

Abstract
The search for inhibitors of viral replication is dependent on understanding the events taking place at the molecular level during viral infection. All the essential steps during the viral life cycle are potential targets for antiviral drugs. Classical inhibitors of herpesvirus replication cause chain termination during viral DNA replication. Similarly, the HIV reverse transcriptase is the major target of anti-HIV compounds. The broad-spectrum antiviral agent ribavirin affects viral nucleic acid replication by multiple mechanisms. Another major enzyme encoded by many viruses is a protease responsible for the processing of virus-encoded polyproteins. The HIV protease has been very successfully targeted, and hepatitis C virus and rhinovirus protease inhibitors are being actively developed. The complex series of interactions during virus entry is a rapidly emerging and promising target for inhibitors of HIV and many other viruses. New anti-influenza drugs inhibit virus release from infected cells. Several stages of the viral life cycle remain incompletely characterized and are therefore poorly exploited in antiviral strategies. These include, among others, the RNA capping reactions catalyzed by many viruses, as well as the membrane association of replication complexes which is common to all positive-strand RNA viruses.
Keywords: Human Immunodeficiency Virus, Respiratory Syncytial Virus, West Nile Virus, Severe Acute Respiratory Syndrome, Atazanavir
Introduction
Viral infections remain a major cause of human disease and mortality, and novel infectious agents are periodically transmitted to humans from other species and may become established in human populations (Strauss and Strauss 2002). It has been possible to control many serious human diseases (smallpox, poliomyelitis, yellow fever, tick-borne encephalitis, Japanese B encephalitis, hepatitis A and B) as well as common viral infections of children (mumps, measles, chicken pox and rubella) by safe and effective vaccines. Influenza virus vaccines are also reasonably effective, provided that the vaccine is made against the circulating virus strains. However, there are many virus infections which currently cannot be prevented by vaccination. These include chronic infections which may be difficult targets for the immune system [human immunodeficiency virus (HIV), hepatitis C virus (HCV) and papillomaviruses], infections that are more common in the developing countries (dengue, hepatitis E), and infections which have been newly discovered or are spreading to new areas [West Nile virus, severe acute respiratory syndrome (SARS) virus]. Common respiratory infections, caused by rhinoviruses as well as other virus groups, are an important health and economic burden. Since the rhinoviruses alone include about a hundred different serotypes, these infections cannot be prevented by vaccination.
We will review several virus groups that have been targeted by antiviral drugs (Table 1). However, it is clear that our antiviral arsenal remains inadequate in many ways and that the search for new antiviral compounds and strategies continues to be essential. Nevertheless, significant progress on this front has been achieved during recent years. This may not be surprising, since the “postgenomic era,” during which the nucleotide sequences of entire genomes have been determined, has existed for many virus groups for two decades or even more. It has been possible to produce, purify and crystallize viral proteins, which has allowed structure-based approaches for the design and synthesis of new antiviral drugs. The search for antiviral agents crucially depends on detailed understanding of the reactions and interactions occuring at the molecular level during viral infection. Every essential step in the virus life cycle is a potential site for antiviral intervention, as summarized in Table 2. Virus-encoded protein targets are attractive, because they are not present in uninfected cells, but host targets involved in viral replication can also be considered. In this review, we will focus on recent advances and prospects in small molecule synthetic antivirals against various groups of RNA viruses. We will not consider antibodies, antisense oligonucleotides, ribozymes, RNA interference or general immunomodulatory substances such as interferons, all of which may be promising in some instances.
Table 1.
Classification of viruses which are targets of antiviral drugs. ss Single-stranded, ds double-stranded, CNS central nervous system
| Virus family | Genome | Diseases/syndromes | Remarks |
|---|---|---|---|
| Picornaviridae | Polyprotein strategy | ||
| Rhinoviruses (∼100) | ssRNA(+) 7–8.5 kb | Common cold | |
| Enteroviruses (64) | CNS, rash etc | Vaccines for polio 1-3 | |
| Flaviridae | Polyprotein, envelope | ||
| Flaviruses (∼30) | ssRNA(+) 10–12 kb | CNS | Arthropod vectors |
| Dengue 1-4 | Hemorrhagic fever | >50 million cases/year | |
| West Nile | CNS | Emerging in America | |
| Hepatitis C | Chronic hepatitis | >150 million carriers | |
| Coronaviridae | Polyprotein, envelope, subgenomic mRNAs | ||
| Coronavirus (3) | ssRNA(+) 27–31 kb | Respiratory | |
| SARS-CoV | SARS | Emerging virus | |
| Orthomyxoviridae | Envelope, nuclear replication | ||
| Influenza A | ssRNA(−) 10–15 kb, segmented | Respiratory | Pandemics, vaccines available |
| Influenza B | Respiratory | Epidemics, vaccines available | |
| Influenza C | Respiratory | Milder infections | |
| Paramyxoviridae | Envelope, mRNA/each protein | ||
| RSV | ssRNA(−) 10–15 kb | Respiratory | |
| Filoviridae | Envelope, paramyxo-like | ||
| Marburg-like | ssRNA(−) 19 kb | Severe systemic infection | Emerging, fatal infections |
| Ebola-like | Hemorrhagic fever | Mortality 50–90% | |
| Bunyaviridae | Envelope, G1 & G2 from polyprotein | ||
| Hantavirus | ssRNA(−) 12–23 kb, segmented | Hemorrhagic fever, renal & pulmonary syndromes | |
| Herpesviridae | Envelope, complex biogenesis | ||
| HSV 1, 2 | dsDNA 120–220 kb | Skin eruptions, CNS etc | Person to person, neonatal infections, typically latent infections with occasional activation |
| VZV | Varicella & herpes-zoster | ||
| CMV | Mononucleosis, congenital | ||
| Poxviridae | Envelope, complex biogenesis | ||
| Smallpox | dsDNA 130–375 kb | Severe systemic infection | Eradication completed in 1979, putative bioterrorist weapon |
| Retroviridae | Envelope, reverse transcription | ||
| HIV-1 | ssRNA(+) 7–10 kb, reverse transcription | Chronic infection with immunodeficiency, AIDS | World-wide, fatal infection |
Table 2.
