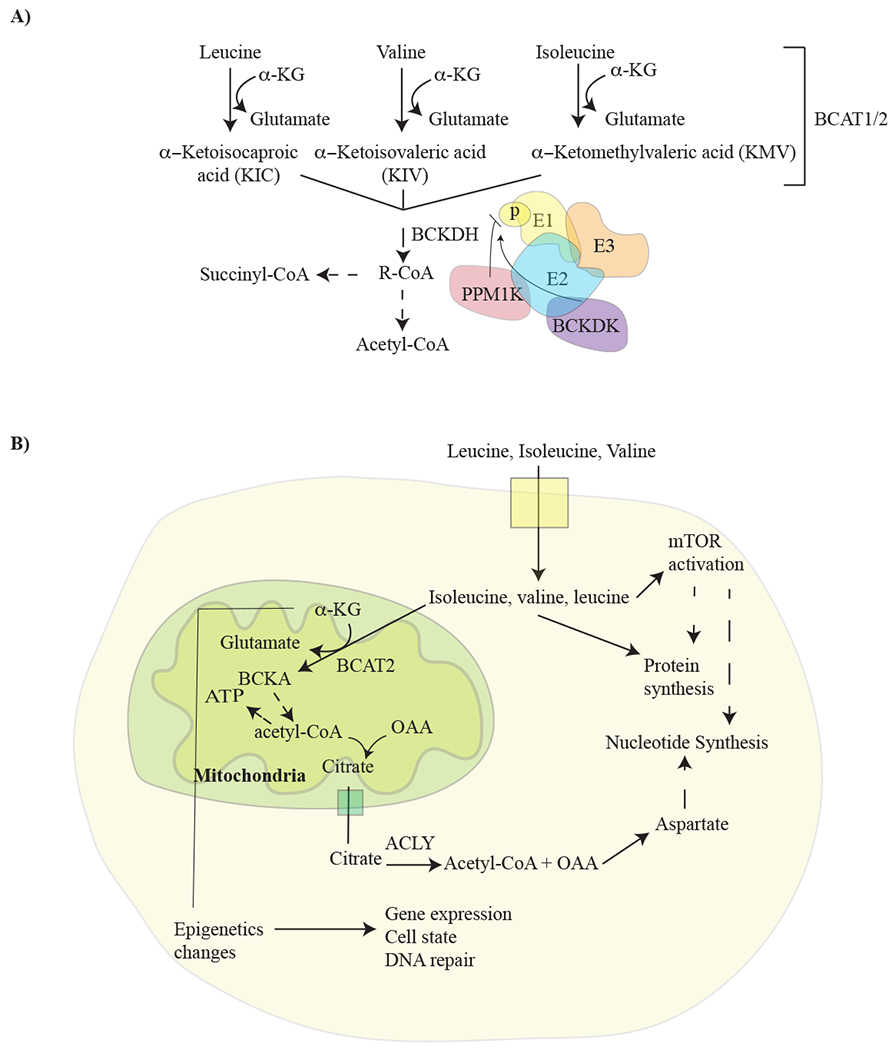
A) Pathway illustrating how branched chain amino acids (BCAA) (leucine, isoleucine, and valine) are broken down, and the regulation of BCAA breakdown by BCKDH. Branched chain amino acid transaminase 1 and 2 (BCAT1/2) transfers nitrogen to α-ketoglutarate (αKG) to produce glutamine and the specified branched chain keto acid (BCKA). These keto acids are then metabolized by branched chain alpha-keto acid dehydrogenase complex (BCKDH), to produce a branched chain acyl-CoA (R-CoA) that can be further metabolized in several steps to the TCA cycle intermediates acetyl-CoA and/or succinyl-CoA (from isoleucine and valine). BCKDH is comprised of three subunits; E1, E2, and E3, and activity of this enzyme is negatively regulated by phosphorylation as shown. Phosphorylation state of BCKDH is determined by the activities of branched chain keto acid dehydrogenase kinase (BCKDK) and PPM1K-protein phosphatase, Mg2+/Mn2+ dependent IK. B) Schematic showing how BCAA breakdown is related to signaling events and anabolic pathways that can play a role in cancer. ACLY, ATP-citrate lyase; OAA, Oxaloacetate.
BCAAs can be an important nutrient source. Leucine is among the most abundant amino acids in protein (Kamphorst et al., 2015), and as such is a major product of protein catabolism from the diet or from the breakdown of tissue protein stores. The end products of complete BCAA oxidation vary based on the specific amino acid; only isoleucine and valine are able to provide carbon precursors that can be used for synthesis of glucose, or any other molecule that is derived from a precursor other than acetyl-CoA. Nevertheless, in principle, BCAA catabolism can satisfy several requirements that are important for cancer cells (Figure 1). First, BCAAs breakdown can provide carbon for synthesis of other molecules. This also allows BCAA oxidation to fuel TCA cycle metabolism and oxidative phosphorylation to provide energy for cells. Second, they can supply nitrogen for de novo nucleotide and amino acid synthesis. Third, BCAA metabolism can alter the levels of metabolite-derived co-factors that influence the epigenome. Finally, these amino acids can affect protein synthesis, either by acting directly as proteinogenic amino acids, or by signaling the nutritional state of cells (Ananieva and Wilkinson, 2018).
Role of BCAA in sensing nutritional status
Cells sense intracellular amino acid availability and use this information to coordinate nutrient availability with processes such as ribosome biogenesis and protein synthesis (Figure 1B). Intracellular leucine is sensed via binding to Sestrin2, a negative regulator of mammalian target of rapamycin complex 1 (mTORC1), a protein kinase that is an important regulator of cell growth (Wolfson et al., 2016). When leucine binds Sestrin2, it disrupts the association between Sestrin2 and Gator2, a positive regulator of mTORC1 activity, to enable kinase activation (Wolfson and Sabatini, 2017; Wolfson et al., 2016). Moreover, leucine derived acetyl-CoA can positively regulate mTORC1 through acetylation of its interacting partner, Raptor (Son et al., 2019). Thus, leucine plays a central role in amino acid sensing by mTORC1, and mTORC1 signaling can also influence lipid and nucleotide synthesis, providing a link between amino acid sensing and other anabolic processes (Ben-Sahra and Manning, 2017). Despite the well characterized role of leucine in regulating the mTORC1 pathway, inappropriate activation of the pathway is widely observed in cancer. This is applicable to a wide range of tumors, including those that experience both nutrient excess and nutrient limitation (Sullivan et al., 2019).
Reactions for BCAA breakdown
In cells, free BCAA can undergo transamination and transfer nitrogen to α-ketoglutarate (αKG) to produce glutamate and their respective branched chain ketoacids (BCKAs; alpha-keto-beta-methylvalerate (KMV, ketoisoleucine), alpha-ketoisocarproate (KIC, ketoleucine), and alpha-ketoisovalerate (KIV, ketovaline)) (Figure 1A). This reaction is catalyzed by the compartment specific BCAA transaminases, BCAT1 and BCAT2. BCAT1 is present in the cytosol, while BCAT2 is localized to the mitochondria. BCAT1 and BCAT2 are both highly active and reversible enzymes that act on all three BCAAs and their corresponding branched chain keto acids (BCKAs). Therefore, the BCAT enzymes can transfer nitrogen from glutamate back to a BCKA and regenerate the BCAA and αKG. Thus, nitrogen exchanges rapidly between the BCAAs, BCKAs, glutamate, and αKG in cells even when net catabolism of BCAAs is minimal, complicating methods to trace BCAA-derived nitrogen in cells and tissues (Harper, 1984).