Antiviral drugs inhibiting different steps in the virus cycle. Bolded agents have been approved for clinical use. AZT Zidovudine, ddI didanosine, ddC zalcitabine, d4T stavudine, ABC abacavir, FTC emtricitabine, PMPA tenofovir disoproxil, 3TC lamivudine, ACV acyclovir, VACV valaciclovir, PCV penciclovir, FCV famciclovir, GCV ganciclovir, VGCV valganxiclovir, CDV cidofovir, IDU idoxuridine, TFT trifluridine, BVDU bridvudin, C-c3Ado carbocyclic 3-deazaadenosine
| Virus cycle | Antiviral agents | Target viruses |
|---|---|---|
| Entry and uncoating | ||
| Plasma membrane | sICAM-1 | Rhinoviruses |
| SCH-C, SCH-D, UK-427, 857 | HIV | |
| Enfuvirtide | HIV-1 | |
| VP-14637, BMS-433771 | RSV | |
| Endosomes | Amantadine, rimantadine | Influenza A |
| Pleconaril, pirodavir | Rhinoviruses | |
| BTA39, BTA188 | Picornaviruses | |
| Genome replication | ||
| Reverse transcription | Chain terminators: | |
| AZT, ddI, ddC, d4T | HIV-1,2 | |
| ABC, FTC, PMPA | ||
| 3TC | HIV-1, 2, HBV | |
| Adefovir-dipivoxil | HBV | |
| Other RT inhibitors: | ||
| Nevirapine, delavirdine | HIV-1 | |
| Efavirenz, capravirine | ||
| Integration | S-1360 | HIV |
| DNA replication | Chain terminators: | |
| ACV, VACV, PCV, FCV | HSV-1, 2, VZV | |
| GCV, VGCV | CMV | |
| CDV | HSV-1, 2, VZV, papova, pox, adeno | |
| Other inhibitors: | ||
| IDU, TFT | HSV-1 | |
| BVDU | HSV-1, VZV | |
| Foscarnet | HSV-1, 2, VZV, CMV | |
| RNA/DNA replication | Ribavirin, viramidine | RSV, HCV, pox, hantaan |
| C-c3Ado, EICAR | Ebola, pox | |
| Enviroxime, 7B | Picorna | |
| VX-497 | HCV | |
| Polyprotein processing | Saquinavir, ritonavir, indinavir, amprenavir, lopinavir, atazanavir, tipranavir | HIV-1, 2 |
| Nelfinavir | HIV-1, 2 (SARS-CoV) | |
| BILN 2061, VX-950 | HCV | |
| Ruprintrivir | Rhino | |
| Virus release | Oseltamivir, zanamivir | Influenza A, B |
Antiviral agents inhibiting the herpesviruses date back to the 1950s and 1960s [idoxuridine (IDU) and trifluorothymidine (TFT)]. As these drugs have toxic side effects, they have only been used topically. Adenine arabinoside (Ara A) is less toxic and can be used systemically in severe herpes simplex virus infections. However, a breakthrough was made when acyclovir (ACV) was synthesized in 1977. This compound is phosphorylated to ACV-monophosphate by the herpes virus thymidine kinase only, followed by further phosphorylations to ACV-triphosphate by cellular kinases, whereafter it is incorporated into DNA causing chain termination. From the 1980s on, ACV has been followed by the improved derivatives ganciclovir, penciclovir, famciclovir and valaciclovir, with wider applications against several herpesviruses (Table 2) (reviewed in De Clercq 2004).
HIV infection
The global spread of the HIV epidemic continues unabated. HIV may be considered to be a success story in antiviral drug development, as a formidable effort has transformed this lethal infection to a disease, which can be controlled to some extent (Pomerantz and Horn 2003). The virus was isolated in 1983, and in 1987 the first specific treatment was approved. This was zidovudine (also known as azidothymidine or AZT), an inhibitor of the viral polymerase, called reverse transcriptase. Zidovudine belongs to the drug class of nucleoside reverse transcription inhibitors (NRTIs). Additional NRTIs have been developed since, and the number of nucleoside or nucleotide analogues approved against HIV now totals eight, the most recent being emtricitabine (Table 2; Fig. 1). However, it was soon realized that HIV rapidly becomes resistant to zidovudine and other compounds given as monotherapy due to mutations that appear in the reverse transcriptase. Major breakthroughs were achieved in the mid-1990s, when non-nucleoside reverse transcriptase inhibitors (NNRTIs) and inhibitors of the viral protease were approved for treatment (Table 2). These developments led to the now prevalent triple combination therapies, which most commonly include a protease inhibitor or NNRTI together with two NRTIs. However, even these regimens, known as highly active antiretroviral therapy, cannot completely eradicate HIV from the body. There is some residual replication of the virus, and resistance to the drugs used eventually develops. Other problems include the considerable side effects caused by the drugs, as well as a large economic burden associated with the combination therapy. Thus, there is considerable incentive for developing compounds with fewer side effects, improved efficacy and better pharmacokinetic properties.
Fig. 1.
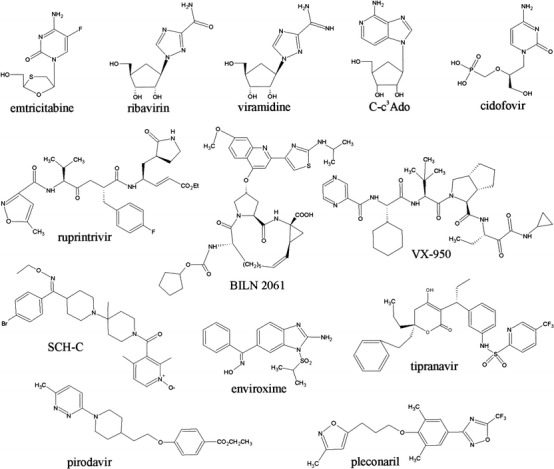
Structures of selected antiviral compounds with their names indicated beneath each compound. The upper row shows various antiviral nucleotide analogues inhibiting viral nucleic acid replication. In the middle are shown peptidomimetic protease inhibitors active against picornaviruses (ruprintrivir) and HCV (BILN 2061 and VX-950), as well as the nonpeptidic tipranavir, which inhibits HIV protease. SCH-C, pirodavir and pleconaril inhibit HIV and rhinovirus entry, respectively, whereas enviroxime appears to target the picornavirus RNA replication complex
The most recently approved protease inhibitor, atazanavir, can be taken once daily. It appears to have a unique resistance profile compared with the other protease inhibitors (Colonno et al. 2004) which means that viruses that have become resistant to atazanavir should still be susceptible to many of the other protease inhibitors. Furthermore, atazanavir does not induce changes in serum lipid levels (Sanne et al. 2003). Changes in lipid metabolism have been a significant problem associated with protease inhibitor treatments. Yet another protease inhibitor, tipranavir, is in phase III clinical trials (Plosker and Figgitt 2003). Tipranavir is the first efficacious non-peptidic inhibitor of the HIV protease (Fig. 1). It appears to have an improved resistance profile in that multiple mutations in the protease are required for high level resistance. Therefore, HIV strains resistant to the previously developed inhibitors still appear to be susceptible to tipranavir (Larder et al. 2000), and tipranavir might be primarily used as a salvage therapy for patients in which other protease inhibitors have failed. In the NNRTI class, capravirine is in phase III clinical trials. It may have activity against mutants resistant to other NNRTIs, although only limited peer-reviewed information is available. In contrast to the polymerase and protease inhibitors, compounds affecting the third enzyme encoded by HIV, namely the integrase, are still in relatively early development. It appears that specific and non-toxic inhibitors of integrase are difficult to develop, since integrase shares properties with other enzymes interacting with nucleic acid substrates. However, some very potent compounds have been reported recently (Hazuda et al. 2004).
A new highly promising class of HIV antivirals are the inhibitors of viral entry, which may act at multiple steps during the complex cell attachment and fusion pathway of HIV (Moore and Doms 2003). The first entry inhibitor has already been approved. This is a 36-amino acid synthetic peptide known as enfuvirtide (Matthews et al. 2004). It is identical in sequence to a portion of the HIV envelope glycoprotein gp41, and acts by forming a complex with a fusion-activated conformer of gp41, thereby inhibiting the fusion of viral and cellular membranes. Due to its chemical nature, enfuvirtide is very expensive to manufacture and it has to be administered by frequent injections. Many other agents targeting the entry process are being developed (Moore and Doms 2003). Several small molecule compounds bind to the HIV co-receptors CCR5 or CXCR4 and prevent HIV from using these proteins in the entry process. The first of these was known as SCH-C and it specifically targeted CCR5 (Strizki et al. 2001) (Fig. 1). SCH-C has subsequently been displaced by another compound, SCH-D. CCR5 is the co-receptor used by most HIV strains. It is not essential for normal development in mice, suggesting that it may be blocked without major side effects. Thus, many companies are targeting CCR5, and promising compounds include UK-427857, developed by Pfizer (Table 2).
Influenza, rhinoviruses and other respiratory infections
Influenza
Influenza is considered by many to be the most significant viral threat to humans. Annual influenza epidemics swipe the globe and occasional new virulent strains cause pandemics of great destructive potential. In the event of a pandemic strain, it may be impossible to manufacture the required quantities of the specific vaccine in time. Amantadine and rimantadine have been available for influenza treatment for a long time, but they have not been extensively used due to their side effects and the rapid development of resistant virus. The influenza virion surface proteins hemagglutinin (which binds cell surface neuraminic acid during virus entry) and neuraminidase (which removes neuraminic acid residues from glycoproteins to allow efficient virus release) have for a long time been the major focus of anti-influenza drug development. Their structures have been known since the early 1980s. The target first preferred was hemagglutinin (HA). However, all attempts failed due to the unfavorable geometry of the sialic acid binding site within the HA protein (Wade 1997). In contrast, neuraminidase has been successfully targeted by small neuraminic acid derivatives designed on the basis of protein structure (von Itzstein et al. 1993; Kim et al. 1997). These compounds, inhaled zanamivir and orally administered oseltamivir, have now been approved for prophylactic and therapeutic treatment for influenza virus infection (Gubareva et al. 2000). Even under optimal conditions these two drugs cannot replace vaccination. However, they offer a means to alleviate the serious symptoms of influenza and in prophylactic use may even prevent the disease (Aoki et al. 2003; Hayden et al. 2004). Neuraminidase inhibitors together with amantadine are the only means against a putative rapidly spreading new pandemic influenza infection. Therefore, the unnecessary use of these drugs should be restricted to a minimum to avoid the creation of drug-resistant influenza virus strains (Gubareva 2004).
The intracellular phase of the influenza virus life cycle also presents several interesting targets. In addition to the RNA-dependent RNA polymerase (RdRp) possessed by all RNA viruses, influenza encodes a unique cap-dependent endonuclease. This enzyme cuts cellular mRNAs to generate short (13–15 nt) cap-containing primers which are subsequently utilized by the polymerase in the initiation of RNA synthesis. Endonuclease inhibitors effective against the virus have been described (Hastings et al. 1996; Parkes et al. 2003).
Other agents
Other viruses that cause significant respiratory infections include the positive-strand RNA rhinoviruses and coronaviruses, as well as respiratory syncytial virus (RSV), a negative-strand RNA virus of the paramyxovirus family. Interesting inhibitors of virus fusion have been described for RSV. The most recently identified compounds (BMS-433771, VP-14637) are orally bioavailable and inhibit virus replication in mouse models (Cianci et al. 2004; Douglas et al. 2003). It has been speculated that the RSV fusion protein can bind several different classes of small molecules, and that the fusion process, with its attendant changes in protein conformation provides multiple targets for inhibition. Together with the successful targeting of the HIV fusion protein, this may indicate that viral fusion proteins in general could be promising targets for drug development (see also Plemper et al. 2004). The different approaches for the treatment of paramyxovirus infections have been reviewed recently (Saladino et al. 2003).
As a result of the recent appearance of the severe acute respiratory syndrome (SARS) virus, coronaviruses now have attained a prominent position in studies of antiviral strategies. Previously they were recognised as a major cause of mild respiratory infections in humans, for which there was no significant need for treatment. Within a year of the recognition of the SARS virus and the sequencing of its genome, several structures of viral replicase proteins have already been determined, and inhibitor studies are in progress. The coronavirus genome encodes several types of enzymes (Snijder et al. 2003), all of which may be considered potential targets for inhibition. Ribavirin, given intravenously to patients suffering from SARS-CoV infection during the outbreak in 2003, was shown to be ineffective in a carefully conducted study (Sung et al. 2004). Existing drugs, as well as large compound libraries, are being screened in large-scale efforts to find inhibitors of SARS-CoV that could be used in case the lethal virus reappears (Wu et al. 2004). The major challenge in the treatment of respiratory infections is that the causative agent need to be accurately and very rapidly identified after the onset of symptoms. As the infection has already progressed significantly when the symptoms appear, antiviral treatment must be initiated prophylactically, or immediately after appearance of symptoms, in order to have any effect. This is a factor that has limited the use of the current anti-influenza virus drugs (Gubareva et al. 2000), and is likely to be an even more significant handicap in treating milder respiratory infections.