BCKAs are also exchanged with the circulation as part of normal physiology but can be further catabolized by some cells/tissues using the multimeric branched chain ketoacid dehydrogenase enzyme complex (BCKDH). BCKDH is comprised of three subunits (E1, E2, and E3) that are important for enzyme catalysis (Figure 1A). The E1 subunit is subject to regulation by branched chain ketoacid dehydrogenase kinase (BCKDK) and protein phosphatase, Mg2+/Mn2+ dependent 1K (PPM1K). The relative activity of BCKDK and PPM1K alters the phosphorylation state of E1 to modulate an inhibitory phosphorylation of BCKDH E2 and affect flux through the pathway (White et al., 2018). Of note, the BCKDH reaction is essentially irreversible in mammalian cells, and it is the fact that animals lack enzymes to synthesize BCKAs de novo that renders BCAAs essential amino acids (Shimomura et al., 2006).
Systemic BCAA metabolism
The interactions between BCAAs and systemic metabolism are not completely understood, but clues can be gleaned from recent studies showing that increased levels of BCAAs in circulation can be associated with cancer as well as metabolic disease in patients. At the organismal level, BCAA metabolism involves multiple tissues, and levels of BCAAs in the plasma can vary based on both dietary intake and whole-body protein turnover (Brosnan and Brosnan, 2006; Mayers et al., 2014) (Figure 2A). Most amino acids are catabolized in the liver, but BCAT is not expressed in the liver and thus BCAAs are a notable exception. A consequence of this is that BCAAs absorbed in the gut have access to the circulation, and levels of BCAAs in diet have a direct impact on blood levels (Brosnan, 2003; Harper, 1984). Excess amino acids, including BCAAs, are stored in protein. Liver and skeletal muscle protein serve as major amino acids reservoirs for mammals and are accessed during periods of fasting. Following liberation from protein, catabolism of BCAA begins in many peripheral tissues, including skeletal muscle, that express BCAT enzymes. This results in net BCKA generation and release into circulation. The liver is a site of circulating BCKA metabolism, because even though this tissue lacks BCAT expression to initiate BCAA breakdown, it has active BCKDH to access BCKA carbon for gluconeogenesis, ketogenesis, or fatty acid synthesis (Neinast et al., 2018). The abundance of leucine in protein may explain in part why this particular amino acid is sensed by mTORC1, and the fact that levels of this amino acid in blood fluctuate based on food intake could be an extension of this principle for BCAAs to also serve as markers of organismal nutritional status.
Figure 2: Systemic metabolism of BCAAs.
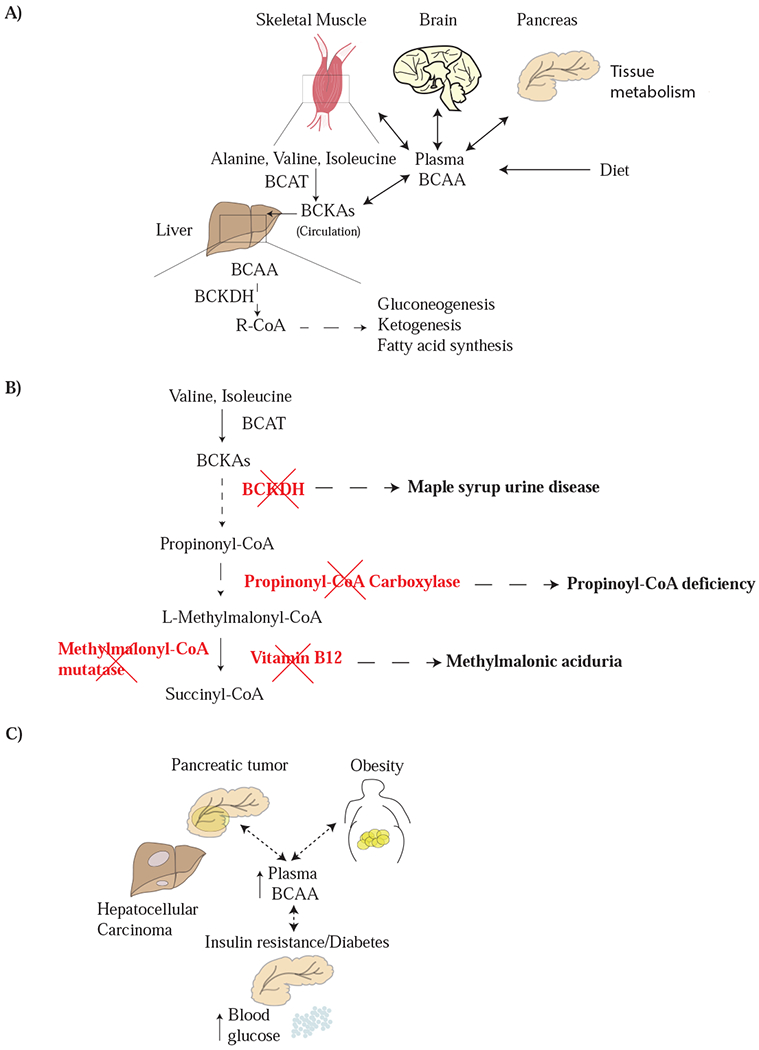
A) Dietary BCAA intake, as well as the balance between BCAA uptake and breakdown by tissues, combine to determine plasma BCAA levels. The liver is deficient in BCAT activity, and thus does not directly metabolize BCAAs. However, the liver can act on circulating branched chain keto acids (BCKAs), which are largely derived from the action of BCAT on protein derived BCAAs in skeletal muscle. This allows the liver to catabolize BCAAs or use them as a source of carbon to produce other molecules for the body such as fatty acids, glucose, and ketones. B) Deficiencies in the BCAA metabolism are responsible for inborn errors of metabolism. The relationship between each enzyme deficiency involving BCAA catabolism and the related disease is shown. C) Systemic metabolic diseases including insulin resistance and diabetes, obesity, and some cancer can all lead to changes in plasma BCAA levels, although the mechanistic underpinnings of these alterations remain an ongoing area of investigation.
Individual cells and tissues must respond to systemic fluctuations in nutrient availability and levels of BCAAs can influence metabolism in ways that are independent of mTOR signaling. BCAAs can influence systemic glucose metabolism by regulating insulin secretion and the sensitivity of peripheral tissues to insulin, helping to coordinate amino acid and carbohydrate metabolism across tissues in an organism (Muoio, 2014; Newgard et al., 2009). Metabolic characterization of mice harboring whole body deletion of mitochondrial Bcat2 suggests that suppression of BCAA breakdown and/or accumulation of excess BCAA affects protein turnover and glucose homeostasis (She et al., 2007a). BCAA breakdown is influenced by insulin levels and disruption of normal physiological functions of the pancreas could have overall consequences on BCAA breakdown by skeletal muscle and on circulating levels of BCAAs. While many studies compare the biological effects of BCAA metabolism on tumor growth, systemic BCAA metabolism might also be indirectly affected by tumors arising in various tissues.