Rhinoviruses
Considerable work has been carried out with inhibitors of rhinovirus replication (Table 2), since the rhinoviruses are the major cause of the common cold, and are therefore among the most prevalent virus infections. The determination of high resolution 3D structures of two picornaviruses, poliovirus type 1 (Hogle et al. 1985) and rhinovirus 14 (Rossmann et al. 1985) initiated the design of a new type of viral inhibitors. In both viruses, one of the structural proteins (VP1) displays a narrow 25 Å deep cleft or canyon encircling the 12 five-fold vertexes of the viral icosahedron. The cleft leads to a hydrophobic pocket below the surface of the virion. The authors suggested that the cleft, surrounded by conserved amino acid residues and spatially protected from antibodies, might be the binding site of the cellular receptors. This hypothesis was confirmed by the determination of the structure of rhinovirus 14 complexed with its intercellular adhesion molecule-1 (ICAM-1) receptor molecule.
As most rhinoviruses use ICAM-1 as their receptor, an obvious idea was to use this molecule to prevent virus attachment to the target cells. ICAM-1 is an integral membrane glycoprotein containing five extracellular immunglobulin-like domains. Since rhinoviruses bind to the two amino-terminal domains, a soluble sICAM-1 comprising these domains was used in competition experiments. The protein was produced in chinese hamster ovary (CH0) cells, which secrete and properly glycosylate human proteins. sICAM-1 could inhibit infection of several rhinoviruses in vitro (Crump et al. 1994; Ohlin et al. 1994). However, in a human trial, intranasal application of soluble sICAM-1 prior to virus challenge of volunteers reduced the symptoms, but did not prevent the infection (Turner 2001).
The early observation that some previously synthesized rhinovirus inhibitors (e.g. disoxaril or WIN 51711) bound to the hydrophobic pocket of VP1 started the development of drugs preventing the adsorption and/or uncoating of the virus (McKinlay et al. 1992). In phase I clinical trials disoxaril turned out to be too toxic. New compounds were developed and have been tested against the rhinoviruses and some other picornaviruses. Compound WIN 54954 entered phase II clinical trials, but showed low activity, whereas the third generation agent pleconaril (Fig. 1) has been evaluated in two extensive trials, both consisting of over 1,000 individuals with symptoms of the common cold which began less than 24 h before beginning of the treatment (Hayden et al. 2003a). These studies showed that pleconaril was well tolerated, but caused only a slight reduction in the severity of the symptoms. The FDA did not give permission to use pleconaril (“Picovir”) as an antiviral drug to treat the common cold, since there was some evidence that the drug interfered with the action of contraceptives. Since pleconaril inhibits a wide variety of picornaviruses, it has been used also for the treatment of severe neonatal coxsackie B virus infections, enteroviral hepatitis and enteroviral meningoencephalitis (Bryant et al. 2004). So far, it has not yet been approved by FDA.
Intranasally applied pirodavir (Fig. 1), another early capsid-binding agent, showed some prophylactic potency in phase II trials of volunteers (Hayden et al. 1992). However, the compound is unstable, and new derivatives with oral availability have been developed which inhibit a wide variety of rhinoviruses as well as some enteroviruses in vitro. So far no clinical trials of these compounds (BTA39, BTA188) have been published (Barnard et al. 2004).
The picornavirus genome is translated as a polyprotein, which is autocatalytically cleaved by the protease domains 2A and 3C. As failure of any of these cleavages is lethal for virus replication, the proteases would be excellent targets. Protease inhibitors have been successful in treating HIV, and promising protease inhibitors have also been synthesized against HCV infections, as discussed elsewhere in this review. Since the determination of the 3D structure of the human rhinovirus 3C protease (Matthews et al. 1994), rational drug design has been very active. Hundreds of new compounds with antiviral activity in vitro have been synthesized. A series of peptidomimetic compounds which bind irreversibly to the active site cysteine of the 3C protease have been tested for their properties, and their in vitro and in vivo antiviral activities. So far, one promising compound (AG7088 or ruprintrivir; Fig. 1) (Dragovich et al. 1999) has been tested in a phase II trial. This compound is poorly soluble in water and was therefore applied as an intranasal spray. When given before infection, the drug had clear antiviral activity, but the results were less encouraging when the drug was given 24 h after virus inoculation (Hayden et al. 2003b).
The mode of action of enviroxime (Fig. 1), a compound discovered in late 1970s, turned out to be different from the inhibitors described above. Enviroxime seems to inhibit RNA synthesis by interacting with enterovirus replicase protein 3A (Heinz and Vance 1995; 1996). Although enviroxime could inhibit rhinoviruses and many picornaviruses, including poliovirus, it lacked therapeutic activity in clinical studies. Several new compounds have been synthesized. One of these (7B) inhibits several rhinoviruses and some enteroviruses, but so far no clinical trials have been reported (Hamdouchi et al. 2003).
Hepatitis C virus
Hepatitis C virus (HCV) is a major cause of chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. It was first identified in 1989, and a blood test became available in 1992. The virus establishes a chronic infection in up to 85% of cases, and it is estimated that HCV has infected more than 170 million people worldwide. As a chronic infection with active virus replication, HCV resembles HIV, and a combination of several drugs will probably be required for effective management or eradication of the virus. The current therapy for hepatitis C is a polyethylene glycol (PEG) modified form of interferon-α, used in combination with ribavirin (Fig. 1) (Moradpour et al. 2002). Although the combination therapy can be quite efficient, it is limited by significant toxicity and lack of response in a large number of patients. Fusion of PEG molecules with interferon results in a biologically active drug with a longer half-life and more favorable pharmacokinetics. Ribavirin is a broad-range antiviral agent, but the mechanism(s) of its action is still unclear. It is believed to inhibit inosine monophosphate dehydrogenase (IMPDH), an enzyme that catalyses a rate-limiting step in GTP synthesis. This leads to a decreased pool of GTP levels in the cell, indirectly leading to the suppression of viral RNA synthesis (Tan et al. 2002). It has also been suggested that ribavirin can act as an RNA virus mutagen, forcing viruses into error catastrophe (Crotty et al. 2000).
Problems in the development of anti-HCV therapeutics include the persistence of the virus, genetic diversity generated during replication in the host, and the development of drug-resistant virus mutants. Research on HCV replication has been hampered by the lack of reproducible infectious culture systems and small-animal models (Tan et al. 2002). However, the study of HCV replication and the development of new therapies has been considerably advanced by the recent development of a cell culture model for HCV, the replicon system (Lohmann et al. 1999). The system is based on the stable replication of subgenomic selectable HCV RNAs, which can be propagated efficiently in huh-7 human hepatoma cells. High levels of replication depend on cell culture adaptive mutations that arise in several of the nonstructural proteins. The replicon system does not allow the production of infectious virus particles.