Signaling and cross-talk between the pancreas, brain, and liver can modulate plasma BCAA levels (Shin et al., 2014). Expression of BCKDH in the liver is regulated by insulin effects on brain, arguing that insulin resistance accompanying obesity and diabetes can inhibit liver BCKA breakdown (Shin et al., 2014) and potentially affect BCAA metabolism in other tissues. It is unclear, at the organismal level, how plasma BCAA levels are maintained in a relatively tight range in animals (Neinast et al., 2019), particularly when levels vary based on disease states in a manner that can be detected in diverse human populations (Felig et al., 1969; Mayers et al., 2014; She et al., 2007b; Wang et al., 2011). Whole body analysis of BCAA uptake and breakdown using isotopic tracers suggest tissue specific differences in BCAA metabolism may be involved as some tissues take up and transaminate BCAA rapidly, with BCAA oxidation occurring maximally in skeletal muscle. In contrast, BCAA contribution to protein is highest in tissues such as the liver, pancreas, and skeletal muscle (Neinast et al., 2019), and metabolic alterations in these tissues in disease states, including some cancers, may be responsible for the consistent changes in BCAA levels associated with human disease (Arany and Neinast, 2018).
Insights into systemic BCAA metabolism from human genetic disorders
Systemic BCAA metabolism has historically been understood from studies of individuals with inherited or acquired deficiencies in enzyme or essential cofactors that are needed for breakdown of these amino acids. Blocking some steps in BCAA catabolism leads to toxic buildup of intermediates. For example, maple syrup urine disease (MSUD) is an autosomal recessive disease that is caused by mutations in one the three genes encoding components of the BCKDH complex: BCKDHA, BCKDHB, and DBT (Blackburn et al., 2017), resulting in impaired ability to breakdown BCKAs. Deficiency in BCKDH activity causes elevated urine and plasma BCAA (Han et al., 2018), and alterations in brain white matter (Klee et al., 2013). It is thought a lack of BCKDH activity in the liver drives this phenotype based on the finding that transplantation of healthy livers can suppress plasma BCAA accumulation (Wendel et al., 1999). These findings illustrate the liver involvement in whole body BCAA metabolism.
Vitamin deficiency can also impact BCAA metabolism. Cobalamin (vitamin B12) is required for conversion of valine- and isoleucine-derived methylmalonic acid to succinyl-CoA, which can subsequently be oxidized in the TCA cycle or serve as a precursor for gluconeogenesis. BCAA breakdown accompanies adipocyte differentiation and deficiencies in cobalamin leads to impaired BCAA breakdown and a differentiation block (Green et al., 2016). Deficiency in cobalamin metabolism can also be noted in genetic diseases such as methylmalonic aciduria (Figure 2B). The enzyme methylmalonyl-CoA mutase, encoded by the gene Mut, catalyzes the conversion of methylmalonyl CoA (MMA-CoA) to succinyl-CoA. In the disease methylmalonic aciduria, mutations in Mut leads to accumulation MMA-CoA. Similarly, deficiencies in propionyl-CoA carboxylase (PCC), composed of two subunits encoded by genes PCCA and PCCB, causes a rare disease called propionic acidemia (PA). Enzyme insufficiency can result from mutations in either gene. PCC localizes to the mitochondria and catalyzes the conversion of propionyl-CoA to methylmalonyl-CoA. Increases in the levels of propionyl CoA can inhibit the urea cycle and alter glutathione levels within the cell (Richard et al., 2018). Plasma branched chain amino acid levels are lower in patients with PCCA or PCCB deficiency, particularly with age (Scholl-Bürgi et al., 2010). However, it is unclear whether this is a consequence of the BCAA free diet that is clinically recommended for affected individuals.
Elevated circulating BCAAs in cancer
Elevations in circulating BCAAs occur early during pancreatic cancer disease progression in both humans and mouse cancer models and correlate with tissue wasting (Mayers et al., 2014). The relationship between elevated circulating BCAAs and breakdown of protein in peripheral tissues can be explained by normal systemic BCAA metabolism. It appears that the breakdown of peripheral tissue protein in pancreatic cancer can exceed the systemic requirement for BCAAs leading to increased levels in blood (Holmstrom and Olive, 2014; Mayers et al., 2014).
Relevant to patients, the increase in circulating BCAAs has some predictive power for pancreatic cancer diagnosis, and may represent a marker that, in combination with other tests, can contribute to earlier diagnosis (Danai et al., 2018; Mayers et al., 2014). In addition, one underlying cause of peripheral tissue wasting in pancreatic cancer can be exocrine insufficiency caused by transformation of normal pancreatic tissue (Danai et al., 2018). This results in a starvation-like response in mouse models of pancreatic cancer that can be reversed by pancreatic enzyme replacement (Danai et al., 2018). Interestingly, reversing tissue wasting associated with exocrine insufficiency in mice with pancreatic cancer worsens outcomes, although muscle wasting is less affected by this intervention. Nevertheless, elevated BCAA levels in human plasma do not predict worse survival outcomes in patients, even though it is reflective of peripheral tissue loss (Danai et al., 2018). Other metabolic disorders such as obesity and type II diabetes are also associated with an increased risk of pancreatic cancer, and high levels of insulin and related growth factors can promote growth of some tumors (Zhang et al., 2019). These metabolic states are also associated with elevated BCAAs in plasma (Pothuraju et al., 2018; Wang et al., 2011), but the association between elevated BCAAs and pancreatic cancer is independent from an association with obesity or type II diabetes (Mayers et al., 2014). Instead, increased protein breakdown with release of BCAAs in excess of whole-body demand may also be present in obesity, diabetes, and/or other insulin resistant states, reflecting a systemic change in metabolism that is also found in pancreatic cancer. Regardless, these studies illustrate how changes in BCAA levels in circulation can be a marker of cancer and other disease states.
BCAAs as a fuel for cancer growth
BCAA metabolism is altered in many solid tumor contexts including melanoma, nasopharyngeal carcinoma, and breast cancer. In melanoma, oncogenic activation of Ras-MEK signaling can sustain activation of mTOR; however, hyperactivation of Ras-MEK signaling fails to suppress mTOR when leucine is deprived resulting in attenuation of autophagy and enhanced cell death (Sheen et al., 2011). A recent study also identified a positive correlation between c-Myc overexpression and enhanced BCAT1 transcript levels, which supported cell invasion (Zhou et al., 2013). Finally, in breast cancer BCAT1 levels are elevated relative to normal breast tissue and promotes mitochondrial biogenesis in an mTOR dependent manner to support proliferation (Zhang and Han, 2017). While a general pro-growth program is activated in response to mTOR signaling, in part due to elevated BCAA levels in this context, it is unclear mechanistically how BCAT1 expression drives enhanced mitochondrial biogenesis. One possibility from studies of breast cancer and hematopoietic cells is that elevated BCAAs can act via mTOR to regulate translation of a subset of mRNAs that encode mitochondrial subunits (Liu et al., 2017; Morita et al., 2013).