HCV has a 9,600-nt positive-sense RNA genome, and it belongs to the family Flaviviridae (Table 1). The genome encodes a single polyprotein of about 3,000 amino acids, which is co- and post-translationally cleaved at several sites by the host signal peptidase and two HCV-encoded proteases. This results in the production of at least three structural and six non-structural proteins. In addition to the proteases, the helicase and polymerase enzymes are coded within the nonstructural proteins. So far, the NS3 protease and NS5B RNA-dependent RNA polymerase have been the major targets in drug development. Unfortunately, the mechanism of viral entry and the receptors for HCV are still poorly understood, as these would offer other promising targets.
BILN 2061 (Fig. 1) is a novel HCV serine protease inhibitor with great therapeutic potential against HCV replication (Lamarre et al. 2003). It is a small molecule that is biologically available through oral ingestion. The ability of BILN 2061 to inhibit NS3 protease activity in human liver cells was tested using the replicon system. In human trials BILN 2061 showed very promising results. The suppression of the viral load in patients treated with BILN 2061 was significantly greater than in IFN-treated patients. VX-950 (Fig. 1) is another small molecule NS3 protease inhibitor that was identified through a structure-based drug design approach, evolving from the natural NS5A-NS5B substrate. The inhibitory effect of VX-950 on polyprotein processing was tested using the HCV replicon system (Lin et al. 2004). The action of VX-950 was compared to that of BILN 2061, and the data suggest that these molecules have different mechanisms of inhibition. Distinct drug-resistant mutations were identified for both protease inhibitors, and mutants that are resistant to BILN 2061 remain fully sensitive to VX-950 (Lin et al. 2004).
VX-497 (merimepodib) is an orally administered small molecule inhibitor of IMPDH. A recent study using the replicon system demonstrated that IMPDH inhibitors such as VX-497 enhance the antiviral activity of ribavirin in vitro (Zhou et al. 2003). The results showed that combination treatment with ribavirin and an IMPDH inhibitor increased the replicon error rate. This data is consistent with the suggestion that ribavirin can act as a mutagen. The results of the phase II clinical trials showed that triple combinations of merimepodib, PEG-interferon and ribavirin were well tolerated and led to a statistically significant increase in the proportion of patients with undetectable levels of hepatitis C virus after 24 weeks of treatment. Viramidine (Fig. 1) is a prodrug of ribavirin, which is converted by adenosine deaminase to ribavirin in liver cells. Compared to ribavirin, there is less uptake of viramidine into red blood cells due to the positive charge of the molecule. This results in the reduction of hematological toxicity (one of the side effects of ribavirin). Phase II clinical trials have shown that viramidine may be as effective as ribavirin against hepatitis C, but can decrease the patients’ risk of developing anemia (Tan et al. 2002).
Flavivirus infections
In addition to HCV, dengue virus diseases represent a global problem (Mairuhu et al. 2004). The disease is already endemic in more than a hundred countries and it is estimated to cause 50 to 100 million infections annually, with about half a million cases of dengue hemorrhagic fever. There are four serotypes of this mosquito-transmitted flavivirus. Even though primary infection by one serotype may be a mild febrile disease, a second infection often leads to a serious hemorrhagic fever. Ribavirin, 6-azauridine and glycyrrhizin inhibit dengue and some other viruses in cell cultures (Crance et al. 2003). Systematic in vivo studies with ribavirin have not been encouraging. Similarly to HCV, the viral protease should be a promising target. The 3D structure of the dengue virus NS3 protease domain has been determined (Murthy et al. 2000).
Another important problem caused by a flavivirus is the recent spread of West Nile virus in Northern America. This mosquito-transmitted virus caused close to 10,000 human cases with 230 deaths during 2003 alone. The virus originated from Africa, and is transmitted by a large variety of mosquito species. Serious cases have been treated with ribavirin plus interferon-α2 combination, but with poor results (reviewed in Gould and Fikrig 2004).
Emerging virus infections
Ebola virus first appeared in 1976 and several outbreaks have been reported in Africa since. The origin of this negative-strand RNA virus is still obscure, but together with the similar Marburg virus, Ebola is one of the most contagious and pathogenic viruses known, as more than half of the infections may be fatal. Mouse-adapted Ebola virus has been used as model for the testing of antiviral drugs. Non-toxic dose of an S-adenosyl- homocysteine hydrolase inhibitor, carbocyclic 3-deaza-adenosine (C-c3Ado) (Fig. 1) resulted in the survival of most of the animals, when given 1 or 2 days after infection (Bray et al. 2000). Inhibitors of this enzyme have been previously shown to inhibit rhabdo- and paramyxoviruses (De Clercq 1998). One possibility is that by lowering the concentration of S-adenosyl-homocysteine in cells, these drugs interfere with the methylation step of viral RNA capping (see below). Hantaan viruses, which can cause severe renal hemorrhagic syndrome, have been treated with some success with intravenous ribavirin (Huggins et al. 1991). A recent in vitro study suggests that ribavirin causes an error catastrophe in Hantaan virus RNA replication (Severson et al. 2003).
The possibility that smallpox (variola) virus may become available for bioterrorists has become a concern for authorities in many countries. Since the complete genome sequences of many poxviruses are available, it would be possible to reconstruct the variola virus. The previously used vaccines are not available in quantities required to vaccinate the general population, and they also occasionally cause severe complications. For these reasons, existing antiviral drugs against poxviruses, and especially against variola, have been re-evaluated. One of the earliest agents was the thiosemicarbazone derivative methisazone (Marboran), which was used with success in the prophylaxis of smallpox in India in 1960s and in the treatment of vaccination complications. Due to its side effects it was not accepted for wider use. It still may be a good lead for drug development. Many drugs developed against other viruses have been shown to inhibit the replication of the vaccinia virus, including agents affecting nucleotide metabolism (e.g. Ribavirin, C-c3Ado, EICAR) and analogues inhibiting DNA synthesis [e.g. 3’C-methylAdo; Ara-C, Cidofovir (Fig. 1) and its derivatives; cHPMPC and HDP-HPMPC) (Neyts and De Clercq 2003). Recently, 24 different antiviral agents were tested for their inhibitory activity on the replication in vitro of several poxviruses. Cidofovir and ribavirin may provide the best choices for general use in case of urgency, as both have been accepted previously for clinical use, although for different purposes (Baker et al. 2003). Plans for large and systematic efforts to look for anti-smallpox agents have been presented (Harrison et al. 2004).
Possible new targets for antiviral drugs
Most viruses synthesize capped RNAs, which are efficiently recognised by the cellular ribosomes for protein synthesis, although some viruses such as the picornaviruses use alternative mechanisms to enhance translation. RNA capping is essential for viruses and involves several enzymatic reactions: RNA triphosphatase, guanylyltransferase and methyltransferase. In several cases, viruses encode their own RNA capping enzymes which may act by mechanisms different from those of the cellular RNA capping apparatus. These differences in specificity should make them attractive targets. Virus-specific capping enzymes have been best characterized in the alphavirus-like superfamily of positive-strand RNA viruses, which includes the arthropod-borne alphaviruses and their distant relatives, the rubella virus and the hepatitis E virus (Ahola and Kääriäinen 1995; Magden et al. 2001; Kääriäinen and Ahola, 2002). Various cap analogues have been tested for inhibitory activity (Lampio et al. 1999). Although some viruses steal caps from cellular RNAs, as described above for influenza endonuclease, other groups of positive and negative strand RNA viruses should possess capping enzymes, many of which remain uncharacterized. As the first example from RNA viruses, the structure of the cap methyltransferase has been described for the flavivirus dengue (Egloff et al. 2002).
All positive-strand RNA viruses replicate their RNA in membrane-bound complexes (Salonen et al. 2004) and therefore inhibition of membrane association could be a generally applicable target for these viruses. However, in many cases the mechanisms of membrane association remain poorly understood (Salonen et al. 2004). Alphaviruses represent the first example of structurally characterized membrane binding. In the middle of the replicase protein nsP1, there is a 20-amino acid peptide, which is responsible for the binding of the whole replication complex to the cytoplasmic side of plasma membrane and endosomes. The peptide forms an amphipathic alpha helix, with hydrophobic residues embedded within the fatty acid tails while positively charged residues interact with negatively charged polar groups of phospholipids (Lampio et al. 2000; Kääriäinen and Ahola, 2002).
Concluding remarks
Antiviral drugs have been screened and tested over the last 50 years. Many of the early drugs were nucleoside and nucleotide analogues, some of which turned out to be specific for viruses, e.g. acyclovir and its derivatives. Modern virology has provided the tools to study the details of virus replication at the molecular level. The enormous challenge caused by the appearance of HIV has shown that new drugs can be developed, once the scientific community makes a concerted effort to solve the details of the virus replication cycle. Similarly to HIV, the replication of HCV is becoming very well characterized and this knowledge is likely to allow the delivery of drugs which can be used for the treatment of patients in the near future. Systematic studies of the influenza hemagglutinin and neuraminidase proteins with their sialic acid ligand have yielded compounds which can be used in disease treatment and prevention. The rational approach has also been used in the development of rhinovirus inhibitors for almost 20 years, but without a clear breakthrough, showing the inherent difficulties in this field. Newly emerging and threatening viruses are another area actively targeted in studies of virus inhibitors.
Acknowledgements
We thank Dr. Ilkka Kilpeläinen for help in drawing the chemical formulas. We wish to acknowledge the financial support of the Academy of Finland (grant no. 201687), University of Helsinki Research Funds, the Sigrid Jusélius Foundation, and the European Union 5th Framework Programme
References
- Ahola T, Kääriäinen L. Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc Natl Acad Sci USA. 1995;92:507–511. doi: 10.1073/pnas.92.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki FY, MacLeod MD, Paggiaro P, Carewicz O, ElSawy A, Wat C, Griffiths M, Waalberg E, Ward P, on behalf of the IMPACT Study Group Early administration of oral oseltamivir increases the benefits of influenza treatment. J Antimicrob Chemother. 2003;51:123–129. doi: 10.1093/jac/dkg007. [DOI] [PubMed] [Google Scholar]
- Baker RO, Bray M, Huggins JW. Potential antiviral therapeutics for smallpox, monkeypox and other orthopoxvirus infections. Antiviral Res. 2003;57:13–23. doi: 10.1016/S0166-3542(02)00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard DL, Hubbard VD, Smee DF, Sidwell RW, Watson KG, Tucker SP, Reece PA. In vitro activity of expanded-spectrum pyridazinyl oxime ethers related to pirodavir: novel capsid-binding inhibitors with potent antipicornavirus activity. Antimicrob Agents Chemother. 2004;48:1766–1772. doi: 10.1128/AAC.48.5.1766-1772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M, Driscoll J, Huggins JW. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor. Antiviral Res. 2000;45:135–147. doi: 10.1016/S0166-3542(00)00066-8. [DOI] [PubMed] [Google Scholar]
- Bryant PA, Tingay D, Dargaville PA, Starr M, Curtis N. Neonatal coxsackie B virus infection - a treatable disease? Eur J Pediatr. 2004;163:223–228. doi: 10.1007/s00431-004-1408-y. [DOI] [PubMed] [Google Scholar]
- Cianci C, Yu KL, Combrink K, Sin N, Pearce B, Wang A, Civiello R, Voss S, Luo G, Kadow K, et al. Orally active fusion inhibitor of respiratory syncytial virus. Antimicrob Agents Chemother. 2004;48:413–422. doi: 10.1128/AAC.48.2.413-422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonno R, Rose R, McLaren C, Thiry A, Parkin N, Friborg J. Identification of I50L as the signature atazanavir (ATV)-resistance mutation in treatment-naive HIV-1-infected patients receiving ATV-containing regimens. J Infect Dis. 2004;189:1802–1810. doi: 10.1086/386291. [DOI] [PubMed] [Google Scholar]
- Crance JM, Scaramozzino N, Jouan A, Garin D. Interferon, ribavirin, 6-azauridine and glycyrrhizin: antiviral compounds active against pathogenic flaviviruses. Antiviral Res. 2003;58:73–79. doi: 10.1016/S0166-3542(02)00185-7. [DOI] [PubMed] [Google Scholar]
- Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Crump CE, Arruda E, Hayden FG. Comparative antirhinoviral activities of soluble intercellular adhesion molecule-1 (sICAM-1) and chimeric ICAM-1/immunoglobulin A molecule. Antimicrob Agents Chemother. 1994;38:1425–1427. doi: 10.1128/aac.38.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Carbocyclic adenosine analogues as S-adenosylhomocysteine hydrolase inhibitors and antiviral agents: recent advances. Nucleosides Nucleotides. 1998;17:625–634. doi: 10.1080/07328319808005205. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30:115–133. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Douglas JL, Panis ML, Ho E, Lin K-Y, Krawczyk SH, Grant DM, Cai R, Swaminathan S, Cihlar T. Inhibition of respiratory syncytial virus fusion by the small molecule VP-14637 via specific interactions with F protein. J Virol. 2003;77:5054–5064. doi: 10.1128/JVI.77.9.5054-5064.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragovich PS, Prins TJ, Zhou R, Webber SE, Marakovits JT, Fuhrman SA, Patick AK, Matthews DA, Lee CA, Ford CE, et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements. J Med Chem. 1999;42:1213–1224. doi: 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]
- Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould LH, Fikrig E. West Nile virus: a growing concern? J Clin Invest. 2004;113:1102–1107. doi: 10.1172/JCI200421623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubareva LV. Molecular mechanisms of influenza virus resistance to neuraminidase inhibitors. Virus Res. 2004;103:199–203. doi: 10.1016/j.virusres.2004.02.034. [DOI] [PubMed] [Google Scholar]
- Gubareva LV, Kaiser L, Hayden FG. Influenza virus neuraminidase inhibitors. Lancet. 2000;355:827–835. doi: 10.1016/S0140-6736(99)11433-8. [DOI] [PubMed] [Google Scholar]
- Hamdouchi C, Sanchez-Martinez C, Gruber J, Del Prado M, Lopez J, Rubio A, Heinz BA. Imidazo[1,2-b]pyridazines, novel nucleus with potent and broad spectrum activity against human picornaviruses: design, synthesis, and biological evaluation. J Med Chem. 2003;46:4333–4341. doi: 10.1021/jm020583i. [DOI] [PubMed] [Google Scholar]
- Harrison SC, Alberts B, Ehrenfeld E, Enquist L, Fineberg H, McKnight SL, Moss B, O’Donnell M, Ploegh H, Schmid SL, Walter KP, Theriot J. Discovery of antivirals against smallpox. Proc Natl Acad Sci USA. 2004;101:11178–11192. doi: 10.1073/pnas.0403600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings JC, Selnick H, Wolanski B, Tomassini JE. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob Agents Chemother. 1996;40:1304–1307. doi: 10.1128/aac.40.5.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden FG, Andries K, Janssen PA. Safety and efficacy of intranasal pirodavir (R77975) in experimental rhinovirus infection. Antimicrob Agents Chemother. 1992;36:727–732. doi: 10.1128/aac.36.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden FG, Herrington DT, Coats TL, Kim K, Cooper EC, Villano SA, Liu S, Hudson S, Pevear DC, Collett M, McKinlay M, and the Pleconaril Respiratory Infection Study Group Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: results of two double-blind, randomized, placebo-controlled trials. Clin Infect Dis. 2003;36:1523–1532. doi: 10.1086/375069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden FG, Turner RB, Gwaltney JM, Chi-Burris K, Gersten M, Hsyu P, Patick AK, Smith IIIrd GJ, Zalman LS. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother. 2003;47:3907–3916. doi: 10.1128/AAC.47.12.3907-3916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden FG, Belshe R, Villanueva C, Lanno R, Hughes C, Small I, Dutkowski R, Ward P, Carr J. Management of influenza in households: a prospective, randomized comparison of oseltamivir treatment with and without postexposure prophylaxis. J Infect Dis. 2004;189:440–449. doi: 10.1086/381128. [DOI] [PubMed] [Google Scholar]
- Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, Vacca JP, Handt L, Motzel SL, Klein HJ, et al. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science. 2004;305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- Heinz BA, Vance LM. The antiviral compound enviroxime targets the 3A coding region of rhinovirus and poliovirus. J Virol. 1995;69:4189–4197. doi: 10.1128/jvi.69.7.4189-4197.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz BA, Vance LM. Sequence determinants of 3A-mediated resistance to enviroxime in rhinoviruses and enteroviruses. J Virol. 1996;70:4854–4857. doi: 10.1128/jvi.70.7.4854-4857.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9 A resolution. Science. 1985;229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- Huggins JW, Hsiang CM, Cosgriff TM, Guang MY, Smith JI, Wu ZO, LeDuc JW, Zheng ZM, Meegan JM, Wang QN, et al. Prospective, double-blind, concurrent, placebo-controlled clinical trial of intravenous ribavirin therapy of hemorrhagic fever with renal syndrome. J Infect Dis. 1991;164:1119–1127. doi: 10.1093/infdis/164.6.1119. [DOI] [PubMed] [Google Scholar]
- von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Smythe ML, White HF, Oliver SW, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- Kääriäinen L, Ahola T. Functions of alphavirus nonstructural proteins in RNA replication. Prog Nucleic Acid Res Mol Biol. 2002;71:187–222. doi: 10.1016/S0079-6603(02)71044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CU, Lew W, Williams MA, Liu H, Zhang L, Swaminathan S, Bischofberger N, Chen MS, Mendel DB, Tai CY, Laver WG, Stevens RC. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J Am Chem Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- Lampio A, Ahola T, Darzynkiewicz E, Stepinski J, Jankowska-Anyszka M, Kääriäinen L. Guanosine nucleotide analogs as inhibitors of alphavirus mRNA capping enzyme. Antiviral Res. 1999;42:35–46. doi: 10.1016/S0166-3542(99)00011-X. [DOI] [PubMed] [Google Scholar]
- Lampio A, Kilpeläinen I, Pesonen S, Karhi K, Auvinen P, Somerharju P, Kääriäinen L. Membrane binding mechanism of an RNA virus-capping enzyme. J Biol Chem. 2000;275:37853–37859. doi: 10.1074/jbc.M004865200. [DOI] [PubMed] [Google Scholar]
- Larder BA, Hertogs K, Bloor S, van den Eynde CH, DeCian W, Wang Y, Freimuth WW, Tarpley G. Tipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samples. AIDS. 2000;14:1943–1948. doi: 10.1097/00002030-200009080-00009. [DOI] [PubMed] [Google Scholar]