It has also been reported that in cardiac and skeletal muscle, elevated BCAA breakdown can support mitochondrial biogenesis (D’Antona et al., 2010). During animal aging, mitochondrial biogenesis influenced by BCAA metabolism is attenuated in endothelial nitric oxide synthase null mice, supporting a role for nitric oxide signaling in mediating these effects, but this specificity in enhancing mitochondrial mass and function was not observed in liver or adipose tissue, suggesting differential breakdown and/or regulation of BCAA in these tissues (D’Antona, et. al., 2016). These findings highlight that any effect of BCAA metabolism on mitochondrial biogenesis will vary based on the tissue. Under certain contexts BCAT1 can also synthesize BCAAs from available BCKAs (Gu et al., 2019; Hattori et al., 2017). Thus, BCAT1 may drive mitochondrial biogenesis at least in part via regulation of mTORC1. Likewise BCAT1 supports growth of cancer cells in a glioblastoma model (Tönjes et al., 2013). In addition, loss of BCAT2 delayed tumor onset in mice challenged with lymphoma, arguing for a pro-tumorigenic role of BCAT2 enzyme in this cancer (Ananieva et al., 2018).
Emerging evidence suggests that the regulation of BCAA catabolism can impact the biology of some cancers, and the tissue where a tumor arises is one factor that can influence whether a cancer utilizes BCAAs to fulfill their metabolic needs. For example, when PDAC and non-small cell lung cancer tumors (NSCLC) are initiated by the same genetic perturbations in Kras and p53, they have differential requirement for BCAAs (Mayers et al., 2016). While NSCLC tumors actively catabolize BCAAs to help satisfy nitrogen requirements for nucleotide synthesis, PDAC tumors obtain nitrogen from other sources. The lack of BCAA catabolism in pancreatic tumors contrasts with normal pancreatic tissue, where breakdown of BCAA supports generation of TCA cycle intermediates (Neinast et al., 2019). This use of BCAAs by normal pancreatic tissue is consistent with BCAT2 activity being elevated in pancreatic exocrine cells relative to other tissues in the body (Sweatt et al., 2004). Likewise, leucine breakdown contributes carbon to acetyl-CoA in wild-type pancreatic acinar cells (Carrer et al., 2019). Mechanistically, how and when during the course of pancreatic tumorigenesis BCAA metabolism is lost remains an open question, although restoring BCAT activity has no effect on pancreatic tumor growth arguing that this metabolic activity is neither detrimental nor supportive of tumor growth in this cancer model (Mayers et al., 2016).
Systemic metabolism dysregulation can impact cancer initiation and progression and is more prominent in some cancers, such as pancreatic cancer (Khalaf and Wolpin, 2019; Pothiwala et al., 2009). However, it is unclear whether the altered plasma BCAA levels in pancreatic cancer plays a direct causal role in the disease (Danai et al., 2018). Supplementing animals with increased leucine to bolster protein synthesis can enhance pancreatic tumor growth in some models (Liu et al., 2014), but increased dietary leucine has no effect in others models of pancreatic cancer (Mayers et al., 2016). Furthermore, the contribution of labeled BCAAs to pancreatic tumor tissue arising in an autochthonous mouse model is minimal, even in models where BCAAs are elevated in the circulation (Mayers et al., 2016). Disruption of both BCAT1 and BCAT2 also has no effect on pancreatic tumor growth despite impairing growth of lung tumors derived from a related autochthonous lung cancer model where circulating BCAAs are not elevated (Mayers et al., 2016). These data argue that increased BCAA levels in the blood do not necessarily reflect a cancer cell-intrinsic requirement for BCAA metabolism. Rather, it may be the lack of BCAAs consumption by tumors that contributes to the mismatch between BCAA release from protein breakdown in peripheral tissues and disposal of these amino acids from the circulation. The data also argue against a universal role for BCAA catabolism across all malignancies.
BCAA levels in blood are also elevated in mouse and human models of hepatocellular carcinoma (HCC) and in this cancer elevated BCAA levels correlate with mTOR hyperactivation (Ericksen et al., 2019). Intriguingly, this phenomenon is not observed during liver regeneration following partial hepatectomy even though proliferating hepatocytes also activate a rapid pro-proliferative mTOR signaling program that is coupled with cell cycle progression (Espeillac et al., 2011). Unlike liver tumors, proliferating hepatocytes during liver regeneration do not suppress BCAA breakdown, suggesting that there is selection for suppression of BCAA breakdown in liver tumors (Ericksen et al., 2019). Nevertheless, oral administration of BCAAs in mice protects against development of HCC by halting the progression of non-alcoholic steatohepatitis (Takegoshi et al., 2017), highlighting that BCAAs can have different effects on cancer biology based on the mechanism leading to their elevation in circulation. It also suggests that the regulation of BCAA breakdown is context dependent and may play different roles in the same tissue in pathological versus physiological settings (Goto et al., 1977).
Defective BCAA breakdown downstream of BCAT can lead to excess accumulation of BCKAs. In glioblastoma excess BCKAs are excreted from the cancer cells, and targeting the export of BCKAs by inhibiting the mono-carboxylate transporter 1 (MCT1) impairs tumor growth (Silva et al., 2017). An attractive model is that this represents a symbiotic relationship between cancer cells and other cell types within the tumor microenvironment, such that the BCKAs are further oxidized by non-cancer cells. A metabolic crosstalk between cell types has been previously ascribed as one mechanism by which glutamate is exchanged between neurons and astrocytes in normal brain tissue for the synthesis of neurotransmitters (Daikhin and Yudkoff, 2000), and perhaps a similar crosstalk is at play involving BCAAs and their breakdown products in glioblastoma.
Role of BCAA in epigenetic regulation
Many histone and DNA modifications rely on metabolic substrates as co-factors. Metabolites can also serve as signaling co-factors to alter the epigenome, and products of BCAA metabolism can impact gene expression in this way. For instance, breakdown of BCAAs can be a source of acetyl-CoA. Acetyl-CoA is the source of acetyl-groups for histone acetylation, and thus acetyl-CoA levels can impact the epigenetic landscape of cells (Campbell and Wellen, 2018; Kaelin and McKnight, 2013; Schvartzman et al., 2018). BCAA catabolism generates acetyl-CoA in the mitochondria. In order to affect epigenetic gene regulation, mitochondrial acetyl-CoA must be rederived within the cytoplasmic compartment. Acetyl-CoA is permeable to the nuclear pore; thus, extra-mitochondrial acetyl-CoA impacts nuclear-cytoplasmic acetyl-CoA pools that can then be used for protein and histone acetylation. There is evidence that different cells rely on different nutrients to support acetyl-CoA levels (Pietrocola et al., 2015), but what determines the relative contribution of BCAAs versus other nutrients in cells, and how this affects compartmentalized acetyl-CoA levels, is incompletely understood. Nevertheless, there is emerging evidence that the coordinated regulation of nuclear-cytoplasmic acetyl-CoA pools is affected by BCAA breakdown (White et al., 2018). Cytosolic acetyl-CoA is also the substrate for fatty acid synthesis, and suppression of BCKDK activity to enhance BCAA degradation using a pharmacological inhibitor can shift the source of acetyl-CoA in cells from glucose to fatty acids. Correspondingly, this is associated with decreased phosphorylation of ATP-citrate lyase (ACLY), a key enzyme that can modulate the nuclear-cytoplasmic acetyl-CoA pool (Sivanand et al., 2018). These data illustrate that modulation of BCAA metabolism can impact the distribution of acetyl-CoA in cells. Additional work is needed to determine how compartmentalized pools of metabolites are sensed and the signaling pathways that are engaged to control metabolite levels to affect epigenetic state.