- Lin C, Lin K, Luong YP, Rao BG, Wei YY, Brennan DL, Fulghum JR, Hsiao HM, Ma S, Maxwell JP, Cottrell KM, Perni RB, Gates CA, Kwong AD. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J Biol Chem. 2004;279:17508–17514. doi: 10.1074/jbc.M313020200. [DOI] [PubMed] [Google Scholar]
- Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- Magden J, Takeda N, Li T, Auvinen P, Ahola T, Miyamura T, Merits A, Kääriäinen L. Virus-specific mRNA capping enzyme encoded by hepatitis E virus. J Virol. 2001;75:6249–6255. doi: 10.1128/JVI.75.14.6249-6255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairuhu ATA, Wagenaar J, Brandjes DPM, van Gorp ECM. Dengue: an arthropod-borne disease of global importance. Eur J Clin Microbiol Infect Dis. 2004;23:425–433. doi: 10.1007/s10096-004-1145-1. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Smith WW, Ferre RA, Condon B, Budahazi G, Sisson W, Villafranca JE, Janson CA, McElroy HE, Gribskov CL. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein . Cell. 1994;77:761–771. doi: 10.1016/0092-8674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov. 2004;3:215–225. doi: 10.1038/nrd1331. [DOI] [PubMed] [Google Scholar]
- McKinlay MA, Pevear DC, Rossmann MG. Treatment of the picornavirus common cold by inhibitors of viral uncoating and attachment. Annu Rev Microbiol. 1992;46:635–654. doi: 10.1146/annurev.mi.46.100192.003223. [DOI] [PubMed] [Google Scholar]
- Moore JP, Doms RW. The entry of entry inhibitors: a fusion of science and medicine. Proc Natl Acad Sci USA. 2003;100:10598–10602. doi: 10.1073/pnas.1932511100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradpour D, Brass V, Gosert R, Wolk B, Blum HE. Hepatitis C: molecular virology and antiviral targets. Trends Mol Med. 2002;8:476–482. doi: 10.1016/S1471-4914(02)02395-X. [DOI] [PubMed] [Google Scholar]
- Murthy HM, Judge K, DeLucas L, Padmanabhan R. Crystal structure of Dengue virus NS3 protease in complex with a Bowman-Birk inhibitor: implications for flaviviral polyprotein processing and drug design. J Mol Biol. 2000;301:759–767. doi: 10.1006/jmbi.2000.3924. [DOI] [PubMed] [Google Scholar]
- Neyts J, De Clercq E. Therapy and short-term prophylaxis of poxvirus infections: historical background and perspectives. Antiviral Res. 2003;57:25–33. doi: 10.1016/S0166-3542(02)00197-3. [DOI] [PubMed] [Google Scholar]
- Ohlin A, Hoover-Litty H, Sanderson G, Paessens A, Johnston SL, Holgate ST, Huguenel E, Greve JM. Spectrum of activity of soluble intercellular adhesion molecule-1 against rhinovirus reference strains and field isolates. Antimicrob Agents Chemother. 1994;38:1413–1415. doi: 10.1128/aac.38.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes KEB, Ermert P, Fässler J, Ives J, Martin JA, Merrett JH, Obrecht D, Williams G, Klumpp K. Use of a pharmacophore model to discover a nw class of influenza endonuclease inhibitors. J Med Chem. 2003;46:1153–1164. doi: 10.1021/jm020334u. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. A target site for template-based design of measles virus entry inhibitors. Proc Natl Acad Sci USA. 2004;101:5628–5633. doi: 10.1073/pnas.0308520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plosker GL, Figgitt DP. Tipranavir. Drugs. 2003;63:1611–1618. doi: 10.2165/00003495-200363150-00009. [DOI] [PubMed] [Google Scholar]
- Pomerantz RJ, Horn DL. Twenty years of therapy for HIV-1 infection. Nat Med. 2003;9:867–873. doi: 10.1038/nm0703-867. [DOI] [PubMed] [Google Scholar]
- Rossmann MG, Arnold E, Erickson JW, Frankenberger EA, Griffith JP, Hecht HJ, Johnson JE, Kamer G, Luo M, Mosser AG, Rueckert RR, Sherry B, Vriend G. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature. 1985;317:145–153. doi: 10.1038/317145a0. [DOI] [PubMed] [Google Scholar]
- Saladino R, Ciambecchini U, Nencioni L, Palamara AT. Recent advances in the chemistry of parainfluenza-1 (Sendai) virus inhibitors. Med Res Rev. 2003;23:427–455. doi: 10.1002/med.10036. [DOI] [PubMed] [Google Scholar]
- Salonen Curr Top Microbiol Immunol. 2004;285:139. doi: 10.1007/3-540-26764-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanne I, Piliero P, Squires K, Thiry A, Schnittman S, AI424-007 Clinical Trial Group Results of a phase 2 clinical trial at 48 weeks (AI424-007): a dose-ranging, safety, and efficacy comparative trial of atazanavir at three doses in combination with didanosine and stavudine in antiretroviral-naive subjects. J Acquir Immune Defic Syndr. 2003;32:18–29. doi: 10.1097/00126334-200301010-00004. [DOI] [PubMed] [Google Scholar]
- Severson WE, Schmaljohn CS, Javadian A, Jonsson CB. Ribavirin causes error catastrophe during Hantaan virus replication. J Virol. 2003;77:481–488. doi: 10.1128/JVI.77.1.481-488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ, Gorbalenya AE. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. Viruses and human disease. San Diego: Academic; 2002. [Google Scholar]
- Strizki JM, Xu S, Wagner NE, Wojcik L, Liu J, Hou Y, Endres M, Palani A, Shapiro S, Clader JW, et al. SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:12718–12723. doi: 10.1073/pnas.221375398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JJY, Wu A, Joynt GM, Yuen KY, Lee N, Chan PKS, Cockram CS, Ahuja AT, Yu LM, Wong VW, Hui DSC. Severe acute respiratory syndrome: report of treatment and outcome after a major outbreak. Thorax. 2004;59:414–420. doi: 10.1136/thx.2003.014076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SL, Pause A, Shi Y, Sonenberg N. Hepatitis C therapeutics: current status and emerging strategies. Nat Rev Drug Discov. 2002;1:867–881. doi: 10.1038/nrd937. [DOI] [PubMed] [Google Scholar]
- Turner RB. The treatment of rhinovirus infections: progress and potential. Antiviral Res. 2001;49:1–14. doi: 10.1016/S0166-3542(00)00135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade RC. ‘Flu’ and structure-based drug design. Structure. 1997;5:1139–1145. doi: 10.1016/S0969-2126(97)00265-7. [DOI] [PubMed] [Google Scholar]
- Wu C, Jan J, Ma S, Kuo C, Juan H, Cheng YE, Hsu H, Huang H, Wu D, Brik A, et al. Small molecules targeting severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci USA. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Liu R, Baroudy BM, Malcolm BA, Reyes GR. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology. 2003;310:333–342. doi: 10.1016/s0042-6822(03)00152-1. [DOI] [PubMed] [Google Scholar]