Epigenetic changes can influence diverse cellular processes such as gene expression, cell-cycle progression, and DNA repair. These changes can in turn alter metabolism in proliferating cells; however, there is also evidence for reciprocal regulation of epigenetics by BCAA metabolism. In certain cancers, most notably acute myeloid leukemia (AML), BCAT1 is highly expressed and contributes to disease progression. Because BCAT1 catalyzes the transfer of nitrogen from BCAAs to αKG (Figure 1), this reaction consumes αKG allowing BCAT1 activity to affect αKG levels in AML cells (Raffel et al., 2017). Decreased αKG levels in this setting affects gene expression because αKG is an essential cofactor for the activity of multiple histone and DNA demethylases, including the Ten-eleven Translocation (TET) and Jumonji Domain-containing Histone Demethylase (JHDM) family of demethylases (Lu and Thompson, 2012) (Figure 3). Thus, lower αKG levels can result in a hypermethylated phenotype that affects AML progression by influencing gene transcription and preventing cancer cell differentiation (Hattori et al., 2017; Raffel et al., 2017).
Figure 3: BCAA metabolism can modulate the epigenome.
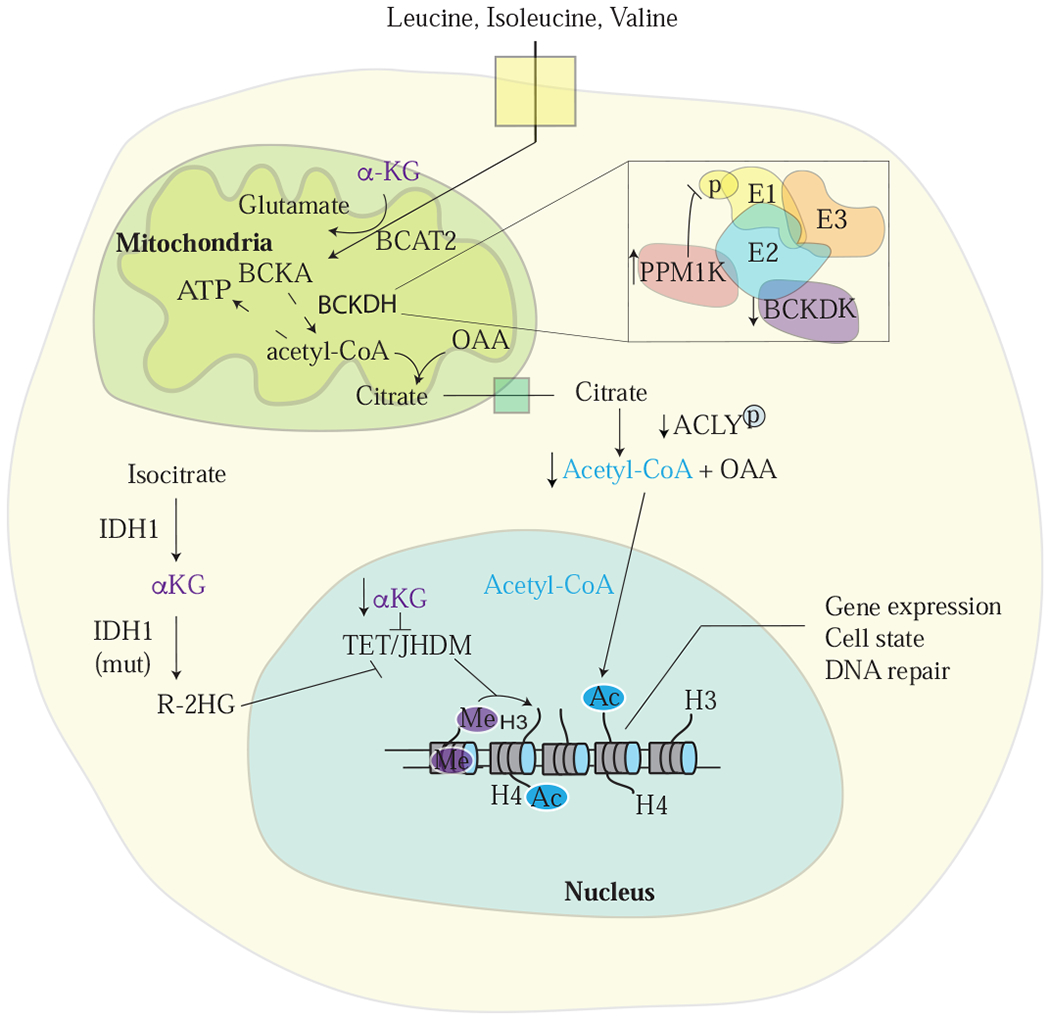
A) Alterations in α-ketoglutarate (αKG) levels, which can be affected by BCAA metabolism, can influence global histone and DNA methylation patterns (as well as cytosine methylation, not shown) via effects on αKG-dependent dioxygenase activity (Gut and Verdin, 2013). Metabolites can also affect transcriptionally regulated expression of enzymes involved in BCAA breakdown. Mitochondrial acetyl-CoA from BCAA catabolism can contribute to cytoplasmic and nuclear pools of this metabolite, which provides the acetyl group for histone acetylation. Phosphorylation of the BCKDH E1 subunit to increase BCAA breakdown can affect ATP-citrate lyase (ACLY) activity as another way to alter acetyl-CoA levels in cells. 2-hydroxyglutarate (2-HG) produced by cancer associated mutations in isocitrate dehydrogenase (IDH1, shown; or IDH2, not shown) can also affect epigenetic regulation of gene expression, as well as inhibit the activity of BCAT1/2.
Branched chain amino acids are essential and cannot be synthesized by mammals, and the output of the BCAT reaction favors BCKA and glutamate generation in most contexts (Hutson et al., 2005) (Figure 1). Historically, this has led to the conclusion that cells in tissues must derive BCAAs from the circulation. However, recent evidence suggests that the reversibility of this reaction enables some cells to synthesize BCAAs from glutamate and circulating BCKAs in pathological contexts such as chronic myeloid leukemia (Gu et al., 2019; Hattori et al., 2017).
Because the BCAT reaction is rapid and reversible, the net production or consumption of BCAAs by these enzymes is likely mediated by substrate and product availability, although it remains possible other regulatory mechanisms have yet to be uncovered. Egl-9 family hypoxia inducible factor 1 (EGLN1) belongs to a class of αKG-dependent dioxygenases that regulates levels of hypoxia-inducible factor (HIFα) proteins in response to oxygen levels. In AML, it was reported that BCAT1-dependent alterations in αKG can regulate HIFα protein levels (Raffel et al., 2017). Moreover, low intracellular cystine levels can cause increases in HIF1α (Briggs et al., 2016). Since, cystine import into cells is coupled to glutamate export, modulation of glutamate levels by BCAT activity may also affect cystine levels and thus impact HIFα.
Mutations in isocitrate dehydrogenase 1/2 (IDH1 or IDH2) represent another mechanism to cause global changes in histone and DNA modifications in select cancers, including AML and glioblastoma (Figueroa et al., 2010). Isocitrate dehydrogenase (IDH1 and IDH2) mutations result in a neomorphic activity to produce (R)-2-hydroxyglutarate (2HG) from αKG (Dang et al., 2009). Accumulation of excess 2HG is another way to inhibit αKG-dependent histone and DNA demethylases, resulting in defective methylation patterns across the genome that can impact cellular function and differentiation in leukemia (Lu et al., 2012). In glioblastoma, transcriptional activation of BCAT1 is observed in tumors harboring wild-type IDH1 (Tönjes et al., 2013). One proposed explanation for this is that mutations in IDH1 affect αKG levels, limiting BCAA transamination and decreasing BCAT1 expression by promoting DNA hypermethylation at the BCAT1 promoter (Tönjes et al., 2013). However, the rate of 2HG production by mutant IDH is slow relative to many other cellular reactions that use αKG (Dexter et al., 2018), and mutant IDH does not change αKG levels in all settings (Dang et al., 2009; Ward et al., 2010), arguing other hypotheses may be needed to explain low BCAT expression in IDH mutant glioma. High 2HG can impair the activity of TET enzymes and contribute to the hypermethylated state. Another possibility is that cells with wildtype IDH1 are more reliant on BCAA metabolism. Breakdown of BCAAs via BCAT1 activity generates glutamate, and suppression of glutamate excretion by modulating BCAT1 activity reduces tumor burden (Tönjes et al., 2013). A relationship between IDH1/IDH2 and redox state has also been noted, where perturbation of glutamate levels enhances susceptibility to oxidative stress (McBrayer et al., 2018). This effect is driven at least in part by 2HG inhibition of BCAT1/2 activity, providing another explanation for why BCAT expression tracks with IDH mutant status in glioma (McBrayer et. al., 2018).
Another frequently dysregulated epigenetic modification in hematologic malignancies is histone H3-lysine 27 methylation (H3K27me), a repressive modification dependent on the histone methyltransferase EZH2 (Knutson et al., 2012). A recent report suggests that, in leukemia, BCAA metabolism is subject to epigenetic regulation of the BCAT1 promoter by EZH2 (Gu et al., 2019). In contrast to normal hematopoietic stem and progenitor cells where BCAT1 expression is epigenetically silenced, mutations or loss in EZH2 function reactivates BCAT1 and sustains BCAA metabolism in AML (Gu et al., 2019).
Role of BCAAs in therapeutic response
Exploiting synthetic lethal interactions between otherwise untargetable genetic events that drive cancer is a paradigm for cancer treatment (Kaelin, 2009), and perturbations in BCAA metabolism that are synthetic lethal with specific genetic mutations may be a strategy for therapeutic intervention.
Exploiting the links between BCAA metabolism and redox state:
Maintaining the correct redox states is critical for cancer progression and can be affected by changes in BCAA metabolism. For example, pancreatic cancers frequently harbor deletions in the chromosomal region containing Smad4. There are two mitochondrial malic enzyme isoforms (ME2 and ME3) with redundant function that can affect redox balance in cells. Co-deletion of ME2 along with Smad4 and genetic perturbation of the isoform ME3, to delete malic enzyme activity, was reported to modulate BCAT2 levels (Dey et. al., 2017). In this context, glutamate generation coupled to the enzymatic activity of BCAT2 was necessary to sustain nucleotide synthesis in pancreatic tumors.
How BCAA availability influences cell redox state is likely context dependent. For example, supplementation with BCAA promotes oxidative stress by enhancing reactive oxygen species production and leading to a pro-inflammatory phenotype of peripheral blood mononuclear cells (Zhenyukh et al., 2017). In contrast, in a rat model of liver disease, BCAA supplementation lowered overall levels of reactive oxygen species, accompanied by prolonged survival (Iwasa et al., 2013).
Response to therapy
A dependence of cells on glutamine to proliferate in culture led to the development of inhibitors, such as CB-839, that target glutaminase (GLS; encoded by GLS1 and GLS2), which is currently in clinical trials to treat cancer (Gross et al., 2014). However, dependence on glutamine metabolism is affected by both genetic and environmental factors (Davidson et al., 2016; Muir et al., 2017), limiting the predictive power of cell culture models to identify a sensitive patient population. One potential way to identify a patient population that will be sensitive to targeting glutamine metabolism arose from studies of BCAA metabolism. Suppression of BCAT activity decreases glutamate biosynthesis with compensatory elevation in GLS activity, providing cells with an alternative source of glutamate (McBrayer et. al, 2018). The fact that IDH mutations are initiating lesions for some gliomas may create metabolic dependencies of these tumors on GLS. In support of this, IDH mutant glioblastoma tumors respond better in vivo a combination of GLS inhibition and radiation therapy (McBrayer et al., 2018; Seltzer et al., 2010).
Emerging evidence suggests that alterations in BCAA metabolism, when considered in combination with known genetic mutations, might be useful to stratify patient subsets for therapy. Epidermal growth factor receptor (EGFR) mutations are found in lung cancer, and a recent screen identified that BCAT1 transcripts are upregulated in response to EGRF inhibitors, resulting in enhanced capacity to tolerate oxidative stress (Wang et al., 2019). In breast cancer, elevated BCAT1 protein levels track with therapy resistance and reduced survival of patients treated with tamoxifen (Thewes et al., 2017), arguing that changes in BCAA metabolism may help stratify patients that are more or less likely to respond to specific treatments.
Conclusion
BCAA metabolism can impact diverse cellular processes ranging from protein synthesis to epigenetic regulation. Dysregulation of BCAA metabolism in cancer can contribute to disease progression by impacting these processes and/or acting as markers of disease state. Further characterization of BCAA metabolism at the organismal level can shed insight into how different tissues handle metabolic demands for amino acids and how this can influence tumor growth. Moreover, identifying requirements for BCAA breakdown in specific cancer types can inform how best to target the metabolism of these amino acids, as well as related metabolic pathways, to improve cancer treatment.
Acknowledgements and Disclosures
We thank Alicia Darnell and Allison Lau for helpful comments. S.S. acknowledges support from the Damon Runyon Cancer Research Foundation. M.G.V.H. acknowledges support from the MIT Center for Precision Cancer Medicine, the Lustgarten Foundation, the Ludwig Center at MIT, the Emerald Foundation, SU2C, the NCI, and a faculty scholars award from HHMI. M.G.V.H. discloses that he is on the scientific advisory board of Agios Pharmaceuticals, Aeglea Biotherapeutics, iTeos, and Auron Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Ananieva EA, and Wilkinson AC (2018). Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 21, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananieva EA, Bostic JN, Torres AA, Glanz HR, McNitt SM, Brenner MK, Boyer MP, Addington AK, and Hutson SM (2018). Mice deficient in the mitochondrial branched-chain aminotransferase (BCATm) respond with delayed tumour growth to a challenge with EL-4 lymphoma. Br. J. Cancer 119, 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, and Neinast M (2018). Branched Chain Amino Acids in Metabolic Disease. Curr. Diab. Rep 3–8. [DOI] [PubMed] [Google Scholar]
- Ben-Sahra I, and Manning BD (2017). mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol 45, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, and Sabatini DM (2014). Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn PR, Gass JM, Pinto e Vairo F, Farnham KM, Atwal HK, Macklin S, Klee EW, and Atwal PS (2017). Maple syrup urine disease: Mechanisms and management. Appl. Clin. Genet 10, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs KJJ, Koivunen P, Cao S, Backus KMM, Olenchock BAA, Patel H, Zhang Q, Signoretti S, Gerfen GJJ, Richardson ALL, et al. (2016). Paracrine Induction of HIF by Glutamate in Breast Cancer: EglNl Senses Cysteine. Cell 166, 126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosnan JT (2003). Interorgan Amino Acid Transport and its Regulation. J. Nutr 133, 2068S–2072S. [DOI] [PubMed] [Google Scholar]
- Brosnan JT, and Brosnan ME (2006). Branched-Chain Amino Acids: Enzyme and Substrate Regulation. J. Nutr 136, 207S–211S. [DOI] [PubMed] [Google Scholar]
- Campbell SL, and Wellen KE (2018). Metabolic Signaling to the Nucleus in Cancer. Mol. Cell 71, 398–408. [DOI] [PubMed] [Google Scholar]
- Carrer A, Trefely S, Zhao S, Campbell SL, Norgard RJ, Schultz KC, Sidoli S, Parris JLD, Affronti HC, Sivanand S, et al. (2019). Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov 9, 416–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antona G, Ragni M, Cardile A, Tedesco L, Dossena M, Bruttini F, Caliaro F, Corsetti G, Bottinelli R, Carruba MO, et al. (2010). Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab 12, 362–372. [DOI] [PubMed] [Google Scholar]
- Daikhin Y, and Yudkoff M (2000). Compartmentation of brain glutamate metabolism in neurons and glia. J. Nutr 130, 1026S–31S. [DOI] [PubMed] [Google Scholar]
- Danai LV, Babic A, Rosenthal MH, Dennstedt EA, Muir A, Lien EC, Mayers JR, Tai K, Lau AN, Jones-Sali P, et al. (2018). Altered exocrine function can drive adipose wasting in early pancreatic cancer. Nature 558, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). Links between metabolism and cancer. Genes Dev 26, 877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger M. a, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter JP, Ward PS, Dasgupta T, Hosios AM, Gunawardena J, and Vander Heiden MG (2018). Lack of evidence for substrate channeling or flux between wildtype and mutant isocitrate dehydrogenase to produce the oncometabolite 2-hydroxyglutarate. J. Biol. Chem 293, 20051–20061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericksen RE, Lim SL, McDonnell E, Shuen WH, Vadiveloo M, White PJ, Ding Z, Kwok R, Lee P, Radda GK, et al. (2019). Loss of BCAA Catabolism during Carcinogenesis Enhances mTORC1 Activity and Promotes Tumor Development and Progression. Cell Metab 29, 1151–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeillac C, Mitchell C, Celton-Morizur S, Chauvin C, Koka V, Gillet C, Albrecht JH, Desdouets C, and Pende M (2011). S6 kinase 1 is required for rapamycin-sensitive liver proliferation after mouse hepatectomy. J. Clin. Invest 121, 2821–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P, Marliss E, and Cahill GF (1969). Plasma amino acid levels and insulin secretion in obesity. N. Engl. J. Med 281, 811–816. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finicle BT, Jayashankar V, and Edinger AL (2018). Nutrient scavenging in cancer. Nat. Rev. Cancer 18, 619–633. [DOI] [PubMed] [Google Scholar]
- Goto M, Shinno H, and Ichihara A (1977). Isozyme patterns of branched-chain amino acid transaminase in human tissues and tumors. Gann, Japanese J. Cancer Res 68, 663–667. [PubMed] [Google Scholar]
- Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP, and Metallo CM (2016). Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol 12, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. (2014). Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther 13, 890–901. [DOI] [PubMed] [Google Scholar]
- Gu Z, Liu Y, Cai F, Patrick M, Zmajkovic J, Cao H, Zhang Y, Tasdogan A, Chen M, Qi L , et al. (2019). Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov 9, 1228–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P, and Verdin E (2013). The nexus of chromatin regulation and intermediary metabolism. Nature 502, 489–498. [DOI] [PubMed] [Google Scholar]
- Han B, Han B, Guo B, Liu Y, and Cao Z (2018). Two novel mutations in the BCKDHB gene that cause maple syrup urine disease. Pediatr. Neonatol 59, 515–519. [DOI] [PubMed] [Google Scholar]
- Harper A (1984). Branched-Chain Amino Acid Metabolism. Annu. Rev. Nutr 4, 409–54. [DOI] [PubMed] [Google Scholar]
- Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, Tayyari F, McSkimming D, Kannan N, Tojo A, et al. (2017). Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 545, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom SR, and Olive KP (2014). Protein breakdown precedes pancreatic tumor development. Nat. Med 20, 1097–1099. [DOI] [PubMed] [Google Scholar]
- Hutson SM, Sweatt AJ, and LaNoue KF (2005). Branched-Chain Amino Acid Metabolism: Implications for Establishing Safe Intakes. J. Nutr 135, 1557S–64S. [DOI] [PubMed] [Google Scholar]
- Iwasa M, Kobayashi Y, Mifuji-Moroka R, Hara N, Miyachi H, Sugimoto R, Tanaka H, Fujita N, Gabazza EC, and Takei Y (2013). Branched-Chain Amino Acid Supplementation Reduces Oxidative Stress and Prolongs Survival in Rats with Advanced Liver Cirrhosis. PLoS One 8, e70309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG (2009). Synthetic lethality: A framework for the development of wiser cancer therapeutics. Genome Med 1, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, and McKnight SL (2013). Influence of metabolism on epigenetics and disease. Cell 153, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalaf N, and Wolpin BM (2019). Metabolic Alterations as a Signpost to Early Pancreatic Cancer. Gastroenterology 156, 1560–1563. [DOI] [PubMed] [Google Scholar]
- Klee D, Thimm E, Wittsack HJ, Schubert D, Primke R, Pentang G, Schaper J, Mödder U, Antoch A, Wendel U, et al. (2013). Structural white matter changes in adolescents and young adults with maple syrup urine disease. J. Inherit. Metab. Dis 36, 945–953. [DOI] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. (2012). A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol 8, 890–896. [DOI] [PubMed] [Google Scholar]
- Liu KA, Lashinger LM, Rasmussen AJ, and Hursting SD (2014). Leucine supplementation differentially enhances pancreatic cancer growth in lean and overweight mice. Cancer Metab 2 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Y, Ni M, Cao H, Signer RAJ, Li D, Li M, Gu Z, Hu Z, Dickerson KE, et al. (2017). Regulation of mitochondrial biogenesis in erythropoiesis by mTORC1-mediated protein translation. Nat. Cell Biol 19, 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, and Thompson CB (2012). Metabolic regulation of epigenetics. Cell Metab 16, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. (2012). IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS, et al. (2014). Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med 20, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM, et al. (2016). Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, Sarosiek KA, Briggs KJ, Robbins AK, Sewastianik T, et al. (2018). Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 175, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Chénard V, Sikström K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, et al. (2013). MTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab 18, 698–711. [DOI] [PubMed] [Google Scholar]
- Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, and Vander Heiden MG (2017). Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elite 6, e27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM (2014). Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 159, 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neinast M, Murashige D, and Arany Z (2018). Branched Chain Amino Acids. Annu. Rev. Physiol. Annu. Rev. Physiol 26, 139–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neinast MD, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ, Li X, Zhan L, White E, Anthony TG, et al. (2019). Quantitative Analysis of the Whole-Body Metabolic Fate of Branched-Chain Amino Acids. Cell Metab 29, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. (2009). A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab 9, 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, and Thompson CB (2015). The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 162, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, and Kroemer G (2015). Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab 21, 805–821. [DOI] [PubMed] [Google Scholar]
- Pothiwala P, Jain SK, and Yaturu S (2009). Metabolic Syndrome and Cancer. Metab. Syndr. Relat. Disord Z 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothuraju R, Rachagani S, Junker WM, Chaudhary S, Saraswathi V, Kaur S, and Batra SK (2018). Pancreatic cancer associated with obesity and diabetes: An alternative approach for its targeting. J. Exp. Clin. Cancer Res 37, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A, et al. (2017). BCAT1 restricts αkG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388. [DOI] [PubMed] [Google Scholar]
- Reeds PJ (2000). Dispensable and Indispensable Amino Acids for Humans. J. Nutr 130, 1835S–40S. [DOI] [PubMed] [Google Scholar]
- Richard E, Gallego-Villar L, Rivera-Barahona A, Oyarzábal A, Pérez B, Rodríguez-Pombo P, and Desviat LR (2018). Altered Redox Homeostasis in Branched-Chain Amino Acid Disorders, Organic Acidurias, and Homocystinuria. Oxid. Med. Cell. Longev 2018, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl-Bürgi S, Sass JO, Heinz-Erian P, Amann E, Haberlandt E, Albrecht U, Ertl C, Sigl SB, Lagler F, Rostasy K, et al. (2010). Changes in plasma amino acid concentrations with increasing age in patients with propionic acidemia. Amino Acids 38, 1473–1481. [DOI] [PubMed] [Google Scholar]
- Schvartzman JM, Thompson CB, and Finley LWS (2018). Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol 217, 2247–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, et al. (2010). Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 70, 8981–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ, and Hutson SM (2007a). Disruption of BCATm in Mice Leads to Increased Energy Expenditure Associated with the Activation of a Futile Protein Turnover Cycle. Cell Metab 6, 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She P, Van Horn C, Reid T, Hutson SM, Cooney RN, and Lynch CJ (2007b). Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. - Endocrinol. Metab 293, E1552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen JH, Zoncu R, Kim D, and Sabatini DM (2011). Defective Regulation of Autophagy upon Leucine Deprivation Reveals a Targetable Liability of Human Melanoma Cells In Vitro and In Vivo. Cancer Cell 19, 613–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura Y, Honda T, Shiraki M, Murakami T, Sato J, Kobayashi H, Mawatari K, Obayashi M, and Harris RA (2006). Branched-Chain Amino Acid Catabolism in Exercise and Liver Disease. J. Nutr 136, 250S–3S. [DOI] [PubMed] [Google Scholar]
- Shin AC, Fasshauer M, Filatova N, Grundell LA, Zielinski E, Zhou JY, Scherer T, Lindtner C, White PJ, Lapworth AL, et al. (2014). Brain insulin lowers circulating bcaa levels by inducing hepatic bcaa catabolism. Cell Metab 20, 898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LS, Poschet G, Nonnenmacher Y, Becker HM, Sapcariu S, Gaupel A, Schlotter M, Wu Y, Kneisel N, Seiffert M, et al. (2017). Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep 18, 2172–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanand S, Viney I, and Wellen KE (2018). Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci 43, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son SM, Park SJ, Lee H, Siddiqi F, Lee JE, Menzies FM, and Rubinsztein DC (2019). Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab 29, 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt AJ, Wood M, Suryawan A, Wallin R, Willingham MC, and Hutson SM (2004). Branched-chain amino acid catabolism: unique segregation of pathway enzymes in organ systems and peripheral nerves. Am. J. Physiol. Metab 286, E64–76. [DOI] [PubMed] [Google Scholar]
- Takegoshi K, Honda M, Okada H, Takabatake R, Matsuzawa-Nagata N, Campbell JS, Nishikawa M, Shimakami T, Shirasaki T, Sakai Y, et al. (2017). Branched-chain amino acids prevent hepatic fibrosis and development of hepatocellular carcinoma in a non-alcoholic steatohepatitis mouse model. Oncotarget 8, 18191–18205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thewes V, Simon R, Hlevnjak M, Schlotter M, Schroeter P, Schmidt K, Wu Y, Anzeneder T, Wang W, Windisch P, et al. (2017). The branched-chain amino acid transaminase 1 sustains growth of antiestrogen-resistant and ERa-negative breast cancer. Oncogene 36, 4124–4134. [DOI] [PubMed] [Google Scholar]
- Tönjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, Pleier SV, Bai AHC, Karra D, Piro RM, et al. (2013). BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med 19, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, et al. (2011). Metabolite profiles and the risk of developing diabetes. Nat. Med 17, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang J, Ren S, Sun D, Huang HY, Wang H, Jin Y, Li F, Zheng C, Yang L, et al. (2019). Branched-Chain Amino Acid Metabolic Reprogramming Orchestrates Drug Resistance to EGFR Tyrosine Kinase Inhibitors. Cell Rep 28, 512–525. [DOI] [PubMed] [Google Scholar]
- Ward PS, and Thompson CB (2012). Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller H. a, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel U, Saudubray JM, Bodner A, and Schadewaldt P (1999). Liver transplantation in maple syrup urine disease. Eur. J. Pediatr 158 Suppl2, S60–4. [DOI] [PubMed] [Google Scholar]
- White PJ, McGarrah RW, Grimsrud PA, Tso SC, Yang WH, Haldeman JM, Grenier-Larouche T, An J, Lapworth AL, Astapova I, et al. (2018). The BCKDH Kinase and Phosphatase Integrate BCAA and Lipid Metabolism via Regulation of ATP-Citrate Lyase. Cell Metab 1281–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, and Sabatini DM (2017). The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab 26, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, and Sabatini DM (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva MO, Fan TWM, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Matés JM, Alonso FJ, Wang C, Seo Y, et al. (2012). The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, and Han J (2017). Branched-chain amino acid transaminase 1 (BCAT1) promotes the growth of breast cancer cells through improving mTOR-mediated mitochondrial biogenesis and function. Biochem. Biophys. Res. Commun 486, 224–231. [DOI] [PubMed] [Google Scholar]
- Zhang Y, and Commisso C (2019). Macropinocytosis in Cancer: A Complex Signaling Network. Trends in Cancer 5, 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang AMY, Magrill J, de Winter TJJ, Hu X, Skovsø S, Schaeffer DF, Kopp JL, and Johnson JD (2019). Endogenous Hyperinsulinemia Contributes to Pancreatic Cancer Development. Cell Metab 30, 403–404. [DOI] [PubMed] [Google Scholar]
- Zhenyukh O, Civantos E, Ruiz-Ortega M, Sánchez MS, Vázquez C, Peiró C, Egido J, and Mas S (2017). High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic. Biol. Med 104, 165–177. [DOI] [PubMed] [Google Scholar]
- Zhou W, Feng X, Ren C, Jiang X, Liu W, Huang W, Liu Z, Li Z, Zeng L, Wang L, et al. (2013). Over-expression of BCAT1, a c-Myc target gene, induces cell proliferation, migration and invasion in nasopharyngeal carcinoma. Mol. Cancer 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, and Thompson CB (2019). Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol 20, 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